

Abstract
Myotonic dystrophy (Dystrophia Myotonica, DM) is the most frequently inherited neuromuscular disease of adult life. It is a multisystemic disease with major cardiac involvement. Core features of myotonic dystrophy are myotonia, muscle weakness, cataract, respiratory failure and cardiac conduction abnormalities. Classical DM, first described by Steinert and called Steinert's disease or DM1 (Dystrophia Myotonica type 1) has been identified as an autosomal dominant disorder associated with the presence of an abnormal expansion of a CTG trinucleotide repeat in the 3' untranslated region of DMPK gene on chromosome 19. This review will mainly focus on the various aspects of cardiac involvement in DM1 patients and the current role of cardiac pacing in their treatment.
Key words: myotonic dystrophy type 1, arrhythmias, cardiac pacing
Introduction
Myotonic dystrophy type 1 (DM1), or Steinert's disease, is the most common muscular dystrophy in the adult life with an incidence of 1 in 8000 births (1-3) and a worldwide prevalence from 2.1 to 14.3:100 000 inhabitants (1, 2). The phenotype is characterized by myotonia and muscle weakness, but a multisystemic involvement with highly variable clinical manifestation (4, 5) is very frequent (6-8). Based on the age of onset and on clinical features, DM1 can be divided into three forms: congenital, classical, and minimal which may occur in the same kindred.
In the classical form, which is the most common, symptoms become evident between the second and the fourth decade of life, showing a slow progression over time (Table 1) (9). The key feature of the disease is myotonia, characterised by delayed relaxation after muscular contraction (Fig. 1) (9); progressive muscular weakness (dystrophy) and wasting are also typical findings; facial, axial, semi-distal, and distal compartments are predominantly involved. Beside muscle involvement, the affected patients can manifest abnormalities of other organs and systems including eye (cataract), endocrine system (diabetes, thyroid dysfunction, hypogonadism), central nervous system (cognitive impairment, mental retardation, attention disorders), gastrointestinal system (dysphagia, constipation, gallbladder stones, pseudo-obstruction), respiratory apparatus (respiratory failure) and heart (Table 2) (9, 10).
Table 1.
Muscle Manifestations of Myotonic Dystrophy (DM1).
| Myotonia | Active and evocated |
| Muscular Dystrophy and Wasting | Facial and masticatory muscles (facies myotonica) |
| Axial Compartment | |
| Semidistal compartment | |
| Distal compartment | |
| Pharyngeal muscles (nasal speech, dysphagia) | |
| Respiratory muscles |
Figure 1.

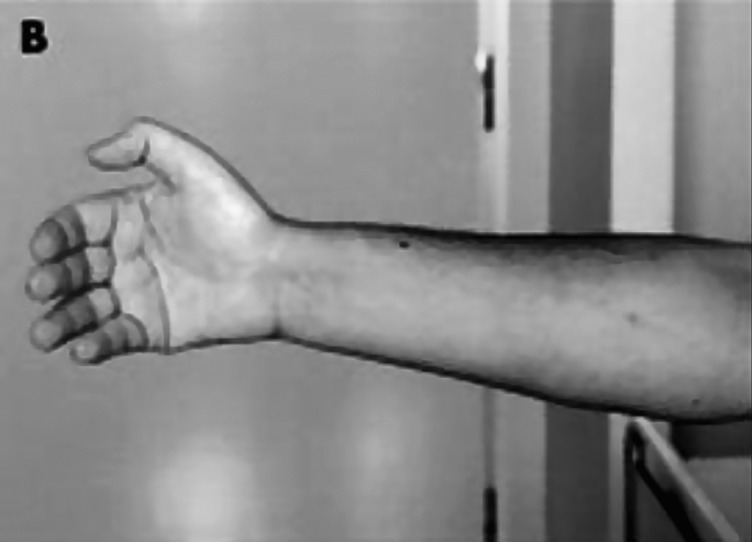
The grip test is a quick and easy way to determine the presence of active myotonia. After contraction of the fist, the patient is unable to relax the muscles of the hand.
Table 2.
Systemic involvement in Myotonic Dystrophy (DM1).
| Eye | Cataract |
| Endocrine System | Diabetes |
| Thyroid dysfunction | |
| Hypogonadism | |
| Gastrointestinal Tract | Dysphagia |
| Constipation | |
| Gallbladder stones | |
| Pseudo-ostruction | |
| Central Nervous System | Cognitive impairment |
| Mental retardation | |
| Attentive disorders | |
| Heart |
Genetics
DM1 is an autosomal dominant disorder with incomplete penetrance and variable phenotypic expression. The genetic basis of DM1 is known to include mutational expansion of a repetitive trinucleotide sequence (CTG) in the 3'-untranslated region of the DMPK (Dystrophia Myotonica Protein Kinase) gene on chromosome 19q13.3. Usually, 5-35 CTG repeats are observed in normal alleles, while their number may reach 50–2000 and more in DM1 (11, 12). The process leading from the abnormal expansion of CTG repeats in a non-coding region of DMPK gene to the cellular dysfunction is still incompletely understood. However, the localisation of DMPK in the heart muscle at the level of intercalated discs, combined with the observation that DMPK reduction, in animal models, compromises the conduction at both the level of the atrioventricular node and the His-Purkinje system (13), suggest the impairment of intercellular impulse propagation as a possible mechanism of disease.
Many attempts have been made to find a possible correlation between the number of CTG repeats and severity of clinical manifestations in DM1. Despite earlier controversial results, evidence is accumulating in favour of a correlation between cardiac involvement and CTG expansion (11-14). Indeed, the number of CTG repeats seems on average to influence the timing of cardiac complications (15), to predict the presence and the progression of ECG abnormalities, and the risk of major cardiac events (16), but it does not predict abnormal findings at electrophysiological studies (EPS) (16).
Cardiac manifestations and sudden death
Cardiac involvement, that often precedes the skeletal muscle one, occurs in 80% of DM1 patients and represents the second most common cause of death, after respiratory causes (17, 18). Endomyocardial biopsies performed on patients with DM1 have shown specific changes, such as perivascular interstitial fibrosis, fatty infiltration, hypertrophy of myocardiocites, and focal myocarditis (8).
Mathieu et al. (9), during a 10 year follow up study on 367 DM1 patients, reported a mortality 7.3 times higher than in an age-matched control population, with a mean age at death of 53 years; furthermore they showed a positive correlation between age at onset of DM1 and age of death. In their series, respiratory failure and cardiovascular disease were the most prevalent causes of death, accounting for about 40% and 30% of fatalities, respectively.
Cardiac mortality usually occurred because of progressive left ventricular dysfunction, ischaemic heart disease, pulmonary embolism, or as a result of unexpected sudden death (SD) (19-23).
Relative contribution of sudden death ranges from about 2-30% in different published series (24-26), according to selection criteria. The hypothesis that cardiac arrhythmias may represent the most prevalent cause of sudden death in DM1 patients is supported by the absence of other causes of sudden death at necropsy studies. Sudden cardiac death may be caused by ventricular asystole, degeneration of ventricular tachycardia (VT), ventricular fibrillation (VF) or electromechanical dissociation (27-29).
Clinical and experimental studies suggested that the basis of the arrhythmic risk and subsequent sudden death could be a heterogeneity of regional and ventricular repolarization (20), a substrate for life-threatening ventricular arrhythmias. Such a heterogeneity is expressed by an increase of QTc and JTc dispersion (29), as reported in other diseases such as hypertrophic cardiomyopathy (24), chronic heart failure (18, 25), myocardial ischemia (26), beta thalassemia major (22) and aortic coarctation (23); however it is not expressed by other parameters such heart rate variability (HRV) or P dispersion (Pd), as it can be observed in other neuromuscular disorders presenting with cardiomyopathy and arrhythmias (28-39).
The evidence of the degeneration of the conduction system in DM generated the hypothesis that bradyarrhythmias might represent the most prevalent mechanism of SD. However, ventricular tachy-arrhythmias are increasingly recognised as a common finding in these patients, possibly explaining some cases of sudden death after pacemaker implantation.
The cardiac manifestations that are seen in DM1 can be grouped into 3 distinct categories: 1) conduction defects; 2) tachyarrhythmias and 3) other manifestations (see Table 3).
Table 3.
Cardiac Involvement in Myotonic Dystrophy (DM1).
| Conduction system | Atrioventricular block, any degree | |
|---|---|---|
| Arrhythmias | Supraventricular | Atrial premature complexes |
| Atrial tachycardia | ||
| Atrial flutter | ||
| Atrial fibrillation | ||
| Ventricular | Ventricular premature complexes | |
| Ventricular tachycardia | ||
| Ventricular fibrillation | ||
| Ventricular function | Systolic function impairment | |
| Diastolic function impairment | ||
| Ischaemic heart disease | ||
| Mitral valve prolapse |
1. Conduction defects
Conduction defects are the most prevalent cardiac abnormalities seen in patients with DM1, occurring in 40% of patients. These defects can affect any part of the conduction system; however, most commonly the His-Purkinje system is affected (8, 27). Minor conduction defects are commonly seen on standard electrocardiograms (ECG) and His-Bundle studies as prolongations of the PR, QRS and HV intervals, also in asymptomatic patients (21, 27). These minor defects can progress to more severe conduction defects often associated with breath shortness, dizziness, fainting, syncope, and sudden death (8).
Because of the progressive nature of DM cardiac disease, a specific protocol for the follow-up of DM1 patients has been designed (40) recommending at least an ECG every 6 months. There is no consensus on the rate of progression of conduction defects. While some authors report a slow progression of conduction defects, others correlate the rate of progression with the size of repeats. Among them, the study conducted by Clarke et al. (15) showed that patients with repeats > 1000 CTG had a more rapid progression. This finding is significant because of risk to develop a complete heart block potentially leading to sudden death (30, 31).
A delayed impulse propagation, along the conduction system, can be associated with a long PR interval (prevalence 20-40% in different studies, depending on patient selection criteria) and/or a wide QRS complex (prevalence 5-25% in different studies, depending on patient selection criteria). Unfortunately, the presence of a long PR interval does not give any clue regarding the site of the conduction delay, as it may occur at any level, from the atrium to the His bundle, through the atrio-ventricular node. However, a wide QRS complex (for right or left bundle branch block), is frequently associated with an infrahissian (below the His bundle) conduction impairment. A prolongation of the HV interval has been observed in about 50% of unselected patients with DM1 (10, 13, 41).
Late potentials are also expression of delayed myocardial activation, usually caused by abnormal tissue (fibrosis or necrosis), and are considered predictors of ventricular arrhythmias. In DM1 it seems that delayed myocardial activation is a consequence of delayed activation along the His-Purkinje system (42) rather than an inhomogeneous conduction, through scattered areas of fibrosis. Abnormal late potentials are therefore expression of a conduction defect, and represent an important noninvasive clue for the presence of a long HV interval. QRS duration > 100 ms and low amplitude signals in the last 40 ms of QRS complex have been shown to predict a prolonged HV interval at the intracardiac electrophysiology study (EPS) with good sensitivity and specificity (80% and 83.3%, respectively) (42, 43).
2. Tachyarrhythmias
In DM1 patients, supraventricular tachyarrhythmias are a common finding on 12 lead ECG or 24 hour Holter monitoring, often asymptomatic. Most common arrhythmias are atrial flutter or fibrillation, observed in up to 25% of patients both as un-sustained and sustained forms (44). Atrial flutter, atrial fibrillation, and atrial tachycardia are also easily inducible at EPS even in the absence of previously documented spontaneous episodes, but the clinical implications of these findings are still uncertain.
Ventricular arrhythmias such as ventricular tachycardia (VT) and ventricular fibrillation may also occur in patients with DM1. The possible mechanisms that may lead to VT are bundle branch re-entry – the most typical cause of VT –, triggered activity, and re-entry around areas of fibro-fatty degeneration of the myocardium (26-27, 45). VT results from the delay of conduction along the Bundle of His, which presents as a prolonged HV interval. The identification of this cause of VT is important because it can be readily treated by a radiofrequency ablation (8). Patients may also benefit from prophylactic pacing devices (40, 43-54). Because of the fatal risk of these arrhythmias as early as the second decade of life (41), it has been suggested that any patient who presents with any clinical symptoms of VT such as presyncope, syncope, shortness of breath, or family history of sudden death should undergo EPS (8, 52-53). Inducible VT is most commonly represented by un-sustained polymorphic VT, but sustained polymorphic VT, VF, and both sustained and un-sustained monomorphic VT have also been reported. Given the risk of sudden death in these patients, the prognostic significance of inducible VTs, even when polymorphic and un-sustained, should be carefully considered, as risk stratification criteria used in other subsets of patients may not apply to DM1 (47).
3. Other cardiac manifestations
Although conduction abnormalities and arrhythmias are the most common heart manifestations of DM1, many other cardiac symptoms have been reported (10). A study performed by Bhakta et al (48) showed that 1,8% of DM1 patients have clinical symptoms of congestive heart failure, maybe also under-represented as a result of their physical inactivity or inability to communicate secondary to mental retardation. Other DM1 associated cardiac manifestations include ischemic heart disease, in the form of stable angina, unstable angina, and myocardial infarction. Mitral valve prolaps because of papillary dysfunction is also found in 25% to 40% of patients and may often be detected on the physical examination.
Implantable cardiac electrical devices
As patients with DM1 are at high risk of sudden cardiac death (SCD), most likely due high-degree atrioventricular (AV) blocks, or ventricular tachyarrhythmias (47), implantation of a pacemaker (PM) or cardioverter defibrillator (ICD) is required in 3-22% of cases (43) to prevent such a fatal event. With this aim, several studies have been done in the past to identify those patients at risk for SCD (28-29, 43). Groh et al. (27) found that DM1 patients – even when asymptomatic – presenting with prolonged PR intervals (≥ 240 milliseconds), wide QRS complex (≥ 120 milliseconds), or atrial tachyarrhythmias were at higher risk when compared to those with normal ECGs. They also showed that SCD was more common in patients with advanced age and a higher muscle impairment score, indicating more severe disease. Another study published by Cudia et al. (52) also reported the advanced age, the male gender, and an advanced muscular disease as risk factors for SCD.
Because the risk of SCD most often appears to be associated to an advanced AV block and asystole, it has become common practice to place a prophylactic permanent pacemaker in DM1 patients when they have severe ECG abnormalities suggestive of advance conduction system disease even if they are asymptomatic. Several studies (27, 43) have shown a reduction in the rate of SCD, adopting this protocol. Lazarus et al. (53) reported a mortality rate of 20%, in a group of 49 DM1 patients, followed for 54 ± 27 month, who showed a HV > 70 milliseconds, and received prophylactic pacemakers. Laurent et al. (43) also found a much lower rate of SCD in high risk patients with DM1 after receiving prophylactic pacemakers. They reported only 10% of deaths among 100 patients – followed for 6 years – equally occurring in patients with and without pacemakers. In their series, there was 1 case of SCD in a patient previously implanted with a pacemaker; however no information was available on the rhythm at the time of death nor through the postmortem interrogation of the pacemaker.
Long-term follow-up of arrhythmias in patients with Myotonic dystrophy (45) has shown that VT/VF can also occur and that permanent pacemakers do not eliminate the risk of SCD. The use of implantable cardioverter defibrillators instead of pacemakers has been so considered to address both brady- and tachy-arrhythmias (47, 49- 50). However, limited data are available on the utility of this approach. Recently Bhakta et al. (54) reported the results of a large prospective study assessing implant rates, indications, characteristics, and outcomes in patients with DM1 receiving a pacemaker or an implantable cardioverter-defibrillator. Device use was evaluated in a prospective, multicenter registry of 406 genetically confirmed adult patients followed for 9.5 ± 3.2 years. Fortysix (11.3%) had or received a pacemaker and 21 (5.2%) received an ICD. Devices were primarily implanted for asymptomatic conduction abnormalities and left ventricular (LV) systolic dysfunction. However, 7 (15.2%) pacemakers were implanted for third-degree atrioventricular block and 6 (28.6%) ICDs were implanted for ventricular tachyarrhythmias (ventricular tachycardia [VT] or fibrillation [VF]). Patients receiving devices were older and more frequently had heart failure, LV systolic dysfunction, atrial tachyarrhythmias, and ECG conduction abnormalities compared to patients with no device. Five (10.9%) pacemaker patients underwent upgrade to an ICD: 3 for LV systolic dysfunction, 1 for VT/VF, and 1 for progressive conduction disease. Seventeen (27.4%) of the 62 patients with devices were pacemaker-dependent at last follow-up. Three (14.3%) ICD patients had appropriate therapies. Twenty-four (52.2%) pacemaker patients died including 13 of respiratory failure and 7 of sudden death. Seven (33.3%) ICD patients died including 2 of respiratory failure and 3 of sudden death. All the patients with ICDs and sudden death had LV systolic dysfunction and 1 death was documented due to inappropriate therapies. The authors concluded that a) DM1 patients commonly receive anti-arrhythmic devices and 2) the risk of VT/VF and sudden death suggests that ICDs rather than pacemakers should be considered for these patients.
This study is the largest to date on the use of implantable PM/ICDs and outcomes in patients with DM1 and the first study questioning whether an ICD placed instead of a PM in patients at high risk for cardiac arrhythmias is of clinical value in preventing SCD.
However some questions remain open and need further investigation:
Whether the use of cardiac resynchronization therapy (CRT) would improve outcomes in DM1 patients.
Whether pacing-induced dyssynchrony lead to heart failure and SCD because of a high percentage of ventricular pacing due to AV block.
Whether CRT pacemakers should prevent progression of ventricular dysfunction and lower the risk of SCD so that ICDs would only be necessary in patients who meet currently accepted indications.
Whether ICDs might be beneficial in terms of improving overall survival, in patients with most advanced conduction system disease often presenting more severe forms of myotonic dystrophy itself.
Site of implant
A peculiar aspect to be considered in DM1 patients is the site of implantation. DM1 patients, who underwent conventional right atrial appendage (RAA) lead implantation, often manifested, during the follow-up period, sensing and pacing defects on the atrial lead. Recent papers have considered an alternative stimulation site, i.e. the interatrial septum, in the region of Bachmann's bundle (BB). They demonstrated that such a region is the atrial site with better sensing and pacing threshold at implantation compared with the RAA, and presents a low rate of sensing and pacing defects in a long term followup (55, 56). Another important aspect to be analyzed is the frequent occurrence of paroxysmal atrial arrhythmias (atrial fibrillation, atrial flutter or atrial tachycardia) in DM1 patients who underwent PMK implantation (17). We demonstrated no significant differences in the prevention of atrial fibrillation, in a group of 40 DM1 patients, followed for 12-month when comparing the conventional RRA with the BB atrial pacing (57).
Recently pacemakers including detailed diagnostic functions (Atrial Preference Pacing algorithm, APP) have been introduced (57-61), that facilitate the diagnosis and management of frequent paroxysmal atrial tachyarrhythmias, often undetected during conventional clinical follow-up.
Atrial Preference Pacing algorithm adapts the atrial pacing rate to a value slightly higher than the intrinsic sinus rate. When the intrinsic atrial activity (sinus rhythm and atrial premature beats) is detected, the algorithm increases the atrial pacing rate by a programmed value above the physiological sinus rate to resume atrial pacing. This can result in suppression and/or prevention of atrial ectopic beats and modify favourably the arrhythmogenic substrate. The efficacy of the automatic atrial overdrive algorithms has been evaluated in several studies, but their outcomes remain controversial. Russo et al. (62) found that the APP significantly reduces the number and the duration of AF episodes in DM1 patients, regardless of the site of atrial stimulation (Bachmann's bundle or RAA). The potential reason of these results is that the high percentage of atrial pacing, warranted by atrial overdrive algorithm, may prevent the relative bradycardia and reduce the number of premature atrial contractions, that initiate the re-entry and predispose to AF.
This aspect – apparently less important than the arrhythmias responsible for sudden death – should be carefully taken into account by cardiologists, because it strongly affects the quality of life of these patients.
Conclusions
Conduction system abnormalities, atrial or ventricular arrhythmias and, less commonly, myocardial dysfunction are observed in patients with DM1 and may occasionally represent the initial manifestations of the disease, even in the absence of overt neuromuscular involvement. Thus, cardiologists should be aware of this diagnosis.
Conversely, in all patients presenting with DM1, a careful clinical and diagnostic evaluation needs to be performed for the identification of patients at risk of major cardiac events, following the protocol established by the group of the RAMYD study (40, 63). An attitude of a low threshold for invasive procedures is suggested, considering the unclear rate of cardiac disease progression and the risk of sudden death in some subsets of patients. However several questions are still unanswered. Future studies are needed to improve the stratification of DM1 patients at high risk of SCD and/or heart failure.
Acknowledgements
This work was in part supported by Telethon grant GUP07013 to LP.
References
- 1.Emery AE. The muscular dystrophies. Lancet. 2002;359:687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- 2.Emery AE. Heritability of common neuromuscular diseases. Neuromuscul Disord. 2010;20:476–476. doi: 10.1016/j.nmd.2010.05.005. [DOI] [PubMed] [Google Scholar]
- 3.Harper PS. Myotonic Dystrophy: some genetic problems. Birth Defects Orig Artic Ser. 1974;10:120–125. [PubMed] [Google Scholar]
- 4.Harley HG, Brook JD, Rundle SA, et al. Expansion of an unstable DNA region and phenotypic variation in myotonic dystrophy. Nature. 1992;355:545–546. doi: 10.1038/355545a0. [DOI] [PubMed] [Google Scholar]
- 5.Walton JN. Clinical examination of the neuromuscular system. In: John Walton., editor. Disorders of Voluntary Muscle. London: Churchill Livingstone; 1981. [Google Scholar]
- 6.Nigro G, Comi LI, Politano L, et al. Cardiomyopathies associated with Muscular Dystrophies. In: Engel A, Franzini-Armstrong C, editors. Myology. New York: Mac Graw-Hill; 2004. pp. 1239–1256. and references therein. [Google Scholar]
- 7.Hawley RJ, Gottdiener JS, Gay JA, et al. Families with myotonic dystrophy with and without cardiac involvement. Arch Intern Med. 1983;143:2134–2136. [PubMed] [Google Scholar]
- 8.Phillips MF, Harper PS. Cardiac disease in myotonic dystrophy. Cardiovasc Res. 1997;33:13–22. doi: 10.1016/s0008-6363(96)00163-0. [DOI] [PubMed] [Google Scholar]
- 9.Mathieu J, Allard P, Potvin L, et al. A 10 year study of mortality in a cohort of patients with myotonic dystrophy. Neurology. 1999;52:1658–1662. doi: 10.1212/wnl.52.8.1658. [DOI] [PubMed] [Google Scholar]
- 10.Pelargonio G, Dello Russo A, Sanna T, et al. Myotonic dystrophy and the heart. Heart. 2002;88:665–670. doi: 10.1136/heart.88.6.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Melacini P, Villanova C, Menegazzo E, et al. Correlation between cardiac involvement and CTG trinucleotide repeat length in myotonic dystrophy. J Am Coll Cardiol. 1995;25:239–245. doi: 10.1016/0735-1097(94)00351-p. [DOI] [PubMed] [Google Scholar]
- 12.Jaspert A, Fahsold R, Grehl, et al. Myotonic dystrophy correlation of clinical symptoms with the size of CTG repeats. J Neurol. 1995;25:239–245. doi: 10.1007/BF00887824. [DOI] [PubMed] [Google Scholar]
- 13.Groh W, Lowe M, Zipes D. Severity of cardiac conduction involvement and arrhythmias in myotonic dystrophy type 1 correlates with age and CTG repeat length. J Cardiovasc Electrophysiol. 2002;13:444–448. doi: 10.1046/j.1540-8167.2002.00444.x. [DOI] [PubMed] [Google Scholar]
- 14.Antonini G, Giubilei F, Mammarella A, et al. Natural history of cardiac involvement in myotonic dystrophy: correlation with CTG repeats. Neurology. 2000;55:1207–1209. doi: 10.1212/wnl.55.8.1207. [DOI] [PubMed] [Google Scholar]
- 15.Clarke NRA, Kelion AD, Nixon J, et al. Does cytosine-thymine- guanine(CTG) expansion size predict cardiac events and electrocardiographic progression in myotonic dystrophy? Heart. 2001;86:411–416. doi: 10.1136/heart.86.4.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lazarus A, Varin J, Ounnoughene Z, et al. Relationships among electrophysiological findings and clinical status, heart function, and extent of DNA mutation in myotonic dystrophy. Circulation. 1999;99:1041–1046. doi: 10.1161/01.cir.99.8.1041. [DOI] [PubMed] [Google Scholar]
- 17.Mankodi A, Thorton C. Myotonic syndrome. Curr Opin Neurol. 2002;15:545–552. doi: 10.1097/00019052-200210000-00005. [DOI] [PubMed] [Google Scholar]
- 18.D'Andrea A, Salerno G, Scarafile R, et al. Right ventricular myocardial function in patients with either idiopathic or ischemic dilated cardiomyopathy without clinical sign of right heart failure: effects of cardiac resynchronization therapy. Pacing Clin Electrophysiol. 2009;32:1017–1029. doi: 10.1111/j.1540-8159.2009.02434.x. [DOI] [PubMed] [Google Scholar]
- 19.Magrì D, Piccirillo G, Bucci E, et al. Increased temporal dispersion of myocardial repolarization in myotonic dystrophy Type 1: Beyond the cardiac conduction system. Int J Cardiol. 2012;156:259–264. doi: 10.1016/j.ijcard.2010.10.132. [DOI] [PubMed] [Google Scholar]
- 20.Zabel M, Portnoy S, Franz MR, et al. Electrocardiographic indexes of dispersion of ventricular repolarization: an isolated heart validation study. J Am Coll Cardiol. 1995;25:746–752. doi: 10.1016/0735-1097(94)00446-W. [DOI] [PubMed] [Google Scholar]
- 21.Sovari AA, Bodine KC, Farokhi F, et al. Cardiovascular manifestations of myotonic dystrophy-1. Cardiol Rev. 2007;15:191–194. doi: 10.1097/CRD.0b013e318070d1a7. [DOI] [PubMed] [Google Scholar]
- 22.Russo V, Rago A, Pannone B, et al. Dispersion of repolarization and beta-thalassemia major: the prognostic role of QT and JT dispersion for identifying the high-risk patients for sudden death. Eur J Haematol. 2011;86:324–331. doi: 10.1111/j.1600-0609.2011.01579.x. [DOI] [PubMed] [Google Scholar]
- 23.Nigro G, Russo V, Rago A, et al. Heterogeneity of ventricular repolarization in newborns with severe aortic coarctation. Pediatr Cardiol. 2012;33:302–306. doi: 10.1007/s00246-011-0132-4. [DOI] [PubMed] [Google Scholar]
- 24.Buja G, Miorelli M, Turrini P, et al. Comparison of QT dispersion in hypertrophic cardiomyopathy between patients with and without ventricular arrhythmias and sudden death. Am J Cardiol. 1993;72:973–976. doi: 10.1016/0002-9149(93)91118-2. [DOI] [PubMed] [Google Scholar]
- 25.Barr CS, Nass A, Freeman M, et al. QT dispersion and sudden unexpected death in chronic heart failure. Lancet. 1994;343:327–329. doi: 10.1016/s0140-6736(94)91164-9. [DOI] [PubMed] [Google Scholar]
- 26.Paventi S, Bevilacqua U, Parafati MA, et al. QT dispersion and early arrhythmic risk during acute myocardial infarction. Angiology. 1999;50:209–215. doi: 10.1177/000331979905000305. [DOI] [PubMed] [Google Scholar]
- 27.Groh WJ, Groh MR, Chandan S, et al. Electrocardiographic abnormalities and sudden death in myotonic dystrophy tipe 1. N Engl J Med. 2008;358:2688–2697. doi: 10.1056/NEJMoa062800. [DOI] [PubMed] [Google Scholar]
- 28.Ducceschi V, Nigro G, Sarubbi B, et al. Autonomic nervous system imbalance and left ventricular systolic dysfunction as potential candidates for arrhythmogenesis in Becker muscular dystrophy. Int J Cardiol. 1997;59:275–279. doi: 10.1016/s0167-5273(97)02933-1. [DOI] [PubMed] [Google Scholar]
- 29.Nigro G, Nigro G, Politano L, et al. Is the value of QT dispersion a valid method to foresee the risk of sudden death? A study in Becker patients. Heart. 2002;87:156–157. doi: 10.1136/heart.87.2.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ammendola E, Russo V, Politano L, et al. Is heart rate variability a valid parameter to predict sudden death in patients with Becker's muscular dystrophy? Heart. 2006;92:1686–1687. doi: 10.1136/hrt.2005.082909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Politano L, Palladino A, Nigro G, et al. Usefulness of heart rate variability as a predictor of sudden cardiac death in muscular dystrophies. Acta Myol. 2008;27:114–122. [PMC free article] [PubMed] [Google Scholar]
- 32.Politano L, Nigro G. Treatment of dystrophinopathic cardiomyopathy: review of the literature and personal results. Acta Myol. 2012;31:24–30. [PMC free article] [PubMed] [Google Scholar]
- 33.Lattanzi G, Benedetti S, Bertini E, et al. Laminopathies: many diseases, one gene. Report of the first Italian Meeting Course on Laminopathies. Acta Myol. 2011;30:138–143. [PMC free article] [PubMed] [Google Scholar]
- 34.Russo V, Rago A, Palladino A, et al. P-wave duration and dispersion in patients with Emery-Dreifuss muscular dystrophy. J Investig Med. 2011;59:1151–1154. doi: 10.2310/JIM.0b013e31822cf97a. [DOI] [PubMed] [Google Scholar]
- 35.Benedetti S, Bernasconi P, Bertini E, et al. The empowerment of translational research: Lessons from laminopathies. Orphanet J Rare Dis. 2012;7:37–37. doi: 10.1186/1750-1172-7-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nigro G, Russo V, Ventriglia VM, et al. Early onset of cardiomyopathy and primary prevention of sudden death in X-linked Emery- Dreifuss muscular dystrophy. Neuromuscul Disord. 2010;20:174–177. doi: 10.1016/j.nmd.2009.12.004. [DOI] [PubMed] [Google Scholar]
- 37.Carboni N, Mura M, Mercuri E, et al. Cardiac and muscle imaging findings in a family with X-linked Emery-Dreifuss muscular dystrophy. Neuromuscul Disord. 2012;22:152–158. doi: 10.1016/j.nmd.2011.09.001. [DOI] [PubMed] [Google Scholar]
- 38.Lancioni A, Rotundo IL, Kobayashi YM, et al. Combined deficiency of alpha and epsilon sarcoglycan disrupts the cardiac dystrophin complex. Hum Mol Genet. 2011;20:4644–4654. doi: 10.1093/hmg/ddr398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rotundo IL, Faraso S, Leonibus E, et al. Worsening of cardiomyopathy using deflazacort in an animal model rescued by gene therapy. PLoS One. 2011;6:e24729–e24729. doi: 10.1371/journal.pone.0024729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dello Russo A, Mangiola F, Della Bella P, et al. Risk of arrhythmias in myotonic dystrophy: trial design of the RAMYD study. J Cardiovasc Med (Hagerstown) 2009;10:51–58. doi: 10.2459/jcm.0b013e328319bd2c. [DOI] [PubMed] [Google Scholar]
- 41.English KM, Gibbs JL. Cardiac monitoring and treatment for children and adolescents with neuromuscular disorders. Dev Med Child Neurol. 2006;48:231–235. doi: 10.1017/S0012162206000491. [DOI] [PubMed] [Google Scholar]
- 42.Morner S, Lindqvist P, Mellberg C, et al. Profound cardiac conduction delay predicts mortality in myotonic dystrophy type 1. J Int Med. 2012;268:59–65. doi: 10.1111/j.1365-2796.2010.02213.x. [DOI] [PubMed] [Google Scholar]
- 43.Laurent V, Pellieux S, Corcia P, et al. Mortality in myotonic dystrophy patients in the area of prophylactic pacing devices. Int J Cardiol. 2011;150:54–58. doi: 10.1016/j.ijcard.2010.02.029. [DOI] [PubMed] [Google Scholar]
- 44.Oloffson B, Forsberg H, Andersson S, et al. Electrocardiographic findings in myotonic dystrophy. Br Heart J. 1988;59:47–52. doi: 10.1136/hrt.59.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chaudhry SP, Frishman WH. Myotonic dystrophies and the heart. Cardiol Rev. 2012;20:1–3. doi: 10.1097/CRD.0b013e31821950f9. [DOI] [PubMed] [Google Scholar]
- 46.Bassez G, Lazarus A, Desguerre I, et al. Severe cardiac arrhythmias in young patients with myotonic dystrophy type 1. Neurology. 2004;63:1939–1941. doi: 10.1212/01.wnl.0000144343.91136.cf. [DOI] [PubMed] [Google Scholar]
- 47.Grigg LE, Chan W, Mond HG, et al. Ventricular tachycardia and sudden death in myotonic dystrophy: clinical, electrophysiologic and pathologicfeatures. J Am Coll Cardiol. 1985;6:254–256. doi: 10.1016/s0735-1097(85)80286-2. [DOI] [PubMed] [Google Scholar]
- 48.Bhakta D, Lowe MR, Groh WJ. Prevalence of structural abnormalities in patients with myotonic dystrophy tipe 1. Am Heart J. 2004;147:224–227. doi: 10.1016/j.ahj.2003.08.008. [DOI] [PubMed] [Google Scholar]
- 49.Curtis AB, Dalal K. The role of implantable cardiac electrical devices in patients with myotonic dystrophy. J Cardiovasc Electrophysiol. 2011;22:1376–1377. doi: 10.1111/j.1540-8167.2011.02221.x. [DOI] [PubMed] [Google Scholar]
- 50.Russo V, Rago A, D'Andrea A, et al. Early onset "electrical" heart failure in myotonic dystrophy type 1 patient: the role of ICD biventricular pacing. Anadolu Kardiyol Derg. 2012;12:517–519. doi: 10.5152/akd.2012.161. [DOI] [PubMed] [Google Scholar]
- 51.Babuty D, Fauchier L, Tena-Carbi D, et al. Significance of late potentials in myotonic dystrophy. Am J Cardiol. 1999;84:1099–1101. doi: 10.1016/s0002-9149(99)00510-x. [DOI] [PubMed] [Google Scholar]
- 52.Cudia P, Bernasconi P, Chiodelli R, et al. Risk of arrhythmia in type I myotonic dystrophy: The role of clinical and genetic variables. J Neurol Neurosurg Psych. 2009;80:790–793. doi: 10.1136/jnnp.2008.162594. [DOI] [PubMed] [Google Scholar]
- 53.Lazarus A, Varin J, Babuty D, et al. Long-term follow-up of arrhythmias in patients with myotonic dystrophy treated by pacing: A multicenter diagnostic pacemaker study. J Am Coll Cardiol. 2002;40:1645–1652. doi: 10.1016/s0735-1097(02)02339-2. [DOI] [PubMed] [Google Scholar]
- 54.Bhakta B, Shen C, Kron J, et al. Pacemaker and implantable cardioverter defibrillator use in a U.S. Myotonic Dystrophy Type 1 population. J Cardiovasc Electrophysiol. 2011;22:1369–1375. doi: 10.1111/j.1540-8167.2011.02200.x. [DOI] [PubMed] [Google Scholar]
- 55.Nigro G, Russo V, Vergara P, et al. Optimal site for atrial lead implantation in myotonic dystrophy patients: the role of Bachmann's Bundle stimulation. Pacing Clin Electrophysiol. 2008;31:1463–1466. doi: 10.1111/j.1540-8159.2008.01210.x. [DOI] [PubMed] [Google Scholar]
- 56.Nigro G, Russo V, Politano L, et al. Right atrial appendage versus Bachmann's bundle stimulation: a two-year comparative study of electrical parameters in myotonic dystrophy type-1 patients. Pacing Clin Electrophysiol. 2009;32:1191–1196. doi: 10.1111/j.1540-8159.2009.02464.x. [DOI] [PubMed] [Google Scholar]
- 57.Nigro G, Russo V, Politano L, et al. Does Bachmann's bundle pacing prevent atrial fibrillation in myotonic dystrophy type 1 patients? A 12 months follow-up study. Europace. 2010;12:1219–1223. doi: 10.1093/europace/euq170. [DOI] [PubMed] [Google Scholar]
- 58.Carlson M, Ip J, Messenger J, et al. A new pacemaker algorithm for the treatment of atrial fibrillation. Results of the Atrial Dynamic Overdrive Pacing Trial (ADOPT) J Am Coll Cardiol. 2003;42:627–633. doi: 10.1016/s0735-1097(03)00780-0. [DOI] [PubMed] [Google Scholar]
- 59.Ogawa H, Ishikawa T, Matsushita K, et al. Effects of right Atrial Pacing Preference in prevention of paroxysmal atrial fibrillation. Atrial Pacing Preference Study (APP Study) Circ J. 2008;72:700–704. doi: 10.1253/circj.72.700. [DOI] [PubMed] [Google Scholar]
- 60.Gold M, Adler S, Fauchier L, et al. Impact of atrial prevention pacing on atrial fibrillation burden: primary results of the Study of Atrial Fibrillation Reduction (SAFARI) trial. Heart Rhythm. 2009;6:295–301. doi: 10.1016/j.hrthm.2008.11.033. [DOI] [PubMed] [Google Scholar]
- 61.Nigro G, Russo V, Rago A, et al. The main determinant of hypotension in nitroglycerine tilt-induced vasovagal syncope. Pacing Clin Electrophysiol. 2012;35:739–748. doi: 10.1111/j.1540-8159.2012.03388.x. [DOI] [PubMed] [Google Scholar]
- 62.Russo V, Rago A, Politano L, et al. The effect of atrial preference pacing on paroxysmal atrial fibrillation incidence in Myotonic Dystrophy type 1 patients: a prospective, randomized, single-bind cross-over study. Europace. 2012;14:486–489. doi: 10.1093/europace/eur373. [DOI] [PubMed] [Google Scholar]
- 63.Task Force on Practice Guidelines: ACC/AHA/HRS 2008 guidelines for device based therapy of cardiac rhythm abnormalities: A report of the American College of Cardiology/American Heart Association Task Force. Circulation. 2008;27:e350–e408. doi: 10.1161/CIRCUALTIONAHA.108.189742. 117. [DOI] [PubMed] [Google Scholar]