

Abstract
Myotonic dystrophy type 2 (DM2) is caused by CCTG-repeat expansions. Occurrence of splicing and mutations in the muscle chloride channel gene CLCN1 have been reported to contribute to the phenotype. To examine the effect of CLCN1 in DM2 in Germany, we determined the frequency of a representative ClC1 mutation, R894X, and its effect on DM2 clinical features. Then, we examined CLCN1 mRNA splice variants in patient muscle functionally expressed the most abundant variant, and determined its subcellular localization. Finally, we established a cellular system for studying mouse clcn1 pre-mRNA splicing and tested effects of expression of (CCUG)18, (CUG)24 and (AAG)24 RNAs. The R894X mutation was present in 7.7% of DM2 families. DM2 R894X-carriers had more myotonia and myalgia than non-carriers. The most abundant CLCN1 splice variant in DM2 (80% of all transcripts) excluded exons 6-7 and lead to a truncated ClC1236X protein. Heterologous ClC1236X expression did not yield functional channels. Co-expression with ClC1 did not show a dominant negative effect, but a slightly suppressive effect. In C2C12 cells, the clc1 splice variants generated by (CCUG)18-RNA resembled those in DM2 muscle and differed from those generated by (CUG)24 and (AAG)24. We conclude that ClC1 mutations exert gene dose effects and enhance myotonia and pain in DM2 in Germany. Additionally, the ClC1236X splice variant may contribute to myotonia in DM2. Since splice variants depend on the types of repeats expressed in the cellular C2C12 model, similar cell models of other tissues may be useful for studying repeatdependent pathogenetic mechanisms more easily than in transgenic animals.
Key words: PROMM, myotonic dystrophy, chloride channel
Introduction
Myotonic dystrophies are dominantly inherited multisystemic disorders. Two types are discriminated clinically and genetically: myotonic dystrophy type 1 (OMIM 160900, DM1) and myotonic dystrophy type 2 (OMIM 602668, DM2). Both forms present with similar features including adult onset, slowly progressive muscle weakness, myotonia, subcapsular cataracts (1,2), cardiac conduction defects (3, 4), and endocrine disorders. DM1 is caused by an expansion of a CTG repeat in the 3'-untranslated region of the dystrophia myotonica-protein kinase (DMPK) gene on chromosome 19q13.3 (5) and DM2 is caused by an expanded CCTG repeat in intron 1 of the zinc finger 9 gene (6).
Due to the phenotypic and genotypic similarities, a common pathogenetic mechanism was assumed (7): RNA transcripts, containing the expanded repeats, accumulate in the cell nuclei as RNA foci (6, 8) and alter the regulation or localization of several RNA-binding proteins including CUG-binding proteins (9) and different forms of muscleblind (8, 10). This is thought to affect nuclear functions such as regulation of pre-mRNA splicing (1).
Splice variants of several genes have been described in DM and the resulting changes of protein function were assumed to explain some of the clinical features, e.g. splice variants of ClC1, encoded by CLCN1, have been suggested to cause the myotonia (11-13). A functional study of 2 typical DM1 ClC1 variants causing early protein truncation has demonstrated a loss of function with dominant-negative effect on full-length channels (14). Additionally, recessive myotonia congenita ClC1 mutations, R894X, have been reported in a genetic studies of e.g. Finish and Finnish-German populations (15-18). Our goal was to further study – genetically and functionally – the significance of ClC1 in DM2, and to establish a cell system to study ClC1 splicing produced by expression of short repeat expansions.
Materials and methods
Clinical and genetic studies.
The families with DM2 were identified through a proband diagnosed as DM based on clinical or electrophysiological myotonia associated with either muscle weakness or bilateral cataracts (19). Patients and family members had blood drawn and underwent neurological examination and EMG as far as they were German residents. For patients referred to us from abroad, clinical examination by one of the authors was not possible. The electrophysiological myotonia was diagnosed according to the criteria of the American Association of Electromyography and Electrodiagnosis (AAEE) (20). For some patients, electrocardiography (ECG) and endocrinological evaluation were further performed. To confirm the DM2 diagnosis, PCR amplification across the DM2 CCTG repeat was performed as previously described. R894X, the most frequent ClC1 mutation in German patients with the recessive inherited type of myotonia, was screened for as described (21).
ClC1 splice variants.
Quadriceps muscle was taken from 3 moderately affected and 1 mildly affected patient (CCTG carriers without R894X mutation) with their informed consent. Control muscle was taken from 5 individuals negative in a malignant hyperthermia testing protocol. Total cellular RNA was extracted from homogenized skeletal muscle tissues and cultured cells using Trizol R (Gibco BRL, USA). Eight pairs of primers were designed to cover the whole cDNA of CLCN1 (Table 1). RT-PCR amplification was carried out in a final volume of 50 μl, using equal amounts (1-2 μg) of total RNA, and 50 pmol upstream and downstream primers with an one step RT-PCR kit (Qiagen, Hilden, Germany). After first strand cDNA synthesis (50°C for 30 min), 35 cycles of amplification were performed, each consisting of 60 sec at 94°C, 60 s at 55°C and 60 s at 72°C, followed by a final 10 min extension at 72°C. The products of amplification were electrophoretically resolved on 2 % agarose gels stained with ethidium bromide (0.5 μg/ml). All variants were confirmed by sequencing. Percent of splicing variant was calculated as (cpm variant band)/(cpm variant band +cmp normal band) x 100 using Scion Image software.
Table 1.
Primer pairs used for RT-PCR.
| Region | Forward primer | Reverse primer | Temp. (°C) | Length (bp) |
|---|---|---|---|---|
| Exon1-4 | CLCN1F ccttggggacagcaagagc |
CLCN1R gcgcgtaggaccacttgtagg |
55 | 486 |
| Exon 3-6 | CLCN2F ggtcagctggagcatggactacg |
CLCN2R gctggcaatgtggacgaagggg |
55 | 328 |
| Exon 5-10 | CLCN3F ggcagtggcatccccgtgggg |
CLCN3R cagctcccaggagcccacag |
55 | 414 |
| Exon 9-13 | CLCN4F gaaggaactaccagcttttgctg |
CLCN4R gggcatagtggtggccacgatgg |
55 | 400 |
| Exon 12-16 | CLCN5F ggattcacccccgggtcaacgttg |
CLCN5R ccatgatgtcctcaacaaagatgg |
55 | 483 |
| Exon 15-18 | CLCN6F gcctgaccttggctggaaccag |
CLCN6R gggtggagagcaagggaaggagg |
55 | 429 |
| Exon 17-22 | CLCN7F ggatgaggatgaggacgaag |
CLCN7R ggccaaggagtgaaaacagg |
55 | 404 |
| Exon 21-23 | CLCN8F gctggtggagcagacaaccctg |
CLCN8R gatgaggacgagctgatcctg |
55 | 486 |
Plasmid construction.
A 519 bp product, with exons 6 and 7 missing, was amplified from DM2 affected persons RNA, using CLCN2F as forward primer and CLCN3R as reverse primer. PCR product was digested with BstEII and the resulting 225 bp fragment used to replace the corresponding fragment into the human skeletal muscle choride channel mammalian expression construct pRc-CMV-hClC1 (22). To obtain a GFP-ClC1 wt and variant fusion constructs, the pRc-CMV-hClC1 wt and variant were digested to completion with KpnI. The coding fragments were gel-purified and subcloned into pEGFP-C1 (Clontech). The sequence of the resulting inframe pEGFP-ClC1 fusion constructs were verified by restriction digestion and sequence analysis.
Functional study.
The pRcCMV constructs containing the full length or the truncated form of the ClC1 channel were transfected into tsA201 cells. Successful expression of the resulting prematurely truncated protein ClC1236X (stop at codon 236) was confirmed by Western blots of total protein of transfected tsA201 cells using rabbit polyclonal primary antibody for ClC1 (1:1,000) and goat anti-rabbit horseradish peroxidase-conjugated secondary antibody (1:5,000), both from Santa Cruz, Biotechnology. Whole-cell patch clamp measurements were performed as previously described (23) using normal and low chloride pipette solutions with composition in mM: 130 CsCl, 2 MgCl2, 5 EGTA and 10 HEPES (pH 7.4) or 14 CsCl, 116 Na-Glutamate, 2 MgCl2, 5 EGTA and 10 HEPES (pH 7.4) respectively and bath solution containing in mM: 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2 and 5 HEPES (pH 7.4). All data were analyzed using a combination of Axon pCLAMP6, Microsoft Excel and Micro- Cal ORIGIN software. Statistical evaluation was done by Student's t-test. Data are shown as mean ± SEM.
Laser microscopy.
Localization of ClC1236X was tested by transfection of the generating GFP-ClC-1 wt and variant fusion constructs into tsA201 cells followed by confocal laser microscopy (Biorad) study. Excitation was performed at 488 nm and emission was collected with a 500 nm long pass filter. Images representing single equatorial planes of 0.5 μm thickness were acquired with a 60x objective and processed with Corel Photopaint 9.0.
Cell system for splicing detection.
C2C12 is a mouse myoblast cell line that can form polynucleated myotubes with a similar expression pattern as regenerating muscle. C2C12 cells were cultivated as previously described (24). These cells were transfected with 25μg of pcDNA3Hygro+ containing either 18 CCTG, 24 CTG, or 24 AAG repeats. RNA was extracted on days 2 or 3 after transfection. Following incubation with DNAase, RT-PCR analysis of the endogenous mouse clcn1 mRNA was performed for the variant excluding exons 6-7. Additionally, to roughly estimate the relative amount of expressed repeat RNA, RTPCR of the repeats themselves was performed on several template dilutions and measured densitometrically.
Results
Genetic and clinical studies.
By clinical and genetic studies, we identified 126 DM2 individuals of 65 families. The recessive ClC1 mutation, R894X, occurred in 5 (7.7%) of these families. One DM2 individual was homozygous and 18 were heterozygous; 10 of 95 (10.5%) unaffected relatives were also heterozygous. To check whether the local population had a higher frequency of R894X, we tested 306 unrelated control samples. The mutation was present only once suggesting a frequency of about 0.3% in the general population.
To examine a possible effect of R894X on the phenotype of CCTG repeat carriers, we reviewed the frequencies of clinical symptoms in 53 DM2 patients, 18 CCTG-R894X (C/X) and 35 CCTG-only carriers (C/R) (Table 2).
Table 2.
Clinical features of DM2 patients with and without R894X mutation. C/X-CCTG and R894X carriers; C/R-CCTG only carriers; F female; M male.
| Feature | DM2 subjects | |||||||
|---|---|---|---|---|---|---|---|---|
| C/X (n = 18 patients) | C/R (n = 35 patients) | |||||||
| Nr. of patients (of 18) | Mean age at onset | F : M | Nr. of families | Nr. of patients (of 35) | Mean age at onset | F : M | Nr. of families | |
| Clinical myotonia | 15 | 33 | 12 : 3 | 5 | 12 | 36 | 7 : 5 | 6 |
| EMG myotonia | 17 | 35 | 14 : 3 | 5 | 24 | 38 | 16 : 8 | 8 |
| Muscle weakness | 8 | 36 | 5 : 3 | 4 | 15 | 38 | 12 : 3 | 7 |
| Proximal muscle pain | 7 | 35 | 4 : 3 | 4 | 5 | 39 | 5 : 0 | 6 |
| Cataracts | 7 | 39 | 6 : 1 | 4 | 14 | 43 | 2 : 1 | 9 |
| Cardiac arrhythmia | 1 | 25 | 1 : 0 | 1 | 2 | 31 | 10 : 5 | 2 |
| Abnormal serum levels of CK, γGT or cholesterol | 8 | 35 | 6 : 2 | 3 | 15 | 37 | 22 : 13 | 13 |
There was no difference on the size of the CCTG repeats between the two groups, the mean size being 1,900 and 1,850 CCTG, respectively. Clinical myotonia was observed in 83% of the C/X carriers compared with 34% (p = 0.0005) of the CCTG-only carrier (C/R); EMG myotonia was found in 94% of the former and in 68% of the latter (p = 0.033); the age at onset of the myotonia was 33 ± 8.1 and 36 ± 13.5, respectively. Muscle weakness was found in 44% C/X and 43% C/R (p = 0.914); the age at onset of weakness was 36 ± 5.8 and 38 ± 16.2 respectively; the muscle pain – typical for DM2 – was observed in 39% C/X, with an age at onset of 35 ± 6.1 years, compared to 14% C/R (p = 0.043), with an onset age of 39±14.3; cataracts were present in 39% C/X and 40% C/R (p = 0.931), and the ages at onset were 39±10.1 and 43±12.3 respectively; cardiac arrhythmia occurred in 6% C/X and 6% C/R(p = 0.981); abnormal serum levels of CK, GT or cholesterol occurred in 46% C/X and 44% C/R (p = 0.914). Women were 78% of C/X compared with 63% C/R (p = 0.271). As women with chloride channel mutations generally show less myotonia than men (25), the higher rate of myotonia in the C/X group containing more women supports that myotonia is enhanced by R894X. Summing it up, the parameters significantly differing between the two groups were myotonia and muscle pain.
CLCN1-RNA splice variants.
By RT-PCR, we identified 5 alternative CLCN1-RNA splice variants, all of which occurred both in DM2 and in control muscles. These were exclusions of exons 2, 9, 11, 6-7, or 2-12, and were predicted to lead to premature truncations and putative loss of protein function. Densitometric measurement of RT-PCR product intensities of DM2 and controls differed only for the variant excluding exons 6-7, termed D6-7, which seemed to be 4 times as abundant as the normal length in DM2 (Fig. 1).
Figure 1.

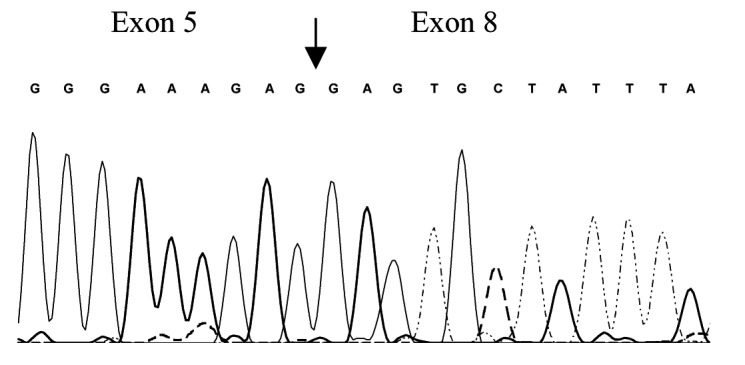
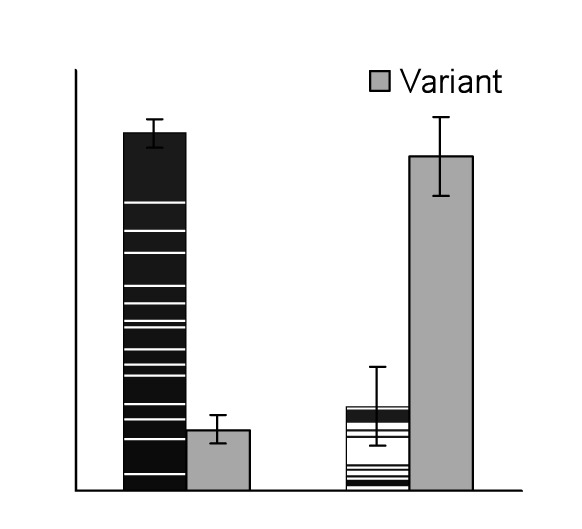
RT-PCR analysis of total RNA from control (C) and moderate DM2 (DM2) skeletal muscle tissue using oligonucleotide complementary to exons 5 and 10 (CLCN3F 5'-ggcagtggcatccccgtgggg-3' and CLCN3R 5'-cagctcccaggagcccacag- 3'). Two bands were distinguished: the standard expected product (S) 414 bp and the product excluding the exons 6 and 7, D6-7 (VAR) 257 bp. Additional discrete visible bands may be due to the RT-PCR condition and false primer annealing and were not ClC1 isoforms, amplicons from different genes as confirmed by subsequent sequencing. B) Sequencing of D6-7 variant showing exclusion of exons 6 and 7. C) Densitometry results suggested that in controls the ratio of normal length-variant vs. D6-7 was 5.7:1 and in DM2 1:4. Error bars represent standard deviation.
Functional expression.
Western blots using a primary antibody directed against the protein N-term confirmed presence of the wt and predicted truncated protein of 60 kDa (Fig. 2A, lanes 1-4) in tsA201 cells transfected with 1-2 μg of plasmid. Chloride currents of the dimeric ClC1 channels were evoked by voltage pulses to levels between -165 and +75 mV lasting 300 ms followed by a voltage pulse to -115 mV lasting 100 ms (representative current traces see Fig. 2B). Under symmetric chloride conditions, full-length ClC1 homodimers exhibited currents, those with ClC1236X (stop at codon 236) homodimers did not (Fig. 2C). To test for a possible dominant-negative effect of ClC1236X on ClC1, we co-transfected in a ratio of 1:1 (0.5μg of wt and variant). The resulting currents did not show biophysical characteristics different from ClC1 homodimers, i.e. the relative open probability curves did not differ: V0.5 for ClC1 and ClC1/ClC1236X–co-expression were -74 ± 5 mV, n = 8 and –76 ± 5 mV, n = 6 respectively (Fig. 2G).
Figure 2.
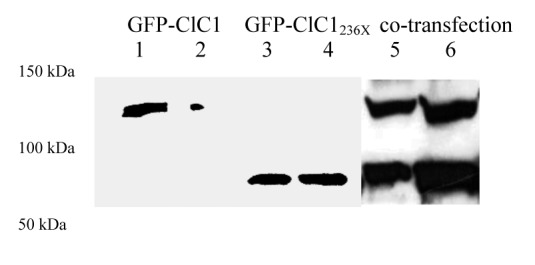
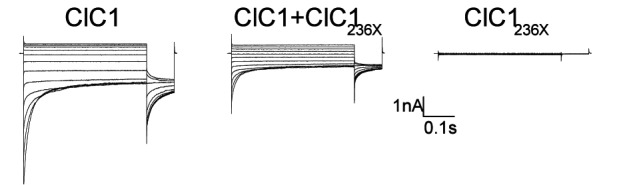
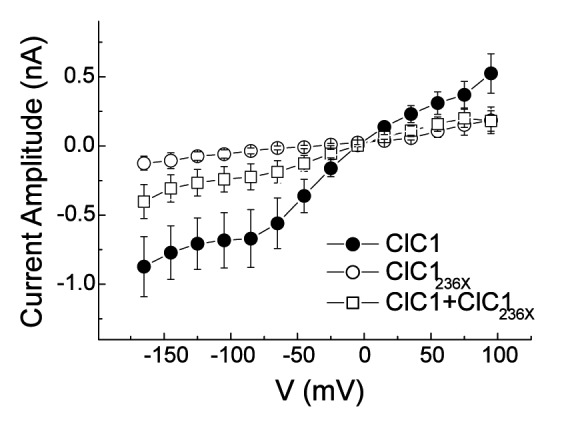
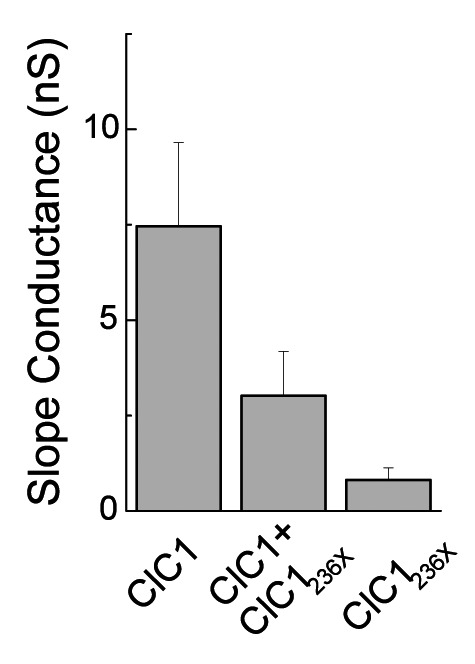
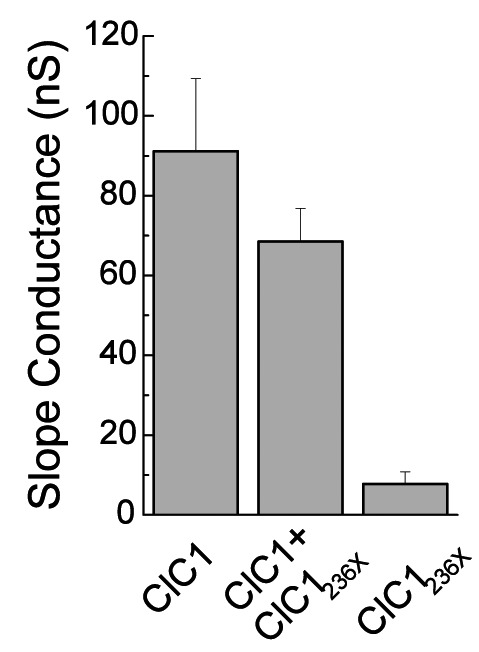
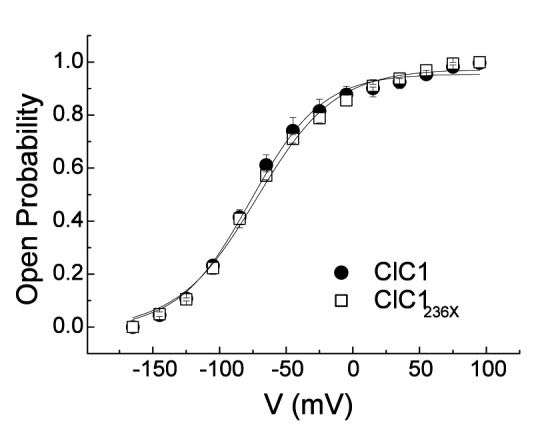

A) Western blots of 30 μg of tsA201 cell protein extracts containing transiently expressed chloride channels. An antibody recognizing the N-terminus of ClC1 detected a ~130 kDa GFP-ClC1 and a truncated ~60 kDa GFP-ClC1236X both corresponding to the theoretically expected lengths. GFP-ClC1 (lanes 1-2), GFP-ClC1236X (lanes 3-4) and GFP-ClC1 1:1 cotransfected with GFP-ClC1236X (lanes 5-6). B) Representative current traces, evoked by 300 ms voltage pulses between –165 and +75 mV followed by a 100 ms pulse to –115mV, for ClC1, variant (ClC1236X) and 1:1 co-transfection experiments with half the amount of each vector. C) Voltage dependence of late current amplitudes. The curves show well distinguishable currents for ClC1 (n=8, 1μg plasmid); no detectable chloride currents for the ClC1236X (n=6, 1μg plasmid) and a slightly more than 50% reduction of current amplitude in 1:1 co-transfection experiments (n=6, 0.5μg + 0.5μg plasmids). D) Voltage dependence of late current amplitudes measured with 16 mM internal Cl- concentration in 1:4 ratio of expressed plasmids; ClC1 n=8, 1μg; ClC1236X n=7, 4μg plasmid; co-transfection n=8, 1μg + 4μg plasmids. E) Normalized slope conductance at 0 mV calculated from the current-voltage relation in Figure 2C; note the insignificant difference between the ClC1+ClC1236X value and half of the ClC1 value. F) Normalized slope conductance at 0 mV calculated from the current-voltage relation in Figure 2D; note the slight difference between ClC1+ClC1236X value and the ClC1 value. G) The relative open probability (Popen), as determined by the tail current amplitude at –115 mV, shows the same biophysical characteristics for ClC1 (n=4) and ClC1236X (n=6). Lines represent fits of the experimental data to a standard Boltzmann function. H) Subcellular localization pattern of GFP-ClC1 and GFP-ClC1236X fusion proteins. The transfected tsA201 cells were imaged using confocal microscopy 24 h following transfection. GFP alone and GFP-ClC1236X were ubiquitous in nucleus and cytoplasm. GFP-ClC1 was localized into the cell membrane. Co-transfection of ClC1 with GFP-ClC1236X mainly exhibited a pattern similar to GFP, however some cells with lower expression rate showed presence in the membrane indicating a possible interaction between GFP-ClC1236X and untagged ClC1. The scale bar is 10 μm.
To test for an effect of ClC1236X on deactivation as previously proposed (12), the kinetics of inward current deactivation were described by fitting a standard exponential function to the time course of tail current decay. The values for the co-expression experiment did not differ significantly from those for ClC1 homodimers: at -115 mV, the time constant was 18±3 ms (n = 6) for ClC1/ ClC1236X–co-expression and 24±4 ms (n = 8) for ClC1 (p = 0.2). However, these currents in the co-expression experiment revealed a reduction of the slope conductance (Fig. 2E) and of late current amplitude at -145 mV: -0.8 ± 0.2 nA for ClC1 (n = 8); -0.30 0.09 nA for ClC1/ ClC1236X–co-expression (n = 6); and -0.11 ± 0.05 nA for ClC1236X (n = 6; Fig. 2C). The late current amplitude for ClC1/ClC1236X–co-expression of -0.3 nA was slightly smaller than 50% of ClC1 -0.8/2 = -0.4 nA. The difference between the two amplitudes was not statistically significant and therefore did not support a clear dominantnegative effect of ClC1236X on ClCl.
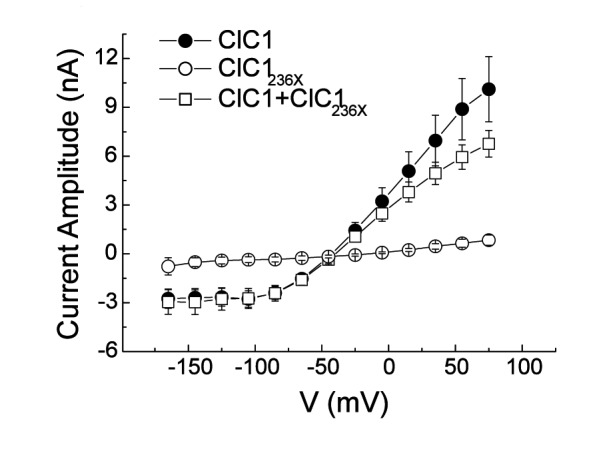
To test for a possible dominant-negative effect in the proportions found in our RT-PCR results in DM2, we performed a second set of experiments with decreased pipette chloride concentration in which we transfected 1 μg of ClC1 alone, 4 μg of ClC1236X alone, and co-transfected 1 μg of ClC1 with 4 μg of ClC1236X. In the co-expression experiment, 1/5*1/5 = 1/25 of channel complexes would be ClC1 homodimers, 2*1/5*4/5 = 8/25 ClC1-ClC1236X heterodimers, and 4/5*4/5 = 16/25 ClC1236X homodimers. If the latter two were non-functional in the sense of a dominant negative effect, the resulting current would be 1/25 of the maximum current. However, as the transfection rate was 5 times larger than the ClC1 expression alone, a current reduction down to 1/5 would already indicate non-functionality of the heterodimers.
The ClC1/ClC1236X-co-expression gave rise to currents with amplitudes similar to those obtained with ClC1 expression alone, especially in the physiologically relevant range around -80mV (Fig. 2D). The slope conductances of ClC1 and ClC1/ClC1236X-co-expression were not significantly different (Fig. 2F). Fitting of current deactivation with two time constants revealed no difference between CLC1 and ClC1/ClC1236X-co-expression being t1 = 10 ± 2 ms and t2 = 90±20 ms for ClC1 and t1 = 9±2 ms and t2 = 87±21 ms for ClC1/ClC1236X-co-expression at -120 mV. This result suggests that either no ClC1- ClC1236X heterodimers are formed or that they are at least partially functional whereby a 50% conductance would be compatible with these results, and this would indicate that one of the 2 channel pores would be functional.
To check for channel localization and indication of heterodimer formation, we performed confocal laser microscopy of GFP fusion proteins (Fig. 2H). TsA201 cells expressing GFP alone or GFP-ClC1236X showed a diffuse fluorescence pattern with uniform intensity throughout cytoplasm and nucleus, GFP-ClC1 alone was localized mostly to the cell membrane. Cells in which ClC1 and GFP-ClC1236X were co-expressed showed mostly the GFP diffuse pattern, however, about 10% of the cells showed a localization of the protein to the cell membrane. This experiment suggested i) that fusion of GFP to ClC1 did not alter its trafficking (Fig. 2H, panel 2); and ii) that ClC1236X can bind to ClC1 forming heterodimers in the membrane (Fig. 2H, panel 5). Western blots of 1:1 co-transfection wt and truncated fusion proteins suggested equal expression rates (Fig. 2A, lanes 5-6). Therefore, the laser microscopy results of panel 4 most likely show over-expression of GFP-ClC1236X which remained diffuse in addition to the heterodimers of GFP-ClC1236X and the untagged ClC1 homodimers in the membrane.
Cell system for splicing detection.
Introduction of a vector generating (CCUG)18-RNA, a non-pathological repeat length, produced alternative splicing of mouse clcn1 mRNA excluding exons 6-7 in our first experiment in 2 of 30 dishes on day 3 after transfection (2 of Nday3 = 30). To
examine the effect of specific repeats, we compared different RNA repeats of 72bp length, (CCUG)18, (CUG)24, and (AAG)24, with respect to splicing on post-transfectional days 2 and 3 during maximum expression. For cells transfected with empty vectors, no splice variants were detected in Nday2 = 15 and Nday3 = 17 dishes. Likewise, for cells expressing (AAG)24, no variants were found in Nday2 = 11 and Nday3 = 19 dishes (Fig. 3A). Aberrant splicing occurred when expressing (CCUG)18: 1 of Nday2 = 16 and 1 of Nday3 = 16 (Figure 3B). Similarly, aberrant splicing occurred when expressing (CUG)24: 0 of Nday2 = 16 and 2 of Nday3 = 16 (Fig. 3C). However, the variants produced by (CCUG)18 and (CUG)24 differed: the former produced exclusion of exons 6-7 twice and once additionally exclusion of exons 6-9, while the latter produced exclusion of exons 6-9 only. Densitometric examination of the relative RNA repeat amount by dilution-RT-PCR suggested that (CCUG)18 and (CUG)24 RNA levels were similar, while (AAG)24 RNA levels were only about 33% of this value (Figure 3D).
Figure 3.




RT-PCR assay of CLCN1 exons 3 to 10, from RNA extracted after expression of different repeat RNAs: (AAG)24 (A), (CCUG)18 (B) and (CUG)24 (C), on days 2 and 3 after transfection. The upper band, S, represents the standard RT-PCR product; • represents skipping of exons 6-9; ** represents skipping of exons 6-7. D) RT-PCR from a series of RNA dilutions of total RNA extracted from transfected C2C12 cells on day 3 after transfection suggested that (CCUG)18 and (CUG)24 RNA levels are similar, while (AAG)24 RNA levels are only about 33% of this value.
Discussion
R894X, the most common ClC1 mutation, was present in 7.7% of our German DM2 families compared with 0.3% of controls of the same geographical region. A possible explanation for our lab to which patients are referred for clarification of myotonic disorders, may be a selection bias towards DM2 with especially prominent myotonic symptoms. This is in agreement with the previously reported Finish studies (15-18) in which additional ClC1 mutations occurred in 5% of DM2 versus 1% of controls, due to a greater need of such patients to consult a doctor because of the myotonia. In contrast, the majority of DM2 patients may seek medical attention for other symptoms of the disease without being aware of their underlying disorder.
However, the presence of the mutation alone cannot explain the myotonia in DM2 as functional co-expression studies for R894X suggested (21). In agreement with this, R894X causes clinical myotonia in the homozygous or compound heterozygous state (21, 26), while it generates latent myotonia, i.e. subclinical myotonia visible in the EMG only, in heterozygous carriers (27). This supports the general idea of a gene dose effect of this mutation that could also be effective in DM patients in which CLCN1- RNA splicing occurs. In such a case, R894X would lead to additional truncation of 50% of the unspliced CLCN1- RNA transcripts which may be sufficient for the latent myotonia to become clinically apparent. According to our data, clinical myotonia was observed in 83% of the C/X carriers compared with 34% of the C/R carriers, suggesting that R894X contributes to the clinical manifestation of myotonia (Table 2).
Muscle pain occurred over twice as often in C/X than in C/R patients. It is a disabling symptom which, because of its aggravation by exercise, cold, and percussion, differs from the pain in other muscular dystrophies (28). Possibly, the myotonic stiffness may contribute to the myalgia comparable to the situation in some patients with myotonia congenita (26).
In DM, CLCN1-RNA splicing changes have been described using two different methods. In both DM1 and DM2, Mankodi et al (11) cloned and sequenced a large number of cDNAs, a method which can capture both rare and frequent variants, but may not representatively reflect the relative frequency of each variant. In DM1, Charlet-B. et al (12) performed RT-PCR, a method which preferentially amplifies the more abundant variants and enables to assess their relative frequency while washing out the rare ones. Figure 4A shows results of these methods for the RNA region between exons 5 and 8. While a direct comparison of these methods must be made with caution, an overall agreement of the results may be found on a certain level. Comparing just the wt with D6/i6b- 7a (variant excluding exon 6 and including exons 6b and 7a) and D6-7 in control samples, Mankodi et al obtained 83:0:3, Charlet et al 85:5:10 and ourselves 85:0:15. For the same variants in DM1, Mankodi et al obtained 56:5:8 in moderately affected, and 2:36:10 in severely affected individuals, Charlet et al 15:80:5 (= 7.5:40:2.5) to a large extent, in agreement with the severely affected cases only. Comparing just wt with D6-7 in DM2, Mankodi obtained 31:10 (= 3.1:1) and ourselves 20:80 (1:4). This raised the question of whether our patients may be more severely affected than Mankodi's ones. To address this, we obtained a biopsy from a young, very mildly affected DM2 patient which yielded 58:42 regardless of the PCR cycle number (Figure 4B). We conclude that the splicing events increase with disease severity and that the D6-7 variant is then increasingly favoured in DM2 and its protein translation to ClC1236X may become functionally relevant.
Figure 4.


Summary of splice variants of CLCN1-RNA in the m-RNA region between exons 5 and 8 comparing different studies with our data (9,10). The positions of the pre-mature stop codons of the splicing variants are indicated. The last line, "other variants", refers to other splicing events affecting the region without being directly displayable in the scheme (e.g. exclusion of exons 4-7 or 2-12 etc). n.d. = not detected. B) RT-PCR analysis of total RNA from a very mild case of DM2 and a control using the same technique as figure 1. The standard expected product (S) of 414 bp and the product excluding the exons 6 and 7, D6-7 (VAR) of 257 bp are shown after 5, 10, 15, 20, 25, 30, and 35 PCR cycles. Densitometry suggested that in mild DM2, the ratio of normal-length variant vs. D6-7 was 58:42 average over all cycle numbers.
In our study, ClC1236X does not seem to exert a truly dominant-negative effect on co-expressed ClC1, but only a slightly suppressive effect when over-expressed. While confocal laser microscopy suggests that a ClC1236X association with ClC1 occurs in the membrane, an additional potential trafficking problem or decreased formation of ClC1-ClC1236X heterodimers cannot be excluded. Even so, our results would be compatible with the idea that ClC1- ClC1236X heterodimers may be 50%-functional and conduct chloride through the pore of the ClC1 part of the dimer. In agreement with this view of the functional effect of the prematurely terminated channel, nonsense mutations of ClC1 resulting in early truncations nearby such as fs231X (29), fs258X (30), or fs289X (31) are all inherited in a recessive and not dominant manner and produce myotonia by a lossof- function mechanism instead of a dominant-negative mechanism. However, in DM1, two splice variants, i) D6/ i6b-7a, resulting in a 256 amino acid protein, and ii) i6b- 7a (variant including exons 6b and 7a), resulting in a 282 amino acid protein, have been studied functionally. They both exert a dominant-negative effect on co-expressed ClC1 channel in Xenopus oocytes (14). Possibly, this effect may be sequence specific as they are the only two truncations containing PVPVLQMSTPLSPVAPHGDRAWAAX, the sequence encoded by exons 6b-7a, a proline rich peptide that might affect the pore of the co-expressed ClC1 wt (32). Therefore, the truncation variants in DM1 may explain why the chloride conductance is more reduced in DM1 than in DM2 and, therefore, why clinical myotonia is more prominent in DM1 than in DM2 (2).
For both types of DM, the clinical variability of myotonia may depend on the degree of nonsense-mediated mRNA decay (NMD) of mRNAs containing premature stop codons. Previous reports have suggested that up to 27% of CLCN1-RNA result in alternatively spliced forms that generate premature termination codons (11, 12) which are subject to NMD; this has been shown especially for CLCN1-RNA variants that contain a premature termination codon in exon 7 (33). The most frequent variants in both DM1 (D6/i6b-7a) and DM2 (D6-7) have their stop codons in this RNA region, being in exons 7a and exon 8 respectively. Therefore, the respective degree of NMD may be similar and contribute to the reduced quantity of CLCN1 mRNA in DM (34). Because chloride current is reduced but not abolished in DM muscle (35), it seems reasonable to assume that at least a portion of transcripts coding for R894X is not degraded and can contribute to reduced chloride conductance and myotonia in DM2.
In the C2C12 cell line, it has been shown that the stable expression of pre-mutation or pathologic DM1 repeats, (CUG)57, (CUG)78, (CUG)100 or (CUG)200, generate nuclear and cytoplasmic RNA foci and affect myotube differentiation (24). In our study, we used C2C12 to transiently express very short DM2 and DM1 repeats of 72bp, (CCUG)18 and (CUG)24. By this experiment, we have shown that i) this system is useful for studying changes of clcn1 pre-mRNA splicing, ii) very short nonpathologic repeats already produce a measurable effect, and iii) the splicing pattern differs depending on the type of repeat expressed. The system would principally allow to dose the repeats by the amount of vector transfected. We assume that the amount of repeats, not its length as suggested formerly (36, 37) is decisive for the effect. In agreement, in congenital DM1, in which the CTG repeats are longest (over 1000 repeats), there is no myotonia at all because few repeats have been accumulated. Over time, many longer repeats will accumulate because they do not degrade as easily (38, 39). For (AAG)24, we therefore cannot exclude a possible effect at higher doses, not reached in our test.
There are slight phenotypic differences between DM2 and DM1 concerning the distribution of weakness (proximal vs. distal, (40), histological findings (fiber type 2 atrophy vs. type 1 atrophy (41), absence of the congenital form in DM2. Additionally, we have shown that the type of the repeat determines the type of CLCN1 mRNA splicing pattern, and that different variants occur in DM2 muscles compared with those described for DM1 muscles. Different variants are also produced by different repeats in the simple C2C12 cell system. Lastly, the resulting ClC1 variants may exert different effects (e.g. not necessarily a dominant negative effect). Taken together, these results suggest that the pathogenetic mechanism in DM2 is different from DM1.
Acknowledgements
We thank Drs. Daniela Schreiber and Nicole Thomasky for their contributions. This work was supported by the German Society for Muscle Diseases (DGM).
References
- 1.Day JW, Ranum LP, et al. Genetics and molecular pathogenesis of the myotonic dystrophies. Curr Neurol Neurosci Rep. 2005;5:55–59. doi: 10.1007/s11910-005-0024-1. [DOI] [PubMed] [Google Scholar]
- 2.Day JW, Ricker K, Jacobsen JF, et al. Myotonic dystrophy type 2: molecular, diagnostic and clinical spectrum. Neurology. 2003;60:657–664. doi: 10.1212/01.wnl.0000054481.84978.f9. [DOI] [PubMed] [Google Scholar]
- 3.Dello Russo A, Mangiola F, Della Bella P, et al. Risk of arrhythmias in myotonic dystrophy: trial design of the RAMYD study. J Cardiovasc Med (Hagerstown) 2009;10:51–58. doi: 10.2459/jcm.0b013e328319bd2c. [DOI] [PubMed] [Google Scholar]
- 4.Schoser BG, Ricker K, Schneider-Gold C, et al. Sudden cardiac death in myotonic dystrophy type 2. Neurology. 2004;63:2402–2404. doi: 10.1212/01.wnl.0000147335.10783.e4. [DOI] [PubMed] [Google Scholar]
- 5.Brook JD, McCurrach ME, Harley HG, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3' end of a transcript encoding a protein kinase family member. Cell. 1992;68:799–808. doi: 10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- 6.Liquori CL, Ricker K, Moseley ML, et al. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–867. doi: 10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 7.Day JW, Ranum LP. RNA pathogenesis of the myotonic dystrophies. Neuromuscul Disord. 2005;15:5–16. doi: 10.1016/j.nmd.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 8.Fardaei M, Rogers MT, Thorpe HM, et al. Three proteins, MBNL, MBLL and MBXL, co-localize in vivo with nuclear foci of expanded- repeat transcripts in DM1 and DM2 cells. Hum Mol Genet. 2002;11:805–814. doi: 10.1093/hmg/11.7.805. [DOI] [PubMed] [Google Scholar]
- 9.Timchenko LT, Timchenko NA, Caskey CT, et al. Novel proteins with binding specificity for DNA CTG repeats and RNA CUG repeats: implications for myotonic dystrophy. Hum Mol Genet. 1996;5:115–121. doi: 10.1093/hmg/5.1.115. [DOI] [PubMed] [Google Scholar]
- 10.Kanadia RN, Johnstone KA, Mankodi A, et al. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–1980. doi: 10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- 11.Mankodi A, Takahashi MP, Jiang H, et al. Expanded CUG repeats trigger aberrant splicing of ClC-1 chloride channel pre-mRNA and hyperexcitability of skeletal muscle in myotonic dystrophy. Mol Cell. 2002;10:35–44. doi: 10.1016/s1097-2765(02)00563-4. [DOI] [PubMed] [Google Scholar]
- 12.Charlet B, Savkur RS, Singh G, et al. Loss of the muscle-specific chloride channel in type 1 myotonic dystrophy due to misregulated alternative splicing. Mol Cell. 2002;10:45–53. doi: 10.1016/s1097-2765(02)00572-5. [DOI] [PubMed] [Google Scholar]
- 13.Wheeler TM, Lueck JD, Swanson MS, et al. Correction of ClC-1 splicing eliminates chloride channelopathy and myotonia in mouse models of myotonic dystrophy. J Clin Invest. 2007;117:3952–3957. doi: 10.1172/JCI33355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berg J, Jiang H, Thornton CA, et al. Truncated ClC-1 mRNA in myotonic dystrophy exerts a dominant-negative effect on the Cl current. Neurology. 2004;63:2371–2375. doi: 10.1212/01.wnl.0000148482.40683.88. [DOI] [PubMed] [Google Scholar]
- 15.Mastaglia FL, Harker N, Phillips BA, et al. Dominantly inherited proximal myotonic myopathy and leukoencephalopathy in a family with an incidental CLCN1 mutation. J Neurol Neurosurg Psychiatry. 1998;64:543–547. doi: 10.1136/jnnp.64.4.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun C, Henriksen OA, Tranebjaerg L. Proximal myotonic myopathy: clinical and molecular investigation of a Norwegian family with PROMM. Clin Genet. 1999;56:457–461. doi: 10.1034/j.1399-0004.1999.560609.x. [DOI] [PubMed] [Google Scholar]
- 17.Suominen T, Schoser B, Raheem O, et al. High frequency of cosegregating CLCN1 mutations among myotonic dystrophy type 2 patients from Finland and Germany. J Neurol. 2008;255:1731–1736. doi: 10.1007/s00415-008-0010-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sun C, Ghelue M, Tranebörg L, et al. Myotonia congenita and myotonic dystrophy in the same family: coexistence of a CLCN1 mutation and expansion in the CNBP (ZNF9) gene. Clin Genet. 2011;80:574–580. doi: 10.1111/j.1399-0004.2010.01616.x. [DOI] [PubMed] [Google Scholar]
- 19.Ashizawa T, Dubel JR, Dunne PW, et al. Anticipation in myotonic dystrophy. II. Complex relationships between clinical findings and structure of the GCT repeat. Neurology. 1992;42:1877–1883. doi: 10.1212/wnl.42.10.1877. [DOI] [PubMed] [Google Scholar]
- 20.Streib EW. AAEE minimonograph #27: differential diagnosis of myotonic syndromes. Muscle Nerve. 1987;10:603–615. doi: 10.1002/mus.880100704. [DOI] [PubMed] [Google Scholar]
- 21.Meyer-Kleine C, Steinmeyer K, Ricker K, et al. Spectrum of mutations in the major human skeletal muscle chloride channel gene (CLCN1) leading to myotonia. Am J Hum Genet. 1995;57:1325–1334. [PMC free article] [PubMed] [Google Scholar]
- 22.Fahlke C, Rüdel R, Mitrovic N, et al. An aspartic acid residue important for voltage-dependent gating of human muscle chloride channels. Neuron. 1995;15:463–472. doi: 10.1016/0896-6273(95)90050-0. [DOI] [PubMed] [Google Scholar]
- 23.Ryan A, Rüdel R, Kuchenbecker M, et al. A novel alteration of muscle chloride channel gating in myotonia levior. J Physiol. 2002;545:345–354. doi: 10.1113/jphysiol.2002.027037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Amack JD, Paguio AP, Mahadevan MS. Cis and trans effects of the myotonic dystrophy (DM) mutation in a cell culture model. Hum Molv Genet. 1999;8:1975–1984. doi: 10.1093/hmg/8.11.1975. [DOI] [PubMed] [Google Scholar]
- 25.Mailänder V, Heine R, Deymeer F, et al. Novel muscle chloride channel mutations and their effects on heterozygous carriers. Am J Hum Genet. 1996;58:317–324. [PMC free article] [PubMed] [Google Scholar]
- 26.Lehmann-Horn F, Rüdel R, Jurkat-Rott K, et al. Nondystrophic myotonias and periodic paralyses. In: Engel AG, Franzini-Armstrong C, editors. Myology. New York: McGraw Hill Medical Publishing Division; 2004. pp. 1257–1300. [Google Scholar]
- 27.Deymeer F, Lehmann-Horn F, Serdaroglu P, et al. Electrical myotonia in heterozygous carriers of recessive myotonia congenita. Muscle Nerve. 1999;22:123–125. doi: 10.1002/(sici)1097-4598(199901)22:1<123::aid-mus20>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 28.George A, Schneider-Gold C, Zier S, et al. Musculoskeletal pain in patients with myotonic dystrophy type 2. Arch Neurol. 2004;61:1938–1942. doi: 10.1001/archneur.61.12.1938. [DOI] [PubMed] [Google Scholar]
- 29.Brugnoni R, Galantini S, Confalonieri P, et al. Identification of three novel mutations in the major human skeletal muscle chloride channel gene (CLCN1), causing myotonia congenita. Hum Mutat. 1999;14:447–447. doi: 10.1002/(SICI)1098-1004(199911)14:5<447::AID-HUMU13>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 30.Esteban J, Neumeyer AM, McKenna-Yasek D, et al. Identification of two mutations and a polymorphism in the chloride channel CLCN-1 in patients with Becker's generalized myotonia. Neurogenetics. 1998;1:185–188. doi: 10.1007/s100480050027. [DOI] [PubMed] [Google Scholar]
- 31.Nagamitsu S, Matsuura T, Khajavi M, et al. A "dystrophic" variant of autosomal recessive myotonia congenita caused by novel mutations in the CLCN1 gene. Neurology. 2000;55:1697–1703. doi: 10.1212/wnl.55.11.1697. [DOI] [PubMed] [Google Scholar]
- 32.Macias MJ, Teijido O, Zifarelli G, et al. Myotonia-related mutations in the distal C-terminus of ClC-1 and ClC-0 chloride channels affect the structure of a poly-proline helix. Biochem J. 2007;403:79–87. doi: 10.1042/BJ20061230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duno M, Colding-Jorgensen E, Grunnet M, et al. Difference in allelic expression of the CLCN1 gene and the possible influence on the myotonia congenita phenotype. Eur J Hum Genet. 2004;12:738–743. doi: 10.1038/sj.ejhg.5201218. [DOI] [PubMed] [Google Scholar]
- 34.Botta A, Vallo L, Rinaldi F, et al. Gene expression analysis in myotonic dystrophy: indications for a common molecular pathogenic pathway in DM1 and DM2. Gene Expr. 2007;13:339–351. doi: 10.3727/000000006781510705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lueck JD, Mankodi A, Swanson MS, et al. Muscle chloride channel dysfunction in two mouse models of myotonic dystrophy. J Gen Physiol. 2007;129:79–94. doi: 10.1085/jgp.200609635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Savkur RS, Philips AV, Cooper TA. Aberrant regulation of insulin receptor alternative splicing is associated with insulin resistance in myotonic dystrophy. Nat Genet. 2001;29:40–47. doi: 10.1038/ng704. [DOI] [PubMed] [Google Scholar]
- 37.Philips AV, Timchenko LT, Cooper TA. Disruption of splicing regulated by a CUG-binding protein in myotonic dystrophy. Science. 1998;280:737–741. doi: 10.1126/science.280.5364.737. [DOI] [PubMed] [Google Scholar]
- 38.Sobczak K, Mezer M, Michlewski G, et al. RNA structure of trinucleotide repeats associated with human neurological diseases. Nucleic Acids Res. 2003;31:5469–5482. doi: 10.1093/nar/gkg766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tian B, White RJ, Xia T, et al. Expanded CUG repeat RNAs form hairpins that activate the double-stranded RNA-dependent protein kinase PKR. RNA. 2000;6:79–87. doi: 10.1017/s1355838200991544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ricker K, Koch MC, Lehmann-Horn F, et al. Proximal myotonic myopathy: a new dominant disorder with myotonia, muscle weakness, and cataracts. Neurology. 1994;44:1448–1452. doi: 10.1212/wnl.44.8.1448. [DOI] [PubMed] [Google Scholar]
- 41.Vihola A, Bassez G, Meola G, et al. Histopathological differences of myotonic dystrophy type 1 (DM1) and PROMM/DM2. Neurology. 2003;60:1854–1857. doi: 10.1212/01.wnl.0000065898.61358.09. [DOI] [PubMed] [Google Scholar]