

Abstract
Previous studies have suggested that muscarinic receptor activation modulates glutamatergic transmission. M-type potassium channels mediate the effects of muscarinic activation in the hippocampus, and it has been proposed that they modulate glutamatergic synaptic transmission. We tested whether M1 muscarinic receptor activation enhances glutamatergic synaptic transmission via the inhibition of the M-type potassium channels that are present in Schaffer collateral axons and terminals. Miniature excitatory postsynaptic currents (mEPSCs) were recorded from CA1 pyramidal neurons. The M1 receptor agonist, NcN-A-343, increased the frequency of mEPSCs, but did not alter their amplitude. The M-channel blocker XE991 and its analogue linopirdine also increased the frequency of mEPSCs. Flupirtine, which opens M-channels, had the opposite effect. XE991 did not enhance mEPSCs frequency in a calcium-free external medium. Blocking P/Q- and N-type calcium channels abolished the effect of XE991 on mEPSCs. These data suggested that the inhibition of M-channels increases presynaptic calcium-dependent glutamate release in CA1 pyramidal neurons. The effects of these agents on the membrane potentials of presynaptic CA3 pyramidal neurons were studied using current clamp recordings; activation of M1 receptors and blocking M-channels depolarized neurons and increased burst firing. The input resistance of CA3 neurons was increased by the application of McN-A-343 and XE991; these effects were consistent with the closure of M-channels. Muscarinic activation inhibits M-channels in CA3 pyramidal neurons and its efferents – Schaffer collateral, which causes the depolarization, activates voltage-gated calcium channels, and ultimately elevates the intracellular calcium concentration to increase the release of glutamate on CA1 pyramidal neurons.
Key points
M-type potassium channels play a key role in modulating neuronal excitability. However, the effects of M-channel activation on synaptic transmission are poorly understood.
This study found that an M1 receptor agonist and M-channel blockers increased action potential-independent glutamate release at Schaffer collateral–CA1 pyramidal neuron synapses in acute hippocampus slices.
This enhancement was dependent on Ca2+ influx from extracellular space but not intracellular calcium stores.
Inhibition of M-channels results in the depolarization of CA3 pyramidal neurons and activated presynaptic voltage-gated P/Q- and N-type calcium channels, which in turn causes Ca2+ influx and increased glutamate release.
Thus, M1 muscarinic agonists modulate action potential-independent glutamatergic synaptic transmission in the hippocampus by inhibition of presynaptic M-channels.
Introduction
Muscarinic acetylcholine receptors (mAChRs) are seven-transmembrane-domain G protein-coupled receptors (GPCRs) that are widely expressed throughout the central nervous system. The M1 subtype is the predominant mAChR in the cortex, hippocampus, striatum and thalamus (Langmead et al. 2008). Muscarinic receptor activation has distinct effects on glutamatergic transmission in different neurons. It inhibits glutamatergic transmission in magnocellular neurons of the basal forebrain (Sim & Griffith, 1996), along with neurons in the basolateral amygdala (Yajeya et al. 2000), striatum (Higley et al. 2009) and spinal cord (Zhang et al. 2007). In the CA3 region of the hippocampus, muscarinic activation inhibits associational-commissural synaptic transmission via presynaptic calcium channel inhibition; however, it enhances mossy fibre–CA3 pyramidal neuron synaptic transmission (Vogt & Regehr, 2001). The activation of muscarinic receptors induces a long-lasting synaptic enhancement at Schaffer collateral–CA1 pyramidal neuron synapses by increasing the release of calcium from postsynaptic endoplasmic reticulum stores both in vivo and in vitro (Fernández de Sevilla et al. 2008). Muscarinic activation also enhances glutamatergic transmission in dentate granule cells (Kozhemyakin et al. 2010). M1 muscarinic receptor activation also inhibits M-type potassium channels (Brown & Adams, 1980; Marrion et al. 1989; Bernheim et al. 1992). M-type potassium channels belong to the Kv7 (KCNQ) K+ channel family (Wang et al. 1998; Selyanko et al. 2002). M-channels activate at a subthreshold membrane potential and do not inactivate, so they generate a steady voltage-dependent outward current near the resting membrane potential (Constanti & Brown, 1981; Delmas & Brown, 2005). Mutations of the KCNQ2 and KCNQ3 genes cause benign familial neonatal convulsions (BFNC) (Biervert et al. 1998; Jentsch, 2000). The expression of KCNQ channels increases during early development in rodent hippocampus (Shah et al. 2002; Geiger et al. 2006; Weber et al. 2006; Safiulina et al. 2008), but the expression of KCNQ2 and KCNQ3 has different developmental pattern in human brain (Kanaumi et al. 2008). It has been suggested that the highest density of KCNQ2 and KCNQ3 immunoreactivity in the CA1 region is in the axon initial segments, where action potentials are generated and the Kv7 channels co-localize with Na+ channels via binding to ankyrin G. This localization allows M-channels to powerfully limit neuronal excitability (Devaux et al. 2004; Chung et al. 2006; Pan et al. 2006) and therefore function as a ‘brake’ on repetitive firing and play a key role in regulating the excitability of various central and peripheral neurons (Yue & Yaari, 2004; Gu et al. 2005; Shen et al. 2005; Brown & Randall, 2009). Other studies have suggested that M-channels are expressed in presynaptic terminals (Cooper et al. 2001; Chung et al. 2006; Garcia-Pino et al. 2010). Physiological studies have suggested that M-channels regulate the release of neurotransmitters. Drugs that block or open M-channels can regulate presynaptic fibre volley and the evoked EPSPs recorded from CA1 pyramidal neurons (Vervaeke et al. 2006). M-channel blocking by XE991 increases the release of noradrenaline, and retigabine, an M-channel opener, decreases noradrenaline release in cultured sympathetic neurons (Hernandez et al. 2008). These drugs also regulate neurotransmitter release from hippocampal synaptosomes (Martire et al. 2004) and cultured hippocampal neurons (Peretz et al. 2007). However, the mechanism by which M-channels regulate neurotransmitter release remains unclear. The current study demonstrates that both M1 muscarinic activation and M-channel inhibition enhance action-potential-independent glutamate release in Schaffer collateral–CA1 pyramidal neuron synapses. This action appears to be dependent on the depolarization of CA3 pyramidal neurons and the activation of voltage-gated calcium channels.
Methods
Slice preparation
All studies were performed according to protocols that were approved by the University of Virginia Animal Use and Care Committee, and the US Army Medical Research and Material Command Animal Care and Use Review Office (ACURO). Adult male (175–250 g) Sprague–Dawley rats were anaesthetized with isoflurane prior to decapitation, which was followed by quick removal of the brain. The removed brains were then sectioned to 300 μm slices using a Leica VT 1200 slicer (Leica Microsystems, Wetzlar, Germany) in ice-cold oxygenated slicing solution. The solution contained the following (in mm): 120 sucrose, 65.5 NaCl, 2 KCl, 1.1 KH2PO4, 25 NaHCO3, 10 d-glucose, 1 CaCl2, and 5 MgSO4. The slices were then incubated for at least 1 h at 32°C in oxygenated ACSF that contained (in mm): 127 NaCl, 2 KCl, 1.1 KH2PO4, 25.7 NaHCO3, 10 d-glucose, 2 CaCl2, and 1.5 MgSO4; the osmolarity in the chamber was 290–300 mosmol l−1. After incubation, the slices were transferred to the recording chamber on the stage of an Olympus Optical BX51 microscope (Olympus, Tokyo, Japan). Unless otherwise stated, all chemicals were obtained from Sigma-Aldrich (St Louis, MO, USA).
Whole-cell recording
Whole-cell patch-clamp recordings were performed under infrared differential interference contrast microscopy (Olympus); a 40× water-immersion objective was used to visually identify CA1 and CA3 pyramidal neurons. The slices were continuously perfused with ACSF solution that was saturated with 95% O2 and 5% CO2 at room temperature. Patch electrodes (final resistances, 3–5 MΩ) were pulled from borosilicate glass (Sutter Instruments, Novato, CA, USA) on a horizontal Flaming-Brown microelectrode puller (Model P-97, Sutter Instruments). For voltage-clamp recordings, the electrode tips were filled with a filtered internal recording solution that consisted of the following components (in mm): 117.5 CsMeSO4, 10 Hepes, 0.3 EGTA, 15.5 CsCl, and 1.0 MgCl2; the pH was 7.3 (with CsOH), and the osmolarity was 310 mosmol l−1. The electrode shank contained (in mm) 4 Mg-ATP salt, 0.3 Na-GTP salt, and 5 QX-314. For current-clamp recording, the pipette solution contained the following (in mm): 135 potassium gluconate, 2.5 NaCl, 10 Hepes, 0.5 EGTA, 4.0 Mg-ATP, 0.4 Na-GTP, 0.1 CaCl2; the pH was 7.3, and the osmolarity was 310 mosmol l−1. Neurons were voltage clamped at −60 mV using a PC-505B amplifier (Warner Instruments, Hamden, CT, USA). Electrode capacitance was electronically compensated. Access resistance was continuously monitored, and if the series resistance increased by 20% at any time, the recording was terminated. Currents were filtered at 2 kHz, digitized using a Digidata 1322 digitizer (Molecular Devices, Sunnyvale, CA, USA), and acquired using Clampex 10.2 software (Molecular Devices). Spontaneous excitatory postsynaptic currents (sEPSCs) were recorded from CA1 pyramidal neurons after blocking the GABAA receptors with the antagonist picrotoxin (50 μm). In preliminary experiments, a combination of 6-cyano-7-nitroquinoxalene-2,3-dione (CNQX) and 2-amino-5-phosphonovaleric acid (APV) blocked all EPSCs. Miniature EPSCs (mEPSCs) were recorded by blocking action potentials with 1 μm TTX (Alomone labs, Jerusalem, Israel). All drugs were bath-applied via a peristaltic pump.
Data analysis
The offline digitized data were analysed with MiniAnalysis (Synaptosoft, Decatur, GA, USA) and Clampfit 10.2 (Molecular Devices). To detect sEPSCs and mEPSCs, a detection threshold was set at three times the root mean square (RMS) of the baseline noise. After detection, the frequency and peak amplitude of EPSCs from individual neurons were analysed. Each detected event from the 20–30 min recording session was visually inspected to remove false detections. The Kolmogorov–Smirnov (K-S) test was used to compare amplitudes and inter-event intervals for continuously recorded EPSCs. The input resistance of CA3 neuron under current-clamp recording was analysed by comparing the slope of current–voltage relationship and measured directly by Clampfit 10.2 at the peak of membrane potentials. The drug effects were compared using a paired Student's t test with a significance level of P < 0.05. Data are expressed as means ± SEM unless otherwise noted.
Results
An M1 muscarinic agonist increases mEPSC frequency in CA1 neurons
Previous studies suggested that muscarinic receptor activation increased presynaptic glutamate release from perforant path to dentate granule cells (Kozhemyakin et al. 2010). In the present study, we investigated the effect of the M1 muscarinic receptor agonist McN-A-343 on glutamate release recorded from Schaffer collateral–CA1 synapses. Application of McN-A-343 (10 μm) increased the frequency of mEPSCs that were recorded from CA1 pyramidal neurons by 69.89 ± 8.28% (0.12 ± 0.03 Hz vs. 0.21 ± 0.05 Hz; n = 7, P < 0.01), but it did not change their amplitudes (16.51 ± 1.14 pA vs. 16.79 ± 1.47 pA; n = 7, P = 0.38, Fig. 1A and B). McN-A-343 caused a significant leftward shift in the cumulative distribution of the inter-event intervals but had no effect on the cumulative distribution of the amplitudes of the mEPSCs (K-S test, Fig. 1C and D). This suggested that the activation of M1 receptors increased action potential-independent glutamate release from the presynaptic terminals of Schaffer collateral–CA1 synapses.
Figure 1. M1 muscarinic agonist increased the frequency of mEPSCs recorded from CA1 pyramidal neurons.
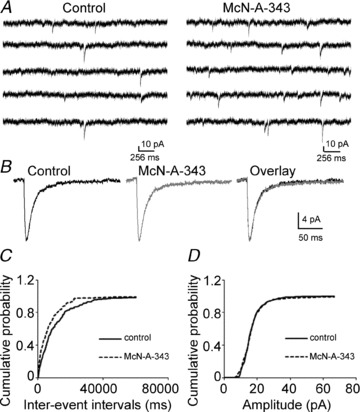
A, representative traces of mEPSCs recorded from CA1 pyramidal neurons before (left) and after (right) the application of 10 μm McN-A-343. B, representative averaged traces of mEPSCs before (black) and during (grey) the application of McN-A-343 along with the overlay of the two traces. C and D, cumulative probability plots of mEPSC frequency (C) and amplitude (D) obtained by pooling data from 7 neurons before (continuous lines) and after (dashed lines) application of McN-A-343.
M-channels regulate presynaptic glutamate release in CA1 neurons
Cholinergic M1 receptor activation inhibits M-channels (Brown & Adams, 1980; Marrion et al. 1989; Bernheim et al. 1992), so we tested whether M-channels regulate glutamate release in these synapses. Blocking M-channels increased the frequency of sEPSCs. After recording sEPSCs for 10 min, M-channel blocker XE991 (10 μm) was applied for 10 min, and it increased the frequency of sEPSCs by 147.56 ± 18.21% (0.11 ± 0.03 Hz vs. 0.30 ± 0.01 Hz, n = 7, P < 0.01), but did not affect their amplitudes (16.91 ± 0.92 pA vs. 16.65 ± 0.80 pA, n = 7, P = 0.50). Blocking M-channels caused a significant leftward shift in the cumulative distribution of sEPSC inter-event intervals but had no effect on the cumulative distribution of their amplitudes (K-S test, data not shown). These data suggested that M-channels modulate action potential-dependent glutamate release from Schaffer collateral–CA1 pyramidal neuron synapses. To study whether the effect of XE991 on release was action potential independent, its effect on mEPSCs was studied. XE991 (10 μm) increased the frequency of mEPSCs by 82.21 ± 7.36% (0.13 ± 0.02 Hz vs. 0.22 ± 0.03 Hz, n = 8, P < 0.01), but had no effect on their amplitudes (17.70 ± 1.26 pA vs. 18.10 ± 1.27 pA, n = 8, P = 0.30, Fig. 2A). XE991 also caused a significant leftward shift in the cumulative distribution of the mEPSC inter-event intervals (Fig. 2B), but had no effect on the cumulative distribution of their amplitudes (Fig. 2C). The frequency of mEPSCs kept increasing after 10 min of XE991 application. The slices were perfused with ACSF to wash out the drug effect. The mEPSC frequency remained elevated for 50 min of washing and returned to baseline at 90 min (data not shown). The effect of XE991 on mEPSC frequency was confirmed by applying linopirdine (10 μm), which also blocks M-channels. Linopirdine increased the frequency of mEPSCs by 68.22 ± 6.05% (0.12 ± 0.01 Hz vs. 0.21 ± 0.03 Hz, n = 6, P < 0.01) but had no effect on their amplitudes (15.35 ± 1.13 pA vs. 15.78 ± 0.94 pA, n = 6, P = 0.23). It also caused a significant leftward shift in the cumulative distribution of the mEPSC inter-event intervals (Fig. 3A). These results suggested that the inhibition of M-channel current enhances the frequency of mEPSCs. Flupirtine (20 μm), an M-channel opener, decreased mEPSC frequency by 25.50 ± 8.04% compared to baseline (0.41 ± 0.08 Hz vs. 0.27 ± 0.04 Hz, n = 8, P < 0.01). Flupirtine caused a significant rightward shift in the cumulative distribution of the inter-event intervals of mEPSCs (Fig. 3B). These data suggested that M-channels are capable of modulating neurotransmitter release independent from action potentials. To confirm that the effect of M1 agonist was mediated by M-channels, we incubated the slice with XE991 for 25 min. After 5 min baseline recording in XE991-containing medium, McN-A-343 was applied, and its effect was blocked. Overall, McN-A-343 did not change the frequency of mEPSCs (baseline 0.53 ± 0.07 Hz, McN-A-343 0.56 ± 0.08 Hz; n = 7, P = 0.38). In two cells, there was a modest (<10%) increase in frequency but the cumulative frequency plot of inter-event intervals from these cells was not shifted (Fig. 3C). This result suggested blocking M-channels prevented McN-A-343 enhancement of mEPSC frequency.
Figure 2. M-channel blocker XE991 increased mEPSC frequency in CA1 neurons.
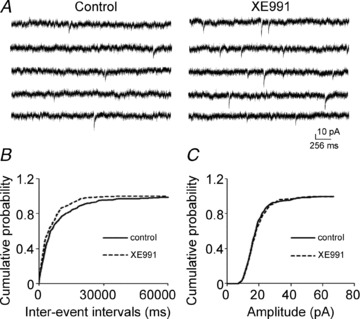
A, representative mEPSC recordings obtained from a CA1 pyramidal neuron before (left) and after (right) the application of 10 μm XE991. B and C, cumulative probability plots of mEPSC frequency (C) and amplitude (D) obtained by pooling data from 8 neurons before (continuous lines) and after (dashed lines) the application of XE991.
Figure 3. The effect of linopirdine and flupirtine, and the occlusion of XE991 to the effect of McN-A-343 on mEPSC in CA1 neurons.
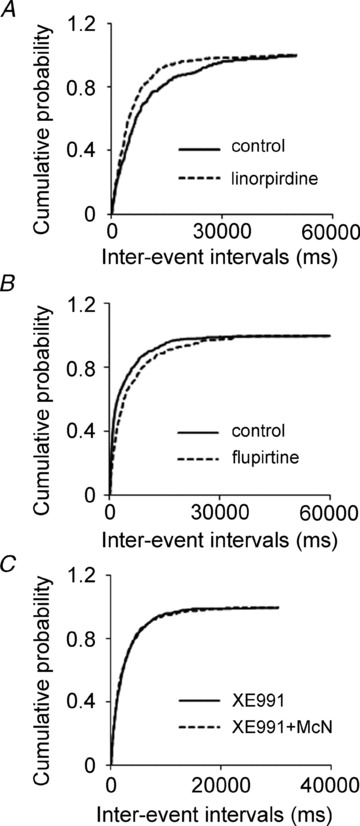
A, and B, cumulative probability plots of mEPSC frequency by pooling data from neurons before (continuous lines) and after (dotted lines) the application of linopirdine (A), flupirtine (B). C, cumulative probability plot of mEPSC frequency by pooling data from neurons before (continuous lines) and after (dashed lines) the application of McN-A-343 after 25 min incubation in XE991.
Calcium influx through voltage-gated calcium channels underlies the effect of XE991 on mEPSC frequency
Calcium plays a critical role in neurotransmitter release. The increase in mEPSC frequency caused by the application of XE991 could be due to Ca2+ release from intracellular stores or the entry of extracellular Ca2+. First, we tested whether release from intracellular stores plays a role in the effect of XE991. Release from intracellular calcium stores was blocked by incubating the slice with 2.5 μm thapsigargin for 20 min. The effect of M-channel blocker XE991 (10 μm) was not affected. XE991 increased the frequency of mEPSCs by 83.88 ± 17.22% (Fig. 4A), which is comparable to the effect of XE991 without thapsigargin incubation (82.21 ± 7.36%, n = 8, P < 0.01); it significantly left shifted the cumulative distribution of inter-event intervals (n = 9, P < 0.01, Fig. 4B). This result suggested that intracellular store release does not contribute to the enhancement of M-channel inhibition on mEPSCs frequency.
Figure 4. The effect of XE991 on mEPSC frequency was eliminated in calcium-free ACSF.
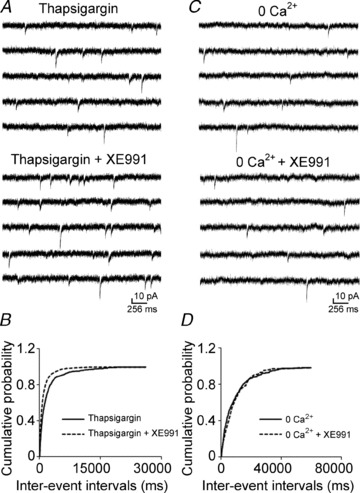
A and C, representative mEPSC recordings obtained from a CA1 pyramidal neuron before (top) and after (bottom) the application of 10 μm XE991 after 20 min incubation in thapsigargin (2.5 μm) (A) or in calcium-free ACSF (C). B and D, cumulative probability plots of the frequency of mEPSCs obtained by pooling data from 8 neurons before (continuous lines) and after (dashed lines) the application of XE991 in thapsigargin incubation (B) or in calcium-free ACSF (D).
Therefore, we tested whether the effect of XE991 on the frequency of mEPSCs was dependent on extracellular Ca2+ by recording mEPSCs in a calcium-free medium. The effect of XE991 on mEPSCs was eliminated in ACSF that lacked calcium. Application of XE991 neither changed the frequency of mEPSCs (0.13 ± 0.02 Hz vs. 0.12 ± 0.01 Hz, n = 8, P = 0.30, Fig. 4C) nor shifted the cumulative distribution of the inter-event intervals of mEPSCs (Fig. 4D). These data suggested that the effect of XE991 on spontaneous glutamate release was dependent on the influx of Ca2+.
We also studied the effect of McN-A-343 in a calcium-free medium. The McN-A-343 enhancement of mEPSCs was blocked in calcium-free medium (baseline, 0.40 ± 0.08 Hz, McN-A-343, 0.41 ± 0.07 Hz, n = 7, P = 0.36). There was no significant shift on the cumulative distribution of inter-event intervals of mEPSCs (data not shown). This suggested that the effect of M1 receptor activation on the frequency of mEPSC is largely mediated by increasing calcium influx from extracellular space. To test whether Ca2+ enters Schaffer collateral terminals by activation of presynaptic voltage-gated calcium channels due to M-channel inhibition, we studied the effect of P/Q- and N-type calcium channel blockers on the XE991-mediated enhancement of mEPSC frequency. These two channels are expressed in the presynaptic terminals of Schaffer collaterals (Wheeler et al. 1994; Qian & Noebels, 2000). Slices were incubated with either ω-agatoxin TK (200 nm) or ω-conotoxin GVIA (1 μm) (Peptides International, Louisville, KY, USA) for 15 min prior to data collection. In the presence of ω-agatoxin TK, which blocks P/Q-type calcium channels, the XE991-mediated enhancement of mEPSC frequency was prevented in approximately half of the cells that were studied. In 5 out of 9 neurons, the application of XE991 did not increase the frequency of mEPSCs (0.23 ± 0.08 Hz vs. 0.20 ± 0.06 Hz, P = 0.46, Fig. 5A and C). In the other four CA1 pyramidal neurons, XE991 still significantly increased the frequency of mEPSCs (0.16 ± 0.03 Hz vs. 0.31 ± 0.07 Hz, P < 0.05, Fig. 5B and D). Blocking N-type channels with ω-conotoxin GVIA also prevented the effect of XE991 on the frequency of mEPSCs in half of the CA1 pyramidal neurons from which recordings were made. In 4 of 8 neurons, the effect of XE991 was eliminated (0.33 ± 0.05 Hz vs. 0.34 ± 0.04 Hz, P = 0.47, Fig. 6A and C). In the other four neurons, XE991 significantly increased the frequency of mEPSCs (0.24 ± 0.04 Hz vs. 0.43 ± 0.07 Hz, P < 0.05, Fig. 6B and D).
Figure 5. P/Q-type calcium channel blocker on the effect of XE991 on mEPSC frequency.
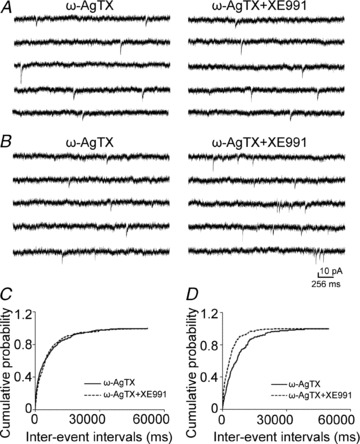
A and B, representative mEPSC recordings obtained from a CA1 pyramidal neuron in which the blocking of P/Q-type of calcium channels by 200 nmω-agatoxin TK did (A) or did not (B) prevent the effect of 10 μm XE991. The left panels include data from recordings in the presence of ω-agatoxin TK before the application of XE991, and the right panels include data from recordings after the application of XE991. C and D, cumulative probability plots of mEPSC frequency obtained by pooling data from CA1 pyramidal neurons before (continuous lines) and after (dashed lines) the application of XE991 in the presence of ω-agatoxin TK that did (C) or did not (D) prevent the effect of XE991.
Figure 6. N-type calcium channel blocker on the effect of XE991 on mEPSC frequency.
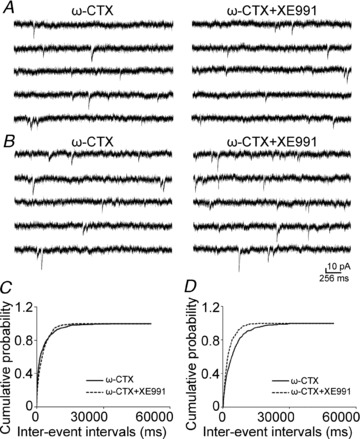
A and B, representative mEPSC recordings obtained from a CA1 pyramidal neuron in which the blocking of N-type calcium channels by ω-conotoxin GVIA (1 μm) did (A) or did not (B) prevent the effects of 10 μm XE991. Data in the left panels are from recordings in the presence of ω-conotoxin GVIA before the application of XE991, and data in the right panels are from recordings after the application of XE991. C and D, cumulative probability plots of mEPSC frequency obtained by pooling data from CA1 pyramidal neurons before (continuous lines) and after (dashed lines) the application of XE991 in the presence of ω-conotoxin GVIA that did (C) or did not (D) prevent the effect of XE991.
Next, we applied the blockers of two channels simultaneously. Slices were incubated in ω-agatoxin TK and ω-conotoxin GVIA for 25–40 min, and after 5 min baseline recording, XE991 was applied. The mean frequency was not significantly increased (baseline, 0.78 ± 0.08 Hz, XE991, 0.82 ± 0.09 Hz; n = 8, P = 0.08, Fig. 7A), and there was no significant shift of cumulative distribution of inter-event intervals for each cell and pooled data (Fig. 7B). This result suggested the enhancement of the frequency of mEPSCs by XE991 was mediated by calcium influx though P/Q- and N-type calcium channels.
Figure 7. Combination of P/Q- and N-type calcium channel blockers on the effect of XE991 on mEPSC frequency.
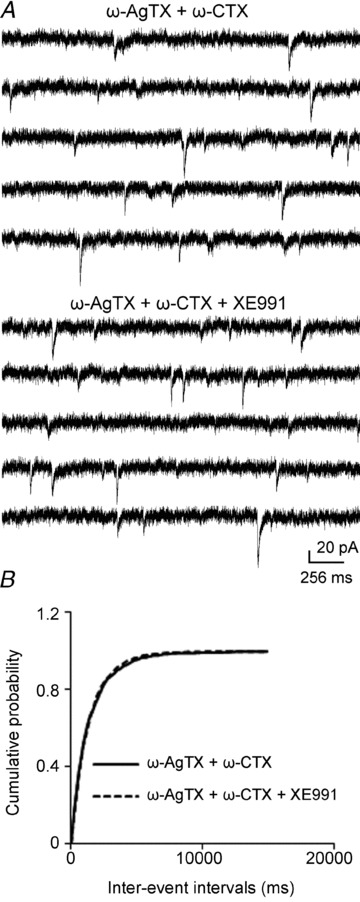
A, representative mEPSC recordings obtained from a CA1 pyramidal neuron in the presence of both ω-agatoxin TK and ω-conotoxin GVIA before the application of XE991 (top), and after the application of XE991 (bottom). B, cumulative probability plots of mEPSC frequency obtained by pooling data from CA1 pyramidal neurons before (continuous lines) and after (dashed lines) the application of XE991 in the presence of ω-agatoxin TK and ω-conotoxin GVIA.
McN-A-343 and XE991 depolarize and increase action potential firing in CA3 neurons
The aforementioned data suggested that inhibition of M-channels resulted in the depolarization of presynaptic terminals of the CA3 pyramidal neuron, which then activated P/Q- or N-type calcium channels, thereby causing calcium influx and ultimately leading to increased glutamate release. We tested the effect of M-channels on the membrane properties of CA3 pyramidal neurons. To study the effects of McN-A-343 and XE991 on CA3 pyramidal neurons, current-clamp recordings of the membrane potentials were obtained from neurons in slices from juvenile rats (24–28 days old). McN-A-343 significantly depolarized CA3 neurons and increased the frequency of action potential firing (0.17 ± 0.08 Hz vs. 2.16 ± 0.72 Hz, n = 6, P < 0.01) of CA3 pyramidal neurons (Fig. 8A and C). To measure the membrane potential more accurately, action potentials were blocked by TTX (1 μm). Application of McN-A-343 depolarized CA3 pyramidal neuron membrane potentials from −61.39 ± 1.49 mV to −49.00 ± 1.78 mV (n = 7, P < 0.01, Fig. 8B and D).
Figure 8. M1 agonist McN-A-343 depolarized and increased action potential firing in CA3 pyramidal neurons.
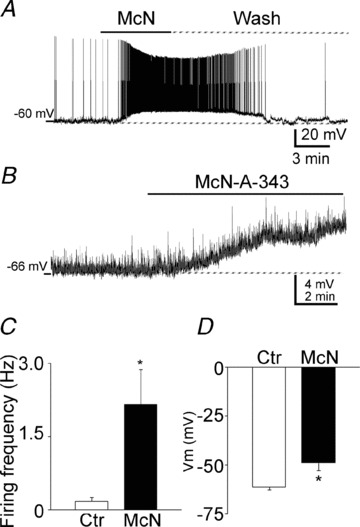
A, representative traces of the membrane potential of a CA3 pyramidal neuron before, during, and after a wash of McN-A-343. Note the profound depolarization and the repetitive action potentials. B, a representative trace of the depolarizing effect of McN-A-343 on the membrane potential of a CA3 pyramidal neuron with action potentials blocked by TTX (1 μm). C and D, quantification of the effect of McN-A-343 on the firing frequency (C) and membrane potential (D) of CA3 pyramidal neurons (means ± SEM).
XE991 had similar effect to that of McN-A-343. It increased the action potential firing frequency (0.42 ± 0.17 Hz vs. 0.79 ± 0.20 Hz, n = 9, P < 0.01, Fig. 9A and D) and depolarized CA3 pyramidal neurons (−61.32 ± 2.45 mV vs. −52.62 ± 2.43 mV, n = 10, P < 0.05, Fig. 9B and E). Furthermore, and consistent with these findings, the M-channel opener flupirtine (20 μm) hyperpolarized CA3 neurons (−58.02 ± 1.61 mV, vs. −62.97 ± 1.36 mV, n = 7, P < 0.01) (Fig. 9C and F).
Figure 9. M-channel blocker and opener on membrane potential and action potential firing in CA3 pyramidal neurons.
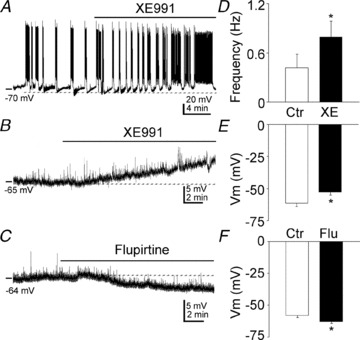
A, representative effect of XE991 on the membrane potential of CA3 pyramidal neurons. B and C, representative recordings of the effects of XE991 (B) and flupirtine (C) on the membrane potential of CA3 pyramidal neurons in the presence of TTX (1 μm). D, quantification of the effect of XE991 on the firing frequency of CA3 pyramidal neurons (means ± SEM). E and F, quantification of the effect of XE991 (E) and flupirtine (F) on the membrane potential of CA3 pyramidal neurons (means ± SEM).
To test whether the inhibition of M-channels increases membrane input resistance in CA3 pyramidal neurons, current–voltage (I–V) relationships were studied. Current was injected from −40 pA to15 pA in 5 pA steps. Exposure to McN-A-343 shifted the I–V curve to a more negative membrane potentials with negative current injections. The slope of the I–V curve increased from 140 MΩ to 197 MΩ (Fig. 10A, n = 6). McN-A-343 increased the input resistance from 156.43 ± 17.51 MΩ to 228.07 ± 17.16 MΩ (n = 6, P < 0.01) analysed by Clampfit. Exposure to XE991 had an effect on membrane input resistance that was similar to that of exposure to McN-A-343 in CA3 pyramidal neurons. The slope of the I–V curve increased from 122 MΩ to 175 MΩ (Fig. 10B, n = 7). XE991 increased the input resistance from 129.75 ± 8.63 MΩ to 194.53 ± 17.92 MΩ (n = 7, P < 0.01) analysed by Clampfit. These results suggested that the M1 agonist McN-A-343 and the M-channel blocker XE991 close M-type potassium channels in CA3 pyramidal neurons.
Figure 10. McN-A-343 and XE991 increased input resistance in CA3 neurons.
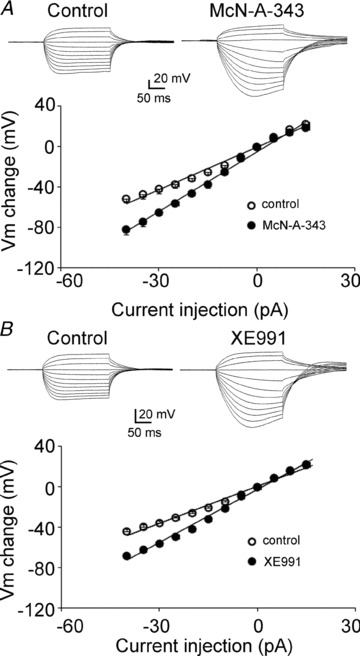
A, top, representative recordings of current injections from −40 pA (in 5 pA steps) before (left) and after (right) the application McN-A-343. Bottom, I–V relationship before (open circles) and after (filled circle) the application of McN-A-343 in CA3 neurons (n = 6). B, top, representative recordings of current injections from −40 pA (in 5 pA steps) before (left) and after (right) the application of XE991. Bottom, I–V relationship before (open circles) and after (filled circles) the application of XE991 in CA3 neurons (n = 7).
Discussion
The present study found that M1 muscarinic activation potentiated glutamate release and that inhibiting M-channels had a similar effect. The application of M-channel blockers increased the frequency of sEPSCs and mEPSCs but had no effect on their amplitudes; the application of an M1 muscarinic receptor agonist had a similar effect to that of the M-channel blockers. The effect of XE991 was dependent on Ca2+ influx through P/Q- and N-type calcium channels. Both the blocking of M-channels and the activation of M1 receptors depolarized CA3 pyramidal neurons. These results suggest that M1 receptor activation inhibits M-type potassium channels and depolarizes CA3 neurons. This causes an influx of Ca2+ through presynaptic voltage-dependent calcium channels and increase spontaneous glutamate release to CA1 neurons. This study demonstrates that M-channels modulate action-potential-independent neurotransmitter release from presynaptic terminals in Schaffer collateral–CA1 pyramidal neuron synapses. These actions occur in addition to the effects on cell soma and axon initial segment, resulting from increased membrane resistance.
M1 receptor agonists and M-channel blockers increase presynaptic glutamate release
In the present study, McN-A-343, a selective agonist for M1receptors, significantly increased the frequency of mEPSCs but had no effect on their amplitudes, which suggests that M1 receptor activation increases presynaptic action potential-independent glutamate release from the Schaffer collaterals. These results are consistent with results from other studies about muscarinic potentiation of glutamatergic transmission in the hippocampus. M1 is the predominant muscarinic receptor in the hippocampus (accounting for approximately 60% of all muscarinic receptors in the hippocampus), whereas M2 and M4 are less abundant (approximately 20%) (Volpicelli & Levey, 2004). The muscarinic agonist carbachol has been associated with increases in presynaptic glutamate release in cultured rat hippocampal neurons and dentate granule cells in slices (Bouron & Reuter, 1997; Kozhemyakin et al. 2010). An M1 agonist has been shown to cause the potentiation of N-methyl-d-aspartate (NMDA) receptor currents in CA1 hippocampal pyramidal neurons (Marino et al. 1998). The activation of muscarinic receptors induces a long-lasting synaptic enhancement at Schaffer collaterals both in vivo and in vitro via an increase in the postsynaptic release of calcium stored in the endoplasmic reticulum (Fernández de Sevilla et al. 2008). A recent study that combined calcium imaging with two-photon laser glutamate uncaging suggests that M1 receptor activation increases glutamate synaptic potentials and Ca2+ transients in CA1 pyramidal neurons (Giessel & Sabatini, 2010). Muscarinic M1 (or at some sites M3 or possibly M5) receptor activation inhibits the M-channel (Brown & Passmore, 2009), which was first named in the 1980s (Brown & Adams, 1980; Constanti & Brown, 1981). Immunochemical studies suggest that M-channels are highly expressed in the axon initial segments of neurons (Chung et al. 2006; Pan et al. 2006; Rasmussen et al. 2007). Thus, M-channels are very important for tuning the firing rates and excitability of various neurons (Yue & Yaari, 2004; Shen et al. 2005; Gu et al. 2005; Shah et al. 2008). Interestingly, M-channel activity also affects the release of neurotransmitters from superior cervical ganglion (SCG) neurons and hippocampal synaptosomes (Martire et al. 2004; Hernandez et al. 2008). Our findings in the current study suggest that M-channels modulate action potential-independent neurotransmitter release from the presynaptic terminals of the Schaffer collaterals. M-channels are active at the resting membrane potential (Constanti & Brown, 1981) and are expressed in presynaptic terminals (Cooper et al. 2001; Chung et al. 2006; Garcia-Pino et al. 2010). The presynaptic potentiation effect of the M-channel blocker XE991 was reversed over a long period of time. This might be due to its binding kinetics. The dissociation of linopirdine, an analogue of XE991, is slow and begins after 60 min (Wolff et al. 2005).
M1 receptor agonist and M-channel blocker depolarize CA3 neurons
An M1 agonist and an M-channel blocker depolarized CA3 pyramidal neurons. This finding is consistent with the location and function of the receptor in these neurons. The M1 receptor is the most commonly expressed muscarinic receptor in the hippocampus in both the pyramidal neuron layer and the stratum radiatum of the CA3 and CA1 regions (Tayebati et al. 2002). A comparable slow, muscarinic receptor-dependent depolarization can be evoked in pyramidal neurons by the direct electrical stimulation of cholinergic afferents in the hippocampus (Cole & Nicoll, 1983; Madison et al. 1987; Segal, 1988; Pitler & Alger, 1990; Morton & Davies, 1997) or medial septal nucleus in a septo-hippocampal slice (Cobb & Davies, 2005). The medial septal nucleus provides the major source of cholinergic innervations to the hippocampus (Dutar et al. 1995) and presents a direct synaptic input to both principal neurons and interneurons (Frotscher & Léránth, 1985; Léránth & Frotscher, 1987). Muscarinic acetylcholine receptors modulate several ionic conductances in addition to the M-current, including IAHP (the Ca2+-activated K+ current that is responsible for slowing action potential discharges) and Ileak (the background leak current) (Halliwell, 1990). Activation of mAChRs also potentiates two mixed cation currents (Ih, the hyperpolarization-activated cation current; and Icat, the Ca2+-dependent non-specific cation current) (Halliwell, 1990; Colino & Halliwell, 1993) and the N-methyl-d-aspartate (NMDA) receptor (Markram & Segal, 1990). This may explain why, in our study, the M1 agonist McN-A-343 had a stronger depolarization effect than the M-channel blocker XE991 in CA3 pyramidal neurons.
Muscarinic receptor and intracellular calcium level
Current study suggested that M1 receptor activation depolarizes presynaptic membrane and activates voltage-gated calcium channel to increase intracellular calcium level and neurotransmitter release by inhibition of M-channels. Previous studies suggested that muscarinic stimulation inhibits voltage-gated calcium channels (Toselli et al. 1989; Qian & Saggau, 1997). However these studies did not classify the subtype of muscarnic receptor that mediates this effect. It was reported that the inhibitory effect of muscarinic receptor activation on calcium channels was mediated by M4 subtype in rat sympathetic neurons (Bernheim et al. 1992) and by M2 subtype in rat magocellular cholinergic basal forebrain neurons (Allen & Brown, 1993). There is no report to suggest that M1 subtype muscarinic receptor inhibits calcium channels.
Muscarinic receptors activate inositol 1,4,5-trisphosphate (IP3) receptor to trigger calcium release from endoplasmic reticulum (ER) stores in CA1 pyramidal neurons, and appears to be action potential independent (Power & Sah, 2002). Another study suggested this effect is mediated by M1 receptor (Fernández de Sevilla et al. 2008). The role of these stores in enhancing synaptic release remains uncertain. Although spontaneous transmitter release (in the absence of action potentials) is largely mediated by calcium release from internal stores (Emptage et al. 2001), our study suggests the modulation of spontaneous transmitter release could be mediated by calcium influx though voltage-gated calcium channels.
Overall, this study demonstrated that M1 muscarinic receptor activation inhibits M-type potassium channels, thereby increasing excitatory glutamate neurotransmitter release from the presynaptic terminals of CA3 neurons by increasing the rate of calcium influx through voltage-dependent calcium channels. This effect of M1 receptor activation could contribute to the generation of seizures.
Acknowledgments
This study was supported by peer reviewed Medical Research Program of the Department of Defense PR 093963 and NINDS (NIH) RO1 NS 40337.
Glossary
- M1
muscarinic type 1
- mEPSCs
miniature excitatory postsynaptic currents
- CNQX
6-cyano-7-nitroquinoxalene-2,3-dione
- K-S
Kolmogorov–Smirnov
- NcN-A-343
(4-hydroxy-2-butynyl)-1-trimethylammonium-3-chlorocarbanilate chloride
- sEPSC
spontaneous excitatory postsynaptic current; XE-991,10,10-bis(4-pyridi-nylmethyl)-9(10H)-anthracenone
Author contributions
J.S. and J.K. designed the study. J.S. performed the experiments and analysed the data. J.S. and J.K. interpreted the data, drafted and revised the article.
References
- Allen TG, Brown DA. M2 muscarinic receptor-mediated inhibition of the Ca2+ current in rat magnocellular cholinergic basal forebrain neurones. J Physiol. 1993;466:173–189. [PMC free article] [PubMed] [Google Scholar]
- Bernheim L, Mathie A, Hille B. Characterization of muscarinic receptor subtypes inhibiting Ca2+ current and M-current in rat sympathetic neurons. Proc Natl Acad Sci U S A. 1992;89:9544–9548. doi: 10.1073/pnas.89.20.9544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biervert C, Schroeder BC, Kubisch C, Berkovic SF, Propping P, Jentsch TJ, Steinlein OK. A potassium channel mutation in neonatal human epilepsy. Science. 1998;279:403–406. doi: 10.1126/science.279.5349.403. [DOI] [PubMed] [Google Scholar]
- Bouron A, Reuter H. Muscarinic stimulation of synaptic activity by protein kinase C is inhibited by adenosine in cultured hippocampal neurons. Proc Natl Acad Sci U S A. 1997;94:12224–12229. doi: 10.1073/pnas.94.22.12224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA, Adams PR. Muscarinic suppression of a novel voltage-sensitive K+ current in a vertebrate neurone. Nature. 1980;283:673–676. doi: 10.1038/283673a0. [DOI] [PubMed] [Google Scholar]
- Brown DA, Passmore GM. Neural KCNQ (Kv7) channels. Br J Pharmacol. 2009;156:1185–1195. doi: 10.1111/j.1476-5381.2009.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JT, Randall AD. Activity-dependent depression of the spike after-depolarization generates long-lasting intrinsic plasticity in hippocampal CA3 pyramidal neurons. The Journal of Physiology. 2009;587:1265–1281. doi: 10.1113/jphysiol.2008.167007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HJ, Jan YN, Jan LY. Polarized axonal surface expression of neuronal KCNQ channels is mediated by multiple signals in the KCNQ2 and KCNQ3 C-terminal domains. Proc Natl Acad Sci U S A. 2006;103:8870–8875. doi: 10.1073/pnas.0603376103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb SR, Davies CH. Cholinergic modulation of hippocampal cells and circuits. J Physiol. 2005;562:81–88. doi: 10.1113/jphysiol.2004.076539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole AE, Nicoll RA. Acetylcholine mediates a slow synaptic potential in hippocampal pyramidal cells. Science. 1983;221:1299–1301. doi: 10.1126/science.6612345. [DOI] [PubMed] [Google Scholar]
- Colino A, Halliwell JV. Carbachol potentiates Q current and activates a calcium-dependent non-specific conductance in rat hippocampus in vitro. Eur J Neurosci. 1993;5:1198–1209. doi: 10.1111/j.1460-9568.1993.tb00974.x. [DOI] [PubMed] [Google Scholar]
- Constanti A, Brown DA. M-currents in voltage-clamped mammalian sympathetic neurones. Neurosci Lett. 1981;24:289–294. doi: 10.1016/0304-3940(81)90173-7. [DOI] [PubMed] [Google Scholar]
- Cooper EC, Harrington E, Jan YN, Jan LY. M-channel KCNQ2 subunits are localized to key sites for control of neuronal network oscillations and synchronization in mouse brain. J Neurosci. 2001;21:9529–9540. doi: 10.1523/JNEUROSCI.21-24-09529.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005;6:850–862. doi: 10.1038/nrn1785. [DOI] [PubMed] [Google Scholar]
- Devaux JJ, Kleopa KA, Cooper EC, Scherer SS. KCNQ2 is a nodal K+ channel. J Neurosci. 2004;24:1236–1244. doi: 10.1523/JNEUROSCI.4512-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutar P, Bassant MH, Senut MC, Lamour Y. The septohippocampal pathway: structure and function of a central cholinergic system. Physiol Rev. 1995;75:393–427. doi: 10.1152/physrev.1995.75.2.393. [DOI] [PubMed] [Google Scholar]
- Emptage NJ, Reid CA, Fine A. Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron. 2001;29:197–208. doi: 10.1016/s0896-6273(01)00190-8. [DOI] [PubMed] [Google Scholar]
- Fernandez de Sevilla D, Núnñez A, Borde M, Malinow R, Buño W. Cholinergic-mediated IP3-receptor activation induces long-lasting synaptic enhancement in CA1 pyramidal neurons. J Neurosci. 2008;28:1469–1478. doi: 10.1523/JNEUROSCI.2723-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frotscher M, Léránth C. Cholinergic innervation of the rat hippocampus as revealed by choline acetyltransferase immunocytochemistry: a combined light and electron microscopic study. J Comp Neurol. 1985;239:237–246. doi: 10.1002/cne.902390210. [DOI] [PubMed] [Google Scholar]
- Garcia-Pino E, Caminos E, Juiz JM. KCNQ5 reaches synaptic endings in the auditory brainstem at hearing onset and targeting maintenance is activity-dependent. J Comp Neurol. 2010;518:1301–1314. doi: 10.1002/cne.22276. [DOI] [PubMed] [Google Scholar]
- Geiger J, Weber YG, Landwehrmeyer B, Sommer C, Lerche H. Immunohistochemical analysis of KCNQ3 potassium channels in mouse brain. Neurosci Lett. 2006;400:101–104. doi: 10.1016/j.neulet.2006.02.017. [DOI] [PubMed] [Google Scholar]
- Giessel AJ, Sabatini BL. M1 muscarinic receptors boost synaptic potentials and calcium influx in dendritic spines by inhibiting postsynaptic SK channels. Neuron. 2010;68:936–947. doi: 10.1016/j.neuron.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu N, Vervaeke K, Hu H, Storm JF. Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J Physiol. 2005;566:689–715. doi: 10.1113/jphysiol.2005.086835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliwell JV( Physiological mechanisms of cholinergic action in the hippocampus. Prog Brain Res. 1990;84:255–272. doi: 10.1016/s0079-6123(08)60910-3. [DOI] [PubMed] [Google Scholar]
- Hernandez CC, Zaika O, Tolstykh GP, Shapiro MS. Regulation of neural KCNQ channels: signalling pathways, structural motifs and functional implications. J Physiol. 2008;586:1811–1821. doi: 10.1113/jphysiol.2007.148304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higley MJ, Soler-Llavina GJ, Sabatini BL. Cholinergic modulation of multivesicular release regulates striatal synaptic potency and integration. Nat Neurosci. 2009;12:1121–1128. doi: 10.1038/nn.2368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch TJ. Neuronal KCNQ potassium channels:physislogy and role in disease. Nat Rev Neurosci. 2000;1:21–30. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- Kanaumi T, Takashima S, Iwasaki H, Itoh M, Mitsudome A, Hirose S. Developmental changes in KCNQ2 and KCNQ3 expression in human brain: possible contribution to the age-dependent etiology of benign familial neonatal convulsions. Brain Dev. 2008;30:362–369. doi: 10.1016/j.braindev.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Kozhemyakin M, Rajasekaran K, Kapur J. Central cholinesterase inhibition enhances glutamatergic synaptic transmission. J Neurophysiol. 2010;103:1748–1757. doi: 10.1152/jn.00949.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead CJ, Watson J, Reavill C. Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol Ther. 2008;117:232–243. doi: 10.1016/j.pharmthera.2007.09.009. [DOI] [PubMed] [Google Scholar]
- Léránth C, Frotscher M. Cholinergic innervation of hippocampal GAD- and somatostatin-immunoreactive commissural neurons. J Comp Neurol. 1987;261:33–47. doi: 10.1002/cne.902610104. [DOI] [PubMed] [Google Scholar]
- Madison DV, Lancaster B, Nicoll RA. Voltage clamp analysis of cholinergic action in the hippocampus. J Neurosci. 1987;7:733–741. doi: 10.1523/JNEUROSCI.07-03-00733.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino MJ, Rouse ST, Levey AI, Potter LT, Conn PJ. Activation of the genetically defined M1 muscarinic receptor potentiates N-methyl-D-aspartate (NMDA) receptor currents in hippocampal pyramidal cells. Proc Natl Acad Sci U S A. 1998;95:11465–11470. doi: 10.1073/pnas.95.19.11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Segal M. Acetylcholine potentiates responses to N-methyl-D-aspartate in the rat hippocampus. Neurosci Lett. 1990;113:62–65. doi: 10.1016/0304-3940(90)90495-u. [DOI] [PubMed] [Google Scholar]
- Marrion NV, Smart TG, Marsh SJ, Brown DA. Muscarinic suppression of the M-current in the rat sympathetic-ganglion is mediated by receptors of the M1-subtype. Br J Pharmacol. 1989;98:557–573. doi: 10.1111/j.1476-5381.1989.tb12630.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martire M, Castaldo P, D’Amico M, Preziosi P, Annunziato L, Taglialatela M. M-channels containing KCNQ2 subunits modulate norepinephrine, aspartate, and GABA release from hippocampal nerve terminals. J Neurosci. 2004;24:592–597. doi: 10.1523/JNEUROSCI.3143-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton RA, Davies CH. Regulation of muscarinic acetylcholine receptor-mediated synaptic responses by adenosine receptors in the rat hippocampus. J Physiol. 1997;502:75–90. doi: 10.1111/j.1469-7793.1997.075bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Z, Kao T, Horvath Z, Lemos J, Sul JY, Cranstoun SD, Bennett V, Scherer SS, Cooper EC. A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J Neurosci. 2006;26:2599–2613. doi: 10.1523/JNEUROSCI.4314-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peretz A, Sheinin A, Yue C, Degani-Katzav N, Gibor G, Nachman R, Gopin A, Tam E, Shabat D, Yaari Y, Attali B. Pre- and postsynaptic activation of M-channels by a novel opener dampens neuronal firing and transmitter release. J Neurophysiol. 2007;97:283–295. doi: 10.1152/jn.00634.2006. [DOI] [PubMed] [Google Scholar]
- Pitler TA, Alger BE. Activation of the pharmacologically defined M3 muscarinic receptor depolarizes hippocampal pyramidal cells. Brain Res. 1990;534:257–262. doi: 10.1016/0006-8993(90)90137-z. [DOI] [PubMed] [Google Scholar]
- Power JM, Sah P. Nuclear calcium signaling evoked by cholinergic stimulation in hippocampal CA1 pyramidal neurons. J Neurosci. 2002;22:3454–3462. doi: 10.1523/JNEUROSCI.22-09-03454.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Noebels JL. Presynaptic Ca2+ influx at a mouse central synapse with Ca2+ channel subunit mutations. J Neurosci. 2000;20:163–170. doi: 10.1523/JNEUROSCI.20-01-00163.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Saggau P. Presynaptic inhibition of synaptic transmission in the rat hippocampus by activation of muscarinic receptors: involvement of presynaptic calcium influx. Br J Pharmacol. 1997;122:511–519. doi: 10.1038/sj.bjp.0701400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen HB, Frøkjær-Jensen C, Jensen CS, Jensen HS, Jørgensen NK, Misonou H, Trimmer JS, Olesen SP, Schmitt N. Requirement of subunit co-assembly and ankyrin-G for M-channel localization at the axon initial segment. J Cell Sci. 2007;120:953–963. doi: 10.1242/jcs.03396. [DOI] [PubMed] [Google Scholar]
- Safiulina VF, Zacchi P, Taglialatela M, Yaari Y, Cherubini E. Low expression of Kv7/M channels facilitates intrinsic and network bursting in the developing rat hippocampus. J Physiol. 2008;586:5437–5453. doi: 10.1113/jphysiol.2008.156257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal M( Synaptic activation of a cholinergic receptor in rat hippocampus. Brain Res. 1988;452:79–86. doi: 10.1016/0006-8993(88)90011-x. [DOI] [PubMed] [Google Scholar]
- Selyanko AA, Delmas P, Hadley JK, Tatulian L, Wood IC, Mistry M, London B, Brown DA. Dominant-negative subunits reveal potassium channel families that contribute to M-like potassium currents. J Neurosci. 2002;22:RC212. doi: 10.1523/JNEUROSCI.22-05-j0001.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Migliore M, Valencia I, Cooper EC, Brown DA. Functional significance of axonal Kv7 channels in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 2008;105:7869–7874. doi: 10.1073/pnas.0802805105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Mistry M, Marsh SJ, Brown DA, Delmas P. Molecular correlates of the M-current in cultured rat hippocampal neurons. J Physiol. 2002;544:29–37. doi: 10.1113/jphysiol.2002.028571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Hamilton SE, Nathanson NM, Surmeier DJ. Cholinergic suppression of KCNQ channel currents enhances excitability of striatal medium spiny neurons. J Neurosci. 2005;25:7449–7458. doi: 10.1523/JNEUROSCI.1381-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim JA, Griffith WH. Muscarinic Inhibition of glutamatergic transmission onto rat magnocellular basal forebrain neurons in a thin-slice preparation. Eur J Neurosci. 1996;8:880–891. doi: 10.1111/j.1460-9568.1996.tb01575.x. [DOI] [PubMed] [Google Scholar]
- Tayebati SK, Amenta F, El-Assouad D, Zaccheo D. Muscarinic cholinergic receptor subtypes in the hippocampus of aged rats. Mech of Ageing Dev. 2002;123:521–528. doi: 10.1016/s0047-6374(01)00353-0. [DOI] [PubMed] [Google Scholar]
- Toselli M, Lang J, Costa T, Lux HD. Direct modulation of voltage-dependent calcium channels by muscarinic activation of a pertussis toxin-sensitive G-protein in hippocampal neurons. Pflugers Arch. 1989;415:255–261. doi: 10.1007/BF00370874. [DOI] [PubMed] [Google Scholar]
- Vervaeke K, Gu N, Agdestein C, Hu H, Storm JF. Kv7/KCNQ/M-channels in rat glutamatergic hippocampal axons and their role in regulation of excitability and transmitter release. J Physiol. 2006;576:235–256. doi: 10.1113/jphysiol.2006.111336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt KE, Regehr WG. Cholinergic modulation of excitatory synaptic transmission in the CA3 area of the hippocampus. J Neurosci. 2001;21:75–83. doi: 10.1523/JNEUROSCI.21-01-00075.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpicelli LA, Levey AI. Muscarinic acetylcholine receptor subtypes in cerebral cortex and hippocampus. Prog Brain Res. 2004;145:59–66. doi: 10.1016/S0079-6123(03)45003-6. [DOI] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, McKinnon D. KCNQ2 and KCNQ3 potassium channel subunits: molecular correlates of the M-channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- Weber YG, Geiger J, Kämpchen K, Landwehrmeyer B, Sommer C, Lerche H. Immunohistochemical analysis of KCNQ2 potassium channels in adult and developing mouse brain. Brain Res. 2006;1077:1–6. doi: 10.1016/j.brainres.2006.01.023. [DOI] [PubMed] [Google Scholar]
- Wheeler DB, Randall A, Tsien RW. Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science. 1994;264:107–111. doi: 10.1126/science.7832825. [DOI] [PubMed] [Google Scholar]
- Wolff C, Gillard M, Fuks B, Chatelain P. [3H]Linopirdine binding to rat brain membranes is not relevant for M-channel interaction. Eur J Pharmacol. 2005;518:10–17. doi: 10.1016/j.ejphar.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Yajeya J, De La Fuente A, Criado JM, Bajo V, Sánchez-Riolobos A, Heredia M. Muscarinic agonist carbachol depresses excitatory synaptic transmission in the rat basolateral amygdala in vitro. Synapse. 2000;38:151–160. doi: 10.1002/1098-2396(200011)38:2<151::AID-SYN6>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Yue C, Yaari Y. KCNQ/M Channels control spike afterdepolarization and burst generation in hippocampal neurons. J Neurosci. 2004;24:4614–4624. doi: 10.1523/JNEUROSCI.0765-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang HM, Chen SR, Pan HL. Regulation of glutamate release from primary afferents and interneurons in the spinal cord by muscarinic receptor subtypes. J Neurophysiol. 2007;97:102–109. doi: 10.1152/jn.00586.2006. [DOI] [PubMed] [Google Scholar]