

Abstract
Chronic alcohol abuse results in decreased bone mineral density (BMD), which can lead to increased fracture risk. In contrast, low levels of alcohol have been associated with increased BMD in epidemiological studies. Alcohol's toxic skeletal effects have been suggested to involve impaired vitamin D/calcium homeostasis. Therefore, dietary vitamin D supplementation may be beneficial in reducing bone loss associated with chronic alcohol consumption. Six-week-old female C57BL/6J mice were pair-fed ethanol (EtOH)-containing liquid diets (10 or 36% total calories) for 78 days. EtOH exposure at 10% calories had no effects on any measured bone or serum parameter. EtOH consumption at 36% of calories reduced BMD and bone strength (P < 0.05), decreased osteoblastogenesis, increased osteoclastogenesis, suppressed 1,25-hydroxyvitamin D3 [1,25(OH)2D3] serum concentrations (P < 0.05), and increased apoptosis in bone cells compared with pair-fed controls. In a second study, female mice were pair-fed 30% EtOH diets with or without dietary supplementation with vitamin D3 (cholecalciferol; VitD) for 40 days. VitD supplementation in the EtOH diet protected against cortical bone loss, normalized alcohol-induced hypocalcaemia, and suppressed EtOH-induced expression of receptor of nuclear factor-κB ligand mRNA in bone. In vitro, pretreatment of 1,25(OH)2D3 in osteoblastic cells inhibited EtOH-induced apoptosis. In EtOH/VitD mice circulating 1,25(OH)2D3 was lower compared with mice receiving EtOH alone (P < 0.05), suggesting increased sensitivity to feedback control of VitD metabolism in the kidney. These findings suggest dietary VitD supplementation may prevent skeletal toxicity in chronic drinkers by normalizing calcium homeostasis, preventing apoptosis, and suppressing EtOH-induced increases in bone resorption.
Introduction
Chronic alcohol consumption has profound effects on the skeleton, interfering with bone growth, quality, and remodeling. Epidemiological studies in alcoholics of both genders report significant decreases in bone mineral density (BMD) and increased bone fractures and osteoporosis risk compared with nondrinkers (Clark et al., 2003; Malik et al., 2009; Pasoto et al., 2011; Wuermser et al., 2011), which is related to changes in bone turnover, particularly decreased bone formation and increased bone resorption (Dai et al., 2000; Alvisa-Negrín et al., 2009; Callaci et al., 2010; Díez-Ruiz et al., 2010). Previously, we have reported significant decreases in tibial and femoral BMD after chronic ethanol (EtOH) consumption in cycling female rats receiving isocaloric diets via intragastric infusion [total enteral nutrition (TEN)] (Shankar et al., 2006; Chen et al., 2011). In those rats, detailed bone histomorphometric analysis demonstrated decreased osteoblast numbers and increased numbers of mature osteoclasts associated with the bone perimeter, which correlated with elevated biochemical markers of bone resorption. Further analysis demonstrated that EtOH exposure increases reactive oxygen species in bone tissue, which stimulates bone resorption while inhibiting bone formation (Chen et al., 2006, 2010, 2011; Shankar et al., 2006, 2008b).
Chronic EtOH consumption also disrupts calcium homeostasis, resulting in hypocalcaemia, which seems to be caused in part by disturbances in vitamin D metabolism in addition to EtOH's inhibitory effect on intestinal calcium resorption (Krawitt, 1975; Keiver et al., 1996; Sampson, 1997). In humans, although findings have been quite variable depending on study population, age, sex, and drinking status, chronic alcohol abuse has been associated with reduced plasma concentrations of 1,25- hydroxyvitamin D3 [1,25(OH)2D3] and 25-hydroxyvitamin D3 [25OHD3] (Sampson, 1997; González-Reimers et al., 2011). A recent comprehensive study in alcoholic patients reported that nutritional status and low 1,25(OH)2D3 plasma concentrations are independently related to increased prevalence of rib and vertebral fractures (González-Reimers et al., 2011). In rodent models, we and others have reported similar decreases in circulating 1,25(OH)2D3 after chronic EtOH exposure (Turner et al., 1988; Keiver et al., 1996; Shankar et al., 2006). In addition, we have reported that this decrease in serum 1,25(OH)2D3 is associated with a significant increase in renal CYP24A1, the enzyme responsible for the conversion of 1,25(OH)2D3 to inactive 1,24,25-hydroxyvitamin D3 metabolite (Shankar et al., 2008b). It is noteworthy that we and others have also reported increases, decreases, or no change in serum concentrations of 25OHD3 after EtOH consumption in rodents (Turner et al., 1988; Wezeman et al., 2007; Shankar et al., 2008b).
Vitamin D3 (VitD; cholecalciferol) supplementation alone or combined with calcium is well tolerated and has shown great promise in reducing fracture risk in men and women (Dawson-Hughes et al., 1997; Bischoff-Ferrari et al., 2010). Skeletal benefits associated with VitD supplementation are attributed to endocrine actions related to the regulation of calcium homeostasis and/or paracrine/autocrine actions within bone cell populations. The direct effects on bone include stimulation of osteoblast differentiation and bone mass accrual while reducing osteoclast activity and inhibiting bone resorption (Atkins et al., 2007; Kogawa et al., 2010). To our knowledge, only one group has published a study investigating the benefit of vitamin D supplementation in protecting against EtOH-mediated bone loss (Wezeman et al., 2007). In that study, the authors administered a daily subcutaneous injection of cholecalciferol (5000 IU/kg/daily) to male rats in combination with a binge alcohol regimen (3g/kg i.p.) for 3 weeks, which resulted in a significant amelioration of the loss of trabecular bone and bone strength in VitD/EtOH-treated rats compared with EtOH-treated baseline controls. However, no additional mechanistic information was reported.
Contrasting with the clinical findings of chronic alcohol abusers, there is epidemiological evidence supporting a positive association between moderate alcohol consumption (1–2 drinks/day) and increased BMD in premenopausal and postmenopausal women, suggesting a bimodal effect of EtOH on bone health in both men and women (New et al., 1997; Berg et al., 2008). The biological mechanisms underlying the benefits associated with long-term moderate drinking are unclear and require further investigation. In the present study, the Lieber-DeCarli liquid feeding model was used to feed EtOH diets to cycling female C57BL/6J mice at two doses: 10 and 36% of total calories, corresponding, respectively, to moderate and heavy drinking, for 78 days, to determine the effects of moderate alcohol consumption on BMD and bone turnover in comparison with the effects observed with higher EtOH intakes. In addition, a separate study was conducted in which an EtOH diet, 30% of total calories, was supplemented with 2000 IU/kg body weight of cholecalciferol to determine whether normalizing vitamin D homeostasis would protect against chronic EtOH-mediated bone loss.
Materials and Methods
Animals and Experimental Design
All experimental procedures were approved by the Institutional Animal Care and Use Committee at the University of Arkansas for Medical Sciences. Animals were housed in an Association Assessment and Accreditation of Laboratory Animal Care-approved animal facility.
Experiment 1.
Fifty 6-week-old C57BL/6J female mice (The Jackson Laboratory, Bar Harbor, ME) were randomly assigned to five weight-matched diet groups (n = 10/group): standard rodent chow diet, a 10% EtOH Lieber-DeCarli liquid diet, a 36% EtOH Lieber-DeCarli diet, and corresponding pair-fed (PF) controls for each EtOH group. Initially, all mice received the control diet, 35% of energy from fat, 18% from protein, and 47% from carbohydrates (Dyets, Bethlehem, PA). For the EtOH groups, EtOH was added to the Lieber-DeCarli diet by substituting carbohydrate calories with EtOH calories (Dyets). The EtOH concentration in the diet was increased slowly in a stepwise manner until 10 or 36% total calories were reached, which constituted final EtOH concentrations of 1.8 and 6.2% (v/v), respectively, and maintained until sacrifice (78 days) as described previously (Wahl et al., 2006). All groups had access to water ad libitum. Mice given the control diet were isocalorically PF to their corresponding EtOH group based on the diet consumption of the previous day. Animal body weights were measured weekly. On days 0, 48, and 78 of the study, all mice were placed on a La Theta X-ray computed topography (CT) scanner (Aloka Co., Tokyo, Japan), under isoflurane anesthesia to determine body composition as described previously (Shankar et al., 2008a). Densitometric calculations of fat and muscle were performed by using CT software (Aloka Co.) and attenuation number thresholds of −120 to −500 for fat and −120 to 350 for muscle. Indices of percentage of fat mass and percentages of lean mass were calculated. Volumetric BMD was measured by using the Alcoa software from specific regions of interest (ROI). The femur ROI included distal, midshaft, and proximal femur bone, tibia ROI included midshaft to proximal tibia bone, and vertebrae ROI included thoracic vertebrae (T1-T12). Total body is defined as all bones scanned from clavicle to the lumbar vertebrae, which includes, scapula, forelimbs, thoracic vertebrae, pelvic girdle, and hind limbs. At sacrifice, trunk blood was collected, and right femurs were harvested and frozen in saline-soaked gauze at −80°C for mechanical strength testing. Left femurs were harvested, and bone marrow was used in ex vivo osteoblast and osteoclast cultures. Left tibias were fixed in EtOH and embedded in plastic for further analysis.
Experiment 2.
Twenty-two 6-week-old female C57BL/6J mice were assigned to four weight-matched groups: EtOH, the Lieber-DeCarli EtOH diet (Dyets), which contains the National Research Council-recommended level of VitD and, based on diet intake, animals received 400 IU VitD/kg body weight; EtOH/VitD, the same Lieber-DeCarli EtOH diet supplemented with additional VitD where mice were calculated to receive 2000 IU/kg body weight; and corresponding PF and PF/VitD controls (n = 4–6/group). In the EtOH groups, a final EtOH concentration of 5.2% (v/v) was achieved as described in experiment 1 and constituted 30% of total calories consumed. Both PF groups were isocalorically fed to their corresponding EtOH group based on the diet consumptions of the previous day. EtOH and PF diets were administered for 40 days. At sacrifice, trunk blood was collected, kidneys and femurs were frozen and stored at −80°C, and right tibial bones were formalin-fixed for μCT analysis. Left tibial bone was fixed in EtOH and embedded in plastic for further analysis. Blood ethanol concentrations (BECs) were analyzed by using an Analox (Huntington Beach, CA) analyzer as described previously (Shankar et al., 2006).
Serum Analysis of Vitamin D Homeostasis and Bone Turnover Markers
Serum 25OHD3 was measured by using a commercially available enzyme immunoassay kit (Immunodiagnostic Systems, Scottsdale, AZ); detection range was 5 to 380 nM, less than 10% CV for interassay variation and less than 8% CV for intra-assay variation. Serum 1,25(OH)2 D3 was also measured by using a commercially available radioimmunoassay kit (Immunodiagnostic Systems); detection range was 5 to 401 pM, and precision was 19 pM (16% CV), 46 pM (8.8% CV), and 162 pM (8.6% CV) for interassay variation and 20% CV, 13% CV, and 11.9% CV for intra-assay variation, respectively. The intact form of parathyroid hormone (PTH) (Immunotopics, Inc., San Clemente, CA), osteocalcin (Biomedical Technologies, Stoughton, MA), and C-terminal telopeptides (CTXs) of type 1 (Immunodiagnostic Systems) were detected in serum by using commercially available enzyme-linked immunosorbent assay kits. Serum phosphorus and ionized calcium were measured by using colorimetric assay kits (BioVision, Mountain View, CA).
Micro-Computed Tomography Analyses
All μCT analyses were consistent with current guidelines for the assessment of bone microstructure in rodents by using micro-computed tomography (Bouxsein et al., 2010). Formalin-fixed tibiae and femora were imaged by using a MicroCT 40 (Scanco Medical AG, Bassersdorf, Switzerland) and a 12-μm isotropic voxel size in all dimensions. The region of interest selected for analysis comprised 240 transverse CT slices representing the entire medullary volume extending 1.24 mm distal to the end of the primary spongiosa with a border lying 100 um from the cortex. Three-dimensional reconstructions were created by stacking the regions of interest from each two-dimensional slice and then applying a gray-scale threshold and Gaussian noise filter as described previously (Suva et al., 2008) and using a consistent and predetermined threshold with all data acquired at 70 kVp, 114 mA, and 200-ms integration time. Fractional bone volume [bone volume/tissue volume (BV/TV)] and architectural properties of trabecular bone trabecular thickness (mm), trabecular number (mm−1), and trabecular spacing (mm) were calculated by using previously published methods (Suva et al., 2008). Likewise, for cortical bone assessment, μCT slices were segmented into bone and marrow regions by applying a visually chosen, fixed threshold for all samples after smoothing the image with a three-dimensional Gaussian low-pass filter (σ = 0.8; support = 1.0) to remove noise and a fixed threshold (245). Cortical geometry was assessed in a 1-mm-long region centered at the tibial midshaft. The outer contour of the bone was found automatically by using the built-in Scanco contouring tool. Total area was calculated by counting all voxels within the contour, bone area was calculated by counting all voxels that were segmented as bone, and marrow area was calculated as total area − bone area. This calculation was performed on all 25 slices (one slice = ∼12.5 μm), using the average for the final calculation. The outer and inner perimeter of the cortical midshaft was determined by a three-dimensional triangulation of the bone surface of the 25 slices, and cortical thickness and other cortical parameters were determined as described previously (Suva et al., 2008). Parameters assessed were total cross-sectional area (mm2), total diameter, cortical thickness (mm), medullary area (mm2)., periosteal perimeter (mm), and endocortical perimeter (mm).
Mechanical Strength Testing
Whole femur mechanical strength testing was done by three-point bending using a MTS 858 Bionix test system load frame (MTS, Eden Prairie, MN) as described previously (Brown et al., 2002). Loading point was displaced at 0.1 mm/s until failure, and load displacement data were recorded at 100 Hz. Test curves were analyzed by using TestWorks software (MTS) to determine measures of whole-bone strength, which are peak load and stiffness. Load to failure was recorded as the load after a 2% drop from peak load.
Analysis of Gene Expression
Total RNA was isolated from kidney and femur shaft by using TRI reagent (Molecular Research Center, Cincinnati, OH) as described previously (Chen et al., 2008). Total RNA was reverse-transcribed by using IScript cDNA synthesis (Bio-Rad Laboratories, Hercules, CA) according to the manufacturer's instructions. Subsequent real-time PCR analysis was carried out by using SYBR green and an ABI 7500 sequence detection system (Applied Biosystems, Foster City, CA). Gene-specific primers were: renal CYP24A1, forward 5′-GCAAATCGATGAGCTGCGTGATGA-3′ and reverse 5′-AGGGCTTCTTCCTCTGTGTCCTTT-3′; renal CYP27B1, forward 5′-ACTCAGCTTCCTGGCTGAACTCTT-3′ and reverse 5′-ACAAGTGTAGGGTCGGCAACGTAA-3′; osteocalcin, forward 5′-TTGTGCTGGAGTGGTCTCTATGAC-3′ and reverse 5′-CACCCTCTTCCCACACTGTACA-3′; collagen type 1a, forward 5′- AGGGTCATCGTCGCTTCTC-3′ and reverse 5′-CTCCAGAGGGGGCTTGTT-3′; receptor of nuclear factor-κB ligand (RANKL), forward 5′-GGGTTCGACACCTGAATGCT-3′ and reverse 5′- AACTGGTCGGGCAATTCTGG-3′; tartrate-resistant acid phosphatase (TRAP), forward 5′-TGGTCCAGGAGCTTAACTGC-3′ and reverse 5′-GCTAGGAGTGGGAGCCATATG-3′; and cathepsin K, forward 5′-GTGGGTGTTCAAGTTTCTGC-3′ and reverse 5′- GGTGAGTCTTCTTCCATAGC-3′.
Ex Vivo Osteoblast and Osteoclast Cell Cultures
Bone marrow cells were harvested from the left femur of chow-fed, 10% EtOH-treated, 36% EtOH-treated mice, and the corresponding PF controls from experiment 1 (n = 6/group) and plated for osteoblast and osteoclast differentiation as described previously (Chen et al., 2008). Cells were cultured for osteoblastogenesis in osteogenic media (αMEM supplemented with 10% FBS and 1 mM l-ascorbic acid phosphate) for 10 days and stained for alkaline phosphatase. Separate osteoblast cultures were cultured in osteogenic media for 25 days and stained with Von Kossa. Alkaline phosphatase- and Von Kossa-stained cultures were counted under a microscope (Carl Zeiss Microscopy, LLC, Thornwood, NY) at 20× magnification. Nonadherent bone marrow cells were plated at a density of 105 cells per well and cultured in Dulbecco's modified Eagle's medium containing l-glutamine, 10% FBS, 100 U/ml penicillin and streptomycin, and 20 nM 1,25(OH)2D3 for 10 days followed by TRAP staining according to the manufacturer's instructions (Sigma-Aldrich, St. Louis, MO). Mature multinucleated osteoclasts, containing five or more nuclei, were counted under a microscope at 20× magnification.
Measurement of Apoptosis
EtOH-fixed, plastic-embedded tibias from the 36% EtOH and PF groups in experiment 1 and the EtOH and EtOH/VitD groups in experiment 2 were sectioned, and apoptotic cells were visualized by using the ApopTag Peroxidase In Situ Apoptosis Detection Kit (Millipore Corporation, Billerica, MA) and terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) of fragmented DNA and immunoperoxidase staining for qualitative detection of apoptotic cells in vivo as described in the manufacturer's instructions. In addition, neonatal calvaria-derived osteoblastic cells from untreated mice were seeded at a density of 105 cells/well in a six-well plate and maintained in osteogenic media (αMEM supplemented with 10% FBS and 1 mM l-ascorbic acid phosphate) for 20 days, at which time cells were pretreated with 1,25(OH)2D3 for 30 min before the addition of increasing concentrations of EtOH. 1,25(OH)2D3 (Enzo Life Sciences, Plymouth Meeting, PA) was prepared as a stock solution (60 μM) in absolute EtOH and stored at −80°C until use. The 1,25(OH)2D3 stock was diluted into CO2-conditioned media (αMEM supplemented with 10% FBS), which was added to the wells for a final concentration of 150 pM 1,25(OH)2D3. After the preincubation step, 25 to 100 μl/ml of a 1 M EtOH stock solution made in CO2-conditioned media was added to the appropriate plates for final EtOH concentrations of 25 to 100 mM. To prevent EtOH evaporation in the media, all plates, including control plates without EtOH treatment, were wrapped in paraffin and maintained at 37°C and 5% CO2 for 24 h, at which time cells from each treatment group were lysed in radioimmunoprecipitation assay buffer (Thermo Fisher Scientific, Waltham, MA) supplemented with protease and phosphatase inhibitors. EtOH-induced apoptosis in vitro was examined by Western blotting using a mouse monoclonal antibody recognizing the cleaved form of caspase-3 (Cell Signaling Technology, Danvers, MA) followed by anti-mouse secondary antibody conjugated with horseradish peroxidase. Blots were developed by using the chemiluminescence detection method according to the manufacturer's instructions (Thermo Fisher Scientific). Protein bands were quantified by using a densitometer, and band densities were corrected for total protein loaded by staining with 0.1% amido black (Sigma-Aldrich).
Data and Statistical Analysis
Data are presented as means ± S.E.M. Comparison between multiple groups was accomplished by one-way ANOVA, followed by Student Newman-Keuls post hoc analysis. The effect of VitD supplementation and EtOH and the interaction thereof were determined by two-way ANOVA, followed by Student Newman-Keuls post hoc analysis. Statistical significance was set at P < 0.05. SigmaPlot software package 11.0 (Systat Software, Inc., San Jose, CA) was used to perform all statistical tests.
Results
Effects of EtOH on Weight Gain and Body Composition in Experiment 1.
The mean EtOH intake for the 36% EtOH group was 30 g/kg/day, and the mean EtOH intake for the 10% EtOH group was 7.5 g/kg/day. At sacrifice, the mean BEC for the 36% EtOH group was 171.5 ± 27.1 mg/dl (range 104.2–260). This range corresponds to BECs observed in our rat TEN model of chronic alcoholism (Shankar et al., 2006) and falls within the range observed in human alcoholics (Wadstein and Skude, 1979). The mean BEC for the 10% EtOH group was 17.1 ± 1.2 mg/dl. Weights at sacrifice did not differ between the 10% EtOH, its PF control, or ad libitum chow-fed groups (21.1 ± 0.61 versus 21.0 ± 0.58 versus 21.5 ± 0.58 g, respectively), but body weight was significantly decreased in both the 36% EtOH group and its PF control compared with all other groups (17 ± 0.13 versus 19 ± 0.45 g; P < 0.05; Mann-Whitney rank sum test). Diet intakes between the 10% PF and 36% PF control groups differed slightly (11.06 versus 10.36 kcal/day, respectively; P < 0.05; Mann-Whitney rank sum test), which may explain the 10% increase in body weight between the PF controls. As seen in Fig. 1, feeding the control high-fat Lieber-DeCarli diet to mice resulted in an increase in fat accumulation (2- to 3-fold), accompanied by a 25% lower percentage of lean mass in both PF control groups compared with chow-fed mice (P < 0.05). It is noteworthy that chronic consumption of EtOH at 36% of total calories, but not at 10% of total calories, reduced the fat accumulation associated with feeding Lieber-DeCarli liquid diets, resulting in an increase in the percentage of lean mass to baseline values obtained in the chow-fed mice (P < 0.05).
Fig. 1.

Female mice were chronically fed EtOH diets for 78 days. Peak EtOH concentrations for 10% EtOH (7 g/kg/day) and 36% EtOH (30 g/kg/day) were reached on day 25 and maintained for the length of the study. Body composition, lean mass (a), and fat mass (b) were assessed for each group by using in vivo CT scanning encompassing the entire visceral region of the animal as described under Materials and Methods and are expressed as a percentage of total body weight. Statistical significance was determined by one-way ANOVA followed by Student-Newman Keuls post hoc analysis. *, P < 0.05, all groups versus chow-fed animals. #, P < 0.001, 36% EtOH versus corresponding PF control. ^, P ≤ 0.001, 36% EtOH versus 10% EtOH.
Effects of High Alcohol Consumption on BMD and Mechanical Strength.
During the study, mice were anesthetized, and BMD was measured for each group by using the La Theta CT scanner as described under Materials and Methods. As seen in Fig. 2a, baseline scans (study day 0) show no differences in total BMD between all groups (P = 0.520). Once maximal EtOH intakes were maintained for several weeks (study day 48), we observed a 4.5 to 6% decrease in total BMD in the 36% EtOH group compared with PF and chow-fed controls, respectively, but it did not reach statistical significance. At study day 78, both PF groups had decreased total BMD compared with the chow-fed group (P < 0.05). Moreover, at day 78, we observed a decrease in total BMD between 36% EtOH and its PF control (P ≤ 0.05), but no difference between the 10% EtOH group and its PF control (P = 0.9). Further analysis of cortical and trabecular BMD in the femur, tibia, and vertebrae on day 78 showed similar results (Table 1). At both the axial and appendicular skeleton, we observed a 17% decrease in cortical BMD in the 36% EtOH group compared with its PF control group (P < 0.001). In trabecular bone, decreases in BMD in both femur and vertebra were observed, a 12 and 18% decrease, respectively; the greatest effect of alcohol on trabecular bone was observed in tibial bone with a 26% decrease in BMD compared with its PF control group (P < 0.001). Throughout the study, we did not see significant changes in trabecular BMD between the 10% EtOH group and its PF control. Likewise, cortical BMD measurements between 10% EtOH and its PF control did not differ, but both had lower cortical BMD values compared with chow-fed controls (P < 0.05). Mechanical strength testing was also performed on whole femurs taken from EtOH-treated and PF groups. As seen in Fig. 2b, the loss in BMD observed in the 36% EtOH group was associated with a decrease in load to failure and stiffness (Fig. 2c) compared with its PF control and 10% EtOH (P < 0.001).
Fig. 2.

CT scanning was used to assesses total BMD (a) for chow-fed, EtOH-treated, and PF control groups in vivo as described under Materials and Methods on study days 0, 48, and 78. At sacrifice, mechanical strength of whole femurs, peak load (b) and stiffness (c), was assessed by three-point bending. Statistical significance was determined by one-way ANOVA followed by Student-Newman Keuls post hoc analysis. *, P < 0.05, all groups versus chow-fed animals. #, P < 0.001, 36% EtOH versus corresponding PF control. ^, P ≤ 0.001, 36% EtOH versus 10% EtOH.
TABLE 1.
Cortical and trabecular BMD (mg/cm3) as measured by CT scanning in experiment 1
After 78 days of chronic EtOH consumption, BMD measurements were obtained by using whole-body CT scanning as described under Materials and Methods. Statistical differences between treatment groups were determined by one-way ANOVA followed by Student-Newman Keuls post hoc analysis. Data in parentheses are S.E.M.
| Group | Cortical BMD |
Trabecular BMD |
||||
|---|---|---|---|---|---|---|
| Femur | Tibia | Vertebrae | Femur | Tibia | Vertebrae | |
| mg/cm3 | ||||||
| Chow | 638 (8.42) | 604 (4.26) | 433 (3.42) | 256 (13.88) | 318 (10.63) | 286 (15.93) |
| PF to 10% EtOH | 610 (9.68)* | 577 (5.93)* | 415 (6.28)* | 258 (15.15) | 317 (16.35) | 310 (12.78) |
| 10% EtOH | 599 (7.65)* | 556 (5.20)* | 410 (4.63)* | 237 (7.69) | 315 (16.89) | 284 (16.023) |
| PF to 36% EtOH | 591 (7.66)* | 567 (4.02)* | 410 (4.76)* | 216 (9.74) | 296 (13.55) | 295 (19.3) |
| 36% EtOH | 493 (11.00)*#^ | 466 (5.5)*#^ | 370 (3.82)*#^ | 190 (10.95)#^ | 216 (9.57)#^ | 242 (13.72)#^ |
, P < 0.05, all groups versus chow-fed animals.
, P < 0.001, 36% EtOH versus corresponding PF control.
, P ≤ 0.001, 36% EtOH versus 10% EtOH.
EtOH Consumption Disrupts Bone Remodeling.
In experiment 1, chronic EtOH consumption decreased circulating osteocalcin by 2-fold in the 36% EtOH group compared with its PF control, 14.2 ± 3.8 versus 24.6 ± 8.5 ng/ml, respectively (P < 0.001; one-way ANOVA followed by Student-Newman Keuls post hoc analysis). Moderate EtOH consumption had no effect on circulating osteocalcin in the 10% EtOH group compared with its PF control (26.9 ± 6.2 versus 28.8 ± 6.1 ng/ml, respectively). Consistent with these findings, we observed an inhibitory effect on osteoblastogenesis in primary bone marrow cells taken from 36% EtOH-treated femurs cultured ex vivo. The number of alkaline phosphatase-stained preosteoblasts was lower in cells from bone exposed to 36% EtOH in vivo compared with chow-fed, PF, and 10% EtOH cultures maintained in osteogenic media for 10 days (P < 0.05) (Fig. 3A). As a result, the number of differentiated osteoblasts identified by Von Kossa staining after 25 days in culture was also statistically different (P < 0.05) (Fig. 3b). Chronic consumption of 10% EtOH had no stimulatory or inhibitory effect on osteoblastogenesis. In addition, the number of differentiated, multinucleated TRAP-positive osteoclasts was increased in the 36% EtOH group compared with its PF control and 10% EtOH group (P < 0.001) in ex vivo primary bone marrow cultures (Fig. 3c).
Fig. 3.
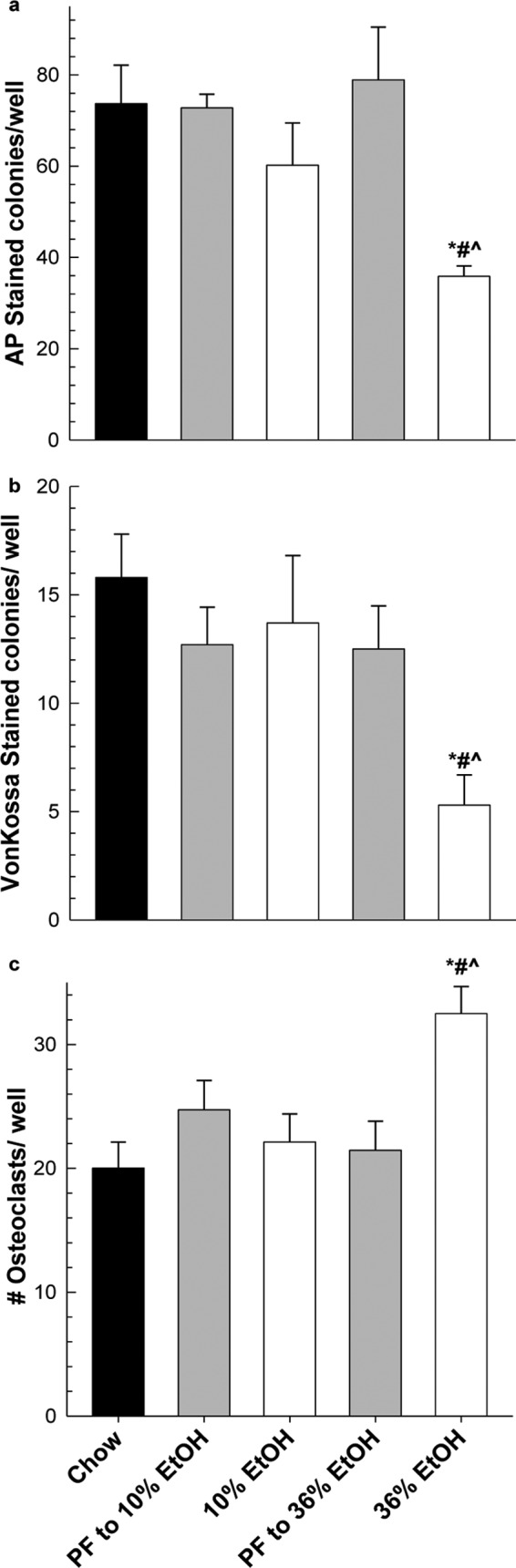
Chronic EtOH consumption alters both osteoblastogenesis and osteoclastogenesis in primary bone marrow cells cultured in osteogenic media for 10 days followed by alkaline phosphatase staining (a), cultured in osteogenic media for 25 days followed by Von Kossa staining (b), or cultured in αMEM containing 10 nM 1,25(OH)2D3 and 15 ng/well recombinant RANKL for 10 days (c). Data represent mean ± S.E.M. for n = 6 animals per group plated in triplicate. Statistical differences were determined by Mann-Whitney rank sum test. *, P ≤ 0.05, all groups versus chow-fed animals. #, P ≤ 0.05, 36% EtOH versus its corresponding PF control. ^, P ≤ 0.05, 36% EtOH versus 10% EtOH.
EtOH Consumption Alters Circulating Vitamin D Metabolites.
As shown in Fig. 4a, serum 1,25(OH)2D3 was drastically reduced after chronic consumption of 36% EtOH compared with its PF control (P < 0.05). Moderate alcohol consumption did not alter 1,25(OH)2D3 concentrations. Despite disruption of vitamin D homeostasis, we did not see significant changes in serum PTH among the EtOH or PF control groups (Fig. 4b), which is consistent with previously published reports demonstrating a lack of development of secondary hyperparathyroidism after EtOH-induced hypocalcaemia (Sampson, 1997).
Fig. 4.

Vitamin D3 and PTH serum concentrations in mice chronically fed EtOH and 25OHD2 (a), 1,25(OH)2D3 (b), or PTH (c). Statistical significance was determined by one-way ANOVA followed by Student-Newman Keuls post hoc analysis. #, P < 0.001, 36% EtOH versus corresponding PF control. ^, P ≤ 0.001, 36% EtOH versus 10% EtOH.
VitD Supplementation Prevents EtOH-Induced Cortical Bone Loss in Experiment 2.
The mean EtOH intake for EtOH and EtOH/VitD groups was 29 g/kg/day, which corresponds to 30% of total caloric intake. At sacrifice, mean BECs were not significantly different and were 218.4 ± 48.3 mg/dl (range 63–329.7) for EtOH and 144.28 ± 28.1 mg/dl (range 71–231) for EtOH/VitD. The amount of diet consumed did not differ between the EtOH and EtOH/VitD groups, but statistical analysis of final body weight using two-way ANOVA followed by Student Newman-Keuls post hoc analysis showed a decrease in body weight in the EtOH group compared with PF control (21 ± 0.45 and 19.2 ± 0.17, respectively) and between the EtOH/VitD and PF/VitD groups (21.1 ± 0.38 and 20.1 ± 0.2, respectively) (P < 0.05). At sacrifice, right femurs from all groups were collected and used for mechanical strength testing. As expected, chronic EtOH feeding resulted in 23 and 32% decreases in load-bearing strength and stiffness in the EtOH group compared with its isocalorically PF control (P < 0.05) (Fig. 5). μCT analysis of left tibial bone in the EtOH-treated group revealed decreases in trabecular BV/TV, number, and thickness (P < 0.05) (Table 2). Trabecular spacing did not change in the EtOH-treated mice compared with PF controls. In cortical bone, EtOH consumption did not change total cross-section area, but did reduce total diameter and thickness and increased medullary area (P < 0.05). VitD supplementation alone reduced trabecular BV/TV and number and increased spacing between PF and PF/VitD controls (P < 0.05). However, the addition of EtOH did not result in further reductions in trabecular BV/TV, number, or increases in spacing between the trabeculae compared with the EtOH/VitD group or the PF/VitD controls. In the cortical compartment, VitD supplementation prevented EtOH-mediated effects on medullary area (P < 0.05), diameter (P = 0.08), and thickness (P = 0.06) comparing EtOH/VitD and EtOH groups. Consistent with the protective effects of dietary VitD supplementation on cortical bone parameters, VitD supplementation prevented the loss of bone load-bearing strength and stiffness produced by EtOH consumption (P < 0.05), but no significant difference was observed in the PF/VitD group relative to the PF group (Fig. 5).
Fig. 5.

Mechanical strength testing of whole femurs from EtOH- and EtOH/VitD-treated mice (n = 7/group) and corresponding PF and PF/VitD controls (n = 4/group). a, peak load. b, stiffness. Statistical significance was determined by two-way ANOVA followed by Student Newman-Keuls post hoc analysis. Values with different letters are significantly different from each other (P < 0.05).
TABLE 2.
The effect of VitD supplementation on bone loss associated with chronic EtOH consumption
After 40 days of chronic EtOH consumption, tibial bone was analyzed by μCT as described under Materials and Methods. Values are mean ± S.E.M. Statistical differences between treatment groups were determined by two-way ANOVA followed by Student-Newman Keuls post hoc analysis. Values with different letter superscripts are significantly different from each other (P < 0.05).
| PF | EtOH | PF/VitD | EtOH/VitD | |
|---|---|---|---|---|
| μCT Trabecular bone parameters | ||||
| BV/TV, % | 6.640 (0.004)a | 4.770 (0.003)b | 4.550 (0.004)b | 4.770 (0.003)b |
| Tb.N, 1/mm | 2.826 (0.168)a | 2.352 (0.098)b | 2.133 (0.194)b | 2.260 (0.146)b |
| Tb.Sp, mm | 0.366 (0.035)a | 0.413 (0.027)a | 0.487 (0.035)b | 0.459 (0.027)a,b |
| Tb. Th, mm | 0.044 (0.001)a | 0.041 (0.001)b | 0.044 (0.001)a,b | 0.042 (0.001)a,b |
| μCT Cortical bone parameters | ||||
| Tt.Ar, mm2 | 0.153 (0.007)a | 0.138 (0.005)a | 0.141 (0.007)a | 0.134 (0.005)a |
| Total diameter, mm | 0.035 (0.002)a | 0.026 (0.001)b | 0.035 (0.002)a | 0.031 (0.001)a,b |
| Ct.Th, mm | 0.173 (0.006)a | 0.149 (0.005)b | 0.179 (0.006)a | 0.165 (0.005)a,b |
| Me.Ar, mm2 | 0.104 (0.006)a | 0.126 (0.006)b | 0.094 (0.006)a | 0.100 (0.005)a |
| Ps.Pm, mm | 1.442 (0.070)a | 1.529 (0.060)a | 1.367 (0.070)a | 1.375 (0.060)a |
| Ec.Pm, mm | 0.567 (0.042)a | 0.647 (0.034)a | 0.533 (0.042)a | 0.563 (0.034)a |
Tb.Th, trabecular bone trabecular thickness; Tb.N, trabecular number; Tb.Sp, trabecular spacing; Tt.Ar, total cross-sectional area; Ct.Th, cortical thickness; Me.Ar, medullary area; Ps.Pm, periosteal perimeter; Ec.Pm, endocortical perimeter.
VitD Supplementation Prevents EtOH-Induced Changes in Biochemical Markers of Bone Remodeling.
Chronic EtOH exposure significantly increased biochemical markers of bone resorption. We observed a 7-fold increase in circulating CTX in the serum of EtOH-treated mice and 2- to 3-fold increases in mRNA expression of RANKL, TRAP, and cathepsin K in the femur shaft of EtOH-treated mice compared with PF controls (P < 0.05) (Fig. 6). In contrast, osteoblast genes associated with bone formation such as osteocalcin and collagen type 1a mRNA expression were decreased in the femur shaft of EtOH-treated mice compared with PF controls (P < 0.05). Serum concentrations of CTX were reduced by 40% in the EtOH/VitD group compared with EtOH alone (P < 0.05). Likewise, mRNA expression of bone resorption biochemical markers was also reduced in the EtOH/VitD group compared with EtOH alone (P < 0.05). In addition, osteocalcin expression increased, but not significantly (P = 0.100), and collagen type 1a mRNA expression increased (P < 0.05) in the EtOH/VitD group compared with EtOH alone.
Fig. 6.

a to d, VitD supplementation to EtOH liquid diet prevents cortical bone loss by reducing EtOH-mediated increases in biochemical markers of bone resorption: circulating CTX in serum (a), RANKL (b), TRAP (c), and cathepsin K (d) mRNA expression in femur shaft. e and f, biochemical markers of bone formation, osteocalcin (e) and collagen type 1a (f) mRNA expression increased in the EtOH/VitD group compared with EtOH alone. Gene expression was measured by real-time PCR and normalized to Hmbs mRNA. Data are expressed as mean ± S.E.M. Statistical significance was determined by two-way ANOVA followed by Student Newman-Keuls post hoc analysis. Values with different letters are significantly different from each other (P < 0.05).
VitD Supplementation Lowers Circulating 1,25(OH)2D3 But Protects against EtOH-Induce Hypocalcaemia.
Dietary supplementation to 2000 IU/kg body weight/day increased serum 25OHD3 concentration by 2.5-fold in PF/VitD mice compared with PF controls (P < 0.05) (Table 3). EtOH alone increased 25OHD3 concentrations compared with PF controls (P < 0.05). EtOH exposure combined with VitD supplementation increased serum 25OHD3 still further compared with EtOH-treated mice (P < 0.01). Concurrently, serum 1,25(OH)2D3 concentrations were decreased by 2-fold in PF/VitD mice compared with PF controls (P < 0.05). EtOH exposure alone reduced 1,25(OH)2D3 concentrations by 30% (P < 0.05). However, EtOH exposure combined with VitD supplementation decreased serum 1,25(OH)2D3 levels still further in the EtOH/VitD group compared with EtOH-treated mice (P < 0.01). Serum PTH levels did not change in response to either EtOH or VitD supplementation alone or in combination. However, serum concentrations of ionized calcium were deceased in the EtOH group compared with PF control mice, (P < 0.05). VitD supplementation alone did not change serum calcium concentrations between PF controls, but calcium concentrations comparable with PF controls were maintained in the EtOH/VitD group compared with EtOH alone (P < 0.05). Serum phosphorous concentrations were not statistically different between any treatment groups.
TABLE 3.
Vitamin D homeostasis parameters in experiment 2
Statistical significance was determined by two-way ANOVA followed by Student Newman-Keuls post hoc analysis. Values with different letter superscripts are significantly different from each other (P < 0.05).
| 25OHD3 | 1,25(OH)2D3 | PTH | Serum Ca2+ | Serum Phosphorous | |
|---|---|---|---|---|---|
| nmol/l | pmol/l | pg/ml | mg/dl | mmol/l | |
| PF | 48.3 (1.09)a | 94.4 (9.6)a | 184.9 (33.2)a | 11.87 (1.48)a | 2.76 (0.163)a |
| EtOH | 99.1 (3.95)b | 66.5 (11)b | 189.0 (21.7)a | 8.00 (1.21)b | 3.17 (0.198)a |
| PF/VitD | 121.5 (2.58)c | 45.3 (2.1)b,c | 162.9 (33.2)a | 11.26 (1.48)a | 3.20 (0.298)a |
| EtOH/VitD | 141.1 (4.1)d | 29.0 (4.1)d | 126.1 (21.7)a | 14.63 (1.48)a | 3.36 (0.232)a |
Increased Circulating 25OHD3 by EtOH Treatment and/or VitD Supplementation Enhances Feedback Regulation of Renal Cytochrome P450 Expression Controlling 1,25(OH)2D3 Synthesis and Degradation.
Chronic EtOH feeding increased renal CYP24A1 mRNA expression 2-fold compared with PF control (P < 0.05) (Fig. 7a). VitD supplementation alone also increased CYP24A1 mRNA expression 2-fold compared with PF controls. Moreover, the addition of EtOH increased CYP24A1 mRNA expression further (63%) compared with either agent alone (P = 0.083). Renal CYP27B1 is the enzyme responsible for the synthesis of 1,25(OH)2D3 from 25(OH)D3 (Christakos et al., 2010). Renal CYP27B1 mRNA also decreased in the EtOH/VitD group (P < 0.05) compared with the EtOH or PF/VitD groups (Fig. 7b). In all groups, we observed no significant changes in bone-specific CYP27B1 mRNA expression in response to EtOH or VitD supplementation alone or in combination (Fig. 7c).
Fig. 7.

VitD supplementation enhances EtOH-induced regulation of renal CYP24A1 (a) and CYP27B1 (b) expression, but not CYP27B1 mRNA expression in femur bone (c). Gene expression was measured by real-time PCR and normalized to Pkg1 mRNA. Data are expressed as mean ± S.E.M. Statistical significance was determined by two-way ANOVA followed by Student Newman-Keuls post hoc analysis. Values with different letters are significantly different from each other (P < 0.05).
Chronic EtOH Consumption Induces Apoptosis in Mouse Tibial Bone Marrow.
Recently, apoptosis has been reported in rat tibia after chronic consumption of EtOH for 17 weeks (Maurel et al., 2011). We observed an increase in apoptosis as measured by qualitative in situ TUNEL staining in tibial bone marrow after chronic EtOH exposure (Fig. 8, a and c). It is noteworthy that VitD supplementation seemed to protect against EtOH-mediated apoptosis in bone marrow cells (Fig. 8d). For a more quantitative analysis of the effects of EtOH and VitD on bone cell apoptosis, we measured EtOH-mediated apoptosis in vitro by using neonatal calvaria-osteoblastic cells derived from control chow-fed C57BL/6J female mice. In cell culture, we observed a dose-dependent increase in active caspase-3 expression in cell lysates after 24 h of EtOH exposure (P < 0.05; 0 < 25 <50 mM). However, pretreating calvaria cells with 150 pM 1,25(OH)2D3 before the addition of EtOH prevented the increase in active caspase-3 expression (Fig. 8e).
Fig. 8.

a to d, representative pictures of in situ TUNEL staining in tibial bone marrow taken from 36% EtOH-treated mice (a) and corresponding PF controls (b), 30% EtOH-treated mice (c), and 30% EtOH/VitD-treated mice (d) as described under Materials and Methods. Arrows indicate apoptotic cells. e, EtOH exposure (0–50 mM) increased cleaved caspase-3 expression in differentiating osteoblastic cells in a dose-dependent manner. Pretreatment with 1,25(OH)2D3 protected against EtOH-induced apoptosis in osteoblastic cells treated with EtOH. Apoptosis was assessed by Western blot with caspase-3 expression quantified by densitometry. Band densities were corrected for total protein loaded by staining with 0.1% amido black. Bars indicate mean ± S.E.M. of triplicate determinations. Statistical differences were determined by one-way ANOVA followed by Student-Keuls post hoc analysis. P < 0.05 for EtOH treatment a < b < c.
Discussion
Consistent with our previously published studies using the rat TEN model (Chen et al., 2006, 2010, 2011; Shankar et al., 2006, 2008b), chronic exposure to high EtOH concentrations significantly decreased trabecular and cortical BMD, which resulted in the loss of bone strength in the female mouse. In experiment 1, bone loss was associated with decreased bone formation and increased osteoclastogenesis. Serum 1,25(OH)2D3 concentrations were also decreased despite normal levels of PTH. EtOH exposure also decreased body weight and fat accrual in these mice, a phenomena that has been reported in heavy and chronic drinkers (Addolorato et al., 2000). Lower body weights may be, in part, explained by disruption of the growth hormone/insulin-like growth factor 1 axis by EtOH as a result of impaired growth hormone secretion (Ronis et al., 2007) or inefficient utilization of EtOH as an energy source (Lieber, 1991). Alternatively, others have reported that EtOH metabolism in white adipose tissue blocks preadipocyte differentiation to mature adipocytes through the inhibition of peroxisome proliferator-activated receptor γ and CCAAT-enhancer-binding protein β expression (Crabb et al., 2011). Chronic EtOH feeding also stimulates lipolysis in white adipose tissue in rats through the increased expression of the major adipose lipases and concurrent down-regulation of genes related to adipose tissue fatty acid uptake. These effects were reported to be reversed by treatment with the peroxisome proliferator-activated receptor γ agonist rosiglitazone (Sun et al., 2012).
It is noteworthy that moderate EtOH exposure (10% EtOH) had no adverse or positive effects on body composition, cortical and trabecular BMD, or bone strength. These findings suggest there is no gain in bone health in response to moderated EtOH exposure in this mouse model. Aside from alcohol, other components found in alcoholic beverages have been reported to have bone anabolic effects, and these, as opposed to EtOH, may contribute to the positive correlation between light-moderate drinking and improved BMD observed in several epidemiological studies (New et al., 1997; Jugdaohsingh et al., 2004; Berg et al., 2008).
In our rat and mouse models of chronic EtOH exposure, heavy alcohol consumption is associated with a significant decrease in serum 1,25(OH)2D3 (Shankar et al., 2006, 2008b). Given the importance of hormonal VitD regulation in maintaining bone density, we conducted an additional experiment to test whether correcting for the loss in 1,25(OH)2D3 through dietary VitD supplementation would provide protection against EtOH-induced bone loss. In experiment 2, μCT analysis of 30% EtOH-treated tibias revealed significant changes in both cortical and trabecular compartments. These changes were associated with decreased bone strength. However, in the EtOH/VitD group, increasing daily VitD intake from 400 to 2000 IU/kg body weight protected against EtOH-dependent losses of cortical bone and bone strength. This protection coincided with decreases in circulating CTX and gene expression of bone resorption markers and increases in the expression of bone formation markers in EtOH/VitD mice compared with EtOH mice. It is noteworthy that VitD supplementation alone reduced trabecular bone in the PF/VitD controls (Table 2). In the literature, it has been reported that VitD supplementation in 10-week-old male rats fed low-calcium, vitamin D-depleted diets produced a positive correlation between trabecular BV/TV and serum 25OHD3 concentrations ≥80 nM (Anderson et al., 2008). However, it has also been reported that dietary VitD supplementation in 6-week-old female mice, significantly decreased trabecular BV/TV through an overall increase in bone turnover in that compartment (Iwamoto et al., 2003). Therefore, we suspect that the reduction trabecular volume observed in the PF/VitD group is a sex- and/or age-dependent, compartment-specific effect associated with vitamin D receptor (VDR) signaling.
Most interesting was that serum calcium in the EtOH/VitD group was normalized to PF controls despite further decreases in serum 1,25(OH)2D3 concentrations. The reduction in serum 1,25(OH)2D3 coincided with a significant reduction in renal CYP27B1 and increased renal CYP24 expression in the EtOH/VitD group relative to the EtOH group. These findings support the current view that endocrine regulation of renal VitD synthesis and metabolism is subject to a feedback loop via serum calcium (Christakos et al., 2010). Our data suggest that circulating 1,25(OH)2D3 is not important in mediating the normalization of serum calcium associated with dietary VitD supplementation in the current model and is consistent with studies suggesting that CYP27B1 in extra-renal tissues is responsible for 1,25(OH)2D3 synthesis and autocrine/paracrine VDR signaling (Heaney et al., 2003; Fleet and Schoch, 2010; Morris and Anderson, 2010; Geng et al., 2011). Although controversial, support for intestinal synthesis of 1,25(OH)2D3 comes from clinical studies by Heaney et al. (2003) in which calcium absorption was increased 25% after 4 weeks of treatment with 50 μg/day of 25OHD3 despite no changes in serum 1,25(OH)2D3 and from studies localizing CYP27B1 in the intestinal villus (Balesaria et al., 2009). Alternatively, 25OHD3 may have direct biological actions to stimulate VDR-dependent signaling in the intestine because it was significantly elevated in the EtOH/VitD group (Rowling et al., 2007; Zhang et al., 2011).
The protection from EtOH-mediated bone loss observed in the EtOH/VitD mice may be secondary to the restoration of calcium homeostasis. In VDR knockout mice, it has been reported that a “rescue diet” containing 2% calcium, 20% lactose, and 1.25% phosphorous is capable of reversing the abnormal mineral homeostasis, rickets, and osteomalacia associated with this genotype (Li et al., 1998). However, in CYP27B1 knockout mice, the rescue diet did not completely restore the impaired bone phenotype, in particular the inhibition of longitudinal bone growth (Dardenne et al., 2003). In these same mice, daily injections of 1,25(OH)2D3 normalized serum calcium and rescued the bone deficiencies, including the aberrant bone growth (Dardenne et al., 2003), which emphasizes the importance of VDR signaling in bone growth and maintenance and the likelihood that a calcium rescue diet in the absence of additional VitD may not completely alleviate EtOH's effect on bone.
In the EtOH/VitD mice, bone turnover is normalized, which could also be related to increased VDR signaling as a result of local conversion of 25OHD3 to 1,25(OH)2D3 in bone cells by CYP27B1 (Geng et al., 2011). In the present study we have shown that in vivo EtOH exposure is associated with the presence of apoptotic cells in the bone marrow (Fig. 8). Moreover, VitD supplementation minimized the in vivo apoptotic response to EtOH exposure, and pretreatment of osteoblastic cells with a physiological relevant dose of 1,25OH2D3 completely suppressed the EtOH-mediated apoptosis in vitro. We suspect that in the EtOH/VitD-treated mice increased VDR signaling prevents or counteracts the effects of EtOH-mediated oxidative stress in osteoblasts.
In summary, dietary VitD supplementation normalizes serum calcium and prevents cortical bone loss and subsequent loss of mechanical strength associated with chronic EtOH exposure. In the intestine, local VitD synthesis may be responsible for increased calcium absorption, which results in the normalization of serum calcium. In bone, the direct actions of locally produced 1,25(OH)2D3 on bone cells may suppress EtOH-induced oxidative stress signaling pathways involved in reducing osteoblastogenesis, increasing bone resorption and stimulating apoptosis. Because the study described here increased VitD in the diet approximately 5-fold, it is plausible that increasing average daily dietary VitD intake by a similar amount would be a feasible approach to preventing the deleterious effects of EtOH on bone in heavy or chronic drinkers.
This work was supported by the National Institutes of Health National Institute on Alcohol Abuse and Alcoholism [Grant RO1 AA18282], the Arkansas Children's Hospital Research Institute Lyons Fund; and the Carl L. Nelson Chair in Orthopaedic Creativity, University of Arkansas for Medical Sciences.
Portions of this work were presented previously: Ronis MJ, Chen JR, Lazarenko O, Badger TM, and Shankar K (2009) Inhibited osteoblastogenesis, enhanced bone resorption and disrupted vitamin D3 homeostasis in female C57BL/6 mice fed alcohol, at the 32nd Annual Scientific Meeting of the Research Society on Alcoholism; 2009 June 20–24; San Diego, CA. Research Society on Alcoholism, Austin, TX.
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
- BMD
- bone mineral density
- EtOH
- ethanol
- TEN
- total enteral nutrition
- VitD
- vitamin D3/cholecalciferol
- 25OHD3
- 25-hydroxyvitamin D3
- 1,25(OH)2D3
- 1,25-hydroxyvitamin D3
- PF
- pair-fed
- CT
- computed topography
- μCT
- micro-CT
- ROI
- regions of interest
- TRAP
- tartrate-resistant acid phosphatase
- CTX
- C-terminal telopeptide
- BEC
- blood ethanol concentration
- CV
- coefficient of variation
- PTH
- parathyroid hormone
- BV/TV
- bone volume/tissue volume
- VDR
- vitamin D receptor
- RANKL
- receptor of nuclear factor-κB ligand
- TUNEL
- terminal deoxynucleotidyl transferase dUTP nick-end labeling
- αMEM
- α-minimal essential medium
- FBS
- fetal bovine serum
- ANOVA
- analysis of variance
- PCR
- polymerase chain reaction.
Authorship Contributions
Participated in research design: Mercer, Chen, Badger, and Ronis.
Conducted experiments: Mercer, Wynne, Lazarenko, and Hogue.
Contributed new reagents or analytic tools: Mason.
Performed data analysis: Mercer, Suva, Chen, and Ronis.
Wrote or contributed to the writing of the manuscript: Mercer, Lumpkin, Suva, Badger, and Ronis.
References
- Addolorato G, Capristo E, Marini M, Santini P, Scognamiglio U, Attilia ML, Messineo D, Sasso GF, Gasbarrini G, Ceccanti M. (2000) Body composition changes induced by chronic ethanol abuse: evaluation by dual energy X-ray absorptiometry. Am J Gastroenterol 95:2323–2327 [DOI] [PubMed] [Google Scholar]
- Alvisa-Negrín J, González-Reimers E, Santolaria-Fernández F, García-Valdecasas-Campelo E, Valls MR, Pelazas-González R, Durán-Castellón MC, de Los Angeles Gómez-Rodríguez M. (2009) Osteopenia in alcoholics: effect of alcohol abstinence. Alcohol Alcohol 44:468–475 [DOI] [PubMed] [Google Scholar]
- Anderson PH, Sawyer RK, Moore AJ, May BK, O'Loughlin PD, Morris HA. (2008) Vitamin D depletion induces RANKL-mediated osteoclastogenesis and bone loss in a rodent model. J Bone Miner Res 23:1789–1797 [DOI] [PubMed] [Google Scholar]
- Atkins GJ, Anderson PH, Findlay DM, Welldon KJ, Vincent C, Zannettino AC, O'Loughlin PD, Morris HA. (2007) Metabolism of vitamin D3 in human osteoblasts: evidence for autocrine and paracrine activities of 1α,25-dihydroxyvitamin D3. Bone 40:1517–1528 [DOI] [PubMed] [Google Scholar]
- Balesaria S, Sangha S, Walters JR. (2009) Human duodenum responses to vitamin D metabolites of TRPV6 and other genes involved in calcium absorption. Am J Physiol Gastrointest Liver Physiol 297:G1193–G1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg KM, Kunins HV, Jackson JL, Nahvi S, Chaudhry A, Harris KA, Jr, Malik R, Arnsten JH. (2008) Association between alcohol consumption and both osteoporotic fracture and bone density. Am J Med 121:406–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischoff-Ferrari HA, Shao A, Dawson-Hughes B, Hathcock J, Giovannucci E, Willett WC. (2010) Benefit-risk assessment of vitamin D supplementation. Osteoporos Int 21:1121–1132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouxsein ML, Boyd SK, Christiansen BA, Guldberg RE, Jepsen KJ, Müller R. (2010) Guidelines for assessment of bone microstructure in rodents using micro-computed tomography. J Bone Miner Res 25:1468–1486 [DOI] [PubMed] [Google Scholar]
- Brown EC, Perrien DS, Fletcher TW, Irby DJ, Aronson J, Gao GG, Hogue WJ, Skinner RA, Suva LJ, Ronis MJ, et al. (2002) Skeletal toxicity associated with chronic ethanol exposure in a rat model using total enteral nutrition. J Pharmacol Exp Ther 301:1132–1138 [DOI] [PubMed] [Google Scholar]
- Callaci JJ, Himes R, Lauing K, Roper P. (2010) Long-term modulations in the vertebral transcriptome of adolescent-stage rats exposed to binge alcohol. Alcohol Alcohol 45:332–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JR, Haley RL, Hidestrand M, Shankar K, Liu X, Lumpkin CK, Simpson PM, Badger TM, Ronis MJ. (2006) Estradiol protects against ethanol-induced bone loss by inhibiting up-regulation of receptor activator of nuclear factor-κB ligand in osteoblasts. J Pharmacol Exp Ther 319:1182–1190 [DOI] [PubMed] [Google Scholar]
- Chen JR, Lazarenko OP, Shankar K, Blackburn ML, Badger TM, Ronis MJ. (2010) A role for ethanol-induced oxidative stress in controlling lineage commitment of mesenchymal stromal cells through inhibition of Wnt/β-catenin signaling. J Bone Miner Res 25:1117–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JR, Lazarenko OP, Shankar K, Blackburn ML, Lumpkin CK, Badger TM, Ronis MJ. (2011) Inhibition of NADPH oxidases prevents chronic ethanol-induced bone loss in female rats. J Pharmacol Exp Ther 336:734–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JR, Shankar K, Nagarajan S, Badger TM, Ronis MJ. (2008) Protective effects of estradiol on ethanol-induced bone loss involve inhibition of reactive oxygen species generation in osteoblasts and downstream activation of the extracellular signal-regulated kinase/signal transducer and activator of transcription 3/receptor activator of nuclear factor-κB ligand signaling cascade. J Pharmacol Exp Ther 324:50–59 [DOI] [PubMed] [Google Scholar]
- Christakos S, Ajibade DV, Dhawan P, Fechner AJ, Mady LJ. (2010) Vitamin D: metabolism. Endocrinol Metab Clin North Am 39:243–253, table of contents [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark MK, Sowers MF, Dekordi F, Nichols S. (2003) Bone mineral density and fractures among alcohol-dependent women in treatment and in recovery. Osteoporos Int 14:396–403 [DOI] [PubMed] [Google Scholar]
- Crabb DW, Zeng Y, Liangpunsakul S, Jones R, Considine R. (2011) Ethanol impairs differentiation of human adipocyte stromal cells in culture. Alcohol Clin Exp Res 35:1584–1592 [DOI] [PubMed] [Google Scholar]
- Dai J, Lin D, Zhang J, Habib P, Smith P, Murtha J, Fu Z, Yao Z, Qi Y, Keller ET. (2000) Chronic alcohol ingestion induces osteoclastogenesis and bone loss through IL-6 in mice. J Clin Invest 106:887–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dardenne O, Prudhomme J, Hacking SA, Glorieux FH, St-Arnaud R. (2003) Rescue of the pseudo-vitamin D deficiency rickets phenotype of CYP27B1-deficient mice by treatment with 1,25-dihydroxyvitamin D3: biochemical, histomorphometric, and biomechanical analyses. J Bone Miner Res 18:637–643 [DOI] [PubMed] [Google Scholar]
- Dawson-Hughes B, Harris SS, Krall EA, Dallal GE. (1997) Effect of calcium and vitamin D supplementation on bone density in men and women 65 years of age or older. N Engl J Med 337:670–676 [DOI] [PubMed] [Google Scholar]
- Díez-Ruiz A, García-Saura PL, García-Ruiz P, González-Calvin JL, Gallego-Rojo F, Fuchs D. (2010) Bone mineral density, bone turnover markers and cytokines in alcohol-induced cirrhosis. Alcohol Alcohol 45:427–430 [DOI] [PubMed] [Google Scholar]
- Fleet JC, Schoch RD. (2010) Molecular mechanisms for regulation of intestinal calcium absorption by vitamin D and other factors. Crit Rev Clin Lab Sci 47:181–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng S, Zhou S, Glowacki J. (2011) Effects of 25-hydroxyvitamin D3 on proliferation and osteoblast differentiation of human marrow stromal cells require CYP27B1/1α-hydroxylase. J Bone Miner Res 26:1145–1153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- González-Reimers E, Alvisa-Negrín J, Santolaria-Fernández F, Candelaria Martín-González M, Hernández-Betancor I, Fernández-Rodríguez CM, Viña-Rodríguez J, González-Díaz A. (2011) Vitamin D and nutritional status are related to bone fractures in alcoholics. Alcohol Alcohol 46:148–155 [DOI] [PubMed] [Google Scholar]
- Heaney RP, Dowell MS, Hale CA, Bendich A. (2003) Calcium absorption varies within the reference range for serum 25-hydroxyvitamin D. J Am Coll Nutr 22:142–146 [DOI] [PubMed] [Google Scholar]
- Iwamoto J, Yeh JK, Takeda T, Ichimura S, Sato Y. (2003) Comparative effects of vitamin K and vitamin D supplementation on prevention of osteopenia in calcium-deficient young rats. Bone 33:557–566 [DOI] [PubMed] [Google Scholar]
- Jugdaohsingh R, Tucker KL, Qiao N, Cupples LA, Kiel DP, Powell JJ. (2004) Dietary silicon intake is positively associated with bone mineral density in men and premenopausal women of the Framingham Offspring cohort. J Bone Miner Res 19:297–307 [DOI] [PubMed] [Google Scholar]
- Keiver K, Herbert L, Weinberg J. (1996) Effect of maternal ethanol consumption on maternal and fetal calcium metabolism. Alcohol Clin Exp Res 20:1305–1312 [DOI] [PubMed] [Google Scholar]
- Kogawa M, Findlay DM, Anderson PH, Ormsby R, Vincent C, Morris HA, Atkins GJ. (2010) Osteoclastic metabolism of 25(OH)-vitamin D3: a potential mechanism for optimization of bone resorption. Endocrinology 151:4613–4625 [DOI] [PubMed] [Google Scholar]
- Krawitt EL. (1975) Effect of ethanol ingestion on duodenal calcium transport. J Lab Clin Med 85:665–671 [PubMed] [Google Scholar]
- Li YC, Amling M, Pirro AE, Priemel M, Meuse J, Baron R, Delling G, Demay MB. (1998) Normalization of mineral ion homeostasis by dietary means prevents hyperparathyroidism, rickets, and osteomalacia, but not alopecia in vitamin D receptor-ablated mice. Endocrinology 139:4391–4396 [DOI] [PubMed] [Google Scholar]
- Lieber CS. (1991) Perspectives: do alcohol calories count? Am J Clin Nutr 54:976–982 [DOI] [PubMed] [Google Scholar]
- Malik P, Gasser RW, Kemmler G, Moncayo R, Finkenstedt G, Kurz M, Fleischhacker WW. (2009) Low bone mineral density and impaired bone metabolism in young alcoholic patients without liver cirrhosis: a cross-sectional study. Alcohol Clin Exp Res 33:375–381 [DOI] [PubMed] [Google Scholar]
- Maurel DB, Jaffre C, Rochefort GY, Aveline PC, Boisseau N, Uzbekov R, Gosset D, Pichon C, Fazzalari NL, Pallu S, et al. (2011) Low bone accrual is associated with osteocyte apoptosis in alcohol-induced osteopenia. Bone 49:543–552 [DOI] [PubMed] [Google Scholar]
- Morris HA, Anderson PH. (2010) Autocrine and paracrine actions of vitamin D. Clin Biochem Rev 31:129–138 [PMC free article] [PubMed] [Google Scholar]
- New SA, Bolton-Smith C, Grubb DA, Reid DM. (1997) Nutritional influences on bone mineral density: a cross-sectional study in premenopausal women. Am J Clin Nutr 65:1831–1839 [DOI] [PubMed] [Google Scholar]
- Pasoto SG, Yoshihara LA, Maeda LC, Bernik MM, Lotufo PA, Bonfa E, Pereira RM. (2011) Osteoporotic hip fractures in non-elderly patients: relevance of associated co-morbidities. Rheumatol Int http://dx.doi.org/10.1007/s00296-011-2154-x [DOI] [PubMed]
- Ronis MJ, Wands JR, Badger TM, de la Monte SM, Lang CH, Calissendorff J. (2007) Alcohol-induced disruption of endocrine signaling. Alcohol Clin Exp Res 31:1269–1285 [DOI] [PubMed] [Google Scholar]
- Rowling MJ, Gliniak C, Welsh J, Fleet JC. (2007) High dietary vitamin D prevents hypocalcemia and osteomalacia in CYP27B1 knockout mice. J. Nutr 137:2608–2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson HW. (1997) Alcohol, osteoporosis, and bone regulating hormones. Alcohol Clin Exp Res 21:400–403 [DOI] [PubMed] [Google Scholar]
- Shankar K, Harrell A, Liu X, Gilchrist JM, Ronis MJ, Badger TM. (2008a) Maternal obesity at conception programs obesity in the offspring. Am J Physiol Regul Integr Comp Physiol 294:R528–R538 [DOI] [PubMed] [Google Scholar]
- Shankar K, Hidestrand M, Haley R, Skinner RA, Hogue W, Jo CH, Simpson P, Lumpkin CK, Jr, Aronson J, Badger TM, et al. (2006) Different molecular mechanisms underlie ethanol-induced bone loss in cycling and pregnant rats. Endocrinology 147:166–178 [DOI] [PubMed] [Google Scholar]
- Shankar K, Liu X, Singhal R, Chen JR, Nagarajan S, Badger TM, Ronis MJ. (2008b) Chronic ethanol consumption leads to disruption of vitamin D3 homeostasis associated with induction of renal 1,25-dihydroxyvitamin D3-24-hydroxylase (CYP24A1). Endocrinology 149:1748–1756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Tang Y, Tan X, Li Q, Zhong W, Sun X, Jia W, McClain CJ, Zhou Z. (2012) Activation of peroxisome proliferator-activated receptor-γ by rosiglitazone improves lipid homeostasis at the adipose tissue-liver axis in ethanol-fed mice. Am J Physiol Gastrointest Liver Physiol 302:G548–G557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suva LJ, Hartman E, Dilley JD, Russell S, Akel NS, Skinner RA, Hogue WR, Budde U, Varughese KI, Kanaji T, et al. (2008) Platelet dysfunction and a high bone mass phenotype in a murine model of platelet-type von Willebrand disease. Am J Pathol 172:430–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RT, Aloia RC, Segel LD, Hannon KS, Bell NH. (1988) Chronic alcohol treatment results in disturbed vitamin D metabolism and skeletal abnormalities in rats. Alcohol Clin Exp Res 12:159–162 [DOI] [PubMed] [Google Scholar]
- Wadstein J, Skude G. (1979) Serum ethanol, hepatic enzymes and length of debauch in chronic alcoholics. Acta Med Scand 205:317–318 [DOI] [PubMed] [Google Scholar]
- Wahl EC, Liu L, Perrien DS, Aronson J, Hogue WR, Skinner RA, Hidestrand M, Ronis MJ, Badger TM, Lumpkin CK., Jr (2006) A novel mouse model for the study of the inhibitory effects of chronic ethanol exposure on direct bone formation. Alcohol 39:159–167 [DOI] [PubMed] [Google Scholar]
- Wezeman FH, Juknelis D, Himes R, Callaci JJ. (2007) Vitamin D and ibandronate prevent cancellous bone loss associated with binge alcohol treatment in male rats. Bone 41:639–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuermser LA, Achenbach SJ, Amin S, Khosla S, Melton LJ., 3rd (2011) What accounts for rib fractures in older adults? J. Osteoporos 2011:457591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang ZL, Ding XF, Tong J, Li BY. (2011) Partial rescue of the phenotype in 1α-hydroxylase gene knockout mice by vitamin D3 injection. Endocr Res 36:101–108 [DOI] [PubMed] [Google Scholar]