

Abstract
A recently discovered neurotransmitter system, consisting of neuropeptide S (NPS), NPS receptor, and NPS-expressing neurons in the brain stem, has received considerable interest due to its modulating influence on arousal, anxiety and stress responsiveness. Comparatively little is known about the properties of NPS-expressing neurons. Therefore in the present study, a transgenic mouse line expressing enhanced green fluorescent protein (EGFP) in NPS neurons was used to characterize the cellular and functional properties of NPS-expressing neurons located close to the locus coeruleus. Particular emphasis was on the influence of corticotropin-releasing factor (CRF), given previous evidence of stress-related activation of the NPS system. Upon acute immobilization stress, an increase in c-fos expression was detected immunocytochemically in brain stem NPS-EGFP neurons that also expressed the CRF receptor 1 (CRF1). NPS-EGFP neurons were readily identified in acute slice preparations and responded to CRF application with a membrane depolarization capable of triggering action potentials. CRF-induced responses displayed pharmacological properties indicative of CRF1 that were mediated by both a reduction in membrane potassium conductance and an increase in a non-specific cation conductance different from the hyperpolarization-activated cation conductance Ih, and involved protein kinase A signalling. In conclusion, stress exposure results in activation of brain stem NPS-expressing neurons, involving a CRF1-mediated membrane depolarization via at least two ionic mechanisms. These data provide evidence for a direct interaction between the CRF and the NPS system and thereby extend previous observations of NPS-modulated stress responsiveness towards a mechanistic level.
Key points
Neuropeptide S (NPS) and its cognate receptor represent a recently discovered transmitter system in the brain modulating anxiety- and stress-related behaviour.
Using a transgenic NPS-EGFP-expressing mouse line, the present study shows that NPS-expressing neurons are situated in close proximity to corticotropin-releasing factor (CRF)-containing fibres at the locus coeruleus in the brain stem and express the CRF receptor 1 (CRF1).
CRF depolarizes NPS neurons via activation of the CRF1 receptor through two different ionic mechanisms (a decrease in potassium and an increase in cation conductance) involving the cAMP signalling pathway.
After acute immobilization stress, NPS neurons display an increased expression of c-fos.
This study identifies a mechanism by which stress-related CRF release might activate NPS neurons in the brain stem, thereby triggering NPS release in target areas such as the amygdala, and functioning as a negative feedback control to buffer stress responsiveness.
Introduction
The neuropeptide S system consists of the 20-amino-acid neuropeptide S (NPS) and its G protein-coupled receptor, NPSR. Experimental NPSR activation by central NPS injection affects food intake (Smith et al. 2006), the sleep–wake cycle, states of arousal (Xu et al. 2004), general anxiety (Xu et al. 2004; Jüngling et al. 2008; Meis et al. 2008; Rizzi et al. 2008), extinction of conditioned fear responses (Jüngling et al. 2008), and consolidation of aversive and neutral memories (Okamura et al. 2011). In rats, central administration of NPS enhances dopamine release in the medial prefrontal cortex, but leaves serotonergic transmission unaffected (Si et al. 2010). Furthermore, recent human studies show that polymorphisms of NPSR are linked to personal fear reactions and to panic disorders (Okamura et al. 2007; Donner et al. 2010; Raczka et al. 2010; Domschke et al. 2011). Adding to this evidence on the involvement of the NPS system in anxiety-related behaviour, recent studies have demonstrated that forced swimming stress in rodents results in an increase in extracellular levels of NPS in the basolateral amygdala (Ebner et al. 2011), implying activation of the NPS system upon stress exposure.
In the mouse, NPS-expressing neurons are located in two areas in the brain stem. One is positioned between the locus coeruleus (LC) and Barrington's nucleus, close to the 4th ventricle, and the second is located between the lateral parabrachial nucleus (LPB) and the Kölliker fuse nucleus (Clark et al. 2011). Recent immunohistochemical and in situ hybridization studies revealed dense projections of NPS-positive fibres and NPSR mRNA expression in a number of brain areas involved in fear and anxiety (e.g. amygdala), learning and memory (e.g. amygdala and subiculum), arousal and stress responses (e.g. anterior paraventricular thalamic nucleus), which together are considered adequate for mediating the modulatory effects of NPS (Clark et al. 2011). In keeping with this, short-term swim stress and prolonged restraint stress in mice have been shown to activate immediate early genes in NPS-expressing neurons in the brain stem (Liu et al. 2011).
While the NPS system thus seems to be in an important position for modulating anxiety, arousal and stress responses, there are no data yet on the physiological properties of NPS-expressing neurons in the brain stem and their modulation by transmitter systems relating to stress responsiveness.
Therefore the present study has been undertaken to characterize the basic physiological and morphological properties of NPS-expressing neurons in the brain stem, and to identify cellular mechanisms contributing to their stress-related activation. The experimental strategy was (i) to use a transgenic mouse line expressing EGFP under the control of the natural NPS-promotor sequence (Liu et al. 2011), allowing reliable identification of NPS-expressing neurons in the brain stem; (ii) to assess stress-induced activation of NPS neurons through monitoring immediate early gene activation and corticotropin-releasing factor receptor 1 (CRF1) expression upon acute immobilization stress (AIS); and (iii) to identify mechanisms of CRF1 stimulation in NPS-expressing neurons using single cell RT-PCR combined with electrophysiological and pharmacological techniques in acute slice preparations in vitro.
Methods
Animals
Heterozygous NPS-EGFP mice (transgenic line E16) were bred with C57BL/6 mice and offspring were genotyped by PCR as described previously (Liu et al. 2011). Mice were kept in a temperature- (21°C) and humidity-controlled (50–60% relative humidity) animal facility with access to food and water ad libitum and a 12 h:12 h light–dark cycle with lights on at 06.00 h. All animal experiments were carried out in accordance with national regulations on animal experimentation (European Committees Council Directive 86/609/EEC; National Research Council of the National Academies) and protocols were approved by the local authorities (Bezirksregierung Münster, AZ 50.0835.1.0, G 53/2005; Institutional Animal Care and Use Committee, University of California Irvine).
Electrophysiological recordings in vitro
Five- to eight-week-old transgenic NPS-EGFP mice (transgenic NPS-EGFP mouse line E16; Liu et al. 2011) of either sex were anaesthetized with Forene (isoflurane; 1-chloro-2,2,2-trifluoroethyl-difluoromethylether; 2.5%) and killed by decapitation. Horizontal slices (300 μm thick) containing the LC were prepared. Whole-cell patch-clamp recordings (in voltage- or current-clamp mode) were performed as described previously (Jüngling et al. 2008). Briefly, we used patch pipettes made of borosilicate glass (GC150T-10, Harvard Apparatus, Edenbridge, UK), pulled on a vertical puller (PA-10, E.S.F. Electronic, Göttingen, Germany). The intracellular solution used to analyse the intrinsic properties of NPS-EGFP neurons contained (in mm): NaCl 10, potassium gluconate 105, potassium citrate 20, Hepes 10, BAPTA 3, MgCl2 1, MgATP 3, and NaGTP 0.5. The pH was adjusted to 7.25. Artificial cerebrospinal fluid (ACSF) was used as extracellular solution and contained (in mm): NaCl 120, KCl 2.5, NaH2PO4 1.25, MgSO4 2, CaCl2 2, and glucose 20. The pH was adjusted to 7.3 by gassing with carbogen (95% O2, 5% CO2). The liquid junction potential was corrected for (10 mV).
All experiments were performed at 30–32°C. Gabazine (25 μm), CGP55845 (10 μm), d-(–)-2-amino- 5-phosphonopentanoic acid (AP5, 50 μm), and 6,7-dinitroquinoxaline-2,3-dione (DNQX, 10 μm) were added to the bathing solution as required to block glutamatergic and GABAergic postsynaptic currents. In some experiments, tetrodotoxin (TTX, 1 μm) was added to decrease network activity (toxins were purchased from Biozol Diagnostica Vertrieb GmbH, Germany). Electrophysiological data were acquired with an EPC10 double amplifier (HEKA, Germany) at a sampling rate of 10 kHz and analysed offline with Clampfit10 software (Molecular Devices Corporation, Sunnyvale, CA, USA).
The active and passive membrane properties were assessed during whole-cell current-clamp recordings at a membrane potential of −60 mV. Hyper- and depolarizing currents were injected for 500 ms (injected currents from −50 pA to +140 pA; ΔI: +10 pA). Active membrane properties were analysed during depolarizing current injections of +80 pA to +120 pA. The input resistance of the recorded neurons was calculated by: Rinput = ΔV/I. ΔV was measured under steady-state conditions at the end of an injected hyperpolarizing current pulse (I = −50 pA) with a duration of 500 ms. The capacitance was calculated by: C = τ/R, whereby the membrane time constant τ was obtained by a monoexponential fit of the membrane potential shift induced by a current injection of −50 pA and a duration of 500 ms. The resting membrane potential was measured immediately after establishing the whole-cell configuration. The after-hyperpolarizing potential (AHP) was measured after the first action potential (AP). The frequency adaptation index (FAI) was calculated by: FAI = frequency of the last two APs/frequency of the first two APs. The amplitude adaptation (AA) was calculated by: AA = amplitude of last AP/amplitude of first AP. The AP half-width was measured from the first AP. Spontaneous action potential generation was recorded in the cell-attached configuration with a patch-pipette filled with extracellular solution (see Perkins, 2006).
A subset of NPS-EGFP-expressing neurons (n = 16) was filled with 1 mg ml−1 neurobiotin (Sigma) for at least 45 min. Slices were fixed with 4% paraformaldehyde over night and then permeabilized with 0.5% Triton X-100 in PBS. Unspecific binding sites were blocked by 5% bovine serum albumin (BSA) in PBS. The cells were stained with streptavidin-DyLight549 conjugate (1:500; Vector Laboratories, Burlingame, CA, USA).
Drug testing during current-clamp recordings
NPS-EGFP neurons were recorded in the current-clamp mode at a membrane potential of ∼−70 mV. Data acquisition started after a minimal equilibration time of about 5 min. CRF (250 nm) and the CRF1-specific agonist Stressin I (250 nm, Sigma) were bath-applied for 5–7 min. The substance-induced shift of the membrane potential was analysed at the end of the substance application. To analyse changes in input resistance, the membrane potential was manually set to baseline values by adjusting a DC offset to exclude changes of the input resistance induced by voltage-dependent conductances. The CRF1-specific antagonist NBI27914 (10 μm, Sigma) was bath-applied 5 min prior to CRF application. 2-Aminoethoxydiphenyl borate (2-APB; 100 μm; Tocris) and ZD 7288 (30 μm; Tocris) were bath-applied at least 5 min prior to CRF to block transient receptor potential (TRP) channels or hyperpolarization-activated cation conductances (Ih), respectively. The protein kinase A (PKA) antagonist H89 (10 μm; Sigma) was used in the intracellular recording solution. 8-Br-cAMP (100 μm; Sigma) was included in the internal pipette solution during current-clamp recordings. The adenylyl cyclase activator forskolin (20 μm; Sigma) was bath-applied.
All recordings were done in the presence of DNQX, gabazine, CGP55845, AP5 and phentolamine hydrochloride (20 μm; Sigma). In some experiments, 1 μm TTX was added to the extracellular solution to minimize network activity.
Voltage-clamp ramps
For voltage-clamp ramp experiments, NPS-EGFP neurons were recorded in the voltage-clamp mode at a holding potential of −60 mV. Depolarizing voltage-clamp ramps (from −120 mV to −20 mV; 40 mV s−1) were repeated at least three times during baseline conditions (interstimulus interval: 75 s) and in the presence of 250 nm CRF or 20 μm forskolin. The injected holding current was monitored constantly during baseline recordings and CRF application. As an intracellular solution we used either the potassium gluconate-based (K-gluc) solution (described above) or a caesium methanesulfonate-based solution (Cs-meth), containing (in mm): 4-AP 5, CsMeSO4 120, EGTA 1, Hepes 10, TEA-Cl 20, MgCl2 2, CaCl2 0.5, Na-ATP 2, Na-GTP 0.5, to block potassium conductances. The extracellular solution contained TTX (1 μm), DNQX (10 μm), AP5 (50 μm), gabazine (25 μm), CGP55845 (10 μm), and phentolamine hydrochloride (20 μm) to reduce network activity. Additionally, 50 μm CdCl2 was added to the extracellular solution to minimize Ca2+ inward currents during ramp experiments. The CRF-induced current was calculated by subtracting the ramp during baseline recordings from ramps recorded in the presence of CRF (5 min after CRF application). Reversal potentials of the CRF-induced currents were analysed by plotting the injected current against the respective membrane potential.
Single-cell PCR
Cell harvesting and single-strand cDNA synthesis was performed as previously reported (Sosulina et al. 2008). A multiplex two-round single-cell PCR was carried out for simultaneous detection of hypoxanthine-guanine phosphoribosyltransferase (HPRT, which was considered to be a housekeeping gene), CRF1 and CRF2. The primers for the amplification of CRF1 were identical to those used in Jasoni et al. 2005. For the amplification of HPRT and CRF2 the following primers were used. HPRT (GenBank accession number NM-013556.2), sense: GCAGTCCCAGCGTCGTGA (position 157), antisense: CAAGGGCATATCCAACAACAAACT (position 726); CRF2 (NM-009953.3), sense: AGTGGCTTTTCCT CTTCATTG (position 859), antisense: CGCGCACCTCT CCATTG (position 1290). For multiplex amplification 45 cycles were performed as described previously (Sosulina et al. 2010). An aliquot (3 μl) of PCR product was used as a template for the second PCR (35 cycles; annealing at 60°C). The nested primers for the amplification of CRF1 and HPRT were identical to those used in Jasoni et al. 2005, and Jüngling et al. 2008, respectively. The following nested primers for amplification of CRF2 were used. Sense: CATTCCCTGCCCTATCATCAT (position 887), antisense: GTAGAAAACGGACACAAAGAAACC (position 1265). The predicted sizes (in base pairs) of the PCR-generated fragments were: 353 (HPRT), 162 (CRF1), 379 (CRF2). The presence of the amplified fragments was identified by electrophoresis in an agarose gel (1.6%) and visualized by ethidium bromide staining, using a molecular weight marker (pUC19, Carl Roth, Karlsruhe, Germany). Negative controls, omission of the reverse transcriptase in the RT step or using a bath solution instead of the collected neurons, did not render any PCR-generated products.
Acute immobilization stress
Male mice (8–10 weeks, n = 3–4 per group) were subjected to 20 min restraint stress, as described previously (Liu et al. 2011). Unstressed transgenic mice served as controls. Two hours after the end of the stress protocol mice were anaesthetized and perfused as described below. Brains were removed and processed for immunohistochemical analysis of EGFP, CRF1, and c-fos staining (as a marker of neuronal activation). In each brain stem section, the total number of EGFP-positive neurons was counted. Numbers of cells double stained for EGFP and CRF1, or triple stained for EGFP, CRF1 and c-fos were determined using appropriate laser illumination and percentages of multi-stained neurons were calculated.
Immunohistochemistry
Immunohistochemical detection of the EGFP transgene and the intrinsic NPS expressed by NPS neurons in the LC region was carried out as described previously (Liu et al. 2011). Briefly, mice were deeply anaesthetized by intraperitoneal injection of a mixture of ketamine (100 mg ml−1 in isotonic saline) and xylazine (20 mg ml−1), then perfused transcardially with saline (0.9% NaCl), followed by 4% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4. Brains were removed and postfixed in the same fixative overnight at 4°C. Brains were cryoprotected in 20% sucrose in 0.1 m phosphate buffer, pH 7.4, at 4°C overnight and then stored at −80°C. Cryostat sections (40 μm) were prepared for free-floating slices. Brain slices were processed as described (Liu et al. 2011). Slices were incubated with primary antisera at optimized dilutions (chicken anti-GFP, 1:1000, ab13970, Abcam; goat anti-CRF1, 1:1000, EB08035, Everest Biotech; rabbit anti-c-fos, 1:500, sc-52, Santa Cruz Biotechnology) in blocking buffer (PBS, 0.3% Triton X-100, 5% normal donkey serum) at 4°C for 48 h. Afterwards, brain slices were washed 3 times with PBS for 5 min before incubation with appropriate fluophore-conjugated secondary antibodies (purchased from Jackson ImmunoResearch Lab, West Grove, PA, USA) in blocking buffer for 1.5 h at optimized dilutions (DyLight 488 AffiniPure donkey anti-chicken IgY (IgG) (H+L), 1:400; Alexa Fluor 568 donkey anti-goat IgG (H+L), 1:500; DyLight 649 AffiniPure donkey anti-rabbit IgG (H+L), 1:400) at room temperature. Slices were washed again 3 × 5 min with PBS before mounting on glass slides with Citifluor mounting media (Ted Pella, Redding, CA, USA) containing 4′,6-diamidino-2-phenylindole (DAPI) to stain cell nuclei. Triple-immunostained brain slices were analysed under a confocal laser-scanning microscope (Zeiss LSM 710 Meta, Zeiss, Thornwood, NY, USA) with single-photon excitation at 488, 543 and 633 nm. Raw image files were adjusted for colour balance, evenness of illumination and contrast using Adobe Photoshop.
For immunohistochemical stainings of corticotropin-releasing factor (CRF) peptide, 50 μm-thick horizontal slices containing the LC were cut. After permeabilization in 0.25% Triton X-100 for 20 min, unspecific binding sites were blocked with 5% BSA and 5% normal goat serum (NGS; in PBS) for 1 h at room temperature. The primary antibody (rabbit anti-CRF, 1 mg ml−1, C-5348, Sigma) was applied at a dilution of 1:500 in PBS containing 0.01% Triton X-100, 2% BSA, and 2% NGS for 24 h at 4°C. The secondary antibody (goat anti-rabbit-Cy3 conjugated; Dianova; cat. no. 111-165-003) was applied at a dilution of 1:400 in PBS for 90 min at room temperature. To control the specificity of the primary antibody against CRF, in some experiments the antibody solution was preabsorbed with 2.5 μm CRF for 40 min. Stained slices were analysed with a laser scanning confocal microscope (Nikon eC1 plus) using an Achro LWD ×16/0.8w objective (Nikon). To detect fluorescence, lasers of 488 nm and 543 nm have been used with adequate emission filters (515/30 and 605/75 nm).
Statistics
All data sets were tested for statistically significant outliers using the Grubbs’ test (significance level P < 0.05). Prior to statistical comparison, the data were tested for normal distribution using the Shapiro–Wilk test. Within-group comparisons were done using Student's t test (significance level *P < 0.05; **P < 0.01). To analyse differences between different groups, a one-way ANOVA followed by a Bonferroni post hoc test was used (significance level *P < 0.05; **P < 0.01). Fractions of double- or triple-stained neurons in the LC area were normalized to the total number of EGFP-positive neurons per section. Percentages per mouse brain were averaged and analysed by two-way ANOVA comparing staining data from stressed and unstressed animals with treatment and gene expression as variables, followed by Bonferroni's post hoc test wherever appropriate.
Results
NPS-EGFP neurons and the CRF system in the brain stem
In acute horizontal slice preparations from transgenic NPS-EGFP mice, EGFP-expressing neurons located between the locus coeruleus (LC) and Barrington's nucleus (BN) were readily visualized by their green fluorescence (Fig. 1A–C). A major population of EGFP-expressing neurons appeared as a thin band of cells on the rostro-caudal axis between LC and BN (Fig. 1A). Immunocytochemical studies verified that EGFP-labelled neurons are also NPS-immunopositive (Fig. 1B). Therefore we refer to them as ‘NPS-EGFP neurons’ in the present study. A subset of these NPS-EGFP neurons (n = 16) was intracellularly filled with neurobiotin and counterstained with streptavidin DyLight549 (Fig. 1C and D). NPS-EGFP neurons possessed cell bodies of various shapes, including spindle-like and multipolar forms, and sparse spine-like protrusions on their dendrites (Fig. 1D).
Figure 1. NPS-EGFP-expressing neurons in the LC region.
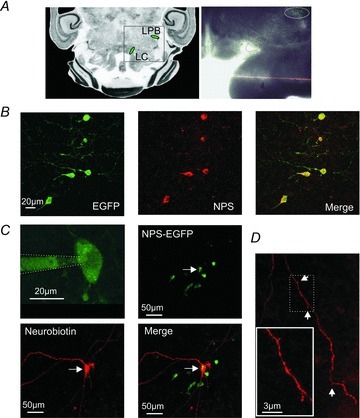
A, scheme of a horizontal slice containing NPS-EGFP neurons close to the LC and the LPB (left; modified from mbl.org). Representative horizontal slice preparation (right). Clusters of NPS-EGFP neurons (green) are marked. B, co-expression of endogenous NPS (red) and the EGFP transgene (green) in neurons of the LC region verified by immunohistochemical staining. C, confocal-microscopy image of an NPS-EGFP-expressing neuron (upper left) in the LC region near the 4th ventricle during a whole-cell patch-clamp recording. Note the diffusion of EGFP into the pipette solution during the whole-cell configuration. NPS-EGFP neuron (green; upper right) filled with neurobiotin and stained with streptavidin-DyLight594 (red; lower panel). D, neurites of neurobiotin-filled NPS-EGFP neurons show only sparse spine-like protrusions.
Immunocytochemical stainings revealed CRF-positive fibres co-localized with NPS-EGFP neurons in the LC area (Fig. 2A and B). At higher magnification, fibres apparently containing synaptic bouton-like structures were visible (Fig. 2B). CRF-positive structures seemed to be localized at the soma and/or proximal dendritic components of the NPS-EGFP neurons. To prove the specificity of the used antibody against CRF, some slices were treated with a CRF antibody that was preabsorbed with 2.5 μm CRF. In these control slices, no fibre-like structures were visible and the overall fluorescent signal was largely reduced to autofluorescence of the preparation (Fig. 2C). Furthermore, the CRF antibody detected CRF-expressing neurons located in the paraventricular nucleus of the hypothalamus (PVN) close to the third ventricle (Fig. 2D).
Figure 2. Co-localization of NPS-EGFP neurons and CRF/CRF1 in the LC region.
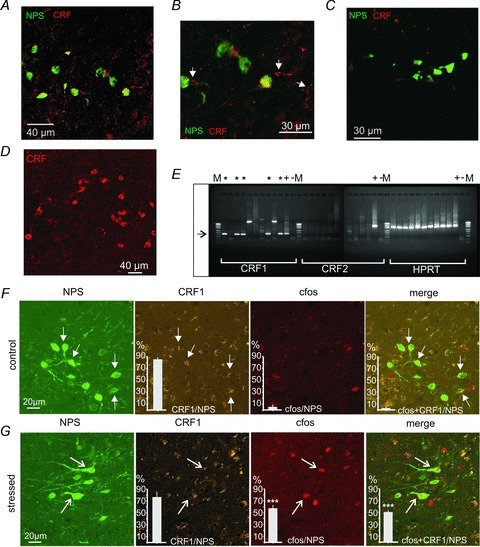
A, immunohistochemical staining for CRF (red) in horizontal brain slices. Fibre-like CRF-positive structures can be detected in the vicinity of NPS-EGFP neurons (green). B, inset of A in higher magnification. Note the presence of fibre-like structures positive for CRF (arrows), which co-localize with NPS-EGFP neurons. C, negative control staining with preabsorbed (2.5 μm CRF for 40 min) anti-CRF antibody. D, staining of CRF-expressing neurons in the PVN prove antiserum specificity. E, single-cell RT-PCR for CRF1, CRF2 and the house-keeping gene HPRT. Five of ten collected samples of individual NPS-EGFP neurons were positive for CRF1 mRNA (*). In contrast, all samples were negative for CRF2 mRNA. (M, marker; +, whole-brain lysate as positive control; –, ACSF from the recording chamber as negative control). F, triple-immunohistochemical stainings in coronal slices for NPS-GFP (green), CRF1 (yellow), and c-fos (red) in control mice that were not subjected to AIS. Arrows denote CRF1 co-expression in NPS-positive neurons. G, triple-immunohistochemical stainings for NPS-EGFP (green), CRF1 (yellow), and c-fos (red) in mice that were exposed to 20 min AIS. Note the presence of c-fos expression in NPS-EGFP neurons, which is absent in controls. Arrows denote cells showing positive staining for NPS, CRF1 and c-fos. Quantification of LC area EGFP-positive neurons co-staining for CRF1 and c-fos in mice subjected to immobilization stress or unstressed control animals. n = 3 mice per group. Total number of EGFP-positive neurons analysed: 60 in control mice, 63 in stressed animals. ***P < 0.001 stress vs. no stress controls after positive two-way ANOVA and Bonferroni's post hoc test.
Next, cytoplasm was collected from individual NPS-EGFP neurons (n = 10) near the LC in acute slice preparations, and single-cell RT-PCR was performed to detect CRF1 and CRF2 mRNA (Fig. 2E). HPRT was used as housekeeping gene, and whole-brain lysate and ACSF collected from the recording chamber were taken as positive (+) and negative (–) controls, respectively. In five of the ten collected cytoplasms, CRF1 transcripts could be detected (*; Fig. 2E). In two of the samples, amplification products of different molecular weight were detected and, thus, considered negative. No collected samples contained CRF2 transcripts (Fig. 2E). These data indicate that at least a subpopulation of NPS-synthesizing neurons expresses the CRF1.
In order to assess stress-related activation of CRF1-expressing NPS-EGFP neurons, transgenic mice were exposed to AIS and triple immunohistochemical stainings were performed to visualize NPS-EGFP, CRF1 and c-fos. In non-stressed control mice, about 80% of NPS-EGFP neurons co-expressed CRF1, and no detectable up-regulation of c-fos was observed in NPS-EGFP neurons or neurons expressing both EGFP and CRF1 (Fig. 2F). In mice that were exposed to 20 min AIS, CRF1 was similarly detected in about 75% NPS-expressing neurons, and more than half of the EGFP/CRF1-positive neurons demonstrated c-fos expression, indicating stress-induced activation of these neurons (Fig. 2G). Two-way ANOVA revealed significant interaction of stress treatment × marker protein expression (F(2,12) = 27.03, P < 0.0001), with main effects of treatment (F(1,12) = 64.30, P < 0.0001) and expression of marker proteins (F(2,12) = 84.73, P < 0.0001).
Characteristics of CRF-induced responses in NPS-EGFP neurons
The co-localization of NPS-EGFP and CRF1 suggested that NPS-EGFP neurons might be activated by CRF via the CRF1. In order to test this hypothesis, cell-attached recordings of NPS-EGFP neurons were performed, and spontaneous spike firing before and after application of CRF (250 nm) was monitored (Fig. 3A). In a majority of neurons (11/19), CRF significantly increased the frequency of spike firing (Fig. 3B). The mean frequency was 0.21 ± 0.09 Hz at baseline and 0.62 ± 0.14 Hz in the presence of CRF (n = 10; P < 0.05; Fig. 3C). Of note, 8 out of 19 neurons did not show spike firing, and application of CRF had no effect.
Figure 3. CRF increases spontaneous spike firing in NPS-EGFP neurons.
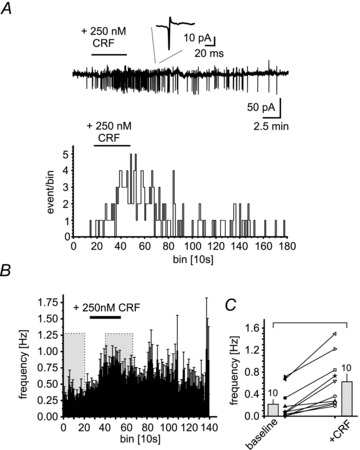
A, example recording of spontaneously generated spikes in a cell-attached configuration (top) during baseline conditions and in the presence of 250 nm CRF. The histogram (recorded events per 10 s bin vs. bin) shows a CRF-induced increase of activity of the recorded NPS-EGFP neuron. B, mean frequencies of generated spikes vs. 10 s bins. The grey boxes indicate the time-points for analysing the frequencies during baseline conditions and in the presence of CRF. Mean frequencies of individual recordings (n = 10) are plotted in C.
In another line of experiments, cellular properties of NPS-EGFP neurons were analysed using whole-cell patch-clamp recordings. Under current-clamp conditions, the membrane potential was set to −60 mV by direct current injection. Hyper- and depolarizing current steps (−50 to +150 pA; +10 pA increments) were applied to investigate electrotonic and electrogenic membrane properties (Fig. 4A). The passive and active membrane properties obtained are summarized in Table 1.
Figure 4. CRF-induced responses in NPS-EGFP neurons.
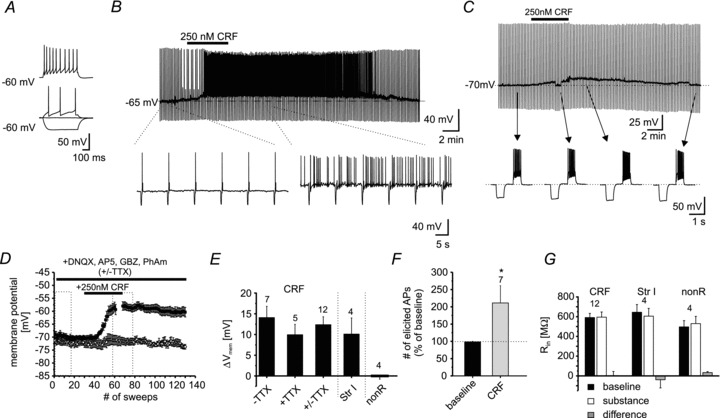
A, example recording of an individual NPS-EGFP neuron in current-clamp mode at a membrane potential of −60 mV. Hyper- and depolarizing currents (depicted are injections of −50, 0, +20 and +70 pA) were injected to analyse passive and active membrane properties. B, example recording of a single NPS-EGFP neuron at a membrane potential of −65 mV. In the presence of 250 nm CRF the neuron depolarized and generated spontaneous action potentials. C, example trace of an NPS-EGFP neuron recorded in current-clamp mode at a membrane potential of −70 mV. The bar indicates the application of 250 nm CRF for 5 min. Hyperpolarizing current injections (−60 pA; 500 ms duration) were done to control the input resistance. Depolarizing currents (+70 pA; 500 ms duration) were injected to elicit action potentials. Magnified examples are shown from the recording at time-points indicated by the arrows. During the CRF-induced depolarization, the membrane potential was re-adjusted to −70 mV to minimize the effects of voltage-dependent conductances on the input resistance. D, time course of the CRF-induced depolarization in NPS-EGFP neurons. Dashed boxes indicate the time intervals taken for quantification. E, quantification of the CRF-induced shift of the membrane potential (−TTX, recording in absence of TTX; +TTX, recording in presence of TTX; +/−TTX, pooled data; Str I, application of 250 nm Stressin I; nonR, non-responsive NPS-EGFP neurons, showing neither a significant de- nor hyperpolarization). F, quantification of the number of elicited action potentials (% of baseline) in response to the depolarizing current injection during baseline recordings and in presence of CRF. G, quantification of the input resistance Rin (CRF, neurons depolarized by CRF; Str I, neurons depolarized by Stressin I; nonR, neurons not affected by CRF). Input resistances are presented for baseline conditions (baseline), during maximal substance effect (substance), and as difference (substance – baseline).
Table 1.
Active and passive membrane properties of NPS-EGFP neurons
| At −60 mV (n = 47) | |
|---|---|
| Passive | |
| Input resistance (MΩ) | 603 ± 20 |
| Capacitance (pF) | 61 ± 2.6 |
| Membrane time const. (ms) | 36.8 ± 1.9 |
| Resting potential (mV) | −71 ± 1 |
| Active | |
| AP threshold (mV) | −43.6 ± 0.5 |
| Amplitude 1st AP (mV) | 70.1 ± 1 |
| AHP amplitude (mV) | 16 ± 0.5 |
| AP half-width (ms) | 1.4 ± 0.05 |
| Frequency adaptation | 0.6 ± 0.03 |
| Amplitude adaptation | 0.71 ± 0.01 |
Input resistance was calculated by R = ΔV/I; with an injected current of −50 pA. Capacitance was calculated by C = τ/R; membrane time constant τ was obtained by a monoexponential fit of the membrane potential shift induced by a current injection of −50 pA. Resting membrane potential was measured immediately after establishing whole-cell configuration. AHP, after-hyperpolarizing potential; measured after the first action potential. Frequency adaptation index calculated by FAI = frequency of the last two APs/frequency of the first two APs. Amplitude adaptation: AA = amplitude of last AP/amplitude of first AP. AP half -width was measured from the first AP.
The effects of CRF (250 nm) were assessed under current-clamp conditions, while membrane input resistance and action potential activity were monitored by repetitive application of hyper- and depolarizing current steps (500 ms duration; −60 pA and +70 to +100 pA), respectively. One subgroup of neurons was tested at membrane resting potential and another subgroup was held at −65 mV through direct current injection. Of these NPS-EGFP neurons, a majority (18/22) responded to application of CRF with a membrane depolarization, which peaked 5–6 min after application and returned to baseline upon discontinuation of CRF application. The membrane input resistance was not significantly different before and during CRF-evoked neuronal activity (591 ± 42 MΩ and 594 ± 53 MΩ, respectively; P = 0.96; n = 12; Fig. 4G). From holding potentials at −65 mV, CRF-induced membrane depolarization typically reached the spike threshold, resulting in generation of action potentials at a mean frequency of 0.89 ± 0.35 Hz (n = 6; Fig. 4B). When tested at membrane resting potential, NPS-EGFP neurons depolarized from −70.1 ± 1 mV at rest to −56.0 ± 1.8 mV in the presence of CRF (Δ+14.1 ± 2.7; n = 7; P < 0.01; Fig. 4D and E). The depolarizing influence of CRF resulted in an increase in action potential generation to 211 ± 50% compared to baseline during depolarizing current injections (n = 7; P < 0.05; Fig. 4F). Finally, a subset of NPS-EGFP neurons was recorded in the presence of TTX (1 μm), in order to minimize contributions of synaptic network activity. Under these conditions, CRF application resulted in a membrane depolarization from −70.4 ± 1 mV to −60.4 ± 2 mV (Δ+10 ± 2.5 mV; n = 5; P < 0.05; Fig. 4E). The shift in membrane potential upon application of CRF was not significantly different (P = 0.31) during recordings with or without the presence of TTX, and the data were thus pooled (baseline: −70.2 ± 0.7 mV, CRF: −57.8 ± 1.5 mV, Δ+12.4 ± 1.9; n = 12; Fig. 4E). Of the recorded NPS-EGFP neurons, four did not show any detectable membrane response to CRF (baseline: −72 ± 1 mV and CRF: −72 ± 1.8 mV; Δ−0.004 ± 0.76 mV; P = 0.99; Fig. 4D and E). These non-responsive neurons had a membrane input resistance of 496 ± 64 MΩ and 529 ± 76 MΩ at the end of substance application (P = 0.78; Fig. 4G).
In view of the immunocytochemical and RT-PCR evidence indicating CRF1 expression in NPS-EGFP neurons, responsiveness to the CRF1-specific agonist Stressin I (250 nm) was tested next. Application of Stressin I induced a depolarization from −71.5 ± 0.7 mV to −61.4 ± 3.4 mV (Δ+10.1 ± 3.9 mV; n = 4; P < 0.025; Fig. 4E), with no concomitant change in membrane input resistance (645 ± 79 MΩversus 605 ± 78 MΩ; P = 0.74; n = 4; Fig. 4G). To further prove that the observed depolarization in NPS-EGFP neurons is due to CRF1 stimulation, the CRF1-specific antagonist NBI27914 (NBI; 10 μm) was used. Recordings were done in the presence of 1 μm TTX while the neurons were held near resting potential and NBI was applied at least 5 min prior to application of CRF. In the presence of NBI, 250 nm CRF failed to induce a depolarization in all tested NPS-EGFP neurons (n = 6; Fig. 5A). Application of NBI induced a small, non-significant hyperpolarization from −72.5 ± 0.3 mV to −74.7 ± 1.1 mV (Δ−2.2 ± 1.4 mV; n = 6; P = 0.08; Fig. 5B and C), and additional application of CRF during the presence of NBI did not result in a further shift in membrane potential (−74.5 ± 1.3 mV; Δ+0.2 ± 0.6 mV; n = 6; P = 0.91; Fig. 5B and C). These data indicate that the observed effects of CRF on NPS-EGFP neurons are mediated via stimulation of CRF1.
Figure 5. CRF effects are mediated by CRF1.
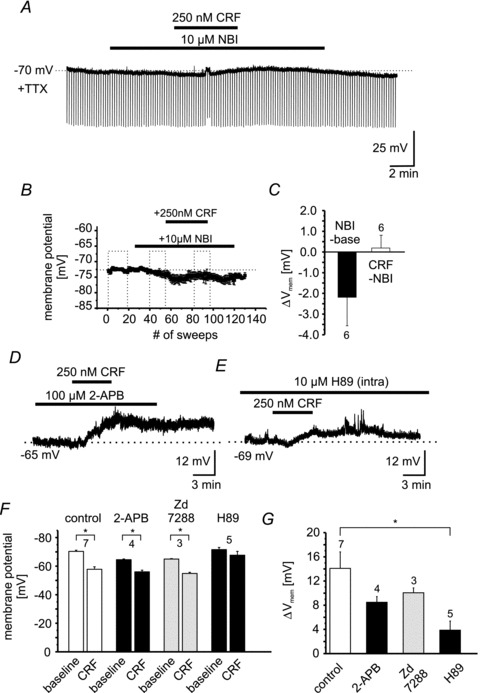
A, example recording of an individual NPS-EGFP neuron in current-clamp mode at a membrane potential of −70 mV. The recording was done in the presence of TTX. Hyperpolarizing currents (−60 pA; 500 ms duration) were injected to monitor the input resistance. Bars indicate the duration of NBI27914 (10 μm) and CRF (250 nm) application. B, scatter plot of the mean membrane potential vs. number of recorded sweeps (sweep duration: 10 s). Dashed boxes indicate the time intervals taken for quantification (baseline; NBI-application; NBI + CRF application). C, quantification of the change in membrane potential induced by NBI (NBI – baseline) or by CRF (CRF – NBI). D, representative current-clamp recording of a single NPS-EGFP neuron. CRF was applied in the presence of 100 μm 2-APB. E, example of a current-clamp recording at −69 mV membrane potential. The intracellular recording solution contained the PKA antagonist H89 (10 μm). Note the reduced depolarization induced by CRF in presence of H89. F, quantification of the membrane potential of NPS-EGFP neurons during baseline conditions and in the presence of CRF during control or in combination with 2-APB, ZD 7288 and H89. G, quantification of the CRF-induced depolarization during control or in the presence of 2-APB, ZD 7288 and H89.
In order to test the possible involvement of TRP-like cation channels, the non-selective TRP channel antagonist 2-APB was tested next (Fig. 5D). 2-APB was applied 5 min prior to CRF in the absence of TTX using a potassium-based internal recording solution. In the presence of 100 μm 2-APB, CRF depolarized NPS-EGFP neurons significantly from −65.5 ± 0.5 mV to −56.0 ± 1.2 mV (Δ+8.5 ± 0.9 mV; n = 4; P < 0.05; Fig. 5F). In addition, in the presence of the Ih-blocker ZD 7288 (30 μm) CRF depolarized NPS-EGFP neurons significantly from −65 ± 0.5 mV to −55 ± 0.8 mV (Δ+10.1 ± 0.8 mV; n = 3; P < 0.05; Fig. 5F). To further assess a possible involvement of the Ih conductance in CRF responses, Ih was analysed under voltage-clamp conditions using a voltage-step protocol (hyperpolarizing voltage steps from −50 mV to −120 mV; −10 mV increments; Budde et al. 2008). The CRF-induced depolarization (ΔVm+7 ± 2 mV; n = 8) of NPS-EGFP neurons was monitored in the current-clamp mode in the presence of TTX. The Ih current was calculated as the difference between the initial current and the steady-state current. The maximal Ih was −20 ± 3 pA at −120 mV during baseline conditions and −19.5 ± 3 pA in the presence of CRF (n = 8; data not shown). The presence of CRF had no effect on Ih magnitude, time course or voltage-dependent activation (data not shown). These results indicate that Ih and TRP-like channels have a limited, if any, contribution to the CRF-induced depolarization in NPS-EGFP neurons.
In view of previous reports on an involvement of cAMP/PKA activity in CRF responses in neocortical and hippocampal neurons (Blank et al. 2003; Hu et al. 2011), the PKA antagonist H89 (10 μm) was added to the intracellular recording solution. These conditions substantially affected CRF responses in that CRF evoked only a small, non-significant shift in membrane potential from −71.6 ± 1.6 mV to −67.7 ± 2.7 mV (Δ+3.9 ± 1.5 mV; n = 5; P = 0.25; Fig. 5F). Of note, H89 did not affect the resting membrane potential (−74 ± 3 mV; n = 5), the input resistance (641 ± 83 MΩ; n = 5), the action potential threshold (−41 ± 1 mV; n = 5), the action potential amplitude (72 ± 1 mV; n = 5), or the AHP (18 ± 1.2 mV; n = 5). To compare the efficacy of CRF to depolarize NPS-EGFP neurons in the absence and presence of 2-APB, ZD 7288 and H89, the ΔVm induced by CRF was compared between the different groups using one-way ANOVA followed by Bonferroni's post hoc test (Fig. 5G). The comparison revealed a significant difference between the four groups (F(3,15) = 4.17; P < 0.05; Fig. 5G). The post hoc analysis revealed a significantly reduced ΔVm by CRF in the presence of H89 compared to CRF alone (P < 0.05; Fig. 5G). According to these findings, PKA activity seems to be involved in CRF-induced depolarization of NPS-EGFP neurons.
Mechanisms of CRF responsiveness in NPS-EGFP neurons
To investigate the mechanisms underlying CRF-induced depolarization in NPS-EGFP neurons, whole-cell voltage-clamp recordings were performed at a holding-potential of −60 mV in the presence of 1 μm TTX. Using a potassium gluconate-based intracellular solution (K-gluc) the mean holding current at −60 mV was 6.3 ± 2.9 pA (n = 11), and application of 250 nm CRF shifted the holding current to −9.1 ± 3.5 pA (P < 0.01; Fig. 6A and B). The mean CRF-induced inward current was −15.5 ± 2.1 pA. To monitor input resistance, brief negative voltage steps were applied (−10 mV; 250 ms; every 10 s). As observed before under current-clamp conditions, no significant change of input resistance could be detected (baseline: 591 ± 42 MΩ and CRF: 595 ± 53 MΩ; Δ+3.7 ± 41 MΩ; n = 11; P = 0.96; Fig. 6C). In the next set of experiments, potassium (K+) conductances were blocked by using a caesium methanesulfonate-based intracellular solution (Cs-meth) containing 4-aminopyridine and tetraethylammonium. Under these conditions, the holding-current was −24.5 ± 2.6 pA during baseline conditions and shifted to −42.9 ± 4.4 pA upon application of CRF (Δ−18.4 ± 2.5 pA; n = 10; P < 0.01; Fig. 6B). These responses were associated with a significant reduction in membrane input resistance from 1057 ± 80 MΩ during baseline conditions to 845 ± 55 MΩ in the presence of CRF (Δ−212 ± 34 MΩ; n = 10; P < 0.05; Fig. 6C). These results indicate that more than one ionic mechanism may be involved in CRF-induced responses in NPS-EGFP neurons.
Figure 6. Conductances underlying the CRF-induced inward current.
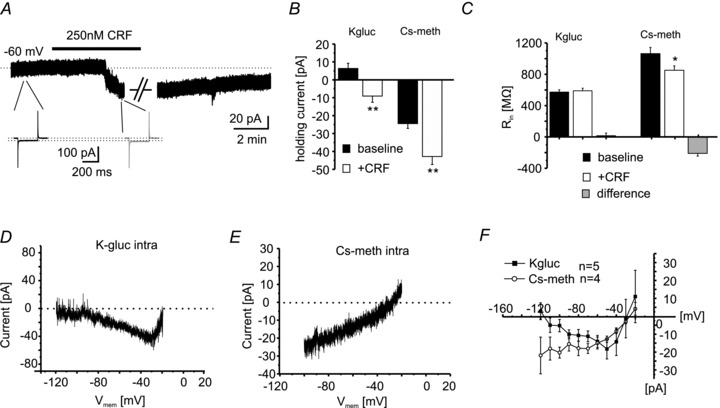
A, voltage-clamp recording of an individual NPS-EGFP neuron at a holding potential of −60 mV. Bar indicates the application of 250 nm CRF. CRF induces a long-lasting inward current, indicated by the shift of the holding current. The input resistance was controlled by brief voltage steps (−10 mV; 250 ms; black: baseline; grey: in presence of CRF). During baseline conditions and during maximal CRF effect (indicated by the gap), depolarizing voltage-clamp ramp experiments were done. (Artifacts have been truncated for clarification.) B, quantification of the holding current at a holding potential of −60 mV during baseline conditions and in the presence of CRF. Potassium gluconate (K-gluc)- and caesium methanesulfonate (Cs-meth; to block potassium conductances)-based intracellular solutions were used. C, quantification of the input resistance recorded with K-gluc- or Cs-meth-based solutions under baseline conditions and in the presence of 250 nm CRF. The change of input resistance was calculated as ResistanceCRF– Resistancebaseline (Difference). D, example of the CRF-induced current calculated from slow depolarizing voltage-clamp ramps recorded under baseline conditions and in the presence of CRF (from −120 to −20 mV; 40 mV s−1; calculated current: RampCRF– Rampbaseline) using a K-gluc-based intracellular solution. E, example of a CRF-induced current using a Cs-meth-based intracellular solution. Same ramp protocols as in D were applied. F, current/voltage relationship of the CRF-induced currents derived from voltage-clamp ramps. Note that using a K-gluc-based solution, the CRF-induced current displays two reversal potentials (∼−110 mV and ∼−25 mV). In contrast, using a Cs-meth-based solution only one reversal potential can be detected (∼−25 mV).
To further analyse the underlying mechanisms, voltage-clamp ramp experiments were performed. Two subgroups of NPS-EGFP neurons were recorded under voltage-clamp conditions with a K-gluc- or Cs-meth-based intracellular solution, respectively, and were depolarized by applying a slow voltage-ramp (40 mV s−1) from a holding potential of −120 mV to a final potential of −20 mV, before and during application of CRF (250 nm). The recordings were performed in the presence of 1 μm TTX and 50 μm CdCl2 to minimize the contribution of voltage-dependent sodium and calcium currents. Current versus voltage (I/V) relationships were obtained by measuring the membrane current at a given voltage level, and the CRF-induced current was calculated by subtracting the ramp I/Vs during baseline conditions from those recorded in the presence of CRF. Examples of the resultant currents are depicted in Fig. 6D and E for K-gluc- and Cs-meth-based solutions, respectively. The CRF-induced inward current shows two apparent reversal potentials during recordings with a K-gluc-based solution (Fig. 6D and F), approaching –109.6 ± 6 mV and −30.4 ± 6.4 mV (n = 5). The calculated equilibrium potential for K+ using the Nernst equation was at −109.4 mV under the present experimental conditions. These results suggested that one component of the CRF-induced inward current involved a reduction in membrane K+ conductance. Indeed, ramp I/Vs obtained during blocked K+ conductances with a Cs-meth-based intracellular solution revealed only one reversal potential of the CRF-induced current, averaging at −26.1 ± 6 mV (n = 4; Fig. 6E and F). These data indicated that the CRF-induced current involves two components: reduction in K+ conductance and activation of a cation conductance.
The CRF1 receptor is known to increase the intracellular concentration of cAMP following its activation. To analyse the effect of increased levels of cAMP on NPS-EGFP neurons, the cAMP-analogue 8-Br-cAMP (100 μm) was included in the internal recording solution during current-clamp recordings, and the membrane potential was monitored immediately after obtaining the whole-cell configuration. Infusion of 8-Br-cAMP resulted in a depolarization which slowly developed from the initial membrane potential −72 ± 1.6 mV within 5–8 min and peaked at +6.7 ± 0.6 mV (n = 10; Fig. 7A and C). Control recordings obtained with no added 8-Br-cAMP did not show significant changes of the membrane potential (0.95 ± 1.1 mV; n = 7; Fig. 7A and C). Furthermore, 8-Br-cAMP occluded any further depolarization by CRF. CRF application following 8-Br-cAMP-induced depolarization shifted the membrane potential from −63.2 ± 0.9 to −62.4 ± 1.2 mV (Δ+0.77 ± 0.46 mV; n = 4). The ΔVm in response to 8-Br-cAMP was significantly different from untreated controls and CRF application in the presence of 8-Br-cAMP (ANOVA: F(2,18) = 18.04; P < 0.01; Bonferroni post hoc test: 8-Br-cAMP vs. control P < 0.01; 8-Br-cAMP vs. cAMP + CRF P < 0.01; control vs. cAMP + CRF P > 0.05).
Figure 7. cAMP-induced depolarization of NPS-EGFP neurons.
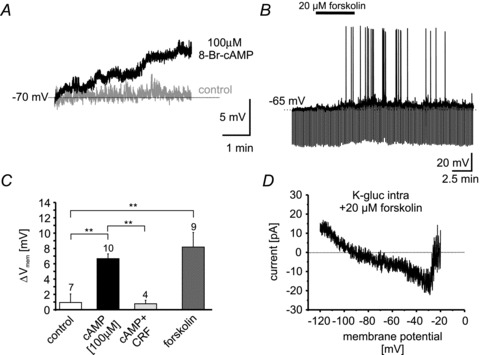
A, example current-clamp recordings at ∼−70 mV membrane potential during control conditions (grey) and with 100 μm 8-Br-cAMP included in the internal pipette solution (black). B, example recording of a NPS-EGFP neuron in the current-clamp mode at ∼−65 mV membrane potential. Bath application of 20 μm forskolin depolarized NPS-EGFP neurons and induced spontaneous action-potential generation. C, quantification of cAMP (100 μm)- or forskolin (20 μm)-induced depolarization compared to untreated control neurons (normal internal solution, no applied drug). Note: the cAMP-induced depolarization occludes CRF-dependent effects (cAMP + CRF). CRF was applied after the cAMP-induced depolarization reached steady-state and was analysed: DepolCRF– DepolcAMP. D, example of the forskolin-induced current calculated from slow depolarizing voltage-clamp ramps recorded under baseline conditions and in the presence of forskolin (from −120 to −20 mV; 40 mV s−1; calculated current: Rampforskolin– Rampbaseline) using a K-gluc-based intracellular solution.
In a different set of experiments, the adenylyl cyclase activator forskolin (20 μm) was bath-applied to NPS-EGFP neurons, and the effect on membrane potential was analysed (Fig. 7B). Forskolin depolarized 9 out of 11 NPS-EGFP neurons from −67.4 ± 0.9 mV to −59.2 ± 1.7 mV (Δ+8.2 ± 1.9 mV; n = 9; Fig. 7C). The shift of the membrane potential induced by forskolin was significantly different from shifts observed in untreated controls but not from 8-Br-cAMP-treated neurons (ANOVA: F(2,23) = 2.86; P < 0.01; Bonferroni post hoc test: forskolin vs. control: P < 0.01; forskolin vs. cAMP: P > 0.05; Fig. 7C). Voltage-ramp experiments in the presence of TTX revealed a forskolin-induced inward current, displaying two reversal potentials at around −87 and −32 mV (n = 2; Fig. 7D) similar to the observed CRF-induced current. These data support the view that CRF1 activation induces depolarizing responses in NPS-EGFP neurons via adenylyl cyclase/cAMP-dependent intracellular pathways.
Discussion
The present study adds the following novel aspects to our understanding of the NPS system. (i) NPS-EGFP neurons are situated in close proximity to CRF-containing fibres and express CRF1 receptors. (ii) AIS results in an increase in c-fos expression in CRF1-positive NPS-EGFP neurons in the LC. (iii) Stimulation of CRF1 in NPS-expressing neurons induces an inwardly directed membrane current mediated via a decrease in K+ and an increase in cation conductance, resulting in a membrane depolarization capable of triggering action potentials. (iv) These data provide evidence for a direct interaction between the CRF and the NPS system and thereby extend previous observations of NPS-modulated stress responsiveness towards a mechanistic level.
Interaction between the NPS and the CRF system in the brain stem
A striking difference between types of cells recorded in the LC region in the present study related to spontaneous spike firing. The population of NPS-EGFP-expressing neurons within the LC lacked spontaneous spike firing (in whole-cell recordings) or showed irregular spike firing at low frequencies (∼0.2 Hz; in cell-attached recordings). By contrast, in NPS-negative neurons encountered in the direct vicinity in the LC, barrages of spontaneous spikes, which rhythmically re-occurred at a low frequency (1.7 Hz), were observed. This type of activity has previously been described as a typical feature of noradrenergic LC neurons in rats and mice (Williams et al. 1984; van den Pol et al. 2002). This slow-oscillatory activity thus seems to be lacking in NPS-expressing neurons, possibly relating to differences in intrinsic membrane properties or lack of tonic transmitter influence.
Among the putative transmitter systems impinging on NPS-expressing neurons, CRF appears to be a prime candidate. It has been previously shown that NPS-EGFP neurons display an increase in c-fos expression after AIS or forced swimming (Liu et al. 2011). The up-regulation of c-fos expression is commonly thought to reflect an increase in neuronal activity (VanElzakker et al. 2008), and thus it was concluded that stressful experiences may activate NPS neurons (Liu et al. 2011). The CRF system is one of the most prominent modulatory neurotransmitter/peptide systems involved in stress responses (for review see: Spiess et al. 1998; Perrin & Vale, 1999). CRF-expressing neurons exist in various brain regions related to stress responsiveness, and the paraventricular nucleus of the hypothalamus (PVN) is considered a major source of CRF (Smagin & Dunn, 2000). Furthermore, the central nucleus of the amygdala (CeA) has been shown to send CRF-containing fibres to the LC (Reyes et al. 2008, 2011). In fact in the present study, CRF-containing fibres were found in close proximity to NPS-EGFP neurons within the LC and CRF1 expression in these neurons was verified at the mRNA and protein level, while no evidence was obtained for expression of CRF2. Therefore it appears reasonable to conclude that CRF might act as a transmitter on the NPS neuronal population located in the LC area. In keeping with this, forced swimming or immobilization stress resulted in an increase in c-fos expression in NPS-EGFP neurons co-expressing CRF1, suggesting a stress-mediated activation involving the CRF system.
Ionic mechanisms of CRF influences on NPS-expressing neurons
Application of CRF depolarized NPS-EGFP neurons and increased the number of elicited action potentials within 2–3 min after beginning the bath application. The CRF-induced depolarization was not inhibited by TTX, excluding indirect contributions by increased network activity. The CRF1-specific agonist Stressin I (Rivier et al. 2007) mimicked the CRF-induced depolarization, whereas the CRF1-specific antagonist NBI27914 abolished CRF-mediated activation of NPS-EGFP neurons. From these data it can be concluded that CRF directly activates NPS-EGFP neurons via CRF1. Depolarizing and/or activating effects of CRF have been observed in neuronal populations in various regions of the rodent brain, including neocortex and LC (Valentino et al. 1983; Gallopin et al. 2006). CRF-induced inward currents in NPS-EGFP neurons were insensitive to 50 μm CdCl2, thereby resembling previous observations made in neurons at the LC (Jedema & Grace, 2004). Ca2+ currents thus do not seem to make major contributions to CRF-mediated depolarization of NPS-EGFP neurons, although the influence of CdCl2-insensitive channels or intracellular stores cannot be excluded. In fact, CRF was found to increase the influx of Ca2+ in CeA neurons (Yu & Shinnick-Gallagher, 1998).
In NPS-EGFP neurons, the CRF-induced inward current displays two apparent reversal potentials. One current component approached a reversal potential close to the calculated K+ reversal potential and was absent during recordings using Cs-meth-based intracellular solutions. Therefore it seems feasible to conclude that the CRF-induced current is in part mediated by a reduction of membrane K+ conductance. Neocortical pyramidal neurons show a similar depolarizing response to CRF, which has been related to a reduction in K+ conductance mediated via the cAMP/PKA signalling cascades (Haug & Storm, 2000; Gallopin et al. 2006; Hu et al. 2011). A very similar mechanism involving cAMP following CRF1 activation has been suggested for LC neurons in rats (Jedema & Grace, 2004). In NPS-EGFP neurons, the presence of the PKA antagonist H89 in the intracellular solution significantly reduced the amplitude of the CRF-induced depolarization, while 8-Br-cAMP resulted in a membrane depolarization from rest and occluded further depolarizing responses to CRF. Furthermore, the adenylyl-cyclase activator forskolin mimicked the CRF-induced current displaying two reversal potentials indicative of a cation and K+ conductance. These data strongly support the view that cAMP/PKA signalling is involved in CRF responses in NPS-EGFP neurons in the brain stem.
Another CRF-induced current component in NPS-EGFP neurons was Cs+ resistant and displayed a reversal potential at ∼−25 to −30 mV, which has been described as a typical reversal potential for currents carried by non-specific cation channels (Yang & Ferguson, 2003). Candidate channels underlying the observed current are, for instance, cyclic nucleotide-gated channels or transient receptor potential (TRP) channels. TRP channels have been suggested to carry a cholecystokinin-evoked non-specific cation current in principal neurons in the amygdala, since the TRP channel blocker 2-APB abolished the responses (Meis et al. 2007). In NPS-EGFP neurons, 2-APB had no significant effect on the CRF-induced depolarization, suggesting that 2-APB-sensitive TRP channels are no major contributors. Previous studies reported a CRF-induced increase in a hyperpolarization-activated cation conductance (Ih), which is typically also regulated by the intracellular adenylyl-cyclase/cAMP system (Wanat et al. 2008; Giesbrecht et al. 2010). In NPS-EGFP neurons, CRF failed to significantly modulate the Ih and the persistence of the CFR-induced depolarization during the presence of the Ih-blocker ZD 7288 in NPS-EGFP neurons suggests that Ih is not involved in the CRF-induced depolarization of NPS-EGFP neurons.
Possible functional implications of CRF–NPS interactions in the brain stem
Release of CRF is an initial step of a neuroendocrine cascade during activation of the hypothalamic–pituitary–adrenal (HPA) axis, which typically mediates physiological responses to stress (Turnbull & Rivier, 1997; Bale & Vale, 2004). NPS, besides mediating acute anxiolytic-like effects in various experimental protocols (Xu et al. 2004; Jüngling et al. 2008; Rizzi et al. 2008; Vitale et al. 2008; Fendt et al. 2010), was found to block acute stress-induced changes in physiological parameters such as hyperthermia, oxidative stress damage, release of serotonin and noradrenaline in the frontal cortex (Leonard et al. 2008; Castro et al. 2009), and to buffer stress-impaired fear extinction in the amygdala (Chauveau et al., 2012). In addition, short-term swimming stress and prolonged restraint stress increases c-fos activity in NPS-positive neurons in the brain stem (Liu et al. 2011), and forced swimming results in an increase in extracellular NPS concentration in the basolateral amygdala (Ebner et al. 2011). Taken together these findings support the hypothesis that NPS acts as part of a neurotransmitter system that is stimulated in response to a stressor. The present study adds to this scenario the notion of a direct functional interaction between the CRF and the NPS system at the level of NPS-expressing neurons in the brain stem. The CRF1-mediated depolarization and associated increase in action potential firing may well result in an increase in NPS release from axon terminals in target areas. The rise in extracellular levels of NPS in the amygdala, detected by microdialysis upon both stress exposure and local depolarization (Ebner et al. 2011), is in line with this conclusion. Neuropeptide systems are critically involved in the acute regulation of the HPA axis (Thorsell, 2010) and they have received increasing attention in the investigation of the neurobiological basis of stress and anxiety disorders Okamura et al. 2007; Wittchen et al. 2011). The NPS system, in turn, may act as part of a negative feedback loop, in order for a response to the stressor to be generated that is both adequate and adaptive (Chauveau et al., 2012). In view of the critical involvement of stress in the pathogenesis of affective and anxiety disorders (deKloet et al. 2005), and the association of genetic variations of NPSR with anxiety-related phenotypes and disorders (Okamura et al. 2007; Donner et al. 2010; Domschke et al. 2011), it will be important to elucidate the role of the NPS system in human stress responsiveness.
Acknowledgments
The authors thank Elke Naß, Petra Berenbrock, Birgit Herrenpoth, Angelika Klinge, Elisabeth Boening and Svetlana Kiesling for excellent technical assistance. The project was funded by the German Research Foundation (DFG; SFB-TR58, TPA03 to H.C.P.), IMF (Innovative Medical Research, University Münster; LE210613 to J.L., SO220608 to L.S.), the IZKF (Interdisciplinary Centre for Clinical Research, Münster; PaHC3/003/10 to H.C.P. and K.J.), a Max Planck Research Award (to H.C.P.), a grant from the National Institute of Mental Health (MH-71363, to R.K.R.) and a seed grant from the Council on Research, Computing and Library Resources (University of California Irvine, to R.K.R.).
Glossary
- AHP
after-hyperpolarizing potential
- AIS
acute immobilization stress
- AP
action potential
- CRF
corticotropin-releasing factor
- CeA
central nucleus of the amygdala
- CRF1/CRF2
CRF receptor 1/2
- EGFP
enhanced green fluorescent protein
- LC
locus coeruleus
- LPB
lateral parabrachial nucleus
- NPS
neuropeptide S
- NPSR
neuropeptide S receptor
- PKA
protein kinase A
- PVN
hypothalamic paraventricular nucleus
- TRP channel
transient receptor potential channel
- TTX
tetrodotoxin
Author contributions
All experiments were performed at the Institute of Physiology I, Westfälische Wilhelms-Universität Münster, Germany, and the Department of Pharmaceutical Sciences, University of California Irvine, Irvine, CA, USA. Conception and design of the experiments: K.J., H.C.P., with contributions from P.C., R.K.R. Collection, analysis and interpretation of data: K.J., X.L., J.L., P.C., L.S., R.K.R., H.C.P. Writing the article: K.J., H.C.P., with contributions from R.K.R. All authors approved the final version of the manuscript.
References
- Bale TL, Vale WW. CRF and CRF receptors: role in stress responsivity and other behaviors. Annu Rev Pharmacol Toxicol. 2004;44:525–557. doi: 10.1146/annurev.pharmtox.44.101802.121410. [DOI] [PubMed] [Google Scholar]
- Blank T, Nijholt I, Grammatopoulos DK, Randeva HS, Hillhouse EW, Spiess J. Corticotropin-releasing factor receptors couple to multiple G-proteins to activate diverse intracellular signaling pathways in mouse hippocampus: role in neuronal excitability and associative learning. J Neurosci. 2003;23:700–777. doi: 10.1523/JNEUROSCI.23-02-00700.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budde T, Coulon P, Pawlowski M, Meuth P, Kanyshkova T, Japes A, Meuth SG, Pape HC. Reciprocal modulation of Ih and ITASK in thalamocortical relay neurons by halothane. Pflugers Arch. 2008;456:1061–1073. doi: 10.1007/s00424-008-0482-9. [DOI] [PubMed] [Google Scholar]
- Castro AA, Moretti M, Casagrande TS, Martinello C, Petronilho F, Steckert AV, Guerrini R, Calo' G, Dal Pizzol F, Quevedo J, Gavioli EC. Neuropeptide S produces hyperlocomotion and prevents oxidative stress damage in the mouse brain: a comparative study with amphetamine and diazepam. Pharmacol Biochem Behav. 2009;91:636–642. doi: 10.1016/j.pbb.2008.10.015. [DOI] [PubMed] [Google Scholar]
- Chauveau F, Lange MD, Jüngling K, Lesting J, Seidenbecher T, Pape HC. Prevention of stress-impaired fear extinction through neuropeptide S action in the lateral amygdala. Neuropsychopharmacology. 2012 doi: 10.1038/npp.2012.3. doi: 10.1038/npp.2012.3. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark SD, Duangdao DM, Schulz S, Zhang L, Liu X, Xu Y-L, Reinscheid RK. Anatomical characterization of the neuropeptide S system in the mouse brain by in situ hybridization and immunohistochemistry. J Comp Neurol. 2011;519:1867–1893. doi: 10.1002/cne.22606. [DOI] [PubMed] [Google Scholar]
- de Kloet ER, Joëls M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6:463–475. doi: 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- Domschke K, Reif A, Weber H, Richter J, Hohoff C, Ohrmann P, Pedersen A, Bauer J, Suslow T, Kugel H, Heindel W, Baumann C, Klauke B, Jacob C, Maier W, Fritze J, Bandelow B, Krakowitzky P, Rothermundt M, et al. Neuropeptide S receptor gene – converging evidence for a role in panic disorder. Mol Psychiatry. 2011;16:938–948. doi: 10.1038/mp.2010.81. [DOI] [PubMed] [Google Scholar]
- Donner J, Haapakoski R, Ezer S, Melén E, Pirkola S, Gratacòs M, Zucchelli M, Anedda F, Johansson LE, Söderhäll C, Orsmark-Pietras C, Suvisaari J, Martín-Santos R, Torrens M, Silander K, Terwilliger JD, Wickman M, Pershagen G, Lönnqvist J, Peltonen L, et al. Assessment of the neuropeptide S system in anxiety disorders. Biol Psychiatry. 2010;68:474–483. doi: 10.1016/j.biopsych.2010.05.039. [DOI] [PubMed] [Google Scholar]
- Ebner K, Rjabokon A, Pape HC, Singewald N. Increased in vivo release of neuropeptide S in the amygdala of freely moving rats after local depolarisation and emotional stress. Amino Acids. 2011;41:991–996. doi: 10.1007/s00726-011-1058-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fendt M, Imobersteg S, Burki H, McAllister KH, Sailer AW. Intra-amygdala injections of neuropeptide S block fear-potentiated startle. Neurosci Lett. 2010;474:154–157. doi: 10.1016/j.neulet.2010.03.028. [DOI] [PubMed] [Google Scholar]
- Gallopin T, Geoffroy H, Rossier J, Lambolez B. Cortical sources of CRF, NKB, and CCK and their effects on pyramidal cells in the neocortex. Cereb Cortex. 2006;16:1440–1452. doi: 10.1093/cercor/bhj081. [DOI] [PubMed] [Google Scholar]
- Giesbrecht CJ, Mackay JP, Silveira HB, Urban JH, Colmers WF. Countervailing modulation of Ih by neuropeptide Y and corticotrophin-releasing factor in basolateral amygdala as a possible mechanism for their effects on stress-related behaviors. J Neurosci. 2010;30:16970–16982. doi: 10.1523/JNEUROSCI.2306-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haug T, Storm JF. Protein kinase A mediates the modulation of the slow Ca2+-dependent K+ current, IsAHP, by the neuropeptides CRF, VIP, and CGRP in hippocampal pyramidal neurons. J Neurophysiol. 2000;83:2071–2079. doi: 10.1152/jn.2000.83.4.2071. [DOI] [PubMed] [Google Scholar]
- Hu E, Demmou L, Cauli B, Gallopin T, Geoffroy H, Harris-Warrick RM, Paupardin-Tritsch D, Lambolez B, Vincent P, Hepp R. VIP, CRF, and PACAP act at distinct receptors to elicit different cAMP/PKA dynamics in the neocortex. Cereb Cortex. 2011;21:708–718. doi: 10.1093/cercor/bhq143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasoni CL, Todman MG, Han SK, Herbison AE. Expression of mRNAs encoding receptors that mediate stress signals in gonadotropin-releasing hormone neurons of the mouse. Neuroendocrinology. 2005;82:320–328. doi: 10.1159/000093155. [DOI] [PubMed] [Google Scholar]
- Jedema HP, Grace AA. Corticotropin-releasing hormone directly activates noradrenergic neurons of the locus ceruleus recorded in vitro. J Neurosci. 2004;24:9703–9713. doi: 10.1523/JNEUROSCI.2830-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jüngling K, Seidenbecher T, Sosulina L, Lesting J, Sangha S, Clark SD, Okamura N, Duangdao DM, Xu YL, Reinscheid RK, Pape HC. Neuropeptide S-mediated control of fear expression and extinction: role of intercalated GABAergic neurons in the amygdala. Neuron. 2008;59:298–310. doi: 10.1016/j.neuron.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard SK, Dwyer JM, Sukoff Rizzo SJ, Platt B, Logue SF, Neal SJ, Malberg JE, Beyer CE, Schechter LE, Rosenzweig-Lipson S, Ring RH. Pharmacology of neuropeptide S in mice: therapeutic relevance to anxiety disorders. Psychopharmacology (Berl) 2008;197:601–611. doi: 10.1007/s00213-008-1080-4. [DOI] [PubMed] [Google Scholar]
- Liu X, Zeng J, Zhou A, Theodorsson E, Fahrenkrug J, Reinscheid RK. Molecular fingerprint of neuropeptide S-producing neurons in the mouse brain. J Comp Neurol. 2011;519:1847–1866. doi: 10.1002/cne.22603. [DOI] [PubMed] [Google Scholar]
- Meis S, Bergado-Acosta JR, Yanagawa Y, Obata K, Stork O, Munsch T. Identification of a neuropeptide S responsive circuitry shaping amygdala activity via the endopiriform nucleus. PLoS One. 2008;3:e2695. doi: 10.1371/journal.pone.0002695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meis S, Munsch T, Sosulina L, Pape HC. Postsynaptic mechanisms underlying responsiveness of amygdaloid neurons to cholecystokinin are mediated by a transient receptor potential-like current. Mol Cell Neurosci. 2007;35:356–367. doi: 10.1016/j.mcn.2007.03.012. [DOI] [PubMed] [Google Scholar]
- Okamura N, Garau C, Duangdao DM, Clark SD, Jüngling K, Pape HC, Reinscheid RK. Neuropeptide S enhances memory during the consolidation phase and interacts with noradrenergic systems in the brain. Neuropsychopharmacology. 2011;36:744–752. doi: 10.1038/npp.2010.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura N, Hashimoto K, Iyo M, Shimizu E, Dempfle A, Friedel S, Reinscheid RK. Gender-specific association of a functional coding polymorphism in the neuropeptide S receptor gene with panic disorder but not with schizophrenia or attention-deficit/hyperactivity disorder. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31:1444–1448. doi: 10.1016/j.pnpbp.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Perkins KL. Cell-attached voltage-clamp and current-clamp recording and stimulation techniques in brain slices. J Neurosci Methods. 2006;154:1–18. doi: 10.1016/j.jneumeth.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin MH, Vale WW. Corticotropin releasing factor receptors and their ligand family. Ann N Y Acad Sci. 1999;885:312–328. doi: 10.1111/j.1749-6632.1999.tb08687.x. [DOI] [PubMed] [Google Scholar]
- Raczka KA, Gartmann N, Mechias ML, Reif A, Büchel C, Deckert J, Kalisch R. A neuropeptide S receptor variant associated with overinterpretation of fear reactions: a potential neurogenetic basis for catastrophizing. Mol Psychiatry. 2010;15:1067–1074. doi: 10.1038/mp.2010.79. [DOI] [PubMed] [Google Scholar]
- Reyes BA, Carvalho AF, Vakharia K, Van Bockstaele EJ. Amygdalar peptidergic circuits regulating noradrenergic locus coeruleus neurons: linking limbic and arousal centers. Exp Neurol. 2011;230:96–105. doi: 10.1016/j.expneurol.2011.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes BA, Drolet G, Van Bockstaele EJ. Dynorphin and stress-related peptides in rat locus coeruleus: contribution of amygdalar efferents. J Comp Neurol. 2008;508:663–675. doi: 10.1002/cne.21683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivier J, Gulyas J, Kunitake K, DiGruccio M, Cantle JP, Perrin MH, Donaldson C, Vaughan J, Million M, Gourcerol G, Adelson DW, Rivier C, Taché Y, Vale W. Stressin1-A, a potent corticotropin releasing factor receptor 1 (CRF1)-selective peptide agonist. J Med Chem. 2007;50:1668–1674. doi: 10.1021/jm0613875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzi A, Vergura R, Marzola G, Ruzza C, Guerrini R, Salvadori S, Regoli D, Calo G. Neuropeptide S is a stimulatory anxiolytic agent: a behavioural study in mice. Br J Pharmacol. 2008;154:471–479. doi: 10.1038/bjp.2008.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si W, Aluisio L, Okamura N, Clark SD, Fraser I, Sutton SW, Bonaventure P, Reinscheid RK. Neuropeptide S stimulates dopaminergic neurotransmission in the medial prefrontal cortex. J Neurochem. 2010;115:475–482. doi: 10.1111/j.1471-4159.2010.06947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smagin GN, Dunn AJ. The role of CRF receptor subtypes in stress-induced behavioural responses. Eur J Pharmacol. 2000;405:199–206. doi: 10.1016/s0014-2999(00)00553-7. [DOI] [PubMed] [Google Scholar]
- Smith KL, Patterson M, Dhillo WS, Patel SR, Semjonous NM, Gardiner JV, Ghatei MA, Bloom SR. Neuropeptide S stimulates the hypothalamo-pituitary-adrenal axis and inhibits food intake. Endocrinology. 2006;147:3510–3518. doi: 10.1210/en.2005-1280. [DOI] [PubMed] [Google Scholar]
- Sosulina L, Graebenitz S, Pape HC. GABAergic interneurons in the mouse lateral amygdala: a classification study. J Neurophysiol. 2010;104:617–626. doi: 10.1152/jn.00207.2010. [DOI] [PubMed] [Google Scholar]
- Sosulina L, Schwesig G, Seifert G, Pape HC. Neuropeptide Y activates a G-protein-coupled inwardly rectifying potassium current and dampens excitability in the lateral amygdala. Mol Cell Neurosci. 2008;39:491–498. doi: 10.1016/j.mcn.2008.08.002. [DOI] [PubMed] [Google Scholar]
- Spiess J, Dautzenberg FM, Sydow S, Hauger RL, Rühmann A, Blank T, Radulovic J. Molecular properties of the CRF receptor. Trends Endocrinol Metab. 1998;9:140–145. doi: 10.1016/s1043-2760(98)00037-x. [DOI] [PubMed] [Google Scholar]
- Thorsell A. Brain neuropeptide Y and corticotropin-releasing hormone in mediating stress and anxiety. Exp Biol Med. 2010;235:1163–1167. doi: 10.1258/ebm.2010.009331. [DOI] [PubMed] [Google Scholar]
- Turnbull AV, Rivier C. Corticotropin-releasing factor (CRF) and endocrine responses to stress: CRF receptors, binding protein, and related peptides. Proc Soc Exp Biol Med. 1997;215:1–10. doi: 10.3181/00379727-215-44108. [DOI] [PubMed] [Google Scholar]
- Valentino RJ, Foote SL, Aston-Jones G. Corticotropin-releasing factor activates noradrenergic neurons of the locus coeruleus. Brain Res. 1983;270:363–367. doi: 10.1016/0006-8993(83)90615-7. [DOI] [PubMed] [Google Scholar]
- van den Pol AN, Ghosh PK, Liu RJ, Li Y, Aghajanian GK, Gao XB. Hypocretin (orexin) enhances neuron activity and cell synchrony in developing mouse GFP-expressing locus coeruleus. J Physiol. 2002;541:169–185. doi: 10.1113/jphysiol.2002.017426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanElzakker M, Fevurly RD, Breindel T, Spencer RL. Environmental novelty is associated with a selective increase in Fos expression in the output elements of the hippocampal formation and the perirhinal cortex. Learn Mem. 2008;15:899–908. doi: 10.1101/lm.1196508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale G, Filaferro M, Ruggieri V, Pennella S, Frigeri C, Rizzi A, Guerrini R, Calò G. Anxiolytic-like effect of neuropeptide S in the rat defensive burying. Peptides. 2008;29:2286–2291. doi: 10.1016/j.peptides.2008.08.014. [DOI] [PubMed] [Google Scholar]
- Wanat MJ, Hopf FW, Stuber GD, Phillips PE, Bonci A. Corticotropin-releasing factor increases mouse ventral tegmental area dopamine neuron firing through a protein kinase C-dependent enhancement of Ih. J Physiol. 2008;586:2157–2170. doi: 10.1113/jphysiol.2007.150078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, North RA, Shefner SA, Nishi S, Egan TM. Membrane properties of rat locus coeruleus neurones. Neuroscience. 1984;13:137–156. doi: 10.1016/0306-4522(84)90265-3. [DOI] [PubMed] [Google Scholar]
- Wittchen HU, Jacobi F, Rehm J, Gustavsson A, Svensson M, Jönsson B, Olesen J, Allgulander C, Alonso J, Faravelli C, Fratiglioni L, Jennum P, Lieb R, Maercker A, van Os J, Preisig M, Salvador-Carulla L, Simon R, Steinhausen HC. The size and burden of mental disorders and other disorders of the brain in Europe 2010. Eur Neuropsychopharmacol. 2011;21:655–679. doi: 10.1016/j.euroneuro.2011.07.018. [DOI] [PubMed] [Google Scholar]
- Xu YL, Reinscheid RK, Huitron-Resendiz S, Clark SD, Wang Z, Lin SH, Brucher FA, Zeng J, Ly NK, Henriksen SJ, de Lecea L, Civelli O. Neuropeptide S: a neuropeptide promoting arousal and anxiolytic-like effects. Neuron. 2004;43:487–497. doi: 10.1016/j.neuron.2004.08.005. [DOI] [PubMed] [Google Scholar]
- Yang B, Ferguson AV. Orexin-A depolarizes nucleus tractus solitarius neurons through effects on nonselective cationic and K+ conductances. J Neurophysiol. 2003;89:2167–2175. doi: 10.1152/jn.01088.2002. [DOI] [PubMed] [Google Scholar]
- Yu B, Shinnick-Gallagher P. Corticotropin-releasing factor increases dihydropyridine- and neurotoxin-resistant calcium currents in neurons of the central amygdala. J Pharmacol Exp Ther. 1998;284:170–179. [PubMed] [Google Scholar]