

Abstract
The catalytic domain, known as light chain (Lc), of the most poisonous botulinum neurotoxins (BoNTs), possesses endoprotease activity that triggers the ultimate poisonous effect to animals and humans. X-ray crystallographic structure of Lc of several BoNT serotypes has identified at least four small ligands at or near the respective active sites. They are sulfate ions in LcA, LcB, and LcE; an acetate ion in LcA; a calcium ion in LcB; and a potassium ion in LcD. Roles of these ligands on the structure and function of the proteins are not known. We have investigated the roles of sulfate, acetate, and calcium on the catalytic activities of LcA, LcB, and LcE using 17-35-residue synthetic peptide substrates. All three ligands inhibited all Lc activities. For LcA and LcB, the order of inhibition effectiveness was calcium>sulfate>acetate. The inhibition effectiveness expressed as IC50, did not correlate with the occurrence or proximity of the ions to the active site. Moreover, addition of acetate or sulfate to LcA did not affect the near-UV circular dichroism spectra, tryptophan, and tyrosine fluorescence spectra, and mid points of thermal denaturation of LcA. Our results suggest that acetate, sulfate, and calcium nonspecifically interact with BoNT Lc, and their occurrence in the crystal structures could have been due to opportunistic binding to complementary pockets.
Keywords: Botulinum neurotoxin, endopeptidase, protease, active site, inhibitor, small molecule
Introduction
Small ubiquitous ions such as sodium and potassium often get associated with larger molecules, both in the cellular or in the extracellular environments. Depending on site of interaction, they may remain inert or impart essential function to the macromolecule. In situations when a ligand is somewhat larger and is not as ubiquitous, serious attention is warranted to establish or disprove a specific role of the ligand on the structure and/or function of a protein. The catalytic endoprotease domains of the most toxic botulinum neurotoxin (BoNT1) are few such proteins where at least four ligands have been detected in the three-dimensional structures.
BoNT produced by strains of Clostridium botulinum, are the deadliest of all toxins [1]. There are seven distinct serotypes, (A-G), of which BoNT/A afflicts humans most frequently. The latter is also the most stable in neurons causing long-term paralysis. The toxins are 150 kDa proteins comprising a 50 kDa zinc-endopeptidase light chain (LC) domain and a 100 kDa binding-translocating heavy chain (HC) domain. Upon entry to animal body, the toxins travel to peripheral neurons where HC binds and translocates LC into the neurons. After dissociating from HC [2], LC cleaves at specific sites on one of three SNARE proteins, thereby blocking exocytosis of acetylcholine into the neuromuscular junction and causing muscle paralysis. The paralysis prevents normal respiration and eventually leads to death of the animal. One of the hallmark features of BoNT/A intoxication is the extreme persistence of symptoms with neuroparalysis, often lasting in excess of 3 to 4 months. Currently, there is no effective postex posure therapeutic available against botulism. Because of extreme toxicity, BoNT is a potential biowarfare and bioterrorism threat, and both Centers for Disease Control and Prevention and the U.S. Department of Agriculture have placed it as a ‘category A’ threat agent.
Since the publication of the first X-ray crystallographic determination of BoNT/A structure in 1998 [3], structures of BoNT/B, BoNT/E, and those of LC of all the serotypes are now available. At least in two cases, sulfate and acetate have been found to be tightly bound at or near the active site of LcA and LcB, respectively. For example, sulfate ion was found tightly bound 3Å from the zinc ion at the active site of BoNT/B [4]. While it is possible that the sulfate was picked up by BoNT/B during purification, its coordination with the active site zinc by replacing the nucleophilic water may suggest some functional role. In addition, Ca++ was bound near the active site of BoNT/B Lc [5], and a potassium ion was bound to LcD (PDB: 2FPQ) [6]. The presence of a sulfate at the catalytic site has also been reported in the BoNT/E [7]. Similarly in an “unliganded” LcA structure, an acetate molecule replaced the nucleophilic water that otherwise coordinates with the active site zinc [8]. In addition to acetate, two sulfate ions were also identified in the “unliganded” and several active site inhibitor liganded LcA structures of its co-crystals [8]. One of these sulfate-oxygens makes weak interaction with guanidino nitrogen of the P1 arginine of the substrate [8,9]. Location of these ligands and their interactions with protein residues are shown in Figure 1. Isolation and purification of LcA used in these studies, however, did not involve deliberate exposure of the protein to acetate or sulfate. Their tightly bound occurrence at and near the active sites, thus might also signify a functional role.
Figure 1.

Coordination of acetate, sulfate, and calcium ions with only the active-site and near-active site residues in BoNT light chains. A: LcA-acetate complex (PDB ID 3BWI), B: LcA-SO4-2 complex (PDB ID 3BWI), C: LcA-SO4-2 complex (PDB ID 3BWI), D: LcB-SO4-2 complex (PDB ID 1EPW, E: LcB-Ca+2 complex (PDB ID 1SOB) and F: LcE-SO4-2 complex (PDB ID 1ZL6). The figures were generated by using RCSB PDB Ligand Explorer 3.9 software freely available at http://www.rcsb.org/pdb/explore.
As part of our ongoing efforts in structurebased design of BoNT/Lc inhibitors, we have reported several tetra-peptides which are among the most efficient inhibitors of the LcA [8-11]. To improve and expand the therapeutic use of these inhibitors and to broaden our understanding on their inhibitions under cellular environment, it is important to delineate the effects of small ions on LcA activity as well as to investigate their roles in other Lcs. Here, we report the effects of sulfate, acetate, and calcium on the catalytic activities of LcA, LcB, and LcE. Our results show that though all of these ions inhibit activities of all three proteins, they are non specific, and their occurrence in the crystal structures could have been opportunistic events.
Materials and methods
Materials
Recombinant LcA and LcB were purified as described [12-14]. Recombinant LcE was from List Biologicals (Campbell, CA). Each light chain (0.5-1 mg/ml) was thawed from -20 °C to room temperature, passed through a PD-10 column and collected in 50 mM Na-phosphate buffer, pH 6.5. LcA (1 ml, 1 mg) was also dialyzed with six changes against 200 ml of 50 mM Tris.HCl buffer, pH 7.5 over a period of 6 days. Purified (95%) sequence-derived substrates from SNAP-25 (SNKTRIDEANQ-RATKML), for LcA, another peptide from SNAP-25 for LcE (MGNEIDTQNRQIDR-IMEKADSNKTRIDEANQRATKML), and one from VAMP for LcB (LSELDDRADALQAGASQFETSAAKLKRKYWWKNLK) [15], all having N-terminus acetylated and C-terminus amidated, were custom-synthesized and purified to >95% by Peptide 2.0 (Chantilly, VA). Sodium acetate, sodium sulfate, calcium chloride were from Sigma (St. Louis, MO).
UV-visible absorption, circular dichroism and fluorescence measurements
To determine protein concentration and to assess purity, UV-visible absorption spectra were recorded at 22°C with a Hewlett-Packard 8452 diode array spectrophotometer. Lc concentration was determined using A0.1% (1-cm light path) value of 1.0 at 278 nm [16] and by BCA assay (Pierce) with BSA as standard.
CD spectra were recorded on a Jasco 718 spectropolarimeter with quartz cuvettes of 1-mm path length at 20 °C. The temperature in the cuvette was regulated with circulating water bath. The protein concentration used to measure CD was 0.02 mg/ml in 50 mm HEPES, pH 7.4. An average of five scans was recorded to increase signal-to-noise ratio at a scan speed of 20 nm/min with a response time of 8 sec. In all measurements, a buffer blank was recorded separately and subtracted from sample recordings. Thermal denaturing curves of BoNT LcA (3.6 mM in Tris-HCl, pH 7.5) in presence of either Na-acetate or Na-sulfate at 0.5 and 10 μM were recorded at 10 °C/min at 0.1C pitch with 8 sec response time. Ellipticity was measured at 222 nm.
Fluorescence emission spectra were recorded at 20°C in a PTI Quanta Master Spectrofluorimeter, Model RTC 2000 equipped with a Peltier controlled thermostat and Felix software package. In all cases, protein concentration was 0.18 mg/ml in 50 mM HEPES, pH 7.4. Emission and excitation slit widths were set at 1 nm. Emission spectra were recorded over a range of 280-400 nm with excitation at 285 from tryptophan and at 280 for tryptophan + tyrosine fluorescence. Each spectrum was an average of five scans.
Enzymatic activity assays
Activity assays were based on UPLC separation and measurement of the cleaved products from a 17-residue SNAP-25 peptide for LcA, 35-residue VAMP peptide for LcB, and a 35-residue SNAP-25 peptide for LcE [17]. A master reaction mixture lacking the Lc was prepared and aliquots were stored at –20 °C. Stocks of 0.05-0.07 mg/ml of Lc in 50 mM Na-HEPES, pH 7.4 containing 0.05% Tween-20 were stored at -20 °C. Before assay, an Lc stock was thawed and diluted further in 50 mM HEPES, pH 7.4 containing bovine serum albumin (BSA). At the time of assay, 5 ml of the diluted Lc was added to 25 ml of the thawed master mix to initiate the enzymatic reaction. Components and final concentration in this 30-ml reaction mixture were 0.9 mM substrate peptide, 0.2 mg/ml BSA, 0.0026 mg/ml LC, and 50 mM Na-HEPES, pH 7.4. After 5-10 min at 37 °C, the reactions were stopped by adding 90 ml of 1% trifluoroacetic acid.
The amounts of uncleaved substrate and the products were measured after separation by a Waters Acquity UPLC™ system equipped with Empower Pro software employing a reverse-phase C18 column (2.1 X 50 mm, 1.7-μm particle size) with 0.1% trifluoroacetic acid as solvent A and 70% acetonitrile/0.1% trifluoroacetic acid as solvent B at a flow rate of 0.5 ml/min [17]. LcA substrate and products were resolved by UPLC with a 0% to 42% gradient of the solvent B over 2 min, followed by column regeneration for 0.7 min. LcB and LcE substrate and products were resolved by UPLC with a 0% to 100% gradient of the solvents over 2 min, held at 100% B for 0.5 min, followed by column regeneration for 0.5 min [17].
Results
Effects of acetate and sulfate on LcA activity
From the facts that acetate, sulfate, and calcium were bound at and near the active sites of LcA, BoNT/B, and LcE, it was expected that these ions will affect catalytic activities of the latter two catalysts. Therefore, we incubated LcA with increasing concentrations of salts of these ions, sodium acetate and sodium sulfate for 5 min followed by the enzymatic activity determinations. pH of the reaction mixtures were routinely checked for any changes after addition of the salts. LcA activity was progressively inhibited by increasing concentrations of acetate and sulfate (data not shown). Results with preincubation of LcA and the salts were same with results when salts were added just prior to start of the reaction. However, six to seven orders of magnitude higher concentrations of sulfate and acetate over LcA concentration were required to observe any substantial inhibition. Such results were unexpected for a tight binding inhibitor. LcA used in these experiments were in its storage buffer of 50 mM Na-phosphate that contained low concentrations of sulfate as a contaminant. Therefore, it is possible that LcA was already saturated with sulfate and/or acetate, and any effect of added salt on its activity was obscured by the already bound ligands on it. We therefore extensively dialyzed LcA against sulfate- and acetate-free, ultrapure Tris-HCl over a period of 1 week in a plastic container to remove any bound acetate and/or sulfate.
Figure 2A shows that increasing concentrations of both sulfate and acetate progressively reduced the activity of LcA, sulfate being much more effective than acetate. For sodium sulfate, IC50 was 13±3 mM, and for sodium acetate the IC50 was 136±8 mM (Table 1). To investigate if the effects of Na-sulfate and Na-acetate on LcA activity are independent of each other, we co-incubated LcA with varying concentrations of the Na-acetate at two fixed concentrations of Na-sulfate, and measured the LcA activity (Figure 2B). At 5 mM Na-sulfate, there was a 25-60% stimulation of inhibition by the addition of 50-100 mM Na-acetate, and at 10 mM Na-sulfate there was a 35-75% stimulation of inhibition over those of the cumulative effects of acetate and sulfate added separately. This was the result with each of the six pairs of acetate and sulfate (Figure 2B). These results suggest two separate binding for acetate and sulfate on LcA that cooperates with each other in increasing their inhibitory effects.
Figure 2.
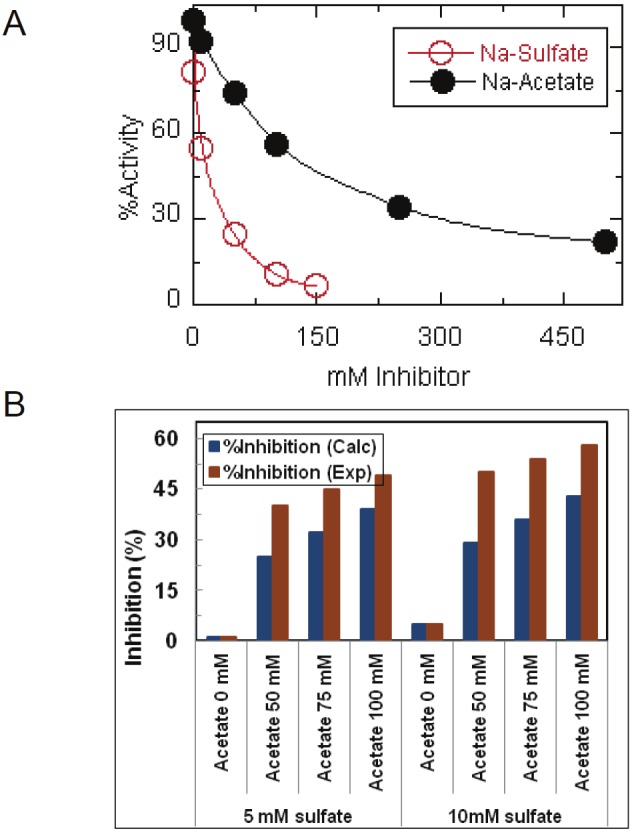
Catalytic activities of LcA in the presence of sodium sulfate and sodium acetate on LcA activity when added separately to the reaction mixtures (A), and when added in combination to the reaction mixtures (B). In B, the blue bars represent the expected %inhibition of the individual inhibition by sulfate and acetate were additive, and the red bars represent the experimentally determined %inhibition for the indicated pairs of sulfate and acetate. Percent LcA acitivity indicates the relative activity in absence of any additions. The data represent an average of five independent assays.
Table 1.
Half maximal inhibitory concentration of salts
| Light Chain | IC50 (Concentration of salt, inhibiting maximum absolute activity by half) (mM) | ||
|---|---|---|---|
| Acetate | Sulfate | Calcium | |
| LcA | 135.7 ± 7.6 | 12.7 ± 2.5 | 5.1 ± 1.7 |
| LcB | 193.9 ± 73.8 | 51.9 ± 11.3 | 9.6 ± 3.6 |
| LcE | n.da. | 50.5 ± 2.9 | n.da. |
The IC50 values are averages of 4-7 data points presented in Figures 2 and 4 computed using the equation IC50 = ([I] X Vi/Vo)/(1- Vi/Vo), where [I] is concentration of inhibitor, Vi and Vo represent reaction velocity in the presence and absence, respectively, of the inhibitor.
not determined.
Although LcA was extensively dialyzed our experiments could not prove that it was free from bound acetate and sulfate. However, because no acetate was found in the active site of LcA-substrate [9] and LcA-tetrapeptide inhibitor [8] complexes, it can be safely argued that during enzymatic reaction acetate gets displaced by the incoming substrate. Sulfate on the other hand might remain bound during catalysis because its presence was detected in the LcA-substrate [9] and LcA-tetrapeptide inhibitor [8] complexes.
If sulfate (and acetate) was indeed a specific substrate binding-site ligand, we expected that some organic sulfate compounds might be better inhibitors than inorganic sulfate. Figure 3 shows that except protamine sulfate, none of the sulfate and acetate compounds (at 1 mM concentration) had any significant effects on LcA activity. We think inhibition by the basic protamine sulfate is an exception in that it inhibits the LcA activity [18] by its interaction with the acidic residues at and near the active site of LcA [8,10].
Figure 3.
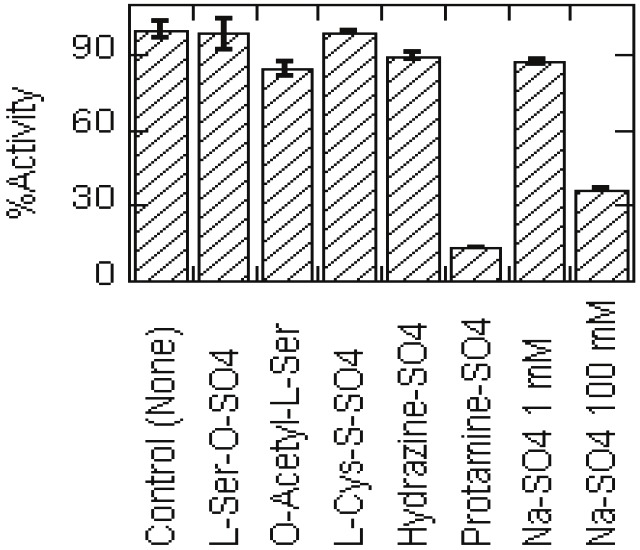
Effects of organic sulfates on LcA activity compared to that of sodium sulfate. Because of solubility issues, each compound was added at 1 mM concentration to the reaction mixtures. For comparison, effect of 100 mM sodium sulfate is also shown. Each data point is an average of 5 assays. Bars indicate the standard deviation.
Effects of calcium and sulfate on LcB activity
One calcium and one sulfate ion was found bound on the catalytic domain of BoNT/B crystal structure of which the latter was coordinated with the active site zinc [4,5]. We determined the effect of sodium sulfate and calcium chloride on the catalytic activity of LcB (Figure 4). For comparison, we also determined the effect of sodium acetate on LcB activity. As with LcA, the inhibitory effects of sodium sulfate were more pronounced than acetate on LcB activity (Figure 4A). Effect of calcium on LcB was much more pronounced than those of either sulfate or acetate. IC50 for calcium chloride, sodium sulfate and sodium acetate were calculated to be 10 mM, 52 mM and 194 mM, respectively (Table 1). Thus, the inhibitory effect of the ions on LcB activity were in the order of calcium > sulfate > acetate. However, sulfate was a much better inhibitor of LcA activity than LcB activity. With very similar IC50 values (130-200 mM), acetate might affect the activities of both LcA and LcB by similar mechanisms.
Figure 4.

Catalytic activities of LcB in the presence of sodium sulfate, sodium acetate, and calcium chloride when added separately to the reaction mixtures (A), and when acetate and sulfate were added in combination to the reaction mixtures (B). In B, the blue bars represent the expected %inhibition if the individual inhibition by sulfate and calcium were additive, and the red bars represent the experimentally determined %inhibition for the indicated pairs of sulfate and calcium. Percent LcB acitivity indicates the relative activity in absence of any additions. The data represent an average of five independent assays. Standard deviation is shown by bars.
When calcium and sulfate was added simultaneously, inhibition of LcB activity was less than their expected cumulative effect (Figure 4B). While this negative combined effect can signify two different binding sites, sequestering of calcium by sulfate in forming less soluble calcium sulfate might also be responsible for the reduced level of inhibition.
To find out if the inhibition by calcium is specific for LcB, we measured the activities of LcA at various CaCl2 concentrations under the conditions described in Figures 2 and 4. Surprisingly, calcium strongly inhibited LcA activity (Table 1) although it was not found associated with any of the 3-dimensional structures described in the literature. The average IC50 of 5.1 mM (Table 1) is very close to the IC50 of 4 mM calculated from the data in an older literature that also showed that LcA activity was inhibited by other divalent metals, such as Mn, Mg, and Ni [19]. Thus, it is likely that inhibition of LcA and LcB by calcium is a non specific phenomenon, although it was found in the crystal structure of BoNT/B [5].
Effect of sulfate on LcE activity
A sulfate ion was also found at the active site in the crystal structure of a mutant form of LcE (Figure 1) [7]. We therefore looked at the effect of sulfate on LcE catalytic activity. Like those in LcA and LcB, activity of LcE too was progressively inhibited by increasing concentrations of sodium sulfate (data not shown). The calculated IC50 of 51 mM is similar to that for LcB but somewhat higher than that for LcA (Table 1).
Secondary and tertiary structures and stability of BoNT/LcA
To determine if the exogenous acetate or sulfate affected secondary and tertiary structures of the LcA, we collected its CD and tryptophan fluorescence spectra, respectively. Extensively dialyzed LcA was incubated with several concentrations of Na-acetate or Na-sulfate at 4 °C for 20h. As shown with 50 mM of the salts (Figure 5A), CD spectra of these samples showed that the spectral shape, and intensity of ellipticity minima at 208 and 223 and positive maxima at 197 did not change from that of the control sample. These results indicated that binding of sulfate or acetate to LcA did not lead to any significant secondary structural changes of the protein. Tryptophan and tyrosine fluorescence spectra of LcA were collected in the absence and presence of sodium sulfate and sodium acetate (0.1 to 13,000 molar excess). No significant differences were observed indicating absence of any effect of the salts on the tertiary structures of LcA as shown for 50 mM of the salts (Figure 5B). These results indicate that both the secondary and tertiary structural features are not affected by the presence of sulfate or acetate ions on LcA. Because no secondary and tertiary structural differences were observed, we followed the thermal denaturation profile of LcA in the presence and absence of 0.4 μM to 50 mM Na-acetate by measuring ellipticity at 222 nm (data not shown). No difference in the midpoint of denaturation (49.5 °C) was observed either.
Figure 5.
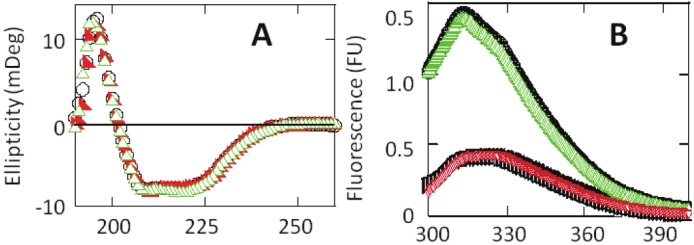
CD spectra (A) and tryptophan and tyrosine fluorescence spectra (B) of LcA (0.18 mg/ml 50 mM tris.HCl, pH 7.4) in the absence (black circle), and presence of 50 mM sodium acetate (green triangle), and 50 mM sodium sulfate (red triangle). In B, tryptophan fluorescence (lower curves) spectrum was collected by exciting at 295 nm, and tyrosine+tryptophan (upper curves) fluorescence spectrum was collected by exciting at 280 nm. The control (no addition) tyrosine spectra is denoted by black circle, and the control (no addition) tryptophan spectrum is denoted by black square. Each spectra represent an average of five scans (1 nm/sec, 8 sec response), collected at 20 °C in 2 mm cuvettes.
Discussions
Because the demonstration of BoNT proteins as zinc-endopeptidases [20], no inorganic or organic cofactor other than the active site zinc has been reported as functionally integral to their protease domains. Catalytic domains of BoNTs contain the conserved Zn-binding motif HEXXH and thus are referred to as Zn-endopeptidases. Zinc participates in BoNT catalysis by stabilizing a transition-state intermediate through the coordination with P1 carbonyl oxygen of the substrate [8,9]. However, analyses of X-ray diffractions of several of these proteins picked up densities corresponding to more than one ion of sulfate, acetate, and calcium at and near the active sites [4,7-9]. Acetate or calcium were not used in purification of the proteins, and ammonium sulfate-precipitated BoNT/B was extensively dialyzed before crystallization [4]. On the other hand, crystallization recipe of LcE contained ammonium sulfate [7]. Overall polypeptide folds and locations of zinc and catalytic residues of LcA, LcB, and LcE are superimposable, in addition to an active site water molecule being displaced by sulfate in LcB and a mutant LcE. It is the location of these ligands at or near the active site of the three proteins that raised questions about their fortuitous presence. Thus, an investigation of the effects of these ions on the enzyme’s catalytic functions was warranted.
Because the commonly used phosphate and HEPES buffers might contain free sulfate as a contaminant, and because phosphate might replace a putative sulfate from the proteins, we extensively dialyzed LcA and LcB in ultrapure tris-HCl to remove any bound small molecule. We hoped that this dialysis removed all bound acetate and sulfate although we are not sure if complete removal was achieved. We have shown that acetate and sulfate inhibited the activity of this extensively dialyzed LcA (Figure 2A). What was more interesting is the stimulation of inhibition by one compound in the presence of the other, as was observed with all six pairs of acetate and sulfate concentrations (Figure 2B). The inhibitory effects of acetate and sulfate thus were synergistic, suggesting at least two potential binding sites on LcA.
The results described in this paper also show that sulfate and acetate inhibited the activities (rates of catalysis) of LcB, and LcE (only sulfate was tested) (Figures 2, 3 and 4). Because acetate was not detected in LcB structure, we did not expect an inhibition of LcB activity by acetate. Contrary to our expectation, we found acetate also inhibited LcB activity (Figure 4), very similarly as it inhibited LcA (Figure 2). These inhibitions, although initially seemed encouraging, the high IC50 values of 10~150 mM suggest that their interactions with the proteins are not strong and specific enough to significantly affect the rates of catalysis. Sulfate was a better inhibitor than acetate for both LcA and LcB (Table 1), but it inhibited LcA more than LcB. The latter result is opposite to the expectation from threedimensional structures in that sulfate was located >10Å away from the active site in LcA [8,9] while it was only 3Å from the active site zinc in LcB [4]. Similarly, calcium inhibited LcA although it was not found at the active site in the three-dimensional structure [5], making it a nonspecific inhibitor of LcB in whose structure it was bound [4].
From our solution studies it is also clear that acetate and sulfate ions do not have any signifi cant structural role on LcA although their electron densities were detected in the threedimensional structures. We failed also to detect any secondary (Figure 5) and tertiary structural changes and the midpoints of thermal denaturation of LcA by the addition of substoichiometric (0.1) to saturating (1000-fold higher) concentrations of acetate and sulfate. The inhibitory effects of the ions we observed therefore, were probably due to their “solvent” effect on the protein’s conformation as has been observed with high concentration of CsCl on bacterial tryptophan synthase [21].
The decrease in activity at high acetate, sulfate, and calcium concentrations may be due to salt-induced rigidity of the LcA, and LcB rather than major alteration of structural orientation because the ions were added as salts, as has been observed in halophilic lactate dehydrogenase [22]. It was reported that at high salt concentration, ions interact with opposite charged groups on protein molecules to form a double layer of ionic groups which decrease electrostatic interaction between protein molecules [23]. The decreased ionic interaction may thus restrict the substrate to reach the active site of LcA that in turn might be responsible for reduced loss of catalytic activity. Because LcE is structurally very similar to LcA, similar phenomenon may also be applicable for the inhibition of its enzymatic activity by acetate and calcium. Inhibition of activity due to low ionic interaction in presence of high salt concentration also has been observed in D-amino acid amino transferase [24].
In conclusion, we have demonstrated that acetate, sulfate, and calcium act as inhibitors, albeit at high concentrations, of LcA without the alteration of any major secondary and tertiary structure. LcA is more readily affected by these ions than LcB and LcE. Although the physiological significance of activity of Lc under the conditions of intracellular concentrations of salts is not known, this study should provide new insight into the effects of salts in the catalytic and structural properties of BoNT/Lcs.
Acknowledgement
We thank Ms. Agatha Macairan for technical assistance in conducting the enzymatic reactions and UPLC™ analyses. This project received support from the Defense Threat Reduction Agency-Joint Science and Technology Office for Chemical and Biological Defense (Grant # JSTOCBD3.10012_06_RD_B to SAA). Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the U.S. Army. We also acknowledge National Research Council (NRC) for their support to RMM through their research associateship program.
Abbreviations
- BoNT
botulinum neurotoxin
- BoNT/A-G
BoNT serotypes A, B, C, D, E, F, G
- Lc or LC
light chain LcA, light chain of serotype A
- LcB
light chain of serotype B
- LcD
light chain of serotype D
- LcE
light chain of serotype E
- SNAP-25
Soluble NSF Attachment Protein of 25 kDa
- VAMP
Vesicle-Associated Membrane Protein
- SNARE
(Soluble NSF Attachment Protein) REceptor
- Tris
tris-2-Amino-2-hydroxymethyl-2-Amino-1-propane-1,3-diol
- HEPES
(4-(2-hydroxyethyl)-1-pipera-zineethanesulfonic acid) DTT, Dithiothreitol
- TMB
3,3’,5,5’-Tetramethylbenzidine
- HRP
horseradish peroxidase
- TFA
Trifluoroacetic acid; BSA, bovine serum albumin
- CD
circular dichroism, Tm, melting temperature
- UPLC
ultra performance liquid chromatography
References
- 1.Simpson LL. Identification of the major steps in botulinum toxin action. Annu Rev Pharmacol Toxicol. 2004;44:167–193. doi: 10.1146/annurev.pharmtox.44.101802.121554. [DOI] [PubMed] [Google Scholar]
- 2.Gul N, Smith LA, Ahmed SA. Light Chain Separated from the Rest of the Type A Botulinum Neurotoxin Molecule Is the Most Catalytically Active Form. PLoS ONE. 2010;5:e12872. doi: 10.1371/journal.pone.0012872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lacy DB, Tepp W, Cohen AC, DasGupta BR, Stevens RC. Crystal structure of botulinum neurotoxin type A and implications for toxicity. Nat Struct Biol. 1998;5:898–902. doi: 10.1038/2338. [DOI] [PubMed] [Google Scholar]
- 4.Swaminathan S, Eswaramoorthy S. Structural analysis of the catalytic and binding sites of Clostridium botulinum neurotoxin B. Nat Struct Biol. 2000;7:693–699. doi: 10.1038/78005. [DOI] [PubMed] [Google Scholar]
- 5.Eswaramoorthy S, Kumaran D, Keller J, Swaminathan S. Role of metals in the biological activity of Clostridium botulinum neurotoxins. Biochemistry. 2004;43:2209–2216. doi: 10.1021/bi035844k. [DOI] [PubMed] [Google Scholar]
- 6.Arndt JW, Chai Q, Christian T, Stevens RC. Structure of botulinum neurotoxin type D light chain at 1.65 A resolution: repercussions for VAMP-2 substrate specificity. Biochemistry. 2006;45:3255–3262. doi: 10.1021/bi052518r. [DOI] [PubMed] [Google Scholar]
- 7.Agarwal R, Binz T, Swaminathan S. Analysis of active site residues of botulinum neurotoxin E by mutational, functional, and structural studies: Glu335Gln is an apoenzyme. Biochemistry. 2005;44:8291–8302. doi: 10.1021/bi050253a. [DOI] [PubMed] [Google Scholar]
- 8.Kumaran D, Rawat R, Ludivico ML, Ahmed SA, Swaminathan S. Structure- and Substrate-based Inhibitor Design for Clostridium botulinum Neurotoxin Serotype A. J Biol Chem. 2008;283:18883–18891. doi: 10.1074/jbc.M801240200. [DOI] [PubMed] [Google Scholar]
- 9.Kumaran D, Rawat R, Ahmed SA, Swaminathan S. Substrate binding mode and its implication on drug design for botulinum neurotoxin A. PLoS Pathog. 2008;4:e1000165. doi: 10.1371/journal.ppat.1000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hale M, Oyler G, Swaminathan S, Ahmed SA. Basic tetrapeptides as potent intracellular inhibitors of type A botulinum neurotoxin protease activity. J Biol Chem. 2011;286:1802–1811. doi: 10.1074/jbc.M110.146464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ludivico M, Smith LA, Ahmed SA. In: Keller JE, editor. Amino Acids and Peptides as Inhibitors of Botulinum Neurotoxin Serotype A Proteolytic Activity; 43rd Annual Meeting of the IBRCC; Silver Spring, MD: CBER, FDA; 2006. p. 67. [Google Scholar]
- 12.Ahmed SA, McPhie P, Smith LA. Autocatalytically fragmented light chain of botulinum a neurotoxin is enzymatically active. Biochemistry. 2003;42:12539–12549. doi: 10.1021/bi030062c. [DOI] [PubMed] [Google Scholar]
- 13.Jensen MJ, Smith TJ, Ahmed SA, Smith LA. Expression, purification, and efficacy of the type A botulinum neurotoxin catalytic domain fused to two translocation domain variants. Toxicon. 2003;41:691–701. doi: 10.1016/s0041-0101(03)00042-4. [DOI] [PubMed] [Google Scholar]
- 14.Gilsdorf J, Gul N, Smith LA. Expression, purification, and characterization of Clostridium botulinum type B light chain. Protein Expr Purif. 2006;46:256–267. doi: 10.1016/j.pep.2005.09.024. [DOI] [PubMed] [Google Scholar]
- 15.Foran P, Shone CC, Dolly JO. Differences in the protease activities of tetanus and botulinum B toxins revealed by the cleavage of vesicle-associated membrane protein and various sized fragments. Biochemistry. 1994;33:15365–15374. doi: 10.1021/bi00255a017. [DOI] [PubMed] [Google Scholar]
- 16.Ahmed SA, Byrne MP, Jensen M, Hines HB, Brueggemann E, Smith LA. Enzymatic autocatalysis of botulinum A neurotoxin light chain. J Protein Chem. 2001;20:221–231. doi: 10.1023/a:1010952025677. [DOI] [PubMed] [Google Scholar]
- 17.Rowe B, Schmidt JJ, Smith LA, Ahmed SA. Rapid product analysis and increased sensitivity for quantitative determinations of botulinum neurotoxin proteolytic activity. Anal Biochem. 2010;396:188–193. doi: 10.1016/j.ab.2009.09.034. [DOI] [PubMed] [Google Scholar]
- 18.Ludivico M, Smith LA, Ahmed SA. Structure-Based Design of Peptide Inhibitors of Botulinum Neurotoxin Serotypes A Proteolytic Activity. The Botulinum Journal. 2009;1:297–308. [Google Scholar]
- 19.Ahmed SA, Smith LA. Light chain of botulinum A neurotoxin expressed as an inclusion body from a synthetic gene is catalytically and functionally active. J Protein Chem. 2000;19:475–487. doi: 10.1023/a:1026549431380. [DOI] [PubMed] [Google Scholar]
- 20.Schiavo G, Rossetto O, Benfenati F, Poulain B, Montecucco C. Tetanus and botulinum neurotoxins are zinc proteases specific for components of the neuroexocytosis apparatus. Ann N Y Acad Sci. 1994;710:65–75. doi: 10.1111/j.1749-6632.1994.tb26614.x. [DOI] [PubMed] [Google Scholar]
- 21.Ruvinov SB, Ahmed SA, McPhie P, Miles EW. Monovalent cations partially repair a conformational defect in a mutant tryptophan synthase alpha 2 beta 2 complex (beta-E109A) J Biol Chem. 1995;270:17333–17338. doi: 10.1074/jbc.270.29.17333. [DOI] [PubMed] [Google Scholar]
- 22.Hecht K, Wrba A, Jaenicke R. Catalytic properties of thermophilic lactate dehydrogenase and halophilic malate dehydrogenase at high temperature and low water activity. Eur J Biochem. 1989;183:69–74. doi: 10.1111/j.1432-1033.1989.tb14897.x. [DOI] [PubMed] [Google Scholar]
- 23.Vojdani F. Solubility. In: Hall GM, editor. Methods of testing protein functionality. New York: Blackie Academic and professional; 1996. pp. 11–60. [Google Scholar]
- 24.Ro HS. Effects of salts on the conformation and catalytic properties of d-amino acid aminotransferase. J Biochem Mol Biol. 2002;35:306–312. doi: 10.5483/bmbrep.2002.35.3.306. [DOI] [PubMed] [Google Scholar]