

Abstract
d-Amino acid oxidase (DAAO) catalyzes the oxidative deamination of d-amino acids including d-serine, a full agonist at the glycine modulatory site of the N-methyl-d-aspartate (NMDA) receptor. To evaluate the significance of DAAO-mediated metabolism in the pharmacokinetics of oral d-serine, plasma d-serine levels were measured in both wild-type mice and transgenic mice lacking DAAO. Although d-serine levels were rapidly diminished in wild-type mice (t½ = 1.2 h), sustained drug levels over the course of 4 h (t½ > 10 h) were observed in mice lacking DAAO. Coadministration of d-serine with 6-chlorobenzo[d]isoxazol-3-ol (CBIO), a small-molecule DAAO inhibitor, in wild-type mice resulted in the enhancement of plasma d-serine levels, although CBIO seems to have only temporary effects on the plasma d-serine levels due to glucuronidation of the key hydroxyl group. These findings highlight the predominant role of DAAO in the clearance of d-serine from the systemic circulation. Thus, a potent DAAO inhibitor with a longer half-life should be capable of maintaining high plasma d-serine levels over a sustained period of time and might have therapeutic implications for the treatment of schizophrenia.
Introduction
Cumulative evidence suggests that allosteric activation of NMDA receptors via the glycine modulatory site might have therapeutic implications for the treatment of schizophrenia (Labrie and Roder, 2010). Both glycine and d-serine, two known endogenous glycine modulatory site agonists, have been shown to ameliorate persistent negative and cognitive symptoms of the disorder (Javitt, 2010), whereas existing antipsychotic medications have failed to provide significant efficacy. d-Serine is particularly promising, because 1) the blood-brain barrier is more permeable to d-serine than to glycine (Oldendorf, 1971), and d-serine exhibits a long half-life in the cortex upon peripheral administration (Hashimoto and Chiba, 2004); 2) d-serine is more potent than glycine at activating the glycine modulatory site of the NMDA receptors (Matsui et al., 1995); and 3) there is no known signal transduction site modulated by d-serine other than the glycine modulatory site, which minimizes the risk of off-target toxicity.
However, clinical development of d-serine could be hampered by the high doses (120 mg/kg) required for the optimal treatment of schizophrenia (Kantrowitz et al., 2010). Furthermore, high doses of d-serine have been reported to cause selective necrosis to the pars recta region of the renal proximal tubules in the rat (Ganote et al., 1974). Moreover, one patient who received high doses (120 mg/kg) of d-serine in an open-label clinical trial showed a nephrotoxic-like pattern (Kantrowitz et al., 2010). Results of studies that used a d-amino acid oxidase (DAAO) inhibitor (Williams and Lock, 2005) and of rats lacking DAAO activity (Maekawa et al., 2005) suggest that the mechanism of d-serine-induced nephrotoxicity is associated with oxidative stress caused by hydrogen peroxide, a by-product of DAAO-mediated metabolism of d-serine in the kidneys (Fig. 1). DAAO (EC 1.4.3.3) is a flavoenzyme that catalyzes the oxidative deamination of d-amino acids, including d-serine, and produces the corresponding α-keto acids, ammonia, and hydrogen peroxide (Dixon and Kleppe, 1965a,b).
Fig. 1.

DAAO-mediated metabolism of d-serine. Small-molecule DAAO inhibitors such as CBIO have the potential to provide a significant improvement to d-serine therapy by increasing d-serine bioavailability and minimizing the formation of hydrogen peroxide, a potential cause of nephrotoxicity.
In mammals, DAAO is present in the kidneys, liver, and brain. It is interesting to note that two recent independent studies revealed that DAAO expression and activity are elevated in schizophrenia (Burnet et al., 2008; Madeira et al., 2008). Because the highest DAAO activity is found in the kidneys (Curti et al., 1992), a substantial amount of orally administered d-serine is metabolized there. This contributes to the rapid clearance of d-serine and, consequently, the high dose required for efficacy. These findings suggest that inhibition of DAAO would exert dual beneficial effects on d-serine therapy by 1) enhancement of d-serine exposure and 2) suppression of hydrogen peroxide generation in the kidneys. Thus, DAAO inhibitors might address the issues associated with clinical use of d-serine and salvage the most clinically efficacious glycine modulatory site agonist. 6-Chlorobenzo[d]isoxazol-3-ol (CBIO) is a potent competitive inhibitor of DAAO and has a Ki value of 100 nM for porcine DAAO (Ferraris et al., 2008). Although its toxicity profile has not been fully established, CBIO has been tested in both mice and rats as treatment for pain and has resulted in no apparent toxicity (Gong et al., 2011; Lu et al., 2012). In another study, oral coadministration of CBIO with d-serine enhanced oral bioavailability of d-serine and its levels in the prefrontal cortex (Ferraris et al., 2008). Subsequent studies revealed that oral coadministration of CBIO with d-serine in a preclinical model of schizophrenia normalized prepulse inhibition deficits induced by (5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine (MK801) at all prepulse intensities to a degree similar to that of 10-fold higher doses of d-serine alone (Hashimoto et al., 2009).
Little is understood, however, as to what extent DAAO-mediated metabolism is involved in the overall clearance of d-serine in vivo. Such information should provide insights into the extent of the benefits provided by DAAO inhibitors as pharmacoenhancers for orally dosed d-serine. In the present study, we compared plasma d-serine levels in mice with or without DAAO activity after oral administration of d-serine. The primary objective of this study was to identify, if any, other d-serine clearance pathways and assess the degree of pharmacoenhancement achieved by DAAO inhibition on oral d-serine pharmacokinetics. In addition, the ability of CBIO to enhance d-serine pharmacokinetics was evaluated in the light of its in vitro metabolic stability in plasma and liver microsomes.
Materials and Methods
Chemicals.
CBIO was obtained from Maybridge (Cornwall, UK). Boric acid, o-phthaldialdehyde (OPA), sodium acetate, and all amino acids except d-serine and d-alanine were obtained from Sigma-Aldrich (St. Louis, MO). d-Serine and d-alanine were obtained from Bachem Bioscience (King of Prussia, PA). Liver microsomes from mice and humans, the NADPH-regenerating system, and UGT Reaction Mix were purchased from BD Biosciences (San Jose, CA). All solvents [high-performance liquid chromatography (HPLC) grade] and N-tert-butyloxycarbonyl-l-cysteine (Boc-l-Cys) were purchased from EMD Biosciences (San Diego, CA) and Novabiochem (a subsidiary of EMD Biosciences; Läufelfingen, Switzerland), respectively. Glacial acetic acid, sodium hydroxide, and ultrapure water were procured from Mallinckrodt Baker, Inc. (Phillipsburg, NJ).
Animals.
All mice were maintained in accordance with Institutional Animal Care and Use Committee regulations at the Johns Hopkins University School of Medicine. To generate the DAOnull targeting vector, recombineering (Liu et al., 2003) was used to replace 1957 base pairs encompassing exons 7 and 8 of Dao (ENSMUSG00000042096) with a neomycin phosphotransferase cassette in a C57BL/6 bacterial artificial chromosome [(BAC) RP23-450E21; Invitrogen, Carlsbad, CA)]. The deletion removes the glycine that is mutated in the naturally occurring DAAO-deficient (ddY) mice and the tyrosine that is thought to be catalytically important. Splicing of exon 6 to exon 9 results in a reading frame shift that leads to a truncated protein with no homology past exon 6. The mutant allele was introduced into 129SvEv mouse embryonic stem cells (TG-ES01-01 ESM07; Eurogentec, Seraing, Belgium) using standard homologous recombination techniques (Joyner, 2000). Quantitative polymerase chain reaction (PCR) of the Dao gene copy number was used to identify targeted clones (DAOF: 5′-CCCATGATCCTAGCCTTGGTATC-3′; DAOR: 5′-CCCCTTGTATGACCTTAGGTCAGT-3′; DAO probe: 5′-AACTCTCCGTACATCATCCCAGGGTAAAACTCC-3′; PPIAF: 5′-GCCAGGGTGGTGACTTTACAC-3′; PPIAR: 5′-GACAAGATGCCAGGACCTGTATG-3′; and PPIA probe: 5′-TGGCGGCAGGTCCATCTACGG-3′). Fluorescent in situ hybridization using the wild-type Dao containing BAC as a probe was used to confirm targeting and the single integration of the targeting vector. Male chimeric mice were generated by injection of the targeted embryonic stem cells into C57BL/6J blastocysts (Nagy et al., 2003). Chimeric mice were bred with 129SvEv mice to produce F1 heterozygotes. Germline transmission was confirmed by PCR analysis (G3: 5′-CAGGGCAAAGGGACTGAATA-3′; G4: 5′-CACTCCACCACCATCGATTA-3′; dNEO2: 5′-ACATAGCGTTGGCTACCCGTGATA-3′). F1 heterozygous males and females were mated to produce F2 wild-type, heterozygous, and homozygous null mutant animals. The colony was maintained on a background of 129SvEv under specific pathogen-free conditions and had unrestricted access to food and water. The mice for experiments were obtained by heterozygous × heterozygous matings. Genotyping was done by Transnetyx, Inc. (Cordova, TN) using automated real-time PCR. Preliminary behavioral characterization of the DAAO-knockout (KO) mice revealed only one significant difference compared with that of the wild-type mice: a decreased center path in the open-field test, which indicates increased anxiety. However, this result was not corroborated by the outcome of the elevated-plus maze test (P. A. Seymour, personal communications).
Animal Study.
Mice (n = 3–6 for each time point for each group except for wild-type mice treated with d-serine and CBIO, wherein n = 2 for T = 120 and 240 min) were dosed orally (10 ml/kg) with either d-serine (30 mg/kg) alone or d-serine (30 mg/kg) in combination with CBIO [30 mg/kg in 10% dimethyl sulfoxide/0.9% saline (w/v)]. The mice were then euthanized 30, 60, 120, or 240 min after dosing. Approximately 1 ml of whole blood was collected from each animal by cardiac puncture and placed into heparinized microcentrifuge tubes, capped, gently inverted a few times, and stored on wet ice until centrifugation (10 min at 800g, 4°C). Thereafter, the top layer of each tube (∼400 μl of plasma) was aspirated via transfer pipette, dispensed into a clean nonheparinized microcentrifuge tube, and stored at −80°C until subsequent analyses. In addition, mouse brains were quickly excised (and the cerebellum and frontal cortex were isolated), weighed, and stored at −80°C until the time of analyses.
Bioanalysis of d-Serine.
Methanol was used to extract amino acids (AAs) from both the plasma and brain samples. Plasma samples were mixed with methanol [20× volumes (v/v)], vortexed briefly, and allowed to stand at room temperature for 2 min. AA-extraction procedures from brain tissues were adapted from previously published methods (Morikawa et al., 2001). Brain tissues were homogenized and sonicated in 20× volumes (w/v) of methanol. Proteins in both matrices were removed after centrifugation at 50,000g for 15 min at 4°C. Aliquots (plasma, 100 μl; brain, 20 μl) of the supernatant were evaporated to dryness by using a vacuum lyophilizer operated at 30°C. Subsequently, the residues were reconstituted in ultrapure water (50 μl) and prepped for AA derivatization.
Amino acid derivatization was carried out on the basis of previously reported methods (Hashimoto et al., 1992). Sodium-borate buffer was made by using 0.4 M boric acid pH-adjusted to 9.0 with sodium hydroxide. On the day of the analysis, 10 mg each of OPA and Boc-l-Cys were dissolved in 1 ml of methanol and 3.5 ml of borate buffer added to the Boc-l-Cys-OPA mixture (derivatization reagent). A 45-μl volume of derivatization reagent was then added to a vial containing 5 μl of either the AA standard or the sample. After 2 min of derivatization at room temperature, an aliquot (10 μl) of the derivatized material was introduced into the HPLC system described below.
The HPLC system consisted of a degasser (DGU-14A; Shimadzu, Columbia, MD), pumps (LC-10ADVP; Shimadzu), an autoinjector (SIL-10ADVP; Shimadzu), a column oven (CTO-10ACVP; Shimadzu, Columbia, MD), and a fluorescence detector (RF-10AXL; Shimadzu). Mobile phase A was made up of 0.1 M sodium acetate buffer (pH 6.0), acetonitrile, and tetrahydrofuran [90:7:3 (v/v/v)], and mobile phase B was made up of 0.1 M sodium acetate buffer (pH 6.0), acetonitrile, and tetrahydrofuran [50:47:3 (v/v/v)]. Amino acids were resolved by using a C18 Nova-Pak analytical column (3.9 × 300 mm, 4 μm; Waters, Milford, MA) maintained at 30°C, with a linear gradient from mobile phase A to B in 120 min, and operated at a constant flow rate of 0.8 ml/min. Fluorescence detection was carried out at 443 nm with excitation at 344 nm. Data were processed by using a system controller from Shimadzu (SCL-10AVP).
Pharmacokinetics Analysis.
Plasma concentrations of d-serine were analyzed by using noncompartmental methods as implemented in the computer software program WinNonlin version 5.2 (Pharsight, Mountain View, CA). The maximum plasma concentration (Cmax) and time to Cmax (Tmax) were the observed values. The half-life (t½) was calculated as 0.693 divided by λz (the elimination rate constant) using a uniform weighting. The area under the plasma concentration time curve (AUC) value was calculated to the last quantifiable sample (AUClast) by using the log-linear trapezoidal rule. The AUC values, wherever applicable, were extrapolated to infinity (AUC∞) by dividing the last quantifiable concentration by the terminal disposition rate constant (λz), which was determined from the slope of the terminal phase of the concentration-time profiles. The value extrapolated was determined by using the equation AUC0–∞ = AUC0–t + Clast/λz, where Clast was the final quantifiable concentration. Acceptance criteria for the model were percentage of AUC extrapolated of ≤25% or the r2 on the λz of ≥ 0.9. The method of Bailer (1988) was used to estimate the variance of AUClast on the basis of the variance of the mean concentration at each time point. To determine whether there was a significant difference between exposure as expressed by AUCs of d-serine in three cohorts (i.e., wild type, wild type with DAAO inhibitor, and the DAAO-KO), a pairwise comparison was performed by using a Z test (Yuan, 1993). The a priori level of significance was P < 0.05.
Metabolic Stability of CBIO in Plasma and Liver Microsomes.
The metabolic stability of CBIO was evaluated by using mouse and human plasma and liver microsomes. For plasma stability, a 5 μM compound was spiked in plasma, and the reaction (150 μl) was stopped at 0, 15, 30, and 60 min by the addition of acetonitrile (300 μl) spiked with internal standard [(IS) 0.1 mM phenyl acetic acid].
Phase I and phase II metabolic stability assays for CBIO were conducted in mouse and human liver microsomes. For phase I metabolism, the reaction was carried out with 100 mM potassium phosphate buffer (pH 7.4) in the presence of an NADPH-regenerating system (1.3 mM NADPH/3.3 mM glucose 6-phosphate/3.3 mM MgCl2/0.4 U/ml glucose-6-phosphate dehydrogenase/50 μM sodium citrate). Reactions in triplicate were initiated by addition of the liver microsomes (mouse or human) to the incubation mixture (compound final concentration was 10 μM; 0.5 mg/ml microsomes). For the phase II glucuronidation reaction, CBIO was added to Tris-HCl buffer [50 mM (pH 7.5)] with microsomes (0.5 mg/ml), along with MgCl2 (8 mM), and alamethicin (25 μg/ml) and preincubated at 37°C. The reaction was initiated (in triplicate) with uridine 5′-diphospho-glucuronic acid (UDPGA) at a final concentration of 2 mM. Control experiments in the absence of NADPH and UDPGA were carried out for both phase I and phase II metabolism, respectively, to determine the specific cofactor-free degradation. At predetermined times (0, 15, 30, and 60 min), aliquots of the mixture were removed and the reaction quenched by the addition of two times the volume of ice-cold acetonitrile spiked with the IS. Compound disappearance was monitored over time by using a liquid chromatography-tandem mass spectrometry (LC/MS/MS) method.
Separation of the analyte from potentially interfering material was achieved by using a Waters X-Terra (50 × 2.1 mm i.d.) column packed with a 3.5-μm C18 stationary phase protected by a guard column packed with 3.5-μm RP18 material. The mobile phase used was composed of acetonitrile/water [70:30 (v/v)] containing 0.1% formic acid delivered isocratically at a flow rate of 0.2 ml/min for a total run time of 5 min. The retention time for CBIO and the IS was 1.2 ± 0.3 min. The column effluent was monitored by using a Waters Micromass Quattro triple-quadrupole mass spectrometric detector, equipped with a Waters HPLC, in the negative-ionization mode. The spectrometer was programmed to allow the [M-1] mass transitions of CBIO at an m/z of 168 > 132 and an m/z of 135 > 90.8 for the IS.
Results
Plasma Pharmacokinetics of d-Serine in Wild-Type and DAAO Knockout Mice.
d-Serine concentration-time profiles in plasma after oral administration of d-serine are shown in Fig. 2, and the pharmacokinetic parameters are summarized in Table 1. The basal plasma levels of d-serine were 3.5 and 10.8 μM in wild-type and DAAO KO mice, respectively. Similar d-serine levels were reported for serum samples from ddY mice with and without DAAO activity (Morikawa et al., 2001). d-Serine was absorbed rapidly in wild-type mice after oral administration (Tmax within 0.7 ± 0.3 h). When d-serine was given in conjunction with CBIO, a potent DAAO inhibitor, the Tmax shifted to 1.0 h, and the Cmax and AUC were increased. In DAAO-KO mice, the Tmax was further delayed to 2.7 ± 0.9 h, and additional increases were observed in the Cmax and AUC values. The terminal half-lives of d-serine in wild-type mice (without or with CBIO) were 1.2 ± 0.1 h and approximately 1.5 h, respectively. In contrast, DAAO-KO mice exhibited the highest Cmax value among the three treatment groups, and there was little sign of d-serine elimination during the period of the measurement (up to 4.0 h). The half-life of d-serine could not be estimated accurately because of insufficient data available for the DAAO-knockout mice and a lack of terminal elimination phase (Fig. 2).
Fig. 2.

Plasma concentrations of d-serine as a function of time in wild-type (WT) and DAAO-KO mice after oral (p.o.) administration of d-serine.
TABLE 1.
Pharmacokinetic parameters of d-serine (30 mg/kg) administered orally to mice
| Cohorts | Dose | Tmax | Cmax | T1/2 | AUClast | AUC0–∞ |
|---|---|---|---|---|---|---|
| mg/kg | h | ng/ml | h | ng · h/ml | ng · h/ml | |
| Wild type | 30 | 0.7 ± 0.3 | 19,609 ± 1338 | 1.2 ± 0.1 | 40,648 ± 6602 | 46,023 ± 8976 |
| Wild type + CBIO* | 30 | 1.0 | 24,277 | 1.5 | 65,778 | 80,899 |
| Knockout | 30 | 2.7 ± 0.9 | 27,791 ± 2019 | >10† | 95,768 ± 6234 | —‡ |
The S.D. of the data cannot be determined because of the insufficient number (2) of data points at 120 and 240 min.
Insufficient data for accurate approximation.
The AUC extrapolated from the AUClast is greater than 25%; hence, the AUC0–∞ value is not reported.
Systemic exposure of d-serine in the three cohorts was evaluated by using the AUClast values. The highest plasma exposure to d-serine was achieved in DAAO-KO mice, with an AUC0–last value of 95,768 ± 6234 h · ng/ml, followed by wild-type mice administered d-serine and CBIO (65,778 h · ng/ml) and wild-type mice treated with d-serine alone (40,648 ± 6602 h · ng/ml) without CBIO. The DAAO-KO mice had approximately 2.4-fold higher exposures compared with those of the wild-type mice and 1.4-fold higher exposures compared with those of the wild-type mice treated with CBIO, all of which were statistically significant differences (P < 0.05). In addition, wild-type mice administered d-serine with CBIO exhibited 1.6-fold higher systemic exposures compared with those of the wild-type mice treated with d-serine alone (P < 0.05). The AUC extrapolated from the AUClast was greater than 25% for DAAO-KO mice; hence, the AUC0–∞ value is not reported.
in Vitro Metabolic Stability of CBIO.
In mouse and human plasma, CBIO was found to be stable over a period of 60 min (data not shown). In mouse and human liver microsomal incubations in the presence or absence of NADPH (Fig. 3), no metabolism of CBIO was observed (∼100% remaining at the end of a 60-min incubation), which indicates that CBIO is not a substrate of cytochrome P450 enzymes. In microsomes fortified with UDPGA, however, CBIO was metabolized rapidly; 10% (mouse) and 22% (human) of the parent compound remained after 60 min of incubation (Fig. 3). Subsequently, the possible formation of CBIO-glucuronide (exact mass: 345.03 Da) was examined in the microsome mixtures (Fig. 4). A new peak with a distinct mass with an m/z of 344.02 (M-1) was detected by LC/MS analysis in both mouse and human microsomes fortified with UDPGA (data not shown).
Fig. 3.
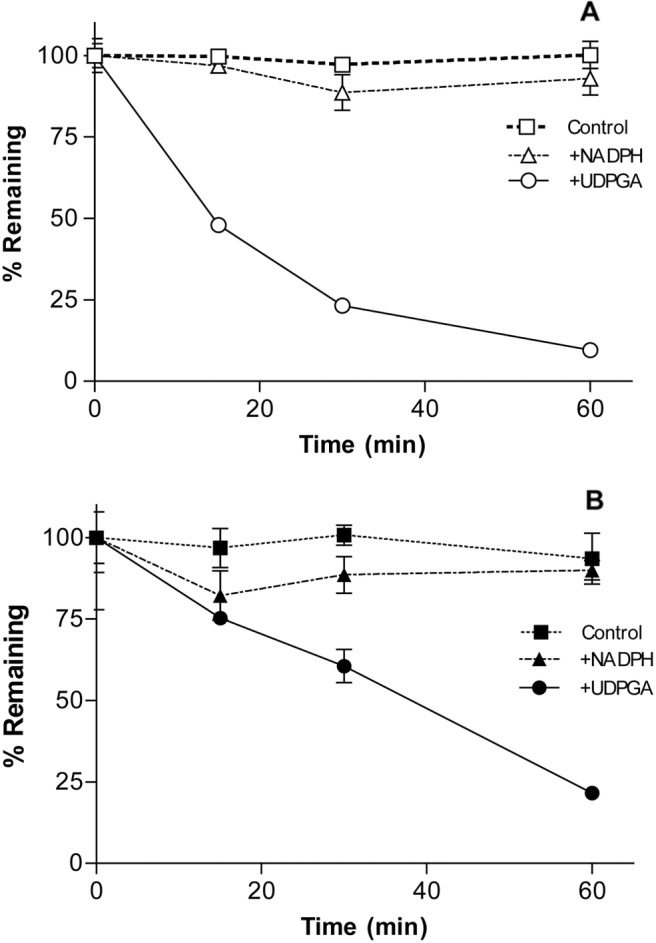
Metabolic stability of CBIO in mouse (A) and human (B) liver microsomes.
Fig. 4.

Putative metabolic pathway of CBIO in liver microsomes in the presence of UDPGA.
Brain Levels of d-Serine.
Levels of d-serine were measured in two regions of the brain (the cerebellum and the cortex) of wild-type and DAAO-KO mice. For the d-serine-treated groups, tissues were taken 1 h after oral administration of d-serine. As shown in Fig. 5A, negligible basal levels of d-serine were detected in the cerebellum of wild-type mice. d-Serine levels did not increase significantly in the cerebellum even in d-serine-treated (no CBIO) wild-type mice 60 min after oral administration. Substantially higher basal quantities of d-serine were found in the cerebellum of DAAO-KO mice (Fig. 5B). d-Serine levels were even higher in DAAO-KO mice treated with d-serine, although the difference was not statistically significant. In the cortex, the basal d-serine levels were nearly identical between the wild-type and DAAO-KO mice. Although both the wild-type and DAAO-KO mice showed higher levels of d-serine after d-serine treatment, neither of them achieved statistically significant increases.
Fig. 5.

d-Serine concentrations in the cerebellum and cortex of wild-type (A) and DAAO-KO (B) mice. For the d-serine-treated groups, tissues were taken 1 h after oral administration.
Discussion
In the present study, we examined the significance of DAAO-mediated metabolism (Fig. 1) in the pharmacokinetics of orally administered d-serine. Plasma levels of d-serine before d-serine treatment were 3.5 and 10.8 μM in wild-type and DAAO-KO mice, respectively. Although genetic deletion of DAAO seems to result in increased plasma levels of d-serine, the origin of plasma d-serine is not well understood and could be attributed to diet (Friedman, 1999), the level formed in the brain by serine racemase (Ohide et al., 2011), or both. When d-serine was given to wild-type 129SvEv mice, d-serine was immediately absorbed and followed by rapid clearance. Coadministration of CBIO, a potent DAAO inhibitor, resulted in a delayed Tmax, longer half-life, and increased AUC. Because DAAO is highly expressed in the kidneys of rodents, the enhanced plasma d-serine exposure can be attributed to the diminished DAAO-mediated metabolism of d-serine caused by CBIO. It is interesting to note that the extent of the increase in plasma d-serine levels was even more profound in DAAO-KO mice treated with d-serine alone. Plasma d-serine levels increased slowly over a period of 2 h (Cmax, 27,264 ng/ml). It is striking that the high levels of d-serine were sustained during the period of the measurement with little indication of plasma clearance. These findings clearly indicate that DAAO plays a predominant role in the overall plasma clearance of d-serine. Although sufficient data are not available to estimate the AUC0–∞ of d-serine in DAAO-KO mice, extrapolation of the given data clearly suggests that the enhancement of d-serine plasma exposures will be much greater than the 2-fold AUClast increase seen over the first 2 h. The much lower clearance value for d-serine in DAAO-KO mice indicates that filtered d-serine is being reabsorbed from the renal proximal tubule, where DAAO is highly expressed (Koibuchi et al., 1995). A similar reabsorption pathway has been proposed for rats (Silbernagl et al., 1999), and it has been reported that the urinary recovery of orally administered d-serine in rats is only 1.2% (Huang et al., 1998), which indicates that d-serine exhibits similar renal pharmacokinetics in mouse and rat kidneys. Thus, it remains unclear why d-serine is nephrotoxic only in rats despite the presence of DAAO in the proximal tubule of both rats (Chan et al., 1979; Le Hir and Dubach, 1981; Usuda et al., 1986) and mice (Koibuchi et al., 1995). The urinary basal levels of d-serine in mice lacking DAAO activity were reported to be significantly higher than those of wild-type mice (Miyoshi et al., 2009). Although these findings seem to suggest a high degree of renal excretion of d-serine, given the low clearance rate of d-serine in DAAO-KO mice, it is more likely a result of a shift in steady-state concentrations of basal d-serine in serum and kidney caused by the loss of DAAO activity.
In contrast to the marked increase in plasma d-serine half-life in DAAO-KO mice, there was little difference in Cmax values between wild-type and DAAO-KO mice. This can be attributed to the significantly lower degree of DAAO activity in the liver compared with that of the kidney in rodents (Burch et al., 1958; Nagata et al., 1988). It is worth noting that humans express DAAO in both the liver and kidney (Holme and Goldberg, 1982) and that inhibition of DAAO in humans is expected to increase not only plasma half-life, as seen in mice, but also the Cmax of oral d-serine by suppressing DAAO-mediated first-pass metabolism in the liver.
The pharmacoenhancing effect of CBIO on d-serine levels was not nearly as drastic as that of genetic DAAO deletion, particularly at the later time points, presumably because of the rapid clearance of CBIO from the circulation. The in vivo pharmacokinetic profile of CBIO was reported recently for rodent species (Lange et al., 2011). As expected from previous studies in which CBIO demonstrated an in vivo pharmacoenhancing effect on d-serine with oral administration (Ferraris et al., 2008), CBIO was reported to be orally available in mice (F = 29%). Although the plasma half-life in mice was not reported, the terminal elimination half-life in the brain was determined to be 1 h, which likely indicates a similarly short half-life of CBIO in plasma. This explains the inability of CBIO to maintain high levels of d-serine plasma levels for a sustained period of time. As expected, CBIO had no effects on plasma levels of coadministered d-serine in DAAO-KO mice (data not shown), which confirms that the ability of CBIO to enhance plasma d-serine levels is predominantly associated with its inhibition of DAAO.
In an attempt to elucidate the mechanism by which CBIO is cleared from the circulatory system, in vitro metabolic stability was measured in plasma and liver microsomes from mice and humans. CBIO was highly stable in mouse and human plasma (>95% remaining after 2 h of incubation; data not shown). In liver microsomes from mice (Fig. 3A) and humans (Fig. 3B), no significant loss of CBIO was detected in the absence or presence of NADPH. The presence of the chloro group seems to make CBIO resistant to CYP450-mediated oxidation. In liver microsomes containing UDPGA, however, CBIO was metabolized substantially; only 10% (mouse) and 21% (human) of the parent compound remained at the end of a 60-min incubation (Fig. 3). The results clearly show that CBIO undergoes phase II glucuronidation in the liver. Because CBIO possesses only a single moiety that is possibly subject to glucuronidation (3-hydroxyl group), the most likely metabolite is CBIO-3-glucuronide (Fig. 4). Indeed, we have detected a new peak corresponding to a mass of CBIO-3-glucuronide with an increasing intensity over time in LC/MS (data not shown).
Unfortunately, the 3-hydroxyl group is essential for the high-affinity binding of CBIO to the DAAO active site (Ferraris et al., 2008). Therefore, removal or masking of this functional group is expected to result in a complete loss of inhibitory potency even though it might circumvent glucuronidation. Further structural optimization of the benzo[d]isoxazol-3-ol scaffold requires careful modulation of the steric and electronic environment surrounding the hydroxyl group in a way that does not compromise the inhibitory potency while minimizing the degree of glucuronidation. It is also important to point out that glucuronidation is only one of many possible metabolic reactions that could take place at this site. Other phase II metabolic reactions, particularly methylation and sulfation, are also common at a free hydroxyl group, and much attention needs to be paid when the structural optimization of CBIO is conducted for the enhancement of metabolic stability. If such improvements do not result in prolonged plasma half-life, the possibility of metabolism by other organs needs to be explored to determine the optimal strategy for further structural optimization.
In the brain, DAAO activity is known to be highest in the cerebellum, whereas relatively low levels of DAAO activity are detected in the cortex. It not surprising that d-serine levels in the brain are inversely correlated with DAAO activity. As shown in Fig. 5A, only negligible basal levels of d-serine were detected in the cerebellum of wild-type mice, whereas substantially higher quantities of d-serine were found in the cortex. This is consistent with the previously reported findings in other strains of mice (Morikawa et al., 2001; Labrie et al., 2009).
Oral administration of d-serine in wild-type mice did not result in increased levels of d-serine in the cerebellum 1 h after administration, presumably because of the rapid metabolism by DAAO. A slight increase in d-serine levels was observed in the cortex of d-serine-treated wild-type mice, although the difference was not statistically significant. It was previously reported that, in rats, a statistically significant increase in cortical d-serine levels was achieved only when ≥320 mg/kg doses of d-serine were given by subcutaneous injection (Smith et al., 2009). In contrast, increases of d-serine levels in cerebrospinal fluid were statistically significant at 160 mg/kg, and nearly 80-fold increases over the basal level were achieved at the highest dose tested (1280 mg/kg). These findings suggest that endogenous d-serine in the brain is mainly confined to the intracellular compartment, probably by the action of the alanine-serine-cysteine (Asc-1) transporter. Added d-serine has much less impact on its levels in homogenized brain tissues containing a substantial quantity of endogenous cytosolic d-serine. Meanwhile, extracellular d-serine concentrations such as those detected in cerebrospinal fluid are boosted more drastically by the added d-serine as a result of the lower basal level of endogenous d-serine.
As shown in Fig. 5B, basal d-serine levels in the cerebellum of DAAO-KO mice were substantially higher than those of wild-type mice. Previous studies that used other strains of mice lacking DAAO activity also had similar results (Morikawa et al., 2001; Labrie et al., 2009). In the cortex, the basal d-serine levels were nearly identical between wild-type and DAAO-KO mice. The results are in good agreement with the negligible DAAO activity detected in the cortex of wild-type rodents (Horiike et al., 1994).
Although both the cerebellum and cortex of DAAO-KO mice had higher levels of d-serine 1 h after d-serine treatment, neither of their increases was statistically significant. We have previously shown by in vivo microdialysis in the mouse frontal cortex that oral d-serine (30 mg/kg) can increase cortical d-serine levels of wild-type ddY mice by up to 4-fold when coadministered with CBIO (Hashimoto et al., 2009). The higher relative increase seen with microdialysis indicates that the impact of added d-serine is more robust in the extracellular compartment of the brain, where the therapeutic target (NMDA receptor) for d-serine is located.
Conclusions
Oral d-serine represents one of the most promising therapeutic agents for treating the negative symptoms and cognitive deficits of schizophrenia, which have been poorly addressed by the existing antipsychotic medications. However, d-serine therapy remains impractical for clinical application because of the high dose required for robust efficacy and the risk of nephrotoxicity. Coadministration of d-serine with a small-molecule DAAO inhibitor might address both of these issues and lead to the development of a clinically viable therapeutic approach based on d-serine. Furthermore, the pharmacoenhancing effects of DAAO inhibition could be more profound in humans, because a substantial amount of DAAO is present in human liver and first-pass metabolism of d-serine can be blocked by DAAO inhibitors. However, DAAO in the human liver might play a crucial role in metabolism and/or detoxification of endogenous/exogenous substances. Therefore, it is critical to assess the potential consequences of liver DAAO inhibition in species known to express DAAO in not only the kidney but also the liver. It is also worth noting that a cross-species comparison of urinary basal d-serine concentrations revealed that much higher levels of d-serine were found in humans compared with those of rats and mice (Huang et al., 1998; Miyoshi et al., 2009), which could be at least partially due to higher rates of renal d-serine excretion in humans. DAAO inhibitors, in theory, are incapable of minimizing the loss of d-serine by renal excretion. Hence, DAAO inhibitors might be less effective in enhancing plasma d-serine levels in humans if renal excretion plays a larger role in human d-serine pharmacokinetics.
Although CBIO has served as a useful pharmacological probe for elucidating the pharmacoenhancing effects of DAAO inhibition on d-serine (Ferraris et al., 2008; Hashimoto et al., 2009), our studies revealed that another improvement to DAAO inhibitors can be made by identifying CBIO analogs that are resistant to glucuronidation. Given the favorable oral d-serine pharmacokinetics in DAAO-knockout mice, a potent DAAO inhibitor with a longer half-life should be capable of maintaining high plasma d-serine levels over a sustained period of time and have therapeutic implications for the treatment of schizophrenia. In our studies, orally administered d-serine had little impact on brain tissue levels of d-serine in both wild-type and DAAO-KO mice. It is conceivable that the added d-serine contributes only a minor portion of d-serine in brain tissues and that microdialysis studies are better suited for examining central nervous system distribution of orally administered d-serine and its therapeutic effects.
This work was supported in part by the National Institutes of Health National Institute of Mental Health [Grant R01-MH091387] (to T.T.); and the Johns Hopkins Brain Science Institute NeuroTranslational Drug Discovery program.
Article, publication date, and citation information can be found at http://dmd.aspetjournals.org.
- NMDA
- N-methyl-d-aspartate
- DAAO
- d-amino acid oxidase
- CBIO
- 6-chlorobenzo[d]isoxazol-3-ol
- OPA
- o-phthaldialdehyde
- HPLC
- high-performance liquid chromatography
- Boc-l-Cys
- N-tert-butyloxycarbonyl-l-cysteine
- KO
- knockout
- AA
- amino acid
- Cmax
- maximum plasma concentration
- Tmax
- time to Cmax
- AUC
- area under the plasma concentration time curve
- UDPGA
- uridine 5′-diphospho-glucuronic acid
- LC/MS/MS
- liquid chromatography-tandem mass spectrometry
- IS
- internal standard
- MK801
- (5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine
- BAC
- bacterial artificial chromosome
- PCR
- polymerase chain reaction.
Authorship Contributions
Participated in research design: Rais, Thomas, Strick, Engle, Brandon, Rojas, Slusher, and Tsukamoto.
Conducted experiments: Rais, Thomas, Wozniak, Wu, Jaaro-Peled, and Engle.
Contributed new reagents or analytic tools: Sawa.
Performed data analysis: Rais, Thomas, and Tsukamoto.
Wrote or contributed to the writing of the manuscript: Rais, Thomas, Jaaro-Peled, Sawa, and Tsukamoto.
References
- Bailer AJ. (1988) Testing for the equality of area under the curves when using destructive measurement techniques. J Pharmacokinet Biopharm 16:303–309 [DOI] [PubMed] [Google Scholar]
- Burch HB, Lowry OH, De Gubareff T, Lowry SR. (1958) Flavin enzymes in liver and kidney of rats from birth to weaning. J Cell Physiol 52:503–510 [DOI] [PubMed] [Google Scholar]
- Burnet PW, Eastwood SL, Bristow GC, Godlewska BR, Sikka P, Walker M, Harrison PJ. (2008) d-Amino acid oxidase activity and expression are increased in schizophrenia. Mol Psychiatry 13:658–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan AW, Perry SG, Burch HB, Fagioli S, Alvey TR, Lowry OH. (1979) Distribution of two aminotransferases and d-amino acid oxidase within the nephron of young and adult rats. J Histochem Cytochem 27:751–755 [DOI] [PubMed] [Google Scholar]
- Curti B, Ronchi S, Simonetta PM. (1992) d- and l-Amino acid oxidases, in Chemistry and Biochemistry of Flavoenzyme (Muller F. ed) pp 69–94, CRC Press, Boca Raton, FL [Google Scholar]
- Dixon M, Kleppe K. (1965a) d-Amino acid oxidase I. Dissociation and recombination of the holoenzyme. Biochim Biophys Acta 96:357–367 [DOI] [PubMed] [Google Scholar]
- Dixon M, Kleppe K. (1965b) d-Amino acid oxidase II. Specificity, competitive inhibition and reaction sequence. Biochim Biophys Acta 96:368–382 [Google Scholar]
- Ferraris D, Duvall B, Ko YS, Thomas AG, Rojas C, Majer P, Hashimoto K, Tsukamoto T. (2008) Synthesis and biological evaluation of d-amino acid oxidase inhibitors. J Med Chem 51:3357–3359 [DOI] [PubMed] [Google Scholar]
- Friedman M. (1999) Chemistry, nutrition, and microbiology of d-amino acids. J Agric Food Chem 47:3457–3479 [DOI] [PubMed] [Google Scholar]
- Ganote CE, Peterson DR, Carone FA. (1974) The nature of d-serine–induced nephrotoxicity. Am J Pathol 77:269–282 [PMC free article] [PubMed] [Google Scholar]
- Gong N, Gao ZY, Wang YC, Li XY, Huang JL, Hashimoto K, Wang YX. (2011) A series of d-amino acid oxidase inhibitors specifically prevents and reverses formalin-induced tonic pain in rats. J Pharmacol Exp Ther 336:282–293 [DOI] [PubMed] [Google Scholar]
- Hashimoto A, Chiba S, Chiba Y. (2004) Effect of systemic administration of d-serine on the levels of d- and l-serine in several brain areas and periphery of rat. Eur J Pharmacol 495:153–158 [DOI] [PubMed] [Google Scholar]
- Hashimoto A, Nishikawa T, Oka T, Takahashi K, Hayashi T. (1992) Determination of free amino acid enantiomers in rat brain and serum by high-performance liquid chromatography after derivatization with N-tert.-butyloxycarbonyl-L-cysteine and o-phthaldialdehyde. J Chromatogr 582:41–48 [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Fujita Y, Horio M, Kunitachi S, Iyo M, Ferraris D, Tsukamoto T. (2009) Co-administration of a d-amino acid oxidase inhibitor potentiates the efficacy of d-serine in attenuating prepulse inhibition deficits after administration of dizocilpine. Biol Psychiatry 65:1103–1106 [DOI] [PubMed] [Google Scholar]
- Holme DJ, Goldberg DM. (1982) Development of a fluorometric assay for amino-acid oxidase activity and its application to the study of human tissues. Biochem Med 28:51–61 [DOI] [PubMed] [Google Scholar]
- Horiike K, Tojo H, Arai R, Nozaki M, Maeda T. (1994) d-Amino-acid oxidase is confined to the lower brain stem and cerebellum in rat brain: regional differentiation of astrocytes. Brain Res 652:297–303 [DOI] [PubMed] [Google Scholar]
- Huang Y, Nishikawa T, Satoh K, Iwata T, Fukushima T, Santa T, Homma H, Imai K. (1998) Urinary excretion of d-serine in human: comparison of different ages and species. Biol Pharm Bull 21:156–162 [DOI] [PubMed] [Google Scholar]
- Javitt DC. (2010) Glutamatergic theories of schizophrenia. Isr J Psychiatry Relat Sci 47:4–16 [PubMed] [Google Scholar]
- Joyner AL. (2000) Gene Targeting: A Practical Approach. Oxford University Press, Oxford, UK [Google Scholar]
- Kantrowitz JT, Malhotra AK, Cornblatt B, Silipo G, Balla A, Suckow RF, D'Souza C, Saksa J, Woods SW, Javitt DC. (2010) High dose d-serine in the treatment of schizophrenia. Schizophr Res 121:125–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koibuchi N, Konno R, Matsuzaki S, Ohtake H, Niwa A, Yamaoka S. (1995) Localization of d-amino acid oxidase mRNA in the mouse kidney and the effect of testosterone treatment. Histochem Cell Biol 104:349–355 [DOI] [PubMed] [Google Scholar]
- Labrie V, Duffy S, Wang W, Barger SW, Baker GB, Roder JC. (2009) Genetic inactivation of d-amino acid oxidase enhances extinction and reversal learning in mice. Learn Mem 16:28–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrie V, Roder JC. (2010) The involvement of the NMDA receptor d-serine/glycine site in the pathophysiology and treatment of schizophrenia. Neurosci Biobehav Rev 34:351–372 [DOI] [PubMed] [Google Scholar]
- Lange JH, Venhorst J, van Dongen MJ, Frankena J, Bassissi F, de Bruin NM, den Besten C, de Beer SB, Oostenbrink C, Markova N, et al. (2011) Biophysical and physicochemical methods differentiate highly ligand-efficient human d-amino acid oxidase inhibitors. Eur J Med Chem 46:4808–4819 [DOI] [PubMed] [Google Scholar]
- Le Hir M, Dubach UC. (1981) The activity pattern of two peroxisomal oxidases in the rat nephron. FEBS Lett 127:250–252 [DOI] [PubMed] [Google Scholar]
- Liu P, Jenkins NA, Copeland NG. (2003) A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res 13:476–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu JM, Gong N, Wang YC, Wang YX. (2012) d-Amino acid oxidase-mediated increase in spinal hydrogen peroxide is mainly responsible for formalin-induced tonic pain. Br J Pharmacol 165:1941–1955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira C, Freitas ME, Vargas-Lopes C, Wolosker H, Panizzutti R. (2008) Increased brain d-amino acid oxidase (DAAO) activity in schizophrenia. Schizophr Res 101:76–83 [DOI] [PubMed] [Google Scholar]
- Maekawa M, Okamura T, Kasai N, Hori Y, Summer KH, Konno R. (2005) d-Amino-acid oxidase is involved in d-serine-induced nephrotoxicity. Chem Res Toxicol 18:1678–1682 [DOI] [PubMed] [Google Scholar]
- Matsui T, Sekiguchi M, Hashimoto A, Tomita U, Nishikawa T, Wada K. (1995) Functional comparison of d-serine and glycine in rodents: the effect on cloned NMDA receptors and the extracellular concentration. J Neurochem 65:454–458 [DOI] [PubMed] [Google Scholar]
- Miyoshi Y, Hamase K, Tojo Y, Mita M, Konno R, Zaitsu K. (2009) Determination of d-serine and d-alanine in the tissues and physiological fluids of mice with various d-amino-acid oxidase activities using two-dimensional high-performance liquid chromatography with fluorescence detection. J Chromatogr B Analyt Technol Biomed Life Sci 877:2506–2512 [DOI] [PubMed] [Google Scholar]
- Morikawa A, Hamase K, Inoue T, Konno R, Niwa A, Zaitsu K. (2001) Determination of free d-aspartic acid, d-serine and d-alanine in the brain of mutant mice lacking d-amino acid oxidase activity. J Chromatogr B Biomed Sci Appl 757:119–125 [DOI] [PubMed] [Google Scholar]
- Nagata Y, Shimojo T, Akino T. (1988) d-Amino acid oxidase in mouse liver–II. Comp Biochem Physiol B 91:503–504 [DOI] [PubMed] [Google Scholar]
- Nagy A, Gertsenstein M, Vintersten K, Behringer R. (2003) Manipulating the Mouse Embryo: A Laboratory Manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- Ohide H, Miyoshi Y, Maruyama R, Hamase K, Konno R. (2011) d-Amino acid metabolism in mammals: biosynthesis, degradation and analytical aspects of the metabolic study. J Chromatogr B Analyt Technol Biomed Life Sci 879:3162–3168 [DOI] [PubMed] [Google Scholar]
- Oldendorf WH. (1971) Brain uptake of radiolabeled amino acids, amines, and hexoses after arterial injection. Am J Physiol 221:1629–1639 [DOI] [PubMed] [Google Scholar]
- Silbernagl S, Völker K, Dantzler WH. (1999) d-Serine is reabsorbed in rat renal pars recta. Am J Physiol 276:F857–F863 [DOI] [PubMed] [Google Scholar]
- Smith SM, Uslaner JM, Yao L, Mullins CM, Surles NO, Huszar SL, McNaughton CH, Pascarella DM, Kandebo M, Hinchliffe RM, et al. (2009) The behavioral and neurochemical effects of a novel d-amino acid oxidase inhibitor compound 8 [4H-thieno [3,2-b]pyrrole-5-carboxylic acid] and d-serine. J Pharmacol Exp Ther 328:921–930 [DOI] [PubMed] [Google Scholar]
- Usuda N, Yokota S, Hashimoto T, Nagata T. (1986) Immunocytochemical localization of d-amino acid oxidase in the central clear matrix of rat kidney peroxisomes. J Histochem Cytochem 34:1709–1718 [DOI] [PubMed] [Google Scholar]
- Williams RE, Lock EA. (2005) Sodium benzoate attenuates d-serine induced nephrotoxicity in the rat. Toxicology 207:35–48 [DOI] [PubMed] [Google Scholar]
- Yuan J. (1993) Estimation of variance for AUC in animal studies. J Pharm Sci 82:761–763 [DOI] [PubMed] [Google Scholar]