

Abstract
Purpose
One of the most important rate-limiting steps in adoptive cell transfer (ACT) is the inefficient migration of T cells to tumors. Since melanomas specifically express the chemokines CXCL1 and CXCL8 that are known to facilitate the CXCR2-dependent migration by monocytes, our aim is to evaluate whether introduction of the CXCR2 gene into tumor-specific T cells could further improve the effectiveness of ACT, by enhancing T-cell migration to tumor.
Experimental Design
In this study, we utilized transgenic pmel-1 T cells which recognize gp100 in the context of H-2Db, that were transduced with luciferase gene to monitor the migration of transferred T cells in vivo. In order to visualize luciferase-expressing T cells within a tumor, a non-pigmented tumor is required. Therefore, we utilized the MC38 tumor model which naturally expresses CXCL1.
Results
Mice bearing MC38/gp100 tumor cells treated with CXCR2/luciferase-transduced pmel-1 T cells showed enhanced tumor regression and survival compared to mice receiving control luciferase transduced pmel-1 T cells. We also observed preferential accumulation of CXCR2-expressing pmel-1 T cells in the tumor sites of these mice using bioluminescence imaging. A similar enhancement in tumor regression and survival was observed when CXCR2-transduced pmel-1 T cells were transferred into mice bearing CXCL1-transduced B16 tumors compared to mice treated with control pmel-1 T cells.
Conclusions
These results implicate that the introduction of the CXCR2 gene into tumor-specific T cells can enhance their localization to tumors and improve antitumor immune responses. This strategy may ultimately enable personalization of cancer therapies based on chemokine expression by tumors.
Keywords: CXCR2, T cell-mediated antitumor immune response, Adoptive cell transfer (ACT), melanoma immunotherapy
Introduction
Given the demonstrated effectiveness of T cells in mediating antitumor immune responses, T cell-mediated immunotherapy is considered an important and promising therapeutic approach against cancer. In particular, adoptive cell transfer (ACT) combined with lymphodepletion has resulted in clinical responses in ~50 - 70% of patients with metastatic melanoma (1, 2). However, the requirement of large numbers of laboratory-expanded T cells (>1 × 1010) makes ACT a costly and labor-intensive treatment. In addition, although some ACT patients achieve long-term disease-free survival, most patients still recur with disease (3). One important limiting factor for ACT is the inefficient migration of T cells into tumor tissue. By labeling T cells prior to ACT, it has been shown that the number of adoptively transferred T cells that migrating to the tumor microenvironment correlates positively with clinical response (4). However, this analysis also demonstrated that the trafficking efficiency of transferred T cells was extremely low (5). Therefore, strategies aimed at improving the migration of T cells to tumor sites are likely to enhance the efficacy of ACT therapy and improve clinical response rates.
Chemokines are secreted proteins that are essential for mediating the trafficking of immune cells to sites of inflammation. Through chemotaxis, cells that express appropriate chemokine receptors can migrate along a chemokine gradient to localize to specific tissues or sites of infection (6). Chemokines also play an important role in T cell-mediated antitumor immune responses. For example, mice lacking CXCR6, the receptor for CXCL16, displayed reduced recruitment of activated effector T cells in breast tumor tissue and impaired tumor regression (7). Affymetrix gene expression analysis of metastatic melanoma specimens has shown that the presence of T cells in tumor tissue is associated with high expression of many chemokines (8, 9), suggesting that their migration to tumors may be regulated by chemokines. Although the majority of human melanoma tumors show high levels of expression of CXCL1 and CXCL8 (10-12), the number of tumor antigen-specific T cells in these tumors is very limited (8, 13). However, melanoma tumors almost invariably show high levels of monocyte/macropage infiltrates that are known to migrate in a CXCR2-dependent fashion towards CXCL1/CXCL8 gradients present in the tumor microenvironment (14, 15). Since T cells do not normally express CXCR2, we hypothesized that genetic modification of tumor antigen-specific T cells with the CXCR2 gene would promote their trafficking to tumor sites, thereby enhancing antitumor immune responses.
To evaluate whether CXCR2 expression in T cells enhances their tumor migration in vivo, we used an ultra-sensitive BLI system based on the detection of optimized firefly luciferase (OFL). We previously demonstrated that BLI using OFL is sensitive enough to track fewer than 10 adoptively transferred T cells in murine models (16), and since it is noninvasive it can allow for the evaluation of individual mice at multiple time points. Using this system, we show here that melanoma antigen-specific T cells transduced to express CXCR2 show improved migration to CXCL1 and gp100-expressing tumors. Furthermore, in two different tumor models tumor-bearing mice treated with CXCR2-expressing T cells showed significantly improved antitumor responses and survival. These data show that enforced CXCR2 expression by tumor-specific T cells can overcome poor migration into tumor sites, thus leading to improved antitumor immune responses.
Materials and Methods
Animals and cell lines
Pmel-1/Thy1.1+ TCR transgenic mice on a C57BL/6 background were kindly provided by Dr. Nicholas Restifo (Surgery Branch, National Cancer Institute, Bethesda, MD). C57BL/6J-Tyr-2J/J albino mice were purchased from the Jackson Laboratory. All mice were maintained in a specific pathogen-free barrier facility at The University of Texas M. D. Anderson Cancer Center. Mice were handled in accordance with protocols approved by the Institutional Animal Care and Use Committee. B16 murine melanoma and MC38 murine colon adenocarcinoma cells were obtained from the National Cancer Institute. All tumor cell lines were maintained in RPMI 1640 with 10% fetal calf serum (FCS) and 100 μg/ml Normocin (Invivogen).
Construction of viral vectors
Full-length murine CXCR2 (mCXCR2) (NP_034039) and mCXCL1 (NP_032202) were amplified from ATCC clones by PCR. The mCXCR2 and mCXCL1 were inserted into the pRV100 and pLV411G viral vector generated in our lab. Optimized firefly luciferase (OFL) was fused with GFP and inserted into pRV100 vectors, as described earlier (16). Full-length human gp100 (gp100) was amplified from the WRG-gp100 vector (gift from Dr. Nicholas Restifo) and subcloned into a lentiviral vector, pLV414G. Sequences of all constructs were verified.
Retrovirus production and transduction of pmel-1 T cells
Retroviral vectors and the packaging vectors were transiently cotransfected into the packaging cell line, Plate-E, using Lipofectamine 2000 (Invitrogen) as previously described (17). Supernatants were harvested 48 h later and concentrated between 25x and 100x using Centricon-Plus 20 tubes (Millipore). Splenocytes from pmel-1 mice were harvested and cultured in RPMI 1640 with 10% FCS, 100 μg/ml Normocin, 250 U/ml hIL-2 (Proleukin; Chiron, Emeryville, CA), and 0.3 μg/ml anti-mouse CD3 (BD Bioscience). After 24 h, the cells were infected with the appropriate virus in the presence of 1.6 μg/ml Polybrene (Sigma) and 2 μg/ml Lipofectamine 2000 under spin conditions at 850 g for 2 h and then incubated overnight at 37°C. The next day, fresh T-cell culture medium was added. Cells were stained with appropriate markers expressed by these viral constructs 3 d after transduction and sorted by a FACSAria (BD Bioscience).
Lentiviral transduction of tumor cells
Lentiviral vectors and packaging vectors, VSVG and Δ8.9, were cotransfected into 293 T cells using lipofectamine 2000, and supernatant was collected after 36 h culture. A total of 1 × 106 tumor cells were preseeded in each well of 6-well plates for 6 h and spun at 850 g for 1 h with 1 ml virus supernatant and 8 μg/ml polybrene. The following day, the supernatant was removed and replaced with growth medium. Infected tumor cells were collected and sorted based on the expression of the reporter gene using a FACSAria.
Immunohistochemistry
Immunohistochemistry was performed on paraffin-embedded melanoma tumor tissue sections. mAbs against human CXCL1 (Proteintech) and CXCL8 (Santa Cruz biotechnology) were used.
Chemotaxis assay
CXCR2-expressing or control T cells (1 × 106) were placed in 0.2 ml of complete medium in the upper chamber (3.0-μm pore size) of a well in a 24-well Transwell plate (Costar, Corning, NY). Medium (0.4 ml) containing chemokine at various concentrations or conditioned medium from the indicated tumor cells was placed in the lower chamber and the plates were incubated for 60 min at 37°C. Conditioned medium was taken from the overnight culture of 2 × 106 tumor cells in 6-well plates (which contained 3 ml of medium per well). The number of cells in the lower chamber was either manually counted, or when OFL-transduced T cells were used for the migration assay, evaluated by measuring luminescence activity after the addition of D-luciferin.
Tumor reactivity assay
To test antitumor reactivity of T cells, we seeded 1 × 105 pmel-1 T cells per well in a 96-well plate and cocultured them with 1 × 104 cells from the indicated tumor lines for 16-18 h. To increase the expression of major histocompatibility complex class (MHC) I molecules, we treated B16 tumor cells with 200 ng/ml of mouse IFN-γ (BD Bioscience) overnight before the assay. T cell responses were measured by quantifying IFN-γ in the culture supernatants by enzyme-linked immunosorbent assay (ELISA; R&D System, Minneapolis, MN), according to manufacturer’s protocols. A 51Cr-release assay was used to determine the cytotoxic tumor killing by pmel-1 T cells. Tumor cells were labeled with 51Cr for 1 h and then cocultured with pmel-1 T cells at various effector/target ratios. CTL activity was calculated as the percentage of specific 51Cr release using the following equation: % specific killing = (sample release - spontaneous release) ÷ (maximal release - spontaneous release) × 100%.
T cell proliferation assay
To evaluate the effect of mCXCL1 on T cell proliferation, we diluted anti-mouse CD3 to 5 μg/ml in PBS and used 100 μl/well to coat the bottoms of wells in a flat-bottom 96-well plate at 4°C overnight. T cells (5 × 104) were seeded into each well with or without mCXCL1 for 60 h. [3H] Thymidine was added for the last 16 h of culture. Cells were harvested and radioactivity counted in a scintillation counter. All experiments were performed in triplicate.
Measurement of CXCL1 in cell culture supernatants
Tumor cells (2 × 106) were cultured in 3 ml RPMI 1640 with 10% FCS for 24 h. The concentration of CXCL1 in the supernatants was determined by ELISA. Each reported value is the mean of triplicate assays.
Bone marrow-derived dendritic cells (DC)
The generation of DC from murine bone marrow cells was performed as previously described (18). Briefly, bone marrow cells were grown at a starting concentration of 1 × 106 cells/ml in complete medium in the presence of 20 ng/ml GM-CSF and 100 ng/ml IL-4 (Peprotech Inc., Rocky Hill, NJ). Fresh DC culture medium was added on days 2 and 4. Loosely adherent cells were transferred to a fresh Petri dish on day 6. The following day, nonadherent cells were resuspended in Opti-MEM media (Invitrogen) at a concentration of 1 × 106 cells/ml and pulsed with 10 μM H-2Db-restricted gp100 peptide (KVPRNQDWL) for 2.5 h at 37°C. Peptide-pulsed DCs were washed 3 times with PBS and immediately injected into mice.
Adoptive transfer, vaccination, and treatment
C57BL/6 albino mice were subcutaneously implanted with either 5 × 105 B16-CXCL1 melanoma cells or 5 × 105 MC38/gp100 tumor cells (day 0). On day 6, lymphopenia was induced by administering a nonmyeloablative dose (350 cGy) of radiation. On day 7, 1 × 106 transduced pmel-1 T cells were adoptively transferred into tumor-bearing mice (n = 5 to 9 per group), followed by intravenous injection of 5 × 105 peptide-pulsed DCs. Recombinant human IL-2 was intraperitoneally administered for 3 d after T cell transfer (1.2 × 106 I.U. once immediately after T cell transfer and 6 × 105 I.U. twice daily for the next 2 d). Tumor sizes were monitored every 2 d. Mice were sacrificed when the tumors exceeded 15 mm in diameter or when ulcers exceeded 2 mm. All experiments were carried out in a blinded, randomized fashion.
In vivo Bioluminescence Imaging (BLI)
Prior to imaging, mice were anesthetized with isoflurane and i.p. injected with 100 μl of 20 mg/ml D-Luciferin (Xenogen Corp., Alameda, CA). After 8 min, animals were imaged using an IVIS 200 system (Xenogen), according to the manufacturer’s manual. Living Image software (Xenogen) was used to analyze data. Regions of interest were manually selected and quantification is reported as the average of photon flux within regions of interest. The bioluminescence signal is represented as photons/s/cm2/sr.
Statistical analysis
The data were represented as mean ± standard error of the mean (SEM). Comparisons of differences in continuous variables between two groups were done using Student’s t test. Differences in tumor size and T cell number among different treatments were evaluated by analysis of variance (ANOVA) repeated-measures function. P-values are based on 2-tailed tests, with P < 0.05 considered statistically significant. The Kaplan-Meier test was used to compare mouse survival between groups by Graph Pad Prism 5.
Results
The presence of CXCR2-cognate chemokines in the tumor microenvironment and the absence of CXCR2 on tumor-infiltrating T cells
We previously analyzed chemokine expression in freshly isolated human melanoma tumors by cDNA microarray analysis. The expression of CXCL1 and CXCL8, two CXCR2-cognate chemokines, was markedly unregulated in the tumor tissue, when compared to levels in normal tissues (10). To confirm the presence of these chemokines in the melanoma tumor microenvironment, we evaluated tissue samples from melanoma patients for the expression of CXCL1 and CXCL8 by immunohistochemistry. Both chemokines were found to be present in the majority of analyzed metastatic lesions, as shown in representative photographs (Figures 1 A). Although a significant amount of CXCL1 and CXCL8 is expressed within the melanoma tumor microenvironment, very few T lymphocytes express the corresponding receptor, CXCR2. Analysis of peripheral blood mononuclear cells (PBMC) from healthy donors and human tumor-infiltrating lymphocyte (TIL) cultures determined that less than 5% of CD4+ or CD8+ T cells express CXCR2 (Fig. 1B and C). Hence, the adoptively-transferred T cells used in ACT therapy are unlikely to be capable of responding to the CXCL1/CXCL8 chemokine gradient present in the melanoma tumor microenvironment. We next explored the hypothesis that introducing the CXCR2 gene into tumor-specific T cells could enhance T-cell migration to tumor.
Figure 1.
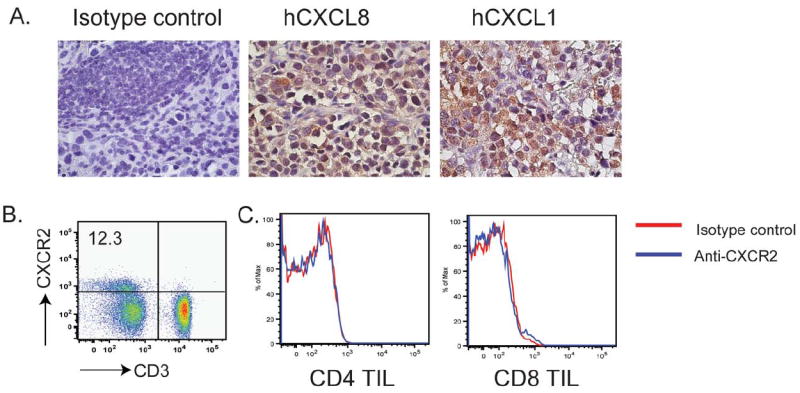
Human melanoma tumors express ligands for CXCR2, but tumor-infiltrating T cells lack CXCR2 expression. (A) Paraffin-embedded, melanoma lymph node metastases were analyzed by immunohistochemical staining for chemokine CXCL8 and CXCL1. Representative staining is shown at 40X magnification. (B) CXCR2 expression on PBMCs, as determined by flow cytometry. PBMCs from five healthy donors were stained with anti-CD3 and anti-CXCR2. (C) FACS analysis of CXCR2 expression on tumor-infiltrating lymphocytes (TILs). TILs isolated from four melanoma patients were stained with anti-CD4, CD8 and CXCR2. Representative results of all stained samples were shown.
Establishment of a murine tumor model to monitor migration and antitumor activity of adoptively transferred T cells
Monitoring T-cell migration to tumor sites has historically been challenging due to poor sensitivity of detection, but we recently described an optimized firefly luciferase gene (OFL) that allows very low numbers of transduced murine T cells to be visualized in vivo (16). To further increase sensitivity of T-cell detection in these studies, we utilized non-pigmented, albino C57BL/6 mice and non-pigmented MC38 tumors transduced to stably express the melanoma tumor antigen gp100 (Fig. 2A). The MC38/gp100 tumor cells were recognized by gp100-specific, TCR-transgenic pmel-1 T cells as shown by IFN-γ production and antigen-specific cytolysis in vitro (Fig. 2B and data not shown). Expression of gp100 did not alter the growth rate of the MC38 tumors in vivo (Fig. 2C), but did cause their specific recognition by adoptively transferred pmel-1 T cells, resulting in suppression of MC38/gp100 tumor growth but not that of parental MC38 tumors (Fig. 2D). More importantly, when we transferred pmel-1 T cells into mice challenged with both MC38 and MC38/gp100 on opposite flanks, the majority of pmel-1 T cells migrated to the MC38/gp100 tumor site, with few cells migrating to the MC38 tumor site (Fig.2E). These results suggest that pmel-1 T cells specifically recognized MC38/gp100 tumor cells, and demonstrated the feasibility of monitoring the migration of tumor-specific T cells at the tumor site using luciferase imaging.
Figure 2.
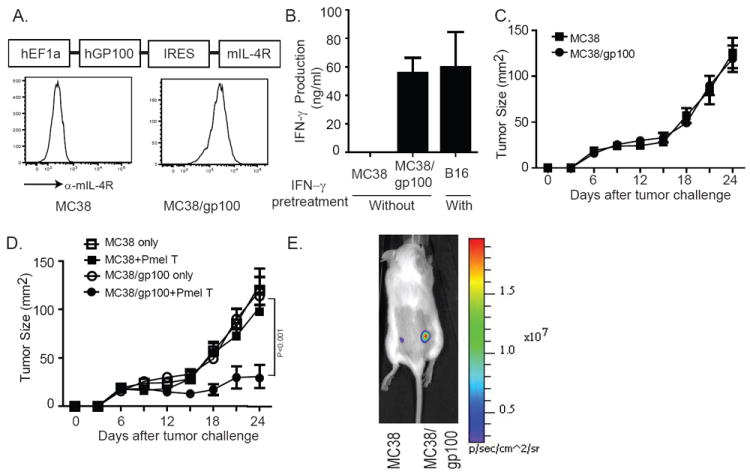
Generation of gp100-expressing MC38 tumor cell lines. (A) Schematic representation of viral vector containing human full-length gp100, an internal ribosomal entry site, and the mIL-4R gene. The construct was transduced into MC38 cells, which were then sorted based on IL-4R expression, as shown in right subpanel. (B) IFN-γ secretion by pmel-1 T cells in response to MC38/gp100 tumors. Pmel-1 T cells were cocultured with the indicated murine tumor cell lines. Twenty-four hours later, IFN-γ concentrations in the supernatants were detected by ELISA. B16 melanoma cells pretreated with IFN-γ were used as a positive control. (C) Growth of MC38/gp100 tumors in C57BL/6 albino mice. C57BL/6 albino mice were s.c. injected with 5 × 105 MC38 or MC38/gp100. Tumor sizes were measured every 3 d. (D) Treatment of MC38/gp100 tumors with pmel-1 T cells. C57BL/6 albino mice were s.c. injected with 5 × 105 MC38 or MC38/gp100. Six days later, all mice were received 350cGy irradiation. Seven days later, 5×106 (5M) pmel-1 T cells were transferred into tumor-bearing mice, along with 5 × 105 gp100 peptide-pulsed DC, both by intravenous injection, and systemic IL-2 treatment. Tumor-bearing mice receiving irradiation only served as the control group. Tumor sizes were measured every 3 d. Pmel-1 T cells suppressed the growth of MC38/gp100 tumors, but not MC38 tumors. Data are plotted as the mean of five mice per group + SEM. Results are representative of two experiments. (E) Pmel-1 T cells migrate specifically to MC38/gp100 tumors. C57BL/6 albino mice were subcutaneously injected with 5×105 MC38/gp100 in the right flank. The same number of MC38 tumor cells was implanted on the left flank. Seven days after tumor implantation, 1×106 sorted luciferase expressing pmel-1 T cells were transferred to tumor-bearing mice. Six days after adoptive transfer, the intensity of luciferase signaling was measured. Data shown were representative of five mice.
CXCR2-transduced T cells migrate specifically towards CXCL1 gradients in vitro
To study chemotactic migration of tumor-specific T cells, we transduced pmel-1 T cells with a retroviral vector containing the murine CXCR2 gene under the control of the MSCV LTR promoter. After retroviral transduction, >70% of the pmel-1 T cells expressed CXCR2 as measured by flow cytometry (Fig. 3A). To monitor the trafficking of transferred pmel-1 T cells in vivo by BLI, pmel-1 T cells were co-transduced with a retroviral vector expressing optimized firefly luciferase and green fluorescent protein (OFL-GFP). Transduced pmel-1 T cells were sorted based on positive GFP and CXCR2 expression to purities of greater than 90% (Fig. 3B). To ensure that introduction of the CXCR2 gene did not alter the effector function of T cells, transduced Pmel-1 T cells were co-cultured with parental MC38 or MC38/gp100 tumor cells, and cytokine production and cytolytic activity were evaluated. In these assays, CXCR2-expressing pmel-1 T cells recognized and responded to cognate antigen presented by MC38/gp100 tumor cells by producing IFN-γ and mediating specific target cell killing at levels comparable to OFL-GFP-transduced control T cells (Fig. 3C and D). As expected, wild type MC38 tumor cells failed to stimulate the activation of CXCR2-expressing pmel-1 T cells. These results demonstrated that retroviral vector-mediated expression of CXCR2 did not alter the antigen specificity of pmel-1 T cells.
Figure 3.
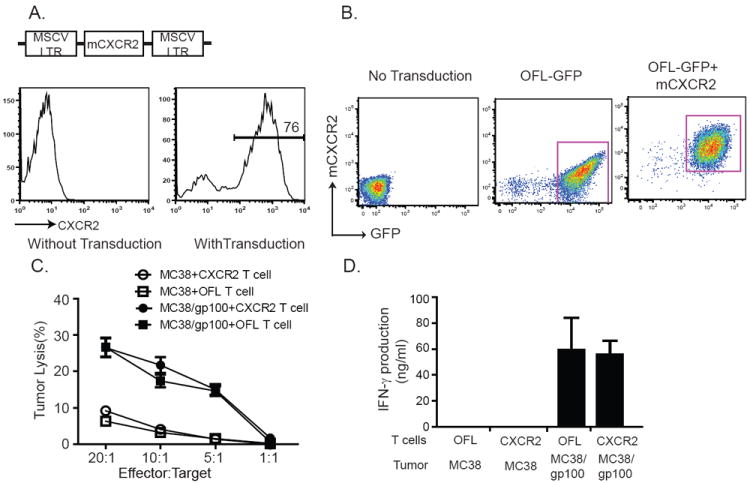
Generation and functional characteristics of CXCR2-expressing pmel-1 T cells. (A) Schematic representation of viral vector containing mCXCR2. Long terminal repeat from the murine stem cell PCMV virus drives high-level, constitutive expression of mCXCR2 in pmel-1 T cells. (B) FACS analysis of transduced pmel-1 T cells. Pmel-1 T cells were transduced with modified OFL-GFP alone or OFL-GFP and mCXCR2. Cells were sorted based on the expression of GFP or CXCR2. Sorted cells were cultured for 3 d and used for ACT. Before transfer, the purity of transferred cells was evaluated by flow cytometry. (C) Cytotoxic reactivity of CXCR2-expressing pmel-1 T cells against MC38/gp100 tumor. (D) IFN-γ secretion by CXCR2-expressing pmel-1 T cells in response to MC38/gp100 tumors.
Although rodents lack a homologue to human CXCL8/IL-8, they do possess CXCL1/KC that can bind to CXCR2 and has been reported to contribute to neutrophil migration (19). Therefore, we next tested whether ectopic expression of CXCR2 in murine T cells could endow them with the capacity to respond to the cognate ligand CXCL1. Using a transwell system, we observed that CXCR2 expression induced pmel-1 T cells to migrate specifically towards CXCL1 in a dose-dependent fashion (Fig. 4A). We also confirmed the ability of CXCR2-expressing pmel-1 T cells to migrate towards CXCL1 produced by two different murine tumor cell lines. Production of CXCL1 was robust and comparable between MC38 and MC38/gp100 tumor cells (Fig. 4B); however, B16 melanoma tumors produced low levels of CXCL1, necessitating the establishment of a stable, CXCL1-expressing B16 tumor cell line (B16-CXCL1) (Fig. 4B). As shown in Fig. 4C, conditioned medium from either MC38 or CXCL1-expressing B16 tumor cells induced enhanced migration of CXCR2-expressing pmel-1 T cells, but not control T cells, in a dose-dependent manner. By contrast, culture medium from wild-type B16 tumor cells did not increase T-cell migration, confirming that the enhanced migration was specific for CXCL1. Furthermore, the expression of CXCL1 did not alter the proliferative capacity of CXCR2-expressing pmel-1 T cells in response to specific T cell receptor (TCR) stimulation (Fig. 4D). T cells expressing CXCR2, but not control T cells, produced low levels of IFN-γ upon CXCL1 stimulation. In contrast, both CXCR2-expressing and non-expressingn T cells produced similar high levels of IFN-γ when treated with both TCR stimulation and CXCL1 (Fig. 4E). Taken together, these results suggest that the introduction of the CXCR2 gene into pmel-1 T cells enhanced their migration along a CXCL1 gradient in vitro in a dose-dependent manner without altering their proliferation or effector function. These results are consistent with those from our previous in vitro study using CXCR2-expressing human T cells (10).
Figure 4.
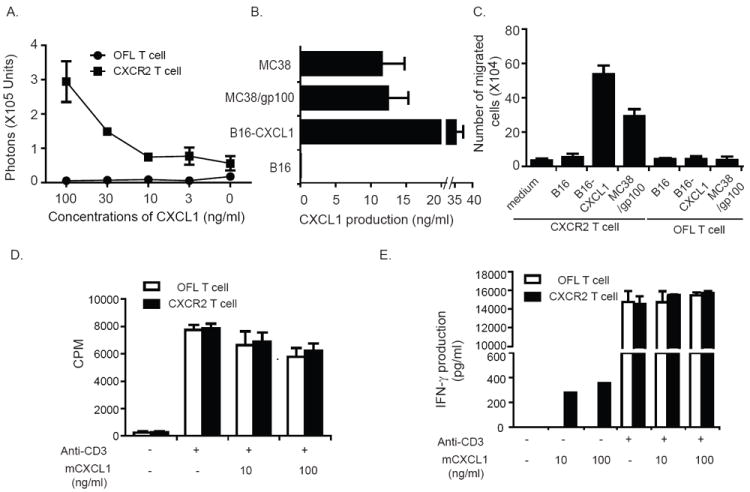
Enhanced migration of CXCR2-expressing T cells along CXCL1 gradients. (A) Migration of CXCR2-expressing T cells along recombinant CXCL1 gradients. C57BL/6 T cells were transduced with either OFL-GFP or OFL-GFP and mCXCR2 and placed into the upper wells of a Transwell system, with CXCL1 in the lower wells at the indicated concentrations. One hour later, D-luciferin was added to the lower wells. T cell migration to the lower well was quantitated by measuring luminescent activity. (B) CXCL1 production by murine tumor lines. Cells (2 × 106) from the indicated murine cell lines were plated in 6-well plates; 24 h later, CXCL1 in supernatants was detected by ELISA. (C) Migration of CXCR2-expressing T cells along tumor cell-derived CXCL1 gradients. C57BL/6 T cells were transduced with either OFL-GFP or OFL-GFP and mCXCR2 and placed into the upper wells of a Transwell system. Conditioned cell culture medium from tumor cells was loaded into the lower wells. One hour after incubation, the cell number in the lower wells was counted. (D) Proliferation of CXCR2-expressing pmel-1 T cells upon TCR stimulation in the presence or absence of CXCL1. CXCR2-expressing T cells or OFL-expressing T cells were stimulated with anti-CD3 and irradiated splenocytes in the presence or absence of recombinant CXCL1. T-cell proliferation was evaluated by 3[H] thymidine incorporation assay. (E) IFN-γ secretion by CXCR2-expressing pmel-1 T cells upon TCR stimulation in the presence or absence of CXCL1. CXCR2-expressing T cells or OFL-expressing T cells were stimulated with anti-CD3 and irradiated splenocytes in the presence or absence of recombinant CXCL1.
CXCR2-expressing T cells demonstrate enhanced trafficking to tumor in vivo
We next analyzed the migration of CXCR2-expressing pmel-1 cells to tumors in vivo. Pmel-1 T cells (1 × 106) expressing OFL-GFP alone or OFL-GFP and CXCR2 were intravenously injected into C57BL/6 albino mice bearing MC38/gp100 tumors (average tumor size = 20 mm2). Mice receiving pmel-1 T cells also received DC vaccination on the day of T-cell transfer, as well as systemic IL-2 treatment for 3 days, as described previously (Supplementary Fig 1A) (20). Luciferase intensity at the tumor site, which reflects the accumulation of adoptively transferred pmel-1 T cells, was monitored on a daily basis. Although the transferred pmel-1 T cells were detected by BLI in tumor sites as early as the second day after ACT, most of the transferred pmel-1 T cells were initially found in the lung and liver (Fig. 5A). Six days after T cell transfer, the majority of the pmel-1 T cells were localized at the tumor site. This pattern of distribution of transferred tumor-specific T cells is consistent with that previously reported in a murine study using a single-photo emission computed tomography (CT)-fusion imaging method for T-cell detection (21). Therefore, we used the luciferase results from mice on day 6 after T-cell transfer as representative data to characterize the tumor migration of transferred pmel-1 T cells. As shown in Fig. 5B, mice that received CXCR2-expressing T cells displayed a stronger luciferase signal at the tumor site than mice that received control T cells on day 6 after T-cell transfer. The average luciferase activity in all mice (n = 8-9) from each group was quantified (Fig. 5C), and showed that treatment with CXCR2-expressing T cells induced more than two-fold higher pmel-1 T-cell infiltration than mice treated with control T cells. By contrast, the percentage of pmel-1 T cells in the peripheral blood remained similar between the two groups of mice (Fig. 5D). In B16 tumors engineered to express CXCL1, we also found an increased percentage of CXCR2-expressing pmel-1 T cells at the tumor site, but not in the spleen, when compared with control T cells (Supplementary Fig 1B). Moreover, the percentages of IFN-γ + pmel T-cells were comparable between mice receiving CXCR2-expressing T cells or control T-cells, further confirming that introducing the CXCR2 gene into tumor-specific T cells does not alter their effector function (Supplementary Fig 1B). These results demonstrate that transduction of T cells with the CXCR2 gene resulted in their preferential accumulation to tumor sites expressing CXCL1.
Figure 5.
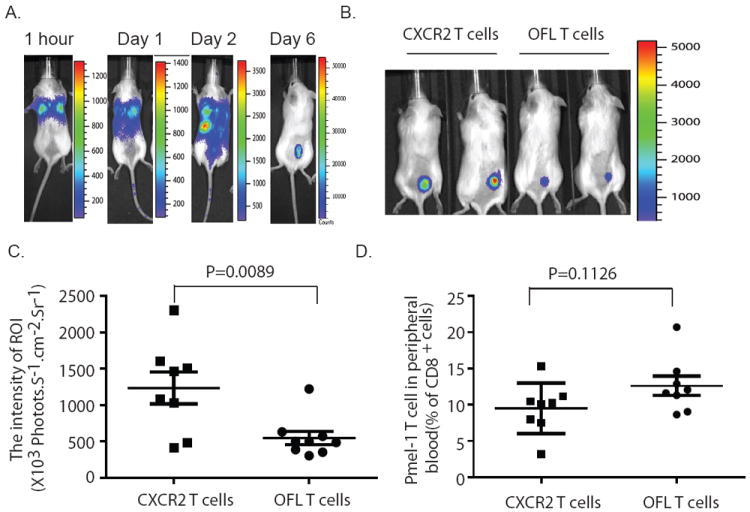
Enhanced migration of CXCR2-expressing pmel-1 T cells to tumor sites. (A) In vivo trafficking of OFL-expressing pmel-1 T cells. OFL-expressing pmel-1 T cells (1 × 106) were transferred into mice bearing established 7-day MC38/gp100 tumors. DC vaccine and IL-2 treatment were performed as previous described. Imaging was performed at indicated time points after T cell transfer. (B) Increased accumulation of CXCR2-expressing pmel-1 T cells at the tumor site. Pmel-1 T cells (1 × 106) expressing either OFL alone or CXCR2 with OFL were transferred into mice bearing established MC38/gp100 tumors. Imaging was performed at day 6 after T cell transfer. Data shown were from representative mice. (C) Quantitative imaging analysis of transferred T cells in tumor-bearing mice. Intensities of the luciferase signal at tumor sites in all tumor-bearing mice are depicted. (D) Percentage of Thy1.1+ pmel-1 T-cells of the CD8+ T cells in the peripheral blood on day 6 after T cell transfer. Thy1.1 is a congenic cell surface marker for transferred pmel-1 T cells. Data for (C) and (D) are plotted as the mean of eight mice per group + SEM. Results are representative of three experiments.
CXCR2-expressing T cells show superior anti-tumor activity in vivo
We next assessed whether CXCR2-mediated accumulation of pmel-1 T cells in tumors would result in more potent antitumor activity. Seven days after challenge with MC38/gp100 tumors, mice were treated with ACT and tumor growth was monitored. As described previously, transfer of 1 × 106 pmel-1 T cells combined with DC vaccination and IL-2 resulted in a significant decrease of MC38/gp100 tumor growth (Fig. 6A) (20). More importantly, tumor growth was significantly reduced in mice receiving CXCR2-expressing pmel-1 T cells compared to mice receiving control (OFL only) pmel-1 T cells (Fig. 6A). The increased tumor suppression by CXCR2-expressing pmel-1 T cells translated into a significant increase in the survival of mice bearing MC38/gp100 tumors (Fig. 6B). In B16-CXCL1 tumor model, tumor growth was also significantly delayed and survival was extended in mice treated with CXCR2-expressing pmel-1 T cells compared with control pmel-1 T cells (Fig. 6C,D), further corroborating our findings in a tumor model that naturally expresses gp100. CXCR2-expressing pmel-1 T cells did not confer better protection in mice bearing wild-type B16 tumors, likely due to the lack of CXCL1 expression by native B16 tumors (Fig.6E). In the B16-CXCL1 model, tumor growth in mice receiving 1 × 106 CXCR2-expressing pmel-1 T cells was comparable to that of mice receiving 5 × 106 control pmel-1 T cells, suggesting a 5-fold increase in antitumor activity (Fig. 6F). These data support our hypothesis that enforced expression of CXCR2 in tumor-specific T cells enhances their therapeutic antitumor activity.
Figure 6.
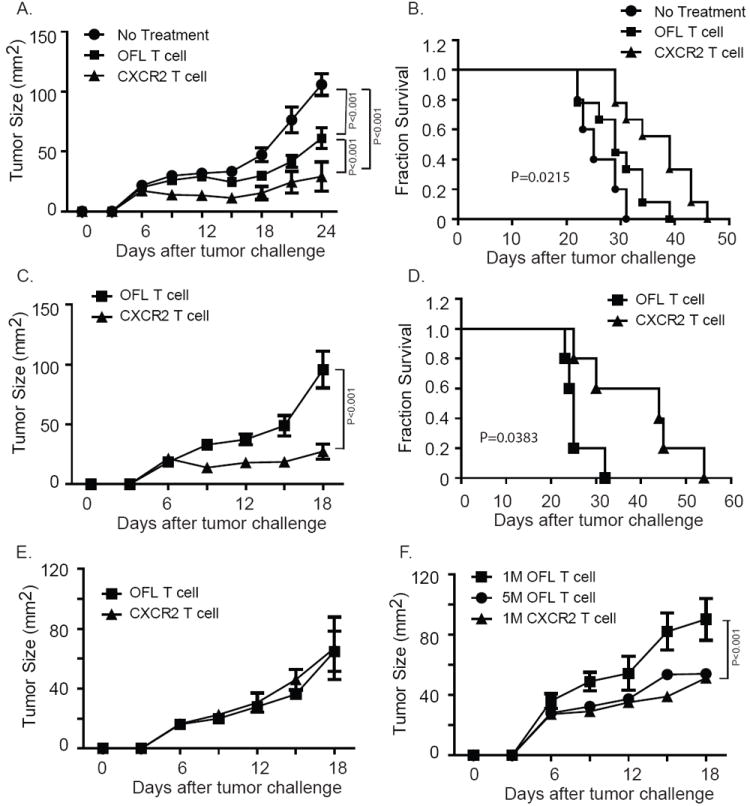
Enhanced suppression of established tumors upon ACT with CXCR2-expressing pmel-1 T cells. Groups of mice were implanted with 5 × 105 tumor cells on day 0, subjected to 350 cGy TBI at day 6, followed on day 7 by ACT with pmel-1 T cells expressing either OFL alone or OFL and CXCR2, along with DC vaccination and systemic IL-2. (A) Tumor growth curve of MC38/gp100 tumor-bearing mice receiving ACT. (B) Kaplan-Meier survival curves of MC38/gp100 tumor-bearing mice treated with ACT. (C) Tumor growth curve and (D) Kaplan-Meier survival curves of B16-CXCL1 tumor-bearing mice treated with ACT with CXCR2-transduced pmel-1 T cells. Tumor growth was monitored every 3 d and plotted as means + SEM. P values in panels (B) and (D) were determined by two-way ANOVA. In both tumor models, CXCR2 transduction enhanced the ability of the T cells to mediate tumor regression and increased survival. Similar results were obtained in repeated experiments. In (A) and (B), N=8-9 for each treatment group; in (C) and (D), n=5 for each treatment group. Results are representative of two experiments. (E) Tumor growth curve of B16 tumor-bearing mice receiving ACT. N=3 for each treatment group. (F) Tumor growth curve of B16-CXCL1 tumor-bearing mice receiving ACT using different numbers of OFL or CXCR2 expressing pmel-1 T cells. Similar protocols for tumor challenge and ACT treatment were followed. Tumor-bearing mice were infused with either 1 × 106 (1M), 5× 106 (5M) OFL expressing T cells, or 1× 106 CXCR2 expressing T cells on day 7 after tumor challenge. N= 3 to 5 mice for each treatment group.
Discussion
Effective immunologic destruction of large and established tumors requires not only a sufficient number of activated T cells that can recognize tumor antigens with high avidity but also the ability of these cells to migrate to sites of malignancy (22). Several lines of evidence support the hypothesis that impaired tumor homing of T cells is often a rate-limiting step in antitumor immunotherapy, particularly the observation in both murine and human studies that tumors frequently continue to grow in the presence of large numbers of circulating, tumor-specific T cells (23, 24). This point is highlighted by the disappointing clinical response rates resulting from immunization with cancer vaccines, despite the successful generation of tumor-specific T cell reactivity in vivo (25). Even with ACT approaches utilizing nonmyeloablative lymphodepletion regimens, where upwards of 75% of the circulating CD8+ T cells can show antitumor activity, approximately half of metastatic melanoma patients still fail to respond to the treatment (26). Monitoring of transferred T cells have shown that the majority of infused T cells localize in the lung, liver, and spleen, while less than 1% of the total transferred T cells migrate to the tumor (5, 27). Finally, extensive T-cell infiltrates are commonly observed in tumors undergoing immunologic rejection, and the presence of such infiltrates correlates well with increased objective response rates (25). In contrast, tumors that are refractory to T cell-based immunotherapies rarely accumulate strong immune cell infiltrates. Compared to the large number of adoptively transferred tumor-specific T cells required for effective treatment (1010-1011), the tumor-infiltrating T cells that presumably mediate tumor rejection in situ represent a small fraction of the total T cells infused (4). There is also growing evidence that suggests further proliferation of T cells in vivo is required for tumor trafficking and regression (20, 28, 29). Collectively, these observations strongly suggest that T-cell homing to tumors is a relatively inefficient process. Furthermore, our own unpublished data also demonstrates that clinical response to ACT positively correlates with the total number of T cells infused, suggesting that providing more cells to the tumor site may improve clinical responses.
CXCR2 is one of the G protein-coupled receptors capable of binding to both CXCL1 and CXCL8 (11). It has been shown to regulate the migration of immune effector cells. For example, Smith et al. found that CXCR2 plays an important role in arresting the rolling of neutrophils to inflamed sites by using CXCR2-deficient mice (30). To take advantage of the high concentration of CXCL1 and CXCL8 in the tumor microenvironment, we transduced pmel-1 T cells with a viral vector expressing the mCXCR2 gene. Our results demonstrated that the expression of CXCR2 by pmel-1 T cells enabled these cells to respond to CXCL1 gradients in the tumor but did not alter their effector functions. These results are consistent with those from our previous study using CXCR2-expressing human T cells (10). Therefore, it is possible that inducing expression of the CXCR2 gene in human tumor-specific T cells could lead these cells to migrate toward CXCL1 and CXCL8 gradients in the melanoma tumor microenvironment in patients.
To further confirm this possibility in vivo, we used the newly established MC38/gp100 tumor model, which allows us to monitor trafficking of transferred tumor-specific pmel-1 T cells in vivo. We found that tumor-specific T cell trafficking to tumor sites was improved following CXCR2 transduction of the T cells. Enhanced trafficking of CXCR2-expressing pmel-1 T cells resulted in delayed tumor growth and extended the survival of tumor-bearing mice. Our results also suggest a positive correlation between increased migration of tumor-specific T cells to the tumor and efficacy of ACT. These findings support our hypothesis that migration of T cells to the tumor is an important and limiting factor for tumor regression in cancer patients. Since the percentage of tumor-specific T cells among the total transferred T cells in patients receiving ACT is much lower than that in most preclinical studies (eg, almost 100% of the transferred pmel-1 T cells were tumor-specific in this study), the recruitment of tumor-specific T cells into tumor sites in the clinical setting may be even more critical for the success of ACT. Therefore, we expect an enhancement in antitumor immune response in patients treated with CXCR2-transduced TIL compared to patients receiving standard ACT.
Utilizing gene-modified T cells to increase the effectiveness of ACT is challenging in cancer patients. However, this approach is feasible. Besides our own studies (31), other groups have also conducted clinical trials with genetically modified T cells. One group conducted a trial using ACT of TIL cells that had been genetically engineered to produce IL-2 and found that transferred TIL persisted better after IL-2 withdrawal (32, 33). In another clinical trial, 11 patients with neuroblastoma were infused with Epstein-Barr virus-specific CTLs transduced with a chimeric antigen receptor that could recognize a tumor-associated antigen expressed by human neuroblastoma cells, disialoganglioside GD2. The engagement of GD2 and these genetically engineered receptors could trigger T-cell activation and infusion of these genetically modified T cells was associated with tumor regression (34). These studies have shown that infusion of genetically modified T cells is feasible and safe and may benefit cancer patients treated with ACT.
While most current strategies to genetically modify tumor-specific T cells are aimed at enhancing T-cell persistence or tumor recognition, our study focuses on an alternate strategy to increase the effectiveness of ACT by promoting T cell homing to the tumor site. This approach may provide an important new avenue in “personalized cancer therapy” to take advantage of defined chemokine signatures within the tumor microenvironment and may further benefit cancer patients undergoing ACT. Based on the results described above, we are currently planning a clinical trial to treat melanoma patients with CXCR2-transduced TILs.
Supplementary Material
Supplementary Figure 1. Increased numbers of CXCR2-expressing T cells at tumor sites in mice bearing B16-CXCL1 tumors. (A) Schematic representation of treatment schedule. (B) Percentage of Thy1.1+ pmel-1 T-cells of CD8+ T cells in tumor and spleen on day 6 after T cell transfer. (C) Percentage of IFN-γ + T-cells of transferred pmel-1 T cells in tumor and spleen on day 6 after T cell transfer. Cells harvested from tumor and spleen were stained with anti-CD8, Thy1.1 and IFN-γ antibodies. For stimulation, cells were treated with irradiated cultured DCs in presence of gp100 peptide in vitro for 4 hours.
Statement of Translational Relevance.
Adoptive T-cell therapy (ACT) has tremendous potential for the treatment of patients with advanced cancers, but a major limitation is the inefficient migration of T cells to the tumor site. In this paper, we report our success in enhancing the migratory ability of T-cell to tumors, further improving the antitumor immune response and survival in tumor-bearing mice in two distinct tumor models, by transduction of tumor-specific T cells with the gene encoding CXCR2. These results suggests that transducing CXCR2 into tumor-specific T cells provides an important avenue in “personalized cancer therapy” to take advantage of defined chemokine signatures within the tumor microenvironment, and is a significant step forward in the improvement of adoptive T-cell therapy. With these results, we planed to utilize the CXCR2 receptor in our ACT-based clinical trials in the next two years here at MD Anderson Cancer Center.
Acknowledgments
Grant support: The University of Texas M. D. Anderson Cancer Center and National Institutes of Health grant R01 CA116206-02 (P. Hwu)
References
- 1.Dudley ME, Wunderlich JR, Yang JC, et al. Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol. 2005;23:2346–57. doi: 10.1200/JCO.2005.00.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wrzesinski C, Paulos CM, Kaiser A, et al. Increased Intensity Lymphodepletion Enhances Tumor Treatment Efficacy of Adoptively Transferred Tumor-specific T Cells. J Immunother. 2009 doi: 10.1097/CJI.0b013e3181b88ffc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosenberg SA, Dudley ME. Adoptive cell therapy for the treatment of patients with metastatic melanoma. Current opinion in immunology. 2009;21:233–40. doi: 10.1016/j.coi.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pockaj BA, Sherry RM, Wei JP, et al. Localization of 111indium-labeled tumor infiltrating lymphocytes to tumor in patients receiving adoptive immunotherapy. Augmentation with cyclophosphamide and correlation with response. Cancer. 1994;73:1731–7. doi: 10.1002/1097-0142(19940315)73:6<1731::aid-cncr2820730630>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 5.Fisher B, Packard BS, Read EJ, et al. Tumor localization of adoptively transferred indium-111 labeled tumor infiltrating lymphocytes in patients with metastatic melanoma. J Clin Oncol. 1989;7:250–61. doi: 10.1200/JCO.1989.7.2.250. [DOI] [PubMed] [Google Scholar]
- 6.Pittet MJ, Mempel TR. Regulation of T-cell migration and effector functions: insights from in vivo imaging studies. Immunological reviews. 2008;221:107–29. doi: 10.1111/j.1600-065X.2008.00584.x. [DOI] [PubMed] [Google Scholar]
- 7.Matsumura S, Wang B, Kawashima N, et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J Immunol. 2008;181:3099–107. doi: 10.4049/jimmunol.181.5.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gajewski TF. Failure at the effector phase: immune barriers at the level of the melanoma tumor microenvironment. Clin Cancer Res. 2007;13:5256–61. doi: 10.1158/1078-0432.CCR-07-0892. [DOI] [PubMed] [Google Scholar]
- 9.Harlin H, Meng Y, Peterson AC, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer research. 2009;69:3077–85. doi: 10.1158/0008-5472.CAN-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kershaw MH, Wang G, Westwood JA, et al. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Human gene therapy. 2002;13:1971–80. doi: 10.1089/10430340260355374. [DOI] [PubMed] [Google Scholar]
- 11.Vandercappellen J, Van Damme J, Struyf S. The role of CXC chemokines and their receptors in cancer. Cancer letters. 2008;267:226–44. doi: 10.1016/j.canlet.2008.04.050. [DOI] [PubMed] [Google Scholar]
- 12.Waugh DJ, Wilson C. The interleukin-8 pathway in cancer. Clin Cancer Res. 2008;14:6735–41. doi: 10.1158/1078-0432.CCR-07-4843. [DOI] [PubMed] [Google Scholar]
- 13.Lizee G, Cantu MA, Hwu P. Less yin, more yang: confronting the barriers to cancer immunotherapy. Clin Cancer Res. 2007;13:5250–5. doi: 10.1158/1078-0432.CCR-07-1722. [DOI] [PubMed] [Google Scholar]
- 14.Dirkx AE, Oude Egbrink MG, Wagstaff J, Griffioen AW. Monocyte/macrophage infiltration in tumors: modulators of angiogenesis. Journal of leukocyte biology. 2006;80:1183–96. doi: 10.1189/jlb.0905495. [DOI] [PubMed] [Google Scholar]
- 15.Nardin A, Abastado JP. Macrophages and cancer. Front Biosci. 2008;13:3494–505. doi: 10.2741/2944. [DOI] [PubMed] [Google Scholar]
- 16.Rabinovich BA, Ye Y, Etto T, et al. Visualizing fewer than 10 mouse T cells with an enhanced firefly luciferase in immunocompetent mouse models of cancer. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:14342–6. doi: 10.1073/pnas.0804105105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Naviaux RK, Costanzi E, Haas M, Verma IM. The pCL vector system: rapid production of helper-free, high-titer, recombinant retroviruses. Journal of virology. 1996;70:5701–5. doi: 10.1128/jvi.70.8.5701-5705.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steinman RM, Inaba K, Turley S, Pierre P, Mellman I. Antigen capture, processing, and presentation by dendritic cells: recent cell biological studies. Human immunology. 1999;60:562–7. doi: 10.1016/s0198-8859(99)00030-0. [DOI] [PubMed] [Google Scholar]
- 19.Czuprynski CJ, Brown JF, Steinberg H, Carroll D. Mice lacking the murine interleukin-8 receptor homologue demonstrate paradoxical responses to acute and chronic experimental infection with Listeria monocytogenes. Microb Pathog. 1998;24:17–23. doi: 10.1006/mpat.1997.0169. [DOI] [PubMed] [Google Scholar]
- 20.Lou Y, Wang G, Lizee G, et al. Dendritic cells strongly boost the antitumor activity of adoptively transferred T cells in vivo. Cancer research. 2004;64:6783–90. doi: 10.1158/0008-5472.CAN-04-1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pittet MJ, Grimm J, Berger CR, et al. In vivo imaging of T cell delivery to tumors after adoptive transfer therapy. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:12457–61. doi: 10.1073/pnas.0704460104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blattman JN, Greenberg PD. Cancer immunotherapy: a treatment for the masses. Science. 2004;305:200–5. doi: 10.1126/science.1100369. [DOI] [PubMed] [Google Scholar]
- 23.Peterson AC, Harlin H, Gajewski TF. Immunization with Melan-A peptide-pulsed peripheral blood mononuclear cells plus recombinant human interleukin-12 induces clinical activity and T-cell responses in advanced melanoma. J Clin Oncol. 2003;21:2342–8. doi: 10.1200/JCO.2003.12.144. [DOI] [PubMed] [Google Scholar]
- 24.Rosenberg SA, Sherry RM, Morton KE, et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J Immunol. 2005;175:6169–76. doi: 10.4049/jimmunol.175.9.6169. [DOI] [PubMed] [Google Scholar]
- 25.Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nature medicine. 2004;10:909–15. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosenberg SA, Dudley ME. Cancer regression in patients with metastatic melanoma after the transfer of autologous antitumor lymphocytes. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(Suppl 2):14639–45. doi: 10.1073/pnas.0405730101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bobisse S, Rondina M, Merlo A, et al. Reprogramming T lymphocytes for melanoma adoptive immunotherapy by T-cell receptor gene transfer with lentiviral vectors. Cancer research. 2009;69:9385–94. doi: 10.1158/0008-5472.CAN-09-0494. [DOI] [PubMed] [Google Scholar]
- 28.Kjaergaard J, Peng L, Cohen PA, Shu S. Therapeutic efficacy of adoptive immunotherapy is predicated on in vivo antigen-specific proliferation of donor T cells. Clinical immunology (Orlando, Fla) 2003;108:8–20. doi: 10.1016/s1521-6616(03)00090-1. [DOI] [PubMed] [Google Scholar]
- 29.Overwijk WW, Theoret MR, Finkelstein SE, et al. Tumor regression and autoimmunity after reversal of a functionally tolerant state of self-reactive CD8+ T cells. J Exp Med. 2003;198:569–80. doi: 10.1084/jem.20030590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Smith ML, Olson TS, Ley K. CXCR2- and E-selectin-induced neutrophil arrest during inflammation in vivo. J Exp Med. 2004;200:935–9. doi: 10.1084/jem.20040424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kershaw MH, Westwood JA, Parker LL, et al. A phase I study on adoptive immunotherapy using gene-modified T cells for ovarian cancer. Clin Cancer Res. 2006;12:6106–15. doi: 10.1158/1078-0432.CCR-06-1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu K, Rosenberg SA. Interleukin-2-independent proliferation of human melanoma-reactive T lymphocytes transduced with an exogenous IL-2 gene is stimulation dependent. J Immunother. 2003;26:190–201. doi: 10.1097/00002371-200305000-00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heemskerk B, Liu K, Dudley ME, et al. Adoptive cell therapy for patients with melanoma, using tumor-infiltrating lymphocytes genetically engineered to secrete interleukin-2. Human gene therapy. 2008;19:496–510. doi: 10.1089/hum.2007.0171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pule MA, Savoldo B, Myers GD, et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nature medicine. 2008;14:1264–70. doi: 10.1038/nm.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Increased numbers of CXCR2-expressing T cells at tumor sites in mice bearing B16-CXCL1 tumors. (A) Schematic representation of treatment schedule. (B) Percentage of Thy1.1+ pmel-1 T-cells of CD8+ T cells in tumor and spleen on day 6 after T cell transfer. (C) Percentage of IFN-γ + T-cells of transferred pmel-1 T cells in tumor and spleen on day 6 after T cell transfer. Cells harvested from tumor and spleen were stained with anti-CD8, Thy1.1 and IFN-γ antibodies. For stimulation, cells were treated with irradiated cultured DCs in presence of gp100 peptide in vitro for 4 hours.