

Abstract
This study investigated the function of each of the hypoxia inducible factor (HIF) prolyl-4-hydroxylase enzymes (PHD1–3) in the first 24 h following transient focal cerebral ischaemia by using mice with each isoform genetically suppressed. Male, 8- to 12-week old PHD1−/−, PHD2+/− and PHD3−/− mice and their wild-type (WT) littermate were subjected to 45 min of middle cerebral artery occlusion (MCAO). During the experiments, regional cerebral blood flow (rCBF) was recorded by laser Doppler flowmetry. Behaviour was assessed at both 2 h and 24 h after reperfusion with a common neuroscore. Infarct volumes, blood–brain barrier (BBB) disruption, cerebral vascular density, apoptosis, reactive oxygen species (ROS), HIF1α, and glycogen levels were then determined using histological and immunohistochemical techniques. When compared to their WT littermates, PHD2+/− mice had significantly increased cerebral microvascular density and more effective restoration of CBF upon reperfusion. PHD2+/− mice showed significantly better functional outcomes and higher activity rates at both 2 h and 24 h after MCAO, associated with significant fewer apoptotic cells in the penumbra and less BBB disruption; PHD3−/− mice had impaired rCBF upon early reperfusion but comparable functional outcomes; PHD1−/− mice did not show any significant changes following the MCAO. Production of ROS, HIF1α staining and glycogen content in the brain were not different in any comparison. Life-long genetic inhibition of PHD enzymes produces different effects on outcome in the first 24 h after transient cerebral ischaemia. These need to be considered in optimizing therapeutic effects of PHD inhibitors, particularly when isoform specific inhibitors become available.
Key points
Cerebral ischaemia results in the activation of multiple pathways that can independently lead to neuronal death. Agents targeting a number of processes at one time are likely to be translated into stroke therapy.
Hypoxia-inducible factor (HIF) is a transcription complex which responds to changes in oxygen. HIF levels are tightly regulated by a group of prolyl hydroxylases (PHDs).
In this study, we investigated the function of each of the HIF-PHDs in the first 24 hours following transient focal cerebral ischaemia by using mice with each isoform genetically suppressed.
We found that the PHD1−/−, PHD2+/−, PHD3−/− mice had different outcomes after inducing ischaemia. In particular, the PHD2+/− mice had an improved rCBF response post-reperfusion with better behavioural scores. The PHD3−/− mice have worse rCBF but no behavioural change.
The information gained enhances understanding of the biological processes involved and informs strategies for therapeutic targeting of the PHD enzymes.
Introduction
Cerebral ischaemia results in the activation of multiple pathways that can independently lead to neuronal death (Won et al. 2002). Use of compounds that effectively target some of these individual pathways has failed to be successfully translated into clinical practice (O’Collins et al. 2006). An alternative to targeting individual pathways is to target entire physiological networks which influence a number of processes at one time and thereby simultaneously suppress both ischaemic and reperfusion damage. Hypoxia-inducible factors (HIF1–3) influence many processes including angiogenesis, vascular remodelling, vascular tone, metabolic regulation and erythropoiesis (Pugh & Ratcliffe, 2003; Kaelin & Ratcliffe, 2008) that protect the organism against hypoxia. It is proposed that regulating HIF induction and accumulation by inhibition of the activity of a family of 2-oxoglutarate (2-OG)-dependent hydroxylase enzymes (prolyl hydroxylase domain 1–3 (PHD1–3) and factor inhibiting HIF (FIH)) is a highly promising therapeutic approach for cerebral ischaemia (Siddiq et al. 2009; Harten et al. 2010; Nagel et al. 2010). The PHDs together regulate the HIF pathway (Epstein et al. 2001), while individual PHDs may regulate other targets (Köditz et al. 2007; Fu et al. 2007; Xie et al. 2009) although these latter findings have yet to be adequately replicated. It has been shown that inhibition of PHDs with different small molecule compounds can confer robust neuroprotection in rodent models of stroke (Siddiq et al. 2005; Puchowicz et al. 2008). To this end, we found that dimethyl-oxalylglycine (DMOG), an esterified oxoglutarate analogue known to inhibit the HIF hydroxylases, protects the brains of rats subjected to focal cerebral ischaemia (Nagel et al. 2011). DMOG treatment induced a number of HIF regulated genes and proteins (e.g. VEGF, eNOS), but the protection observed was not simply related to HIF-1α protein levels present at the time point when the animals were killed (Nagel et al. 2011). DMOG, like many other currently available HIF hydroxylase inhibitors, inhibits most 2-OG-dependent enzymes, and may therefore affect other signalling pathways and important cellular processes, e.g. DNA repair, histone methylation, ankyrin hydroxylation and fatty acid transport (Loenarz & Schofield, 2008).
To investigate these phenomena further, we now use mice in which the PHD alleles have been individually genetically targeted to evaluate the precise role of each enzyme in the early response to cerebral ischaemia. To study the roles of PHD1 and PHD3, we used mice with homozygous deletions. However, this was not possible for PHD2, as complete germline knockdown of this isoform is embryo-lethal due to severe placental and heart defects (Takeda et al. 2006); we therefore used PHD2 heterozygous mice which have no such defects (Mazzone et al. 2009). The information gained enhances understanding of the biological processes involved and informs strategies for therapeutic targeting of the PHD enzymes, in particular, when isoform specific inhibitors become available.
Methods
Animals
Wild-type, PHD1−/−, PHD2+/− and PHD3−/− mice were generated as previously described (Bishop et al. 2008; Aragonés et al. 2008; Mazzone et al. 2009). Mice for PHD1 and PHD3 experiments were bred on mixed Swiss/129SvEv background, whereas those used in the PHD2 experiments were bred on a C57/Bl6 background. Eight- to twelve-week-old male mice were used for the experiments. All procedures were in accordance with the UK Home Office Animals (Scientific Procedures) Act 1986 and Local Ethical Review Procedures (University of Oxford Medical Sciences Division Ethical Review Committee).
Genotyping
Animals were genotyped by PCR using primers specific to transgene sequences (Supplemental Table 1). DNA was extracted from mouse ear clips using an E.Z.N.A. tissue DNA isolation kit (Omega Bio-Tek, Inc., Norcross, GA, USA) according to the manufacturer's protocol, and 1–2 μl of the processed DNA was used for each PCR.
Middle cerebral artery occlusion (MCAO)
Anaesthesia was induced with isoflurane (3% initially, 1% to 1.5% maintenance) in O2 and N2O (1:3). Under the operating microscope (AM-P6, Alltion Co. Wuzhou city, Guangxi province, China), the right common carotid artery (CCA), the right external carotid artery (ECA), and the right internal carotid artery (ICA) were isolated and a 6–0 suture was tied at the origin of the ECA and at the distal end of the ECA. A silicone rubber coated monofilament (Filament size 6–0, diameter 0.09–0.11 mm, length 20 mm; diameter with coating 0.21 ± 0.02 mm, and coating length 5–6 mm, Doccol Co, USA) was pushed up the ICA to occlude the origin of the middle cerebral artery (MCA). The suture remained in situ for 45 min, after which it was removed to allow reperfusion and the ECA was permanently tied. In sham-operated mice, the monofilament was inserted only temporarily into the intracranial portion of ICA and withdrawn immediately to control for direct damage to the arterial intima. The core body temperature of all mice were maintained at 36.5°C during surgery, occlusion, and the immediate reperfusion period using a homeostatic heating blanket linked to a rectal probe (Harvard Apparatus, Edenbridge, UK). The surgeon was unaware of the animal genotype.
Measurement of regional cerebral blood flow (rCBF)
A 0.5 mm diameter microfibre laser-Doppler probe (Oxford Optronix, Oxford, UK) was attached to the skull with cyanoacrylate glue 3 mm lateral to midline and 1 mm posterior to the bregma to measure rCBF. Adequate MCAO was assumed if ≥70% reduction in rCBF occurred immediately after placement of the intraluminal occluding suture; otherwise the experiment was terminated. Data were expressed as a mean percentage of the baseline pre-ischaemic value.
Temperature regulation and activity measurements
Mice were implanted with intra-abdominal radiofrequency probes (TA10TA-F20; Transoma Medical) 7 days before the MCAO. Core temperature was sampled every 20 s using receivers (RLA-1020; Data Science Int., St Paul, MN, USA) interfaced to a computer running ART 2.2 (for statistical comparison and graphical display the mean values of each hour were calculated). This telemetry system minimizes stress and allows temperature monitoring/control in the freely moving animal. Core temperature and activity (counts per 20 s) were monitored for 7 days. In the post-operative recovery period the telemetry probes were also used to activate a heating light over the cage thereby controlling core body temperature. The movement of the animal over the receiver is detected as activity (via changes in the probe's signal strength). Thus activity is a relative measure and does not indicate an exact distance travelled.
Behaviour
Mice were assessed at 2 and 24 h after the MCAO. Behavioural assessment consisted of scoring forelimb flexion, reduced resistance to lateral push, gait toward the paretic side, and rotational behaviour (Bederson et al. 1986). The score used was as follows: 0, normal; 1, consistent forelimb and axial flexion in a direction contralateral to the side of the lesion when lifted by the tail; 2, observations of score 1 with consistently reduced resistance to lateral push and gait toward the paretic side; 3, observation of score 2 plus large or weak circling toward the paretic side; 4, score 2 plus small circling toward the paretic side; and 5, score 2 plus tight circling toward the paretic side. The scoring was performed by a trained individual unaware of the animal genotype or surgical intervention.
Tissue processing
The mice were given sodium pentobarbital (70 mg kg−1 intraperitoneally) 24 h after the MCAO, and were perfused with chilled (4°C) phosphate-buffered saline (PBS), followed by 4% formalin (in PBS). The brains were then removed and stored in chilled (4°C) 10% formalin (in PBS) before embedding in paraffin wax. Representative coronal 10 μm and 6 μm sections were obtained from each 1 mm slice of embedded brain using a microtome.
Infarction volume measurement
To assess infarction volume, 10 μm sections were stained with either Nissl or haematoxylin & eosin (H&E) following standard protocols. Areas of infarction were delineated by a ‘blinded’ investigator using NIS Elements imaging software (Nikon, Kingston upon Thames, Surrey, UK). The infarction volume was calculated as the sum of section volumes made by multiplying the area of infarction on each section by the thickness of the corresponding slice. The infarction volume was corrected for oedema using the following equation: corrected infarct volume = total infarct volume – (right (ipsilateral) hemisphere volume – left (contralateral) hemisphere volume). The corrected infarction volume was presented as a percentage of the volume of the contralateral hemisphere.
Periodic acid schiff (PAS) staining
To visualize the distribution of glycogen in tissue sections, PAS staining was undertaken using a PAS staining kit (Sigma-Aldrich, St Louis, MO, USA) according to the manufacturer's protocol. Briefly, 10 μm sections were routinely de-paraffinized and hydrated in distilled water, and incubated in periodic acid solution for 5 min. After rinsing with distilled water, the sections were incubated in Schiff's reagent for 15 min, followed by washing in running tap water for 5 min. Thereafter, sections were counter-stained with haematoxylin for 90 s, and dehydrated in a graded series of ethanol. After mounting (Surgipath, Peterborough, UK) the PAS staining was observed by a ‘blinded’ investigator using a Nikon Eclipse E110M fluorescence microscope.
Blood–brain barrier (BBB) disruption measurement
BBB disruption was assessed by mouse IgG extravasation detected using immunohistochemistry with a Vectastain Elite ABC Kit (mouse IgG) (Vector Laboratories, Peterborough, UK). Briefly, neighbouring 6 μm sections were de-waxed and rehydrated, and the antigen binding sites were exposed using Target Retrieval solution, pH 6.0 (Dako UK Ltd, Ely, Cambridgeshire, UK) at 95°C for half an hour. Endogenous peroxidase activity and non-specific binding sites were blocked before incubating in biotinylated anti-mouse IgG antibody (1:100; Vector Laboratories) for 2 h at room temperature. Immunohistochemical staining followed the protocol of the ABC staining kit (Vector Laboratories). Photomicrographs were obtained as above. Areas of mouse IgG stain were delineated by a ‘blinded’ investigator using the NIS Elements imaging software (Nikon). The extent of BBB disruption was calculated as the sum of section volumes made by multiplying the area stained with the thickness of the slice. The BBB disruption was corrected for oedema and presented as a percentage of the volume of the contralateral hemisphere.
TUNEL assay for the detection of apoptotic cells
TUNEL assay was performed on neighbouring 6 μm sections using an ApopTag Plus Fluorescein in situ Apoptosis Detection Kit (Millipore, Temecula, CA, USA) according to the manufacturer's protocol. The sections were mounted with medium containing 4,6-diamidino-2-phenylindole (DAPI, Vector Laboratories) as a counter stain. Fluorescence was detected using a Nikon Eclipse E110M fluorescence microscope and images were processed using the NIS Elements imaging software. Four standard non-overlapping high power fields from the ischaemia boundary and two in the ischaemic core were counted by a ‘blinded’ investigator. The ratio of TUNEL positive cells to DAPI positive nuclei was determined.
Immunohistochemistry
Neighbouring 6 μm coronal sections were prepared as above, incubated with primary antibodies: goat anti-8-hydroxy-2-deoxyguanosine (8-OHdG) (1:100; Santa Cruz Biotechnology, Santa Cruz, CA, USA), 4-hydroxy-2-nonenal (4-HNE) antibodies (1:100; Santa Cruz Biotechnology), rabbit anti-mouse CD31 antibody (1:100; Acris Antibodies GmβH, Schillerstrabe 5, Herford, Germany), goat anti-HIF1α antibody (1:100; Santa Cruz Biotechnology) at 4°C overnight, then with appropriate biotinylated secondary antibodies (1:100; Vector Laboratories) for 2 h at room temperature. Immunohistochemical staining and photomicrographs were obtained as above and positive structures per field were counted by a ‘blinded’ investigator.
Statistical analysis
Data were represented as means ± SEM or median with interquartile range where appropriate. Parametric data were analysed using Student's t test for single comparisons or one-way ANOVA followed by Bonferroni's test for multiple comparisons. Non-parametric data were assessed with a Mann–Whitney U test or Kruskall–Wallis test with Dunn's multiple comparison test. Continuous data of body temperature, activity and rCBF were analysed by ANOVA for repeated measurements (rm-ANOVA) with Bonferroni post hoc comparisons for genotype specific differences. Mortality rates were compared with Fisher's exact test. Pearson's correlation was used to assess the relationships among histology data. Statistical analyses were performed using SPSS (for Windows, v. 17). A P value < 0.05 was considered statistically significant.
Results
rCBF measurements
MCAO resulted in a greater than 70% reduction in rCBF in all experimental groups (Fig. 1). There was no significant difference in rCBF at any time point in PHD1−/− mice compared to their WT littermates (Fig. 1A). In both PHD2 and PHD3 comparisons, there was a significant effect of genotype (rm-ANOVA, P < 0.01). Post hoc testing revealed that on reperfusion, in PHD2+/− mice the rCBF returned faster to pre-ischaemic levels than their WT littermates with statistically significant increases of 37%, 95% CI 6–68% and 40%, 95% CI 9–71% at 10 and 15 min post-reperfusion, respectively (P < 0.01; Fig. 1B). In PHD3−/− mice, however, the rCBF returned slower to pre-ischaemic levels than their WT littermates with a statistically significant difference of 31%, 95% CI 8–54% at 15 min post-reperfusion (P < 0.01; Fig. 1C).
Figure 1. Summary of rCBF during MCAO in PHD1−/−, PHD2+/− and PHD3−/− mice and their WT littermates.
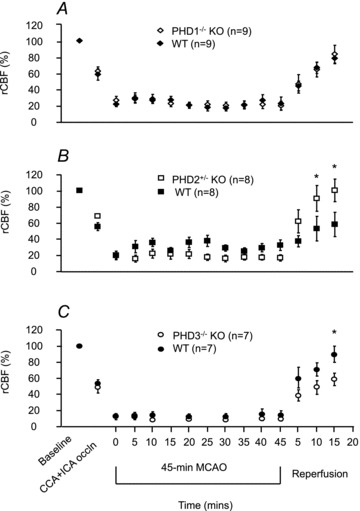
Upon MCAO, rCBF dropped significantly below 30% of baseline levels. On reperfusion, the rCBF in PHD2+/− mice returned faster to preischaemic levels than their WT littermates (B). In contrast, the rCBF in PHD3−/− mice returned slower to baseline levels than their WT littermates (C) whereas there was no difference in rCBF return in PHD1−/− mice and their WT littermates (A) (*P < 0.01, post hoc testing of rm-ANOVA).
Mortality, body weight and temperature homeostasis
No significant differences were observed in mortality following the MCAO, being between 10 and 15% in all the genetically modified mice and their WT littermates (Suppl. Table 2). There was similar weight loss in the knock-out and WT mice 24 h after the MCAO ranging from 10 to 15% (Suppl. Table 2), although PHD2+/− mice were significantly lighter than there WT littermates (24.4 ± 0.8g vs. 27.3 ± 0.6 g, P < 0.05, Suppl. Table 2). All PHD genetically modified mice and their WT littermates had normal diurnal temperature curves at baseline with a significant effect of time in rm-ANOVA (P < 0.01). However, no statistically significant difference due to genotype occurred in post hoc tests (Suppl. Fig. 1). During the surgery for MCAO, body temperatures were regulated between 36 and 37°C through the heating blanket. After the surgery, animals lost their ability to regulate body temperature. Their core temperature tended to fall but was returned to the lower end of normal range (i.e. 36°C) with the aid of the telemetric temperature control system (Suppl. Fig. 1). Body temperatures after the MCAO were similar in all experiments and there were no statistically significant genotype related differences after post hoc testing.
Activity and neuroscores
At baseline, all animals showed similar absolute activity levels and comparable diurnal activity patterns with significant changes over time (P < 0.01, rm-ANOVA), while there were no statistically significant genotype-dependent differences (rm-ANOVA; Suppl. Fig. 2). After the MCAO there was significant reduction of activity in all animals (P < 0.05) and no diurnal activity patterns. Interestingly, the PHD2+/− mice were significantly more active than their WT littermates at 7 of 25 time points (28%) of the observed time after the MCAO (P < 0.05), while at no time points were WT mice significantly more active than the PHD2+/− mice. PHD1−/− or PHD3−/− mice did not show differences in activity compared with their littermates after the MCAO (Suppl. Fig. 2).
Behavioural tests were performed under blinded conditions on all animals at 2 h and 24 h after reperfusion. In keeping with the activity measurements there were no significant differences in neuroscore between PHD1−/− or PHD3−/− mice and their WT littermates at either 2 h or 24 h after reperfusion (Fig. 2A and C), but at both time points the PHD2+/− mice had significantly better neuroscores than their WT littermates (PHD2+/−: 3, 1.25–4 (2 h) and 1.5, 1–3 (24 h) vs. WT: 5, 4.125–5 (2 h) and 5, 4.125–5 (24 h), respectively, P < 0.05, Fig. 2B).
Figure 2. Neuroscores assessed at both 2 and 24 h after MCAO in PHD1−/−(A), PHD2+/− (B), PHD3−/− (C) mice and their WT littermates.
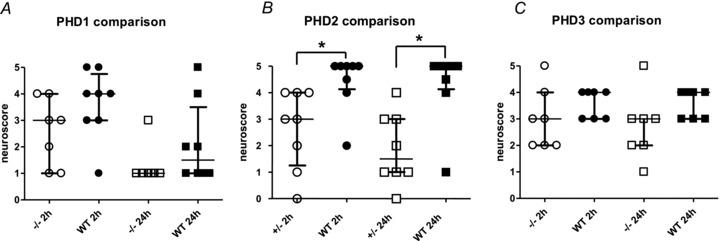
There were no significant difference in neuroscore between PHD1−/− and PHD3−/− mice and their WT littermates at either 2 h or 24 h after reperfusion, but at both time points the PHD2+/− mice had significantly better neuroscores than their WT littermates (median with IQR; *P < 0.05, Kruskall–Wallis test with Dunn's multiple comparison test).
Histological analyses
The quantification of infarct volume was similar using both the Nissl and H&E stains (Fig. 3A and B; Suppl. Fig. 3A). The trends observed towards a reduction in infarct volume in PHD2+/− mice and PHD1−/− mice compared with the corresponding WT littermates did not reach statistical significance with this group size (PHD1−/−: 16 ± 2.3%vs. WT: 24 ± 4.9%, P = 0.215 and PHD2+/−: 26.7 ± 6.7%vs. WT: 40 ± 5.5%, P = 0.154). There was no apparent or statistically significant difference in infarction volume between PHD3−/− and their WT littermates (Fig. 3D).
Figure 3. Histological analysis of infarct volume and the volume of mouse IgG extravasation.
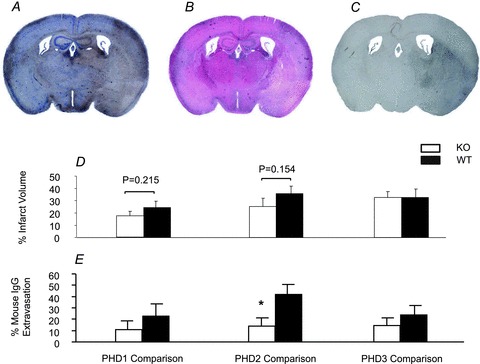
The quantification of infarct volume using the Nissl and H&E stains was similar (A and B). Infarct volumes showed a trend to be smaller in PHD2+/− mice (open bar) and PHD1−/− mice (open bar) compared with the corresponding WT littermates (filled bars). In contrast, there was no difference in infarction volume between PHD3−/− (open bar) and their WT littermates (filled bar) (D). Mouse IgG extravasation located in the ischaemic hemisphere (A–C). The volume of blood–brain barrier disruption assessed by the IgG extravasation was significantly smaller in the PHD2+/− mice (open bar) than their WT littermate (filled bar) whereas this was not seen in either PHD1 or PHD3 comparisons (*P < 0.05, t test, E).
BBB disruption, as estimated by mouse IgG extravasation located in the ipsilateral hemisphere, was linearly related to the infarct volume (Fig. 3A–C; Suppl. Fig. 3B). The volume of BBB disruption in the PHD2+/− mice was significantly smaller than their WT littermates (PHD2+/−: 13.9 ± 7.5%vs. WT: 41.7 ± 8.9%, P < 0.05) whilst no significant difference was found between PHD1−/−, or PHD3−/− mice and their WT littermates (PHD1−/−: 11 ± 7.7%vs. WT: 22.8 ± 11.1%, P = 0.40 and PHD3−/−: 14.3 ± 6.8%vs. WT: 23.5 ± 8.3%, P = 0.41; Fig. 3E).
Following MCAO, apoptotic cells were seen in the ipsilateral hemisphere but not in the contralateral hemisphere. Apoptotic cells were more prominent in the penumbra than in the ischaemic core (Fig. 4B). The apoptotic ratio in the penumbra was significantly less in PHD2+/− mice than in the WT mice (PHD2+/−: 23.8 ± 6.9%vs. WT: 43.5 ± 3.8%, P < 0.05), while the ratio was not different between PHD1−/−, or PHD3−/− and their WT littermates (PHD1−/−: 48.1 ± 5.5%vs. WT: 47.3 ± 6%, P = 0.68 and PHD3−/−: 52.7 ± 7.1%vs. WT: 58.6 ± 8.7%, P = 0.23; Fig. 4C).
Figure 4. Apoptotic cell detection on mouse 6 μm brain sections.
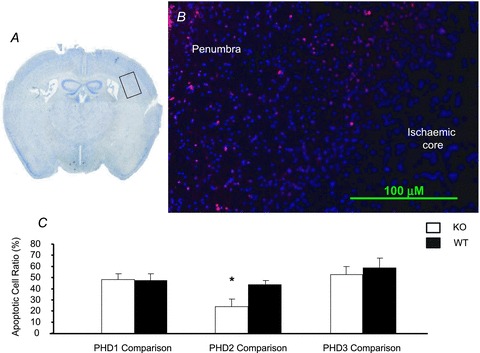
A and B, representative Nissl and ApopTag stains. The box in A indicates the area shown in B. Apoptotic cells were more prominent in the penumbra than in the ischaemic core (B). The ratio of TUNEL positive cells to DAPI positive nuclei in the penumbra of PHD2+/− mice (open bar) was significantly higher than those of the WT mice (filled bar), while the ratio was not different between PHD1−/−, PHD3−/− and their WT littermates (*P < 0.05, t test, C).
Immunohistochemical staining with anti-CD31 antibody labelled capillaries throughout the brain and positively labelled vascular structures in one cortical peri-infarct region and one subcortical ischaemic core region as well as corresponding mirror-image regions were counted by an investigator blinded to the genetic status of the animals. PHD2+/− mice had significantly more cerebral capillaries than their WT littermates (PHD2+/−: 154 ± 8.5 vs. WT: 126.7 ± 9.5 CD31-positive structures/field, P < 0.05), whilst no differences were found between PHD1−/−, or PHD3−/− mice and their WT littermates (PHD1−/−: 150.5 ± 10.1 vs. WT: 134.8 ± 9.3 CD31-positive structures/field, P = 0.22 and PHD3−/−: 120.3 ± 7.8 vs. WT: 124.6 ± 9.5 CD31-positive structures/field, P = 0.48; Fig. 5A). Representative images from the PHD2+/− mice and their WT littermates are shown (Fig. 5B and C, respectively).
Figure 5. CD31 immunostains of mouse brain sections.
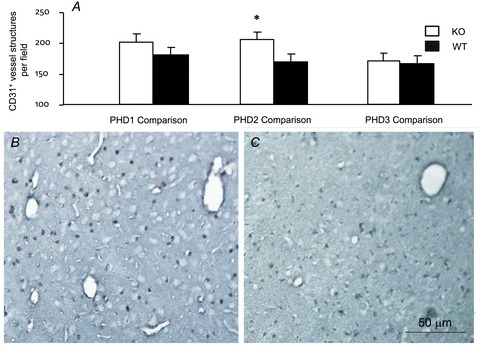
A, the number of CD31-positive structures with the morphology of capillaries was quantified in sections obtained from mice after 45 min MCAO and 24 h reperfusion. The PHD2+/− mice (open bar) had significantly more CD31-positive structures than their WT littermates (filled bar), whereas this was not seen in either PHD1 or PHD3 comparisons (*P < 0.05, t test). B and C, representative CD31 immunohistochemistry images from PHD2+/− (B) and wild-type control (C) mice, respectively.
PAS staining of brain sections taken 24 h after MCAO showed no differences in the distribution or intensity of glycogen storage in either the ipsilateral ischaemic core or contralateral hemisphere between PHD1−/−, PHD2+/− or PHD3−/− mice and their WT littermates (Suppl Fig. 4). 8-OHdG immunoreactivity was present within the ischaemic core and extended to penumbra areas (Suppl Fig. 5A and B). 4-HNE immunoreactivity had a similar staining distribution pattern to 8-OHdG within the core and penumbra (Suppl Fig. 5C and D). No significant differences were seen between PHD1−/−, PHD2+/− or PHD3−/− mice and their WT littermates (PHD1−/− 194 ± 15 vs. WT 208 ± 7 OHdG positive cells/field, P = 0.41; PHD2+/− 207 ± 11 vs. WT 217 ± 12, OHdG positive cells/field, P = 0.56; PHD3−/− 202 ± 8 vs. WT 211 ± 9 OHdG positive cells/field, P = 0.60; PHD1−/− 222 ± 14 vs. WT 216 ± 6 HNE positive cells/field, P = 0.71; PHD2+/− 235 ± 9 vs. WT 238 ± 14, HNE positive cells/field, P = 0.45; PHD3−/− 264 ± 12 vs. WT 239 ± 16 HNE positive cells/field, P = 0.25).
HIF1α was up-regulated not only in the ipsilateral hemisphere but also in the contralateral hemisphere in all the animals after MCAO (Fig. 6A and B), but not in sham-operated control animals (Fig. 6C and D). In the ipsilateral hemisphere, HIF-1α labelling was more prominent in the peri-ischaemic area than in the ischaemic core (Fig. 6A). Under higher magnification, the HIF-1α positivity was confirmed to localize to the nuclei. However, no apparent genotype specific differences were seen in the number of HIF positive cells per field in these regions (PHD1−/− 588 ± 18 vs. WT 609 ± 19 HIF-1α positive cells/field, P = 0.43; PHD2+/− 620 ± 15 vs. WT 601 ± 26, HIF-1α positive cells/field, P = 0.55; PHD3−/− 574 ± 16 vs. WT 592 ± 14 HIF-1α positive cells/field, P = 0.39).
Figure 6. HIF1α detection on 6 μm mouse brain sections from a wild-type animal ipsilateral hemisphere following MCAO (A), contralateral hemisphere following MCAO (B), ipsilateral hemisphere following sham operation (C), and contralateral hemisphere following sham operation (D).
At this time point the sections from PHD1−/−, PHD2+/− and PHD3−/− animals showed similar staining pattern to those of the wild-type animals.
Discussion
The present study indicates that constitutive heterozygote PHD2 deficiency has a beneficial effect on the functional outcome (based on neuroscores) and activity rates (assessed by telemetric monitoring) up to 24 h after MCAO. In contrast, no statistically significant net benefits were seen in either PHD1−/− or PHD3−/− mice at this early time point. These results are in keeping with the role of PHD2 as the most important HIF prolyl hydroxylase in setting the normoxic levels of HIF (Berra et al. 2003; Appelhoff et al. 2004) and also with results showing mice bearing a hypomorphic PHD2 allele are protected against myocardial ischaemia (Hyvärinen et al. 2010). In contrast, complete knockout of the PHD2 gene is deleterious, causing embryonic lethality due to a combination of placental and cardiac defects (Takeda et al. 2006), and even somatic inactivation of this gene leads to premature mortality associated with marked venous congestion and dilated cardiomyopathy (Minamishima et al. 2008). However, the reduced residual enzyme activity in both the heterozygote deficient and the hypomorphic PHD2 mouse lines is sufficient to avoid these deleterious effects whilst providing protection against tissue ischaemia (Mazzone et al. 2009; Hyvärinen et al. 2010). Taken together these observations indicate that partial reduction of PHD2 activity can have beneficial effects that disappear on complete suppression of this enzyme, which emphasizes the need for care in establishing a balance between risks and benefits in therapeutic PHD inhibition.
We estimated infarct volume using two distinct histological approaches and observed a 33.2% reduction in PHD1−/− and a 32.9% reduction of infarct volume in PHD2+/− mice compared with littermate controls. This reduction did not reach statistical significance at this early time point, perhaps because the injury has not fully matured. In this regard it is noteworthy that statistically significant changes in infarct volume were not seen in a study analysing the effects of neuronal HIF-1α deficiency until 3 days after MCAO (Baranova et al. 2007).
Lowering of brain temperature is the one experimental variable found to have protective effects in animal models of cerebral ischaemia that translates into therapeutic benefit in human studies, at least in circumstances of global cerebral hypoxia (The Hypothermia After Cardiac Arrest Study Group, 2002; Bernard et al. 2002; Cabanas et al. 2011). The need for care in maintaining temperature homeostasis was emphasized by our finding that PHD2+/− mice used in these experiments, but not the PHD1−/− or PHD3−/− mice, were lighter than their littermates at the time they were studied, an observation in accord with the reported reduced body weights in PHD2 hypomorphic mice (Hyvärinen et al. 2010) and mice with conditional PHD2 inhibition (Minamishima et al. 2008). We therefore took care to exclude potential confounding effects from differences in body temperature during our study. Firstly, we assessed the thermoregulatory behaviour of PHD deficient mice compared with their littermate controls at baseline but documented no effects of genotype on thermoregulation. Secondly, we used an effective feedback mechanism to restore and maintain core temperature during surgery and the recovery period. We believe these measures ensure that the results seen in our experiments were not due to confounding effects of temperature on outcome.
The HIF prolyl hydroxylases act via the HIF pathways to regulate blood supply, influencing haematocrit, vessel formation and maturity, and vascular tone (Milkiewicz et al. 2004; Nangaku et al. 2007; Huang et al. 2008; Loinard et al. 2009; Chan et al. 2009; Mazzone et al. 2009). The extent of ischaemia and the speed of restoration of blood flow are important determinants of the degree of damage resulting from stroke (Adams et al. 2003; Sutherland et al. 2011). Differences in genetic backgrounds may have marked effects on these features. The C57/Bl6 background of the PHD2+/− mice is known to have an incomplete circle of Willis but greater native pial collaterals and more robust vasodilation in response to acetylcholine than the SV129 background present in the PHD1−/− and PHD3−/− mice (Connolly et al. 1996; Fujii et al. 1997; Zhang et al. 2010). This means that while comparisons between the different PHD deficient mice and their littermates remain legitimate, direct comparison between the PHD1−/−, PHD2+/− and PHD3−/− mice must be treated with caution in this study. Furthermore as in all animal studies we cannot be certain that the effects of PHD deficiency described on one genetic background would apply to all other backgrounds. We showed no effects of PHD1−/− status on cerebral reperfusion following MCAO when compared with WT littermates. The PHD3−/− mice showed impaired restoration of CBF after ischaemia compared with their littermates. This delay occurred despite similar vessel densities, suggesting that it was due to aspects of vessel or circulatory function rather than just number. We have previously shown that in the PHD3−/− mice blood pressure is lower at baseline that their littermates and that they have impaired sympathoadrenal function with blunted baroreceptor reflexes (Bishop et al. 2008). Since reperfusion after vascular occlusion depends on systemic blood pressure (McColl et al. 2004), it is likely that these features explain the impaired reperfusion in the PHD3−/− mice. Importantly, these differences in reperfusion may mask other potentially beneficial effects of PHD3 deficiency on the outcome following MCAO in these mice. The improved outcomes in the PHD2+/− animals compared with their WT littermates correlated with improved restoration of blood flow but were not associated with a change in haematocrit (T.B. and C.W.P. – unpublished data). Our immunohistological analysis showed an increased capillary density in the brains of PHD2+/− mice compared with their WT littermates on the same background, suggesting that pre-existing angiogenesis in PHD2+/− mice may provide a direct explanation for the enhanced reperfusion and improved outcome after focal cerebral ischaemia–reperfusion. A recent study has shown that PHD2+/− mice displayed preformed collateral arteries that preserved perfusion and prevented tissue necrosis after hindlimb ischaemia (Takeda et al. 2011). However, no such differences were reported in the hearts of mice that were hypomorphic for PHD2, even though these mice were protected against myocardial ischaemia–reperfusion injury (Hyvärinen et al. 2010). Taken together these results suggest that effects on the vasculature may be tissue or strain specific, depend on the nature of PHD deficiency and that other mechanisms contribute to cytoprotection in PHD deficient animals.
The philosophy behind the approach of targeting the PHDs was to entrain entire physiological networks that have evolved to protect the organism against hypoxia and ischaemia by regulating a co-ordinated response involving changes in the expression of more than 500 downstream genes (Geiger et al. 2011). In this regard it is interesting that the outcome of the deletion of individual PHDs differ, but it is not surprising that we have not been able to delineate a single downstream mechanism as being responsible for the effects. We examined markers of oxidative damage because PHD1, but not PHD2 and PHD3, knockdown has been reported to prevent oxidative stress-induced neuronal death in primary neuron cultures (Siddiq et al. 2009). However, in our current experiments we did not find any statistically significant benefit of PHD1 deficiency on outcome following MCAO nor any genotype-specific differences in oxidative stress levels as measured by immunostaining for 8-OHdG (a marker of oxidative DNA damage) and 4-HNE (a marker of lipid oxidation) on brain sections prepared 24 h after MCAO. A number of factors may contribute to these discrepancies, including the magnitude and duration of exposure to ROS in the in vivo setting compared with the in vitro experiments, the protective and clearance mechanisms present in vivo and the multitude of cell types in addition to neurons (e.g. endothelial cells, astrocytes, and pericytes) that contribute to outcome in the mouse stroke model (Chen et al. 2010). PHD1−/− status, but not PHD2+/− or PHD3−/− status, has also been reported to confer protection in a hindlimb ischaemia model (Aragonés et al. 2008) and this is attributed to PPAR-α and HIF-2α mediated metabolic adaptation. We used PAS staining to assess glycogen stores in the brain and did not find any changes in this aspect of metabolic adaptation between any of the genotypes. In previous work we showed that PHD3−/− status, but not PHD2+/− or PHD1−/− status, led to reduced apoptosis in sympathetic neurons (Bishop et al. 2008). However, in the MCAO model we report here a significant reduction in the apoptotic ratio in the penumbra in PHD2+/− mice compared with their littermates, but not in the other genotype comparisons. Similarly, BBB disruption was significantly less extensive in the PHD2+/− mice than in littermate controls. Since both reduced apoptosis and BBB disruption may either protect against ischaemic damage or result from reduced ischaemia we cannot resolve whether the observed differences in the PHD2+/− comparison are primary or secondary. Overall these results suggest the beneficial effects we have observed from PHD2+/− status are likely to arise from the co-ordinated integration of minor perturbations in multiple mechanisms and that approaches analysing the effects of knocking-down individual downstream components would require very careful analysis and interpretation.
In this regard it is obviously of interest to examine the HIF response itself in these different genotypes. HIF-1α protein was detected at 24 h after MCAO in the ipsilateral hemisphere, more in the peri-ischaemic area than in the core. The absence of HIF-1α protein in the ischaemic core is likely to result from the loss of cellular activity consequent to the tissue necrosis and cell death in this zone (Yuan, 2009). Intriguingly, in contrast to a previous publication (Baranova et al. 2007), we also saw HIF immunoreactivity in the contralateral hemisphere following the MCAO. This was also seen in all the MCAO animals but was not seen in any sham operated animals, indicating that it occurred as a result of the ischaemia–reperfusion protocol rather than any non-specific effects of anaesthesia or subsequent steps in tissue preparation. No significant genotype specific differences were detected in any of these parameters at the 24 h time point. In our previous study analysing the effects of DMOG treatment, which simultaneously inhibits all the HIF hydroxylase enzyme family, the protection observed was associated with up-regulation of HIF target genes but not simply related to total HIF-1α levels assessed by immunoblotting at the termination of the experiment (Nagel et al. 2011). The absence of a simple relationship between outcome and HIF levels at the end of the experiment probably reflects the complex dynamics of this pathway with its multiple feedback loops (Horak et al. 2010; Kelly et al. 2011; Demidenko & Blagosklonny, 2011). Evidence that the time course of effects on the HIF pathway matters comes from the contrast between reports that long-term neuron-specific knockout of HIF-1α worsens outcomes following MCAO in mice (Baranova et al. 2007) but early RNA interference based HIF inhibition has beneficial effects on the outcome of cerebral ischaemia–reperfusion injury (Chen et al. 2009).
In conclusion, PHD2+/− mice have significantly increased microvessel density, improved CBF on reperfusion, and show significantly better functional outcomes and higher locomotor activity associated with significantly fewer apoptotic cells in the penumbra and significantly less BBB disruption with a trend to reduced infarct volumes 24 h after the MCAO than littermate controls. PHD3−/− mice have lower CBF after reperfusion than WT littermates but comparable functional outcomes, suggesting that deleterious haemodynamic effects are counteracted by alternative neuroprotective benefits in this circumstance. PHD1−/− mice have statistically non-significant reductions in infarct volumes following MCAO compared with their littermates at this time-point in this model. These effects are seen following lifelong PHD deficiency and thus reflect a composite of developmental changes, affecting features such as vessel density and overall sympathetic tone, and perhaps more acute effects amplifying the hypoxic stimuli inherent in this ischaemia–reperfusion model. Only these latter effects can contribute to the observed protection entrained by PHD inhibitor administration acutely at the time of cerebral ischaemia (Nagel et al. 2011). These new results suggest it is possible that chronic pharmacological partial inhibition of PHD2 would stimulate adaptations that provide protection against damage from a subsequent stroke but this potential benefit would have to be weighed against any side effects. The effects of complete and partial PHD gene knock-out in the brain could be further investigated using conditional PHD knockout mice in which the PHD deletion was targeted to specific cell types. However, care would be needed in interpreting the results since some of the likely mediators (e.g. VEGF and erythropoietin) are secreted, whilst others have cell autonomous effects. The use of conditional knockouts could also be extended to allow distinction between acute and chronic effects of PHD deficiency. Further studies are also needed to explore the effects at later time points both within the target tissues and systemically.
Acknowledgments
We are grateful for funding received from the Dunhill Medical Trust, Medical Research Council UK, the Fondation Leducq, the Wellcome Trust and the British Heart Foundation. S.N. was supported by the DFG, Germany. We would like to thank Drs Julie Adam and Keith Brooks for their technical support in generating, breeding and transporting the mice. Also we are grateful to Prof. Margaret Esiri who made expert comments on the histological studies.
Glossary
- BBB
blood–brain barrier
- CCA
common carotid artery
- CI
confidence interval
- DAPI
4,6-diamidino-2-phenylindole
- DMOG
dimethyl-oxalylglycine
- ECA
external carotid artery
- FIH
factor inhibiting HIF
- H&E
hematoxylin and eosin
- HIF
hypoxia inducible factors
- 4-HNE
4-hydrooxy-2-nonenal
- ICA
internal carotid artery
- MCA
middle cerebral artery
- MCAO
middle cerebral artery occlusion
- 2-OG
2-oxoglutarate
- PAS
periodic acid Schiff
- 8-OhdG
8-hydroxy-2′-deoxyguanosine
- rCBF
regional cerebral blood flow
- ROS
oxygen reactive species
Author contributions
This project was conceived by A.M.B., C.W.P. and P.J.R. The experiments in this paper were designed by R.C., S.N., M.P. and C.W.P., and were performed by R.C. and S.N. Data were analysed and interpreted by R.C., S.N., M.P. and C.W.P. Genetically modified mice were generated by T.B., P.P., C.W.P. and P.J.R. The manuscript was prepared by R.C., C.W.P., S.N., M.P. and A.M.B. All authors have read and approved the final copy. All experiments were performed at Nuffield Department of Medicine, University of Oxford.
Disclosure
P.J.R and C.W.P. are scientific co-founders of, and hold equity, in ReOx Ltd, a company with interests in drug discovery relating to the HIF pathway.
Supplementary material
Supplemental Table 1
Supplemental Table 2
Supplemental Figure 1
Supplemental Figure 2
Supplemental Figure 3
Supplemental Figure 4
Supplemental Figure 5
References
- Adams HP, Jr, Adams RJ, Brott T, del Zoppo GJ, Furlan A, Goldstein LB, Grubb RL, Higashida R, Kidwell C, Kwiatkowski TG, Marler JR, Hademenos GJ. Guidelines for the Early Management of Patients With Ischemic Stroke: A Scientific Statement From the Stroke Council of the American Stroke Association. Stroke. 2003;34:1056–1083. doi: 10.1161/01.STR.0000064841.47697.22. [DOI] [PubMed] [Google Scholar]
- Appelhoff RJ, Tian YM, Raval RR, Turley H, Harris AL, Pugh CW, Ratcliffe PJ, Gleadle JM. Differential function of the Prolyl Hydroxylases PHD1, PHD2 and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279:38458–38465. doi: 10.1074/jbc.M406026200. [DOI] [PubMed] [Google Scholar]
- Aragonés J, Schneider M, Van Geyte K, Fraisl P, Dresselaers T, Mazzone M, Dirkx R, Zacchigna S, Lemieux H, Jeoung NH, Lambrechts D, Bishop T, Lafuste P, Diez-Juan A, Harten SK, Van Noten P, De Bock K, Willam C, Tjwa M, Grosfeld A, Navet R, Moons L, Vandendriessche T, Deroose C, Wijeyekoon B, Nuyts J, Jordan B, Silasi-Mansat R, Lupu F, Dewerchin M, Pugh C, Salmon P, Mortelmans L, Gallez B, Gorus F, Buyse J, Sluse F, Harris RA, Gnaiger E, Hespel P, Van Hecke P, Schuit F, Van Veldhoven P, Ratcliffe P, Baes M, Maxwell P, Carmeliet P. Deficiency or inhibition of oxygen sensor Phd1 induces hypoxia tolerance by reprogramming basal metabolism. Nat Genet. 2008;40:170–180. doi: 10.1038/ng.2007.62. [DOI] [PubMed] [Google Scholar]
- Baranova O, Miranda LF, Pichiule P, Dragatsis I, Johnson RS, Chavez JC. Neuron-specific inactivation of the hypoxia inducible factor 1α increases brain injury in a mouse model of transient focal cerebral ischemia. J Neurosci. 2007;27:6320–6332. doi: 10.1523/JNEUROSCI.0449-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bederson JB, Pitts LH, Tsuji M, Nishimura MC, Davis RL, Bartkowski H. Rat middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17:472–476. doi: 10.1161/01.str.17.3.472. [DOI] [PubMed] [Google Scholar]
- Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G, Smith K. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346:557–563. doi: 10.1056/NEJMoa003289. [DOI] [PubMed] [Google Scholar]
- Berra E, Benizri E, Ginouvès A, Volmat V, Roux D, Pouysségur J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1α in normoxia. EMBO J. 2003;22:4082–4090. doi: 10.1093/emboj/cdg392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop T, Gallagher D, Pascual A, Lygate CA, de Bono JP, Nicholls LG, Ortega-Saenz P, Oster H, Wijeyekoon B, Sutherland AI, Grosfeld A, Aragones J, Schneider M, van Geyte K, Teixeira D, Diez-Juan A, Lopez-Barneo J, Channon KM, Maxwell PH, Pugh CW, Davies AM, Carmeliet P, Ratcliffe PJ. Abnormal sympathoadrenal development and systemic hypotension in PHD3-/- mice. Mol Cell Biol. 2008;28:3386–3400. doi: 10.1128/MCB.02041-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabanas JG, Brice JH, De Maio VJ, Myers B, Hinchey PR. Field-induced therapeutic hypothermia for neuroprotection after out-of hospital cardiac arrest: a systematic review of the literature. J Emerg Med. 2011;40:400–409. doi: 10.1016/j.jemermed.2010.07.002. [DOI] [PubMed] [Google Scholar]
- Chan DA, Kawahara TL, Sutphin PD, Chang HY, Chi JT, Giaccia AJ. Tumor vasculature is regulated by PHD2-mediated angiogenesis and bone marrow-derived cell recruitment. Cancer Cell. 2009;15:527–538. doi: 10.1016/j.ccr.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Hu Q, Yan J, Yang X, Shi X, Lei J, Chen L, Huang H, Han J, Zhang JH, Zhou C. Early inhibition of HIF-1α with small interfering RNA reduces ischemic-reperfused brain injury in rats. Neurobiol Dis. 2009;33:509–517. doi: 10.1016/j.nbd.2008.12.010. [DOI] [PubMed] [Google Scholar]
- Chen RL, Balami J, Esiri M, Chen LH, Buchan AM. Stroke in ageing: an overview of evidence. Nat Rev Neurology. 2010;6:256–265. doi: 10.1038/nrneurol.2010.36. [DOI] [PubMed] [Google Scholar]
- Connolly ES, Winfree CJ, Stern DM, Solomon RA, Pinsky DJ. Procedural and strain-related variables significantly affect outcome in a murine model of focal cerebral ischemia. Neurosurgery. 1996;38:523–532. doi: 10.1097/00006123-199603000-00021. [DOI] [PubMed] [Google Scholar]
- Demidenko ZN, Blagosklonny MV. The purpose of the HIF-1/PHD feedback loop: to limit mTOR-induced HIF-1α. Cell Cycle. 2011;10:1557–1562. doi: 10.4161/cc.10.10.15789. [DOI] [PubMed] [Google Scholar]
- Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell. 2001;107:43–54. doi: 10.1016/s0092-8674(01)00507-4. [DOI] [PubMed] [Google Scholar]
- Fu J, Menzies K, Freeman RS, Taubman MB. EGLN3 prolyl hydroxylase regulates skeletal muscle differentiation and myogenin protein stability. J Biol Chem. 2007;282:12410–12418. doi: 10.1074/jbc.M608748200. [DOI] [PubMed] [Google Scholar]
- Fujii M, Hara H, Meng W, Vonsattel JP, Huang Z, Moskowitz MA. Strain-related differences in susceptibility to transient forebrain ischemia in SV-129 and C57black/6 mice. Stroke. 1997;28:1805–1810. doi: 10.1161/01.str.28.9.1805. [DOI] [PubMed] [Google Scholar]
- Geiger K, Leiherer A, Muendlein A, Stark N, Geller-Rhomberg S, Saely CH, Wabitsch M, Fraunberger P, Drexel H. Identification of hypoxia-induced genes in human SGBS adipocytes by microarray analysis. PLoS One. 2011;6:e26465. doi: 10.1371/journal.pone.0026465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harten SK, Ashcroft M, Maxwell PH. Prolyl hydroxylase domain inhibitors: a route to HIF activation and neuroprotection. Antioxid Redox Signal. 2010;12:459–480. doi: 10.1089/ars.2009.2870. [DOI] [PubMed] [Google Scholar]
- Horak P, Crawford AR, Vadysirisack DD, Nash ZM, DeYoung MP, Sgroi D, Ellisen LW. Negative feedback control of HIF-1 through REDD1-regulated ROS suppresses tumorigenesis. Proc Natl Acad Sci U S A. 2010;107:4675–4680. doi: 10.1073/pnas.0907705107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang M, Chan DA, Jia F, Xie X, Li Z, Hoyt G, Robbins RC, Chen X, Giaccia AJ, Wu JC. Short hairpin RNA interference therapy for ischemic heart disease. Circulation. 2008;118(14 Suppl):S226–S233. doi: 10.1161/CIRCULATIONAHA.107.760785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyvärinen J, Hassinen IE, Sormunen R, Mäki JM, Kivirikko KI, Koivunen P, Myllyharju J. Hearts of hypoxia-inducible factor prolyl 4-hydroxylase-2 hypomorphic mice show protection against acute ischemia-reperfusion injury. J Biol Chem. 2010;285:13646–13657. doi: 10.1074/jbc.M109.084855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG, Jr, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30:393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- Kelly TJ, Souza AL, Clish CB, Puigserver P. A hypoxia-induced positive feedback loop promotes hypoxia-inducible factor 1α stability through miR-210 suppression of glycerol-3-phosphate dehydrogenase 1-like. Mol Cell Biol. 2011;31:2696–2706. doi: 10.1128/MCB.01242-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köditz J, Nesper J, Wottawa M, Stiehl DP, Camenisch G, Franke C, Myllyharju J, Wenger RH, Katschinski DM. Oxygen-dependent ATF-4 stability is mediated by the PHD3 oxygen sensor. Blood. 2007;110:3610–3617. doi: 10.1182/blood-2007-06-094441. [DOI] [PubMed] [Google Scholar]
- Loenarz C, Schofield CJ. Expanding chemical biology of 2-oxoglutarate oxygenases. Nat Chem Biol. 2008;4:152–156. doi: 10.1038/nchembio0308-152. [DOI] [PubMed] [Google Scholar]
- Loinard C, Ginouvès A, Vilar J, Cochain C, Zouggari Y, Recalde A, Duriez M, Lévy BI, Pouysségur J, Berra E, Silvestre JS. Inhibition of prolyl hydroxylase domain proteins promotes therapeutic revascularization. Circulation. 2009;120:50–59. doi: 10.1161/CIRCULATIONAHA.108.813303. [DOI] [PubMed] [Google Scholar]
- Mazzone M, Dettori D, Leite de Oliveira R, Loges S, Schmidt T, Jonckx B, Tian YM, Lanahan AA, Pollard P, Ruiz de Almodovar C, De Smet F, Vinckier S, Aragonés J, Debackere K, Luttun A, Wyns S, Jordan B, Pisacane A, Gallez B, Lampugnani MG, Dejana E, Simons M, Ratcliffe P, Maxwell P, Carmeliet P. Heterozygous deficiency of PHD2 restores tumor oxygenation and inhibits metastasis via endothelial normalization. Cell. 2009;136:839–851. doi: 10.1016/j.cell.2009.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl BW, Carswell HV, McCulloch J, Horsburgh K. Extension of cerebral hypoperfusion and ischaemic pathology beyond MCA territory after intraluminal filament occlusion in C57Bl/6J mice. Brain Res. 2004;997:15–23. doi: 10.1016/j.brainres.2003.10.028. [DOI] [PubMed] [Google Scholar]
- Metzen E, Berchner-Pfannschmidt U, Stengel P, Marxsen JH, Stolze I, Klinger M, Huang WQ, Wotzlaw C, Hellwig-Bürgel T, Jelkmann W, Acker H, Fandrey J. Intracellular localisation of human HIF-1α hydroxylases: implications for oxygen sensing. J Cell Sci. 2003;116:1319–1326. doi: 10.1242/jcs.00318. [DOI] [PubMed] [Google Scholar]
- Milkiewicz M, Pugh CW, Egginton S. Inhibition of endogenous HIF inactivation induces angiogenesis in ischaemic skeletal muscles of mice. J Physiol. 2004;560:21–26. doi: 10.1113/jphysiol.2004.069757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minamishima YA, Moslehi J, Bardeesy N, Cullen D, Bronson RT, Kaelin WG., Jr Somatic inactivation of the PHD2 prolyl hydroxylase causes polycythemia and congestive heart failure. Blood. 2008;111:3236–3244. doi: 10.1182/blood-2007-10-117812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel S, Talbot NP, Mecinović J, Smith TG, Buchan AM, Schofield CJ. Therapeutic manipulation of the HIF hydroxylases. Antioxid Redox Signal. 2010;12:481–501. doi: 10.1089/ars.2009.2711. [DOI] [PubMed] [Google Scholar]
- Nagel S, Papadakis M, Chen R, Hoyte LC, Brooks KJ, Gallichan D, Sibson NR, Pugh C, Buchan AM. Neuroprotection by dimethyloxalylglycine following permanent and transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2011;31:132–143. doi: 10.1038/jcbfm.2010.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nangaku M, Izuhara Y, Takizawa S, Yamashita T, Fujii-Kuriyama Y, Ohneda O, Yamamoto M, van Ypersele de Strihou C, Hirayama N, Miyata T. A novel class of prolyl hydroxylase inhibitors induces angiogenesis and exerts organ protection against ischemia. Arterioscler Thromb Vasc Biol. 2007;27:2548–2554. doi: 10.1161/ATVBAHA.107.148551. [DOI] [PubMed] [Google Scholar]
- O’Collins VE, Macleod MR, Donnan GA, Horky LL, van der Worp BH, Howells DW. 1,026 experimental treatments in acute stroke. Ann Neurol. 2006;59:467–477. doi: 10.1002/ana.20741. [DOI] [PubMed] [Google Scholar]
- Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9:677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- Puchowicz MA, Zechel JL, Valerio J, Emancipator DS, Xu K, Pundik S, LaManna JC, Lust WD. Neuroprotection in diet-induced ketotic rat brain after focal ischemia. J Cereb Blood Flow Metab. 2008;28:1907–1916. doi: 10.1038/jcbfm.2008.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiq A, Ayoub IA, Chavez JC, Aminova L, Shah S, LaManna JC, Patton SM, Connor JR, Cherny RA, Volitakis I, Bush AI, Langsetmo I, Seeley T, Gunzler V, Ratan RR. Hypoxia-inducible factor prolyl 4-hydroxylase inhibition. A target for neuroprotection in the central nervous system. J Biol Chem. 2005;280:41732–41743. doi: 10.1074/jbc.M504963200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddiq A, Aminova LR, Troy CM, Suh K, Messer Z, Semenza GL, Ratan RR. Selective inhibition of hypoxia-inducible factor (HIF) prolyl-hydroxylase 1 mediates neuroprotection against normoxic oxidative death via HIF- and CREB-independent pathways. J Neurosci. 2009;29:8828–8838. doi: 10.1523/JNEUROSCI.1779-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland BA, Papadakis M, Chen RL, Buchan AM. Cerebral blood flow alteration in neuroprotection following cerebral ischaemia. J Physiol. 2011;589:4105–4114. doi: 10.1113/jphysiol.2011.209601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Ho VC, Takeda H, Duan LJ, Nagy A, Fong GH. Placental but not heart defects are associated with elevated hypoxia-inducible factor α levels in mice lacking prolyl hydroxylase domain protein 2. Mol Cell Biol. 2006;26:8336–8346. doi: 10.1128/MCB.00425-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda K, Aguila HL, Parikh NS, Li X, Lamothe K, Duan LJ, Takeda H, Lee FS, Fong GH. Regulation of adult erythropoiesis by prolyl hydroxylase domain proteins. Blood. 2008;111:3229–3235. doi: 10.1182/blood-2007-09-114561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda Y, Costa S, Delamarre E, Roncal C, Leite de Oliveira R, Squadrito ML, Finisguerra V, Deschoemaeker S, Bruyère F, Wenes M, Hamm A, Serneels J, Magat J, Bhattacharyya T, Anisimov A, Jordan BF, Alitalo K, Maxwell P, Gallez B, Zhuang ZW, Saito Y, Simons M, De Palma M, Mazzone M. Macrophage skewing by Phd2 haplodeficiency prevents ischaemia by inducing arteriogenesis. Nature. 2011;479:122–126. doi: 10.1038/nature10507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Hypothermia after Cardiac Arrest Study Group. Mild therapeutic hypothermia to improve the neurologic outcome after cardiac arrest. N Engl J Med. 2002;346:549–556. doi: 10.1056/NEJMoa012689. [DOI] [PubMed] [Google Scholar]
- Won SJ, Kim DY, Gwag BJ. Cellular and molecular pathways of ischemic neuronal death. J Biochem Mol Biol. 2002;35:67–86. doi: 10.5483/bmbrep.2002.35.1.067. [DOI] [PubMed] [Google Scholar]
- Xie L, Xiao K, Whalen EJ, Forrester MT, Freeman RS, Fong G, Gygi SP, Lefkowitz RJ, Stamler JS. Oxygen-regulated β2-adrenergic receptor hydroxylation by EGLN3 and ubiquitylation by pVHL. Sci Signal. 2009;2:ra33. doi: 10.1126/scisignal.2000444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan J. Neuroprotective strategies targeting apoptotic and necrotic cell death for stroke. Apoptosis. 2009;14:469–477. doi: 10.1007/s10495-008-0304-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Prabhakar P, Sealock R, Faber JE. Wide genetic variation in the native pial collateral circulation is a major determinant of variation in severity of stroke. J Cereb Blood Flow Metab. 2010;30:923–934. doi: 10.1038/jcbfm.2010.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.