

Abstract
Based on the observation that antipsychotic medications display antagonist properties at dopamine D2-like receptors, aberrant dopamine signaling has been proposed to underlie psychosis in patients with schizophrenia. Thus, it is not surprising that considerable research has been devoted to understanding the mechanisms involved in the antipsychotic action of these compounds. It is important to note that the majority of these studies have been performed in “normal” experimental animals. Given that these animals do not possess the aberrant neuronal information processing typically associated with schizophrenia, the aim of the current study was to examine the dopamine D2 receptor system in a rodent model of schizophrenia. Here, we demonstrate that methylazoxymethanol acetate (MAM)-treated rats display an enhanced effect of quinpirole on dopamine neuron activity and an aberrant locomotor response to D2-like receptor activation, suggesting changes in postsynaptic D2-like receptor function. To better understand the mechanisms underlying the enhanced response to D2-like ligands in MAM-treated rats, we examined the expression of D2, D3, and dopamine transporter mRNA in the nucleus accumbens and ventral tegmental area by quantitative reverse transcription-polymerase chain reaction. MAM-treated rats displayed a significant increase in dopamine D3 receptor mRNA expression in the nucleus accumbens with no significant changes in the expression of the D2 receptor. Taken together, these data demonstrate robust alterations in dopamine D2-like receptor function in a rodent model of schizophrenia and provide evidence that preclinical studies examining the mechanisms of antipsychotic drug action should be performed in animal models that mirror aspects of the abnormal neuronal transmission thought to underlie symptoms of schizophrenia.
Introduction
Schizophrenia is a disease affecting up to 1% of the population (Stilo and Murray, 2010). Although the exact etiology of schizophrenia is not currently known, there is significant evidence for a dysfunction within the mesolimbic dopamine system (Abi-Dargham, 2004). This “dopamine hypothesis” was largely based on the discovery that dopamine D2 receptor antagonists are effective in treating the positive symptoms of the disease (Seeman et al., 1975; Creese et al., 1976). Since that time, extensive research has been performed to better understand the mechanisms underlying the antipsychotic properties of dopamine D2 receptor antagonists (for review see Seeman, 1987, 2002). Clinically, the blockade of D2 receptors generally occurs over 24 to 48 h, depending on the antipsychotic dose and dosing schedule, and the rate of therapeutic improvement is highest in the first days and first week of treatment (Agid et al., 2003). However, full recovery and maximal therapeutic efficacy of antipsychotics typically require weeks of administration (Agid et al., 2003). Thus, it is likely that the antipsychotic effects are not necessarily associated with D2 receptor blockade, per se, but rather reflect secondary changes in dopamine system function.
Dopamine D2-like receptors are expressed in dopamine neurons of the ventral mesencephalon where they function as autoreceptors (Mercuri et al., 1997; Centonze et al., 2002). Thus, the systemic or local administration of D2 agonists has been demonstrated to dose-dependently decrease dopamine neuron activity (Pucak and Grace, 1991; Mercuri et al., 1997; Centonze et al., 2002). This decrease in activity is attributable to a rapid hyperpolarization via activation of K+ channels (Neve et al., 2004). It is noteworthy that whereas the acute systemic administration of D2 antagonists increases dopamine neuron activity (firing rate and burst firing), the chronic administration (21 days) of classic antipsychotic drugs produces a dramatic decrease in the number of spontaneously active dopamine neurons (Grace and Bunney, 1986; Grace, 1992; Grace et al., 1997). It was determined that these neurons were hyperexcited to the point of inactivity, a condition known as depolarization block (Bunney and Grace, 1978; Grace, 1992). In addition, it has been suggested that the induction of depolarization block may be attributable to an interaction between the neuroleptic and anesthetic (Melis et al., 1998); however, given that antipsychotics produce depolarization block in paralyzed rats, this seems unlikely (Bunney and Grace, 1978; Chiodo and Bunney, 1985). Those electrophysiological studies therefore provided significant information regarding the mechanism of action of antipsychotic medications that might explain, at least in part, the delayed maximal efficacy and lack of tolerance observed clinically during antipsychotic drug treatment.
Studies have reassessed the delayed-onset hypothesis by examining the time course of antipsychotic efficacy in patients (Agid et al., 2003). Consistent with a wealth of clinical data, the maximal clinical efficacy took weeks to establish (Agid et al., 2003). However, in contrast to a delayed onset, it was suggested that these patients displayed an initial antipsychotic response, the efficacy of which increased across time. These data questioned the depolarization block hypothesis of antipsychotic drug action that typically requires 3 weeks to observe a physiological response. It is important to note, however, that the initial electrophysiological studies were performed in “normal” experimental animals that do not possess the aberrant neuronal information processing typically associated with schizophrenia. Given that schizophrenia is typically associated with an enduring dopamine hyperfunction (Abi-Dargham, 2004; Lodge and Grace, 2011), it is likely that neuroadaptive changes occur within the dopamine system that have the potential to alter the response to antipsychotic drug administration. For this reason, the aim of the current study was to examine the dopamine D2-like receptor system in a rodent model of schizophrenia that displays augmented dopamine system function (Moore et al., 2006; Lodge and Grace, 2009).
Materials and Methods
All experiments were performed in accordance with the guidelines outlined in the Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources, 1996) and approved by the Institutional Animal Care and Use Committee of the University of Texas Health Science Center.
Animals.
Methylazoxymethanol acetate (MAM) treatments were performed as described previously (Moore et al., 2006). In brief, timed pregnant female Sprague-Dawley rats were obtained at gestational day 16 (Harlan, Indianapolis, IN) and housed individually in plastic breeding tubs. MAM (diluted in saline, 25 mg/kg i.p.) was administered on gestational day 17. Control rats received injections of saline (1 ml/kg i.p). Male pups were weaned on day 21 and housed in groups of two to three with littermates until adulthood, at which time they were used for physiological or behavioral studies. All experiments were performed on multiple litters of MAM- and saline-treated rats.
VTA Dopamine Neuron Extracellular Recordings.
Rats (300–450 g) were anesthetized with chloral hydrate (400 mg/kg i.p.) and placed in a stereotaxic apparatus. Anesthesia was maintained by supplemental administration of chloral hydrate as required to maintain suppression of limb compression withdrawal reflex. A core body temperature of 37°C was sustained by a thermostatically controlled heating pad (David Kopf Instruments, Tujunga, CA). Glass extracellular microelectrodes (impedance 6–14 MΩ) were lowered into the VTA (anterior/posterior, −5.3; medial/lateral, +0.6 mm from bregma and −6.5 to −9.0 mm ventral of brain surface) by using a hydraulic microdrive (David Kopf Instruments). Spontaneously active dopamine neurons were identified with open filter settings (low pass 30 Hz; high pass 30 kHz) by using previously established electrophysiological criteria (Grace and Bunney, 1983).
Once a stable dopamine neuron was identified, baseline neuronal activity was recorded for 5 min. After baseline recordings, dose-response curves were generated for cumulative doses of the dopamine D2-like agonist quinpirole (1.0, 2.0, 7.0, and 20.0 μg/kg i.v. into lateral tail vein), and neuronal activity was measured for 2 min per dose. In a subset of rats, the dopamine D2-like receptor antagonist sulpiride (20 mg/kg i.v.) was administered after the final dose of quinpirole, and neuronal activity was recorded for an additional 3 to 5 min. Only one neuron was recorded per animal.
The effect of selective ligands on the activity of the population of dopamine neurons in the VTA was determined by administering either a selective D2 [3-(4-(4-chlorophenyl-4-hydroxypiperidino)methyl)indole (L-741,626) (Bowery et al., 1996)], D3 [4″-acetyl-N-(4-(4-(2-methoxyphenyl)-1-piperazinyl)butyl)(1,1″-biphenyl)-4-carboxamide (GR 103691) (Audinot et al., 1998)], or D4 [sonepiprazole (Merchant et al., 1996)] antagonist or vehicle [either acidified (0.1% acetic acid) distilled water (D2) or acidified (0.1% acetic acid) 10% dimethyl sulfoxide in distilled water (D3 and D4)]. All drugs demonstrated low nanomolar affinity for their respective receptors (Bowery et al., 1996; Merchant et al., 1996; Audinot et al., 1998) and were administered at 1 mg/kg (into lateral tail vein) before determining the number of spontaneously active DA neurons encountered while making six to nine vertical passes, separated by 200 μm, in a predetermined pattern to sample equivalent regions of the VTA. Once a stable dopamine neuron was identified, baseline neuronal activity was recorded for 3 min.
At the cessation of the experiments, the rats were decapitated, and their brains were removed, fixed for 24 h (3.7% formaldehyde), cryoprotected (10% w/v sucrose in phosphate-buffered saline), and sectioned (25-μm coronal sections) on a cryostat (Leica, Wetzlar, Germany). Sections containing electrode placements were mounted onto gelatin-chrom alum-coated slides and stained with neutral red (0.1%) and thionin acetate (0.01%) for histochemical confirmation of electrode tracks within the VTA (Paxinos and Watson, 1986).
Behavior.
Rats (300–450 g) were placed in an open-field arena (MED Associates, St. Albans, VT) where spontaneous locomotor activity in the x-y plane was determined for 45 min by beam breaks and recorded. Immediately after the 45-min baseline period, rats were injected with quinpirole (0.1, 0.5, or 2.0 mg/kg), and locomotor activity was recorded for an additional 180 min.
Quantitative PCR.
Rats (300–450 g) were deeply anesthetized with sodium pentobarbital (100 mg/kg i.p.) and rapidly decapitated. The nucleus accumbens and VTA were isolated by tissue punch and immediately homogenized in lysis/binding solution (Ambion, Austin, TX). RNA was precipitated and separated by filtration using a commercially available kit (Ambion RNAqueous-4PCR). The concentration of RNA was determined by absorbance at 260 nm with a NanoDrop Spectrometer (Thermo Fisher Scientific, Waltham, MA) and converted to single-stranded cDNA by using a High Capacity cDNA Reverse Transcription Kit (Ambion) and thermal cycler. Real-time PCR was performed with FAM-labeled TaqMan primers targeting either the dopamine D2 receptor (Rn00561126_m1), dopamine D3 receptor (Rn00567568_m1), dopamine D4 receptor (Rn00564071_m1), dopamine transporter SLC6A3 (Rn00562224_m1), tyrosine hydroxylase (Rn00562500_m1), RGS 2 (Rn00584932_m1), RGS 9 (Rn00570117_m1), G protein-coupled inwardly rectifying potassium channel (GIRK) 2 (Rn00755103_m1), or glyceraldehyde-3-phosphate dehydrogenase (Rn01775763_g1). Detection of FAM-labeled DNA was performed by using either a Fluidigm 48.48 Dynamic Array Integrated Fluidic Circuit (Fluidigm, South San Francisco, CA) or a CFX384 Real-Time PCR Detection System (Bio-Rad Laboratories, Hercules, CA).
Analysis.
Electrophysiological analysis of dopamine neuron activity was performed by using commercially available computer software (LabChart version 7.1; ADInstruments Ltd., Chalgrove, Oxfordshire, UK), and dose-response curves were generated and analyzed by using the Prism software program (GraphPad Software Inc., San Diego, CA). Locomotor activity was collected with Activity Monitor (MED Associates), and analysis of FAM-labeled DNA was performed by using commercially available software (Bio-Rad Laboratories and Fluidigm). All data are represented as the mean ± S.E.M. unless otherwise specified.
Materials.
(−)-Quinpirole HCl, (±)sulpiride, chloral hydrate, and artificial cerebrospinal fluid all were purchased from Sigma (St. Louis, MO). L-741626, GR 103691, and sonepiprazole were purchased from Tocris Bioscience (Ellisville, MO). The RNAqueous-4PCR and High Capacity cDNA Reverse Transcription Kits as well as FAM-labeled TaqMan primers and Gene Expression Master Mix were purchased from Applied Biosystems (Foster City, CA). All other chemicals and reagents were of either analytical or laboratory grade and purchased from standard suppliers.
Results
VTA Dopamine Neuron Recordings.
Consistent with previous studies (Lacey et al., 1987; Marinelli et al., 2006), the systemic administration of quinpirole induced a dose-dependent decrease in the average firing rates of VTA dopamine neurons (Fig. 1). MAM-treated rats displayed an enhanced sensitivity to the inhibitory effects of quinpirole. Specifically, a nonlinear regression of the dose-response curves demonstrated a significant difference between saline- and MAM-treated rats [SAL, logIC50 −5.02 ± 0.21 (9.5 μg/kg); MAM, logIC50 −5.42 ± 0.14 (3.8 μg/kg); F = 6.636; p < 0.05; extra sum-of-squares F test; n = 8/group].
Fig. 1.

MAM-treated rats display an augmented electrophysiological response to quinpirole. A, representative dopamine neuron demonstrating a dose-dependent decrease in firing rate in response to quinpirole (cumulative doses indicated above) that is restored by the administration of the D2 antagonist sulpiride. B, group data demonstrating dose-response curves to quinpirole in saline- and MAM-treated rats. Inset in B demonstrates a significant decrease in the logIC50 for quinpirole in MAM-treated rats. †, significant difference from saline; p < 0.05; extra sum-of-squares F test (n = 8 neurons/group).
To better understand the function of the distinct dopamine receptor subtypes in the VTA we examined the effect of selective D2 [L-741,626 (Bowery et al., 1996)], D3 [GR 103691 (Audinot et al., 1998)], or D4 [sonepiprazole (Merchant et al., 1996)] receptor antagonists on dopamine neuron activity (Fig. 2). In saline-treated rats, the D2 and D3 antagonists significantly increased the number of spontaneously active dopamine neurons [Veh, 1.00 ± 0.05; D2, 1.74 ± 0.10; D3, 1.72 ± 0.15; t = 3.881 (D2) and 3.967 (D3); p < 0.05; two-way ANOVA with Holm-Sidak; n = 5–6 rats/group)], whereas a similar trend was observed with the D4 antagonist (Veh, 1.00 ± 0.05; D4, 1.49 ± 0.14; t = 2.571; p = 0.058; two-way ANOVA with Holm-Sidak; n = 5 rats/group) without altering average firing rate (Veh, 3.92 ± 0.35; D2, 3.66 ± 0.42; D3, 3.44 ± 0.23; D4, 4.19 ± 0.27; Fdrug = 1.386; p > 0.05; two-way ANOVA with Holm-Sidak; n = 31–64 neurons/group) or burst firing (Veh, 27.3 ± 4.4; D2, 29.4 ± 3.7; D3, 23.5 ± 3.0; D4, 26.8 ± 3.2; Fdrug = 1.031; p > 0.05; two-way ANOVA with Holm-Sidak; n = 31–64 neurons/group). Consistent with previous observations, MAM-treated rats displayed an increase in dopamine neuron population activity compared with saline-treated rats (SAL/Veh, 1.00 ± 0.05; MAM/Veh, 2.11 ± 0.20; t = 5.821; p < 0.05; two-way ANOVA with Holm-Sidak; n = 5 rats/group) with no change in firing rats (SAL/Veh, 3.92 ± 0.35; MAM/Veh, 4.31 ± 0.29; Fstrain = 0.844; p > 0.05; two-way ANOVA with Holm-Sidak; n = 31–65 neurons/group) or burst firing (SAL/Veh, 27.3 ± 4.4; MAM/Veh, 28.2 ± 3.5; Fstrain = 3.184; p > 0.05; two-way ANOVA with Holm-Sidak; n = 31–65 neurons/group). It is noteworthy that the dopamine receptor-selective ligands produced the opposite response in MAM-treated rats, i.e., a decrease in dopamine neuron population activity [Veh, 2.11 ± 0.20; D2, 1.14 ± 0.14; D3, 1.41 ± 0.09; D4, 1.43 ± 0.13; t = 5.050 (D2), 3.829 (D3), and 3.564 (D4); p < 0.05; two-way ANOVA with Holm-Sidak; n = 5–6 rats/group] without altering average firing rate (Veh, 4.31 ± 0.29; D2, 3.98 ± 0.39; D3, 3.71 ± 0.33; D4, 4.09 ± 0.33; Fdrug = 1.386; p > 0.05; two-way ANOVA with Holm-Sidak; n = 37–65 neurons/group) or burst firing (Veh, 28.2 ± 3.5; D2, 30.7 ± 4.3; D3, 29.3 ± 3.1; D4, 37.3 ± 3.7; Fdrug = 1.031; p > 0.05; two-way ANOVA with Holm-Sidak; n = 37–65 neurons/group).
Fig. 2.
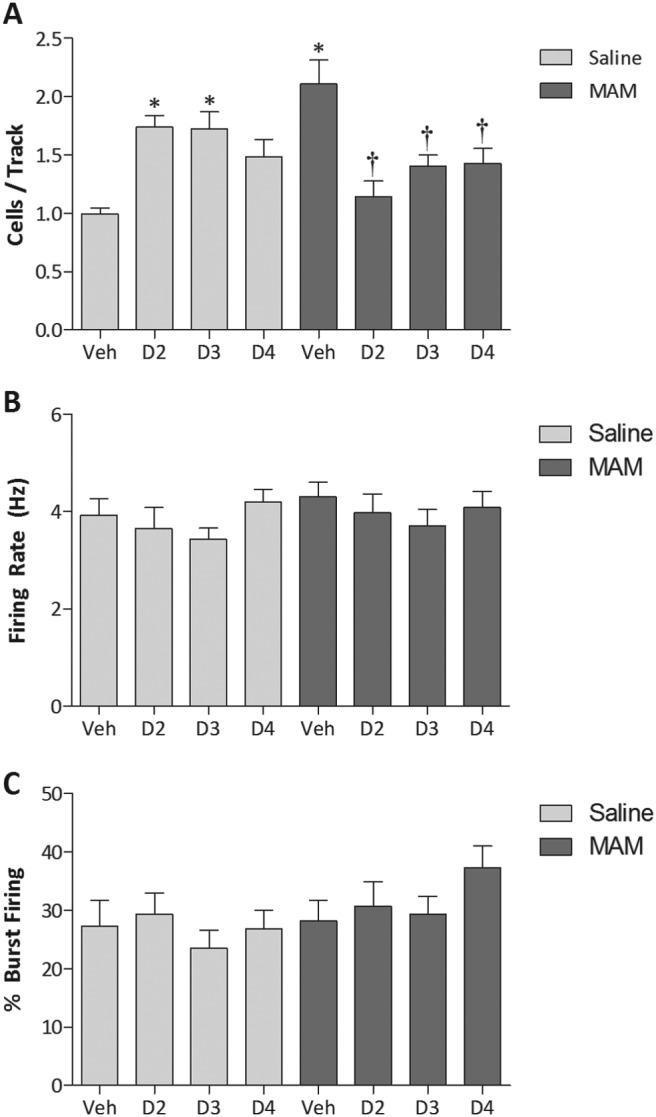
Saline- and MAM-treated rats display qualitatively different responses to the administration of selective dopamine receptor antagonists. Three parameters of activity were recorded; the number of spontaneously active dopamine neurons (A), average firing rate (B), and average percentage of burst firing (C). *, significant difference from saline-vehicle-treated rats; †, significant difference from MAM-vehicle-treated rats; p < 0.05; two-way ANOVA with Holm-Sidak post hoc; n = 5–6 rats/group (A) and n = 31–65 neurons/group (B and C).
Locomotor Activity.
The studies detailed above evaluated D2-like function at the level of the dopamine cell body. To examine whether postsynaptic D2 receptor function was altered in the MAM model, we examined the locomotor response to quinpirole. Consistent with previous observations, quinpirole produced an initial suppression of spontaneous locomotor activity at both low and high doses (Svensson et al., 1994; Szechtman et al., 1994; Li et al., 2010). Increasing doses of quinpirole produced a biphasic response with an initial decrease in ambulation being followed by robust increases in locomotor activity, again consistent with previous observations (Svensson et al., 1994; Szechtman et al., 1994; Li et al., 2010). It is noteworthy that MAM-treated rats were more sensitive to the locomotor-inducing effects of quinpirole, with a majority (75%) of MAM-treated rats displaying robust locomotor activation in response to the 0.1 mg/kg dose (compare with 25% for saline-treated rats; Fig. 3).
Fig. 3.
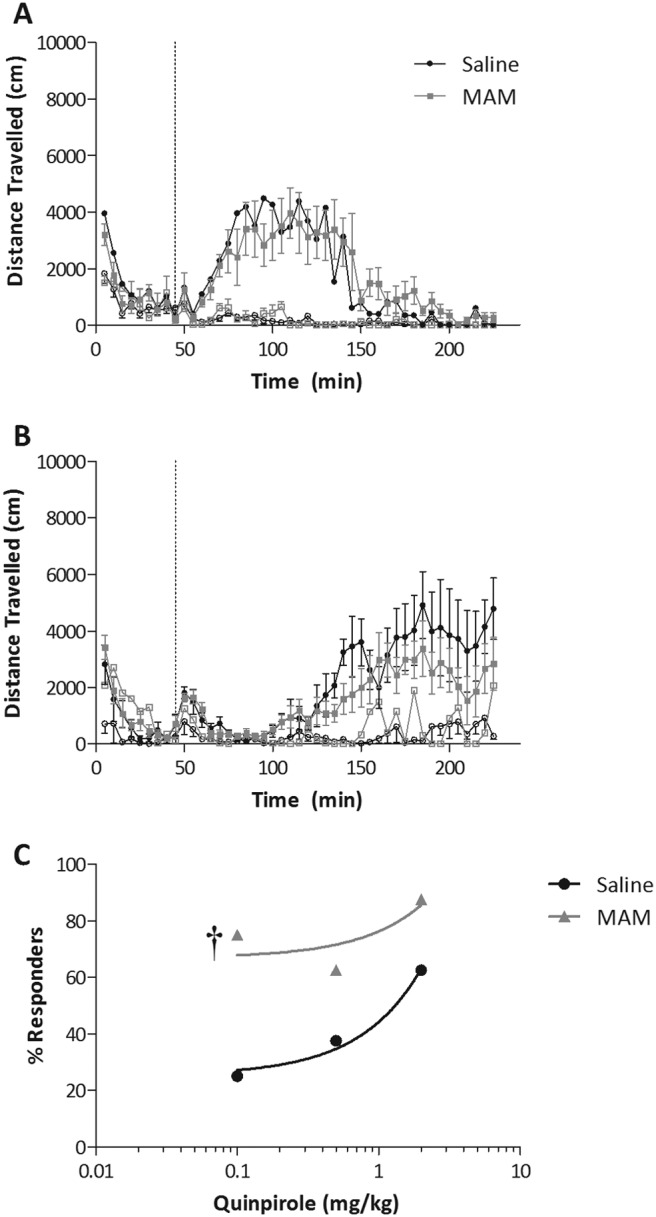
MAM-treated rats display an aberrant locomotor response to quinpirole. A, time course and magnitude to a low dose of quinpirole (0.1 mg/kg i.p.). B, an initial locomotor suppression (∼60 min in duration) followed by locomotor activation in response to a high dose of quinpirole (2.0 mg/kg i.p.). The closed symbols in A and B represent the rats with a significant increase in locomotor activity in response to quinpirole administration (responders). C, the proportion of rats displaying an augmented locomotor to each dose of quinpirole. Linear regression of the dose response demonstrates that MAM-treated rats display a sensitized locomotor activating response to quinpirole compared with control. †, significant difference in intercept from saline; p < 0.05; linear regression (n = 8 MAM or saline rats/dose).
Quantitative RT-PCR.
To examine the molecular mechanisms that may underlie the enhanced response to quinpirole observed in MAM-treated rats, quantitative RT-PCR was performed by using probes for the dopamine D2, D3, and D4 receptors and the dopamine transporter (SLC6A3), tyrosine hydroxylase, and dopamine receptor signaling molecules RGS2, RGS9, and GIRK2. Consistent with previous studies, the mRNA expression of the D3 receptor was at significantly lower levels than the D2 receptor in both the nucleus accumbens (ΔCt, 2.17 compared with 7.78; t = 17.083; p < 0.05; two-way ANOVA with Holm-Sidak) and VTA (ΔCt, 3.86 compared with 8.99; t = 15.642; p < 0.05; two-way ANOVA with Holm-Sidak). Dopamine D4 receptor expression was below the limits of detection for both the Fluidigm and Bio-Rad Laboratories systems, consistent with previous observations demonstrating extremely low levels of D4 mRNA in rodents (Vrana et al., 1995). mRNA expression of the dopamine transporter (ΔCt, 3.12 compare with 13.42; t = 31.360; p < 0.05; two-way ANOVA with Holm-Sidak) and tyrosine hydroxylase (ΔCt, 1.47 compare with 8.71; t = 22.067; p < 0.05; two-way ANOVA with Holm-Sidak) were preferentially expressed in the VTA, as expected. Quantification of the mRNA expression data revealed that MAM-treated rats displayed a significant increase in dopamine D3 mRNA expression in the nucleus accumbens (Sal, 1.04 ± 0.14; MAM, 1.78 ± 0.24; t = 3.071; p < 0.05; two-way ANOVA with Holm-Sidak), but not in the VTA (Sal, 1.08 ± 0.23; MAM, 1.32 ± 0.56; p > 0.05; two-way ANOVA with Holm-Sidak). There were no significant differences in the expression of any of the other genes examined in either brain region (Fig. 4).
Fig. 4.
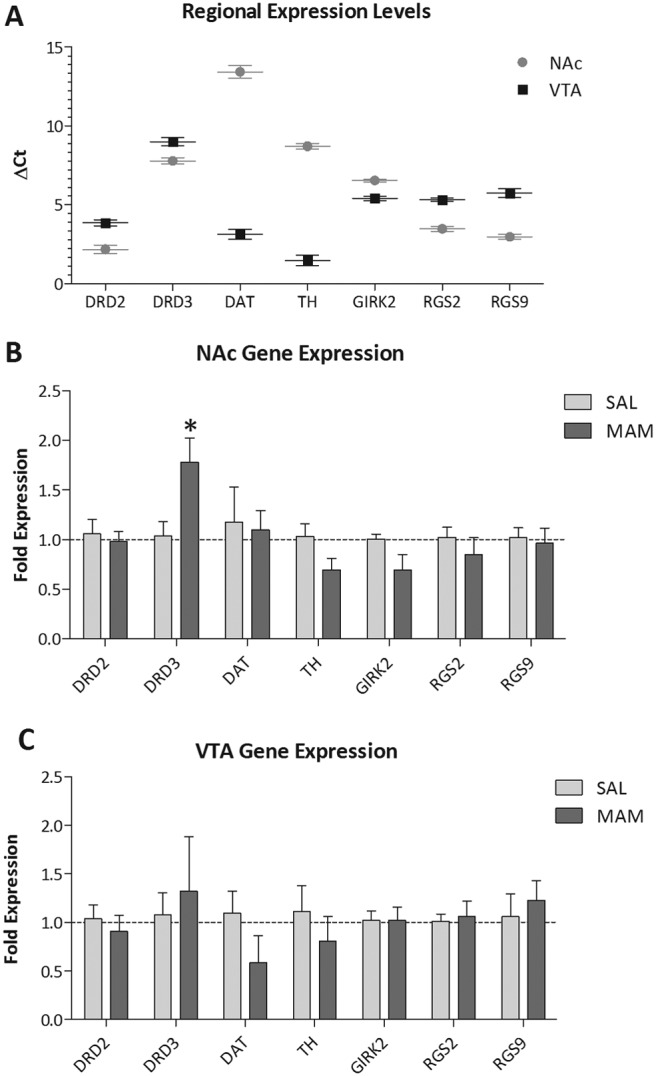
Increased dopamine D3 receptor expression in MAM-treated rats. Quantitative RT-PCR was performed on RNA extracted from the nucleus accumbens and VTA of saline- and MAM-treated rats. A, expression data demonstrate higher mRNA levels of D3 compared with D2 receptor expression throughout both the NAc and VTA. B, MAM-treated rats display a significant increase in the expression of the dopamine D3 receptor throughout the NAc but not VTA. It is noteworthy that there were no significant changes in the mRNA expression of any of the other examined markers. *, significant difference from saline; p < 0.05; two-way ANOVA with Holm Sidak post hoc (n = 5 rats/group).
Discussion
Dopamine D2 receptor antagonists have been used for more than 50 years to treat psychiatric disorders (Kapur and Mamo, 2003; Miyamoto et al., 2005); however, the mechanisms underlying their antipsychotic efficacy are not completely understood. Initial trials in human patients with schizophrenia determined that antipsychotics required weeks of repeated treatment to obtain the maximal therapeutic effects (for review see Agid et al., 2003). Thus, it is clear that the antipsychotic properties are not simply associated with acute dopamine D2 receptor blockade, but rather attributable to alterations secondary to chronic receptor antagonism. Previous studies by Grace and colleagues demonstrated that although acute D2 receptor antagonism increases dopamine neuron activity (via inhibition of tonic autoreceptor activity at the level of the dopamine neuron), chronic administration of haloperidol produces the opposite effect, actually decreasing the number of dopamine neurons firing spontaneously (Grace and Bunney, 1986; Grace, 1992; Grace et al., 1997). This decrease in dopamine neuron population activity was demonstrated to be attributable to a hyperexcitable state of the neuron known as depolarization block (Bunney and Grace, 1978; Grace, 1992). Those studies led to the delayed-onset hypothesis of antipsychotic drug action. Other clinical studies have challenged this model based on data demonstrating a gradual improvement in symptoms that begins soon after the initial treatment has begun (Agid et al., 2003). It is important to note, however, our understanding of the mechanism of antipsychotic drug action is based largely on data obtained from normal rats that may not reflect the effects that these compounds have in disease states such as schizophrenia. Indeed, here, we demonstrate enduring changes in dopamine D2 receptor function in a rodent model of schizophrenia that probably affect the response to antipsychotic medications. Furthermore, a recent study has demonstrated that MAM-treated rats display electrophysiological responses to both first- and second-generation antipsychotics that are qualitatively different from those observed in normal animals (Valenti et al., 2011). Thus, the presence of an augmented dopamine system may actually sensitize the system to facilitate antipsychotic-induced depolarization block of dopamine neurons.
Using in vivo electrophysiology and behavior, we now demonstrate that MAM-treated rats display an augmented response to the D2-like agonist administration. Specifically, quinpirole administration produced a dose-dependent inhibition of spontaneous dopamine neuron activity, previously demonstrated to be attributable to increased K+ conductances (Neve et al., 2004). It is noteworthy that MAM-treated rats displayed a leftward shift of the dose-response curve and a significant decrease in the logIC50 for quinpirole, suggesting an increase in D2 receptor function at the level of the dopamine cell body. It should be noted that an increase in autoreceptor sensitivity may be expected to result in a decrease in dopamine neuron activity; however, MAM-treated rats do not demonstrate decreases in dopamine neuron firing rates (Fig. 2; Lodge and Grace, 2007). To examine the potential mechanisms underlying the enhanced presynaptic response to quinpirole, we performed quantitative RT-PCR for dopamine receptor mRNA expression. It is noteworthy that there were no apparent differences in the expression of D2, D3, DAT, or TH mRNA between MAM- and saline-treated rats. Given that the hyperpolarization and suppression of dopamine neuron excitability D2 receptor are mediated via the activation of GIRK2 (Kuzhikandathil et al., 1998), the coupling efficiency of which is enhanced by RGS2 protein (Lomazzi et al., 2008), we also examined the expression of these mediators of D2 receptor function. Neither GIRK2 nor RGS2 were significantly altered throughout the VTA of MAM-treated rats. Thus, the enhanced response to quinpirole does not seem to be associated with changes in receptor mRNA expression at the level of the dopamine cell body and may reflect an enhanced baseline dopamine tone caused by an increase in dopamine neuron population activity (Fig. 2; Lodge and Grace, 2007), although this remains to be established.
As mentioned above, studies by Grace and colleagues have demonstrated that antipsychotic drugs produce a response in MAM-treated rats that is qualitatively different from that in control rats. To examine the relative contributions of the three distinct D2-like receptors to this response, we examined the effect of antagonists selective for the dopamine D2 (L-741,626; Bowery et al., 1996), D3 (GR 103691; Audinot et al., 1998), or D4 (sonepiprazole; Merchant et al., 1996) receptors. All three drugs were administered at equivalent doses and surprisingly produced qualitatively similar responses. Thus, blockade of either the D2 or D3 receptor produced a significant increase in dopamine neuron population activity, with a similar trend observed with the D4 antagonist. Consistent with recent studies examining the response to haloperidol and sertindole, the administration of a D2, D3, or D4 antagonist produced the opposite response in MAM-treated rats, i.e., a decrease in dopamine neuron population activity. Thus, it seems that an augmented dopamine system facilitates D2-like antagonist-induced depolarization block of dopamine neurons and further that this response is qualitatively similar for antagonists selective for either the D2, D3, or D4 receptor.
The studies detailed above evaluate D2-like function at the level of the dopamine neuron. To examine whether postsynaptic D2 receptor function was altered in the MAM model, we examined the locomotor response to quinpirole. Consistent with previous observations, quinpirole produced an initial suppression of spontaneous locomotor activity in control rats (Svensson et al., 1994; Szechtman et al., 1994; Li et al., 2010). It is noteworthy that MAM-treated rats were more sensitive to the locomotor-inducing effects of quinpirole such that they displayed an opposite behavioral response to low doses of quinporole (i.e., locomotor activation compared with suppression). Thus, it seems that MAM-treated rats display enduring changes in dopamine D2 receptor function both at the level of the dopamine cell body and in the nucleus accumbens. To examine the potential molecular changes associated with the augmented locomotor response to quinpirole, we examined mRNA expression of dopamine D2, D3, D4, and RGS9 (a known regulator of NAc D2 receptor function) (Rahman et al., 2003) by way of quantitative RT-PCR. MAM-treated rats displayed a significant increase in dopamine D3 mRNA expression in the nucleus accumbens with no significant differences in D2 receptor or RGS9 expression. Thus, the functional changes in dopamine receptor function may be attributable, in part, to increased expression of the dopamine D3 receptor at the level of the nucleus accumbens; however, increased D3 receptor turnover caused by enhanced stimulation cannot be discounted. This increase in D3 mRNA expression is consistent with postmortem findings demonstrating increased D3 receptor density throughout the basal ganglia of unmedicated patients with schizophrenia (Gurevich et al., 1997). In addition, this increased D3 receptor density seems to be reduced by antipsychotic medications (Joyce and Gurevich, 1999), which typically have similar affinities for the D2 and D3 receptors (Schotte et al., 1996). Furthermore, increased D2-like binding is observed in patients with schizophrenia after dopamine depletion, demonstrating enhanced baseline occupancy of the D2-like receptors (Abi-Dargham et al., 2000). A further association between the D3 receptor and schizophrenia is observed in genetic studies where polymorphisms of the D3 receptor have been associated with the disease (Urraca et al., 2011; Zhang et al., 2011) as well as antipsychotic efficacy (Hwang et al., 2010; Vehof et al., 2012). For this reason, there have been a number of preclinical studies suggesting that the D3-specific antagonist may represent a more specific antipsychotic profile (Gyertyán et al., 2008; Millan et al., 2008; Kiss et al., 2010). A number of selective D3 receptor ligands [i.e., 2-(3-(4-(2-tert-butyl-6-trifluoromethylpyrimidin-4-yl)piperazin-1-yl)propylsulfanyl)3H-pyrimidin-4-one fumarate (A-437203) and N-[4-[2-(propylamino) ethyl]phenyl]-4-(trifluoromethoxy)-benzenesulfonamide (PNU-177864)] have been investigated as potential antipsychotic medications, but have not proved to be efficacious. Nonetheless, it seems that alterations in the expression of the dopamine D3 receptor is a consistent observation in preclinical models as well as patients with schizophrenia and, although it may not reflect a therapeutic target, it may, nonetheless, contribute to schizophrenia symptoms.
Taken as a whole, these data demonstrate enduring changes in dopamine receptor function in a developmental disruption model of schizophrenia. It is therefore likely that the effects of antipsychotic medications will vary based on the state of the dopamine system and expression of D2-like receptors. Indeed, we demonstrate a qualitatively different physiological response to selective D2-like ligands in the MAM-treated rat, consistent with recent studies (Valenti et al., 2011). Thus, the current results provide critical information regarding changes in dopamine system function in an animal model of schizophrenia and moreover demonstrate the importance of using appropriate animal models when examining potential mechanistic correlates of antipsychotic efficacy.
Acknowledgments
We thank Jennifer Donegan and Dr. Jason O'Connor for valuable assistance with RT-PCR. The Fluidigm Dynamic Array was run at the Core for Advanced Translational Technologies at the University of Texas Health Science Center, San Antonio (director: Dr. Douglas Williamson).
This work was supported by the National Institutes of Health National Institute of Mental Health [Grant MH090067]; and a National Alliance for Research on Schizophrenia and Depression award from the Maltz Family Foundation.
D.J.L. receives consulting fees from Dey Pharmaceuticals.
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
- MAM
- methylazoxymethanol acetate
- ANOVA
- analysis of variance
- Ct
- cycle threshold
- DAT
- dopamine transporter
- FAM
- 5-carboxyfluorescein
- GIRK
- G protein-coupled inwardly rectifying potassium channel
- NAc
- nucleus accumbens
- PCR
- polymerase chain reaction
- RGS
- regulator of G-protein signaling
- RT
- reverse transcription
- SAL
- saline
- TH
- tyrosine hydroxylase
- Veh
- vehicle
- VTA
- ventral tegmental area
- L-741,626
- 3-(4-(4-chlorophenyl-4-hydroxypiperidino)methyl)indole
- GR 103691
- 4″-acetyl-N-(4-(4-(2-methoxyphenyl)-1-piperazinyl)butyl)(1,1″-biphenyl)-4-carboxamide
- A-437203
- 2-(3-(4-(2-tert-butyl-6-trifluoromethylpyrimidin-4-yl)piperazin-1-yl)propylsulfanyl)-3H-pyrimidin-4-one fumarate
- PNU-177864
- N-[4-[2-(propylamino)ethyl]phenyl]-4-(trifluoromethoxy)-benzenesulfonamide.
Authorship Contributions
Participated in research design: Perez and Lodge.
Conducted experiments: Perez and Lodge.
Performed data analysis: Perez and Lodge.
Wrote or contributed to the writing of the manuscript: Perez and Lodge.
References
- Abi-Dargham A. (2004) Do we still believe in the dopamine hypothesis? New data bring new evidence. Int J Neuropsychopharmacol 7 (Suppl 1):S1–S5 [DOI] [PubMed] [Google Scholar]
- Abi-Dargham A, Rodenhiser J, Printz D, Zea-Ponce Y, Gil R, Kegeles LS, Weiss R, Cooper TB, Mann JJ, Van Heertum RL, et al. (2000) Increased baseline occupancy of D2 receptors by dopamine in schizophrenia. Proc Natl Acad Sci U S A 97:8104–8109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agid O, Kapur S, Arenovich T, Zipursky RB. (2003) Delayed-onset hypothesis of antipsychotic action: a hypothesis tested and rejected. Arch Gen Psychiatry 60:1228–1235 [DOI] [PubMed] [Google Scholar]
- Audinot V, Newman-Tancredi A, Gobert A, Rivet JM, Brocco M, Lejeune F, Gluck L, Desposte I, Bervoets K, Dekeyne A, et al. (1998) A comparative in vitro and in vivo pharmacological characterization of the novel dopamine D3 receptor antagonists (+)-S 14297, nafadotride, GR 103,691 and U 99194. J Pharmacol Exp Ther 287:187–197 [PubMed] [Google Scholar]
- Bowery BJ, Razzaque Z, Emms F, Patel S, Freedman S, Bristow L, Kulagowski J, Seabrook GR. (1996) Antagonism of the effects of (+)-PD 128907 on midbrain dopamine neurones in rat brain slices by a selective D2 receptor antagonist L-741,626. Br J Pharmacol 119:1491–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunney BS, Grace AA. (1978) Acute and chronic haloperidol treatment: comparison of effects on nigral dopaminergic cell activity. Life Sci 23:1715–1727 [DOI] [PubMed] [Google Scholar]
- Centonze D, Usiello A, Gubellini P, Pisani A, Borrelli E, Bernardi G, Calabresi P. (2002) Dopamine D2 receptor-mediated inhibition of dopaminergic neurons in mice lacking D2L receptors. Neuropsychopharmacology 27:723–726 [DOI] [PubMed] [Google Scholar]
- Chiodo LA, Bunney BS. (1985) Possible mechanisms by which repeated clozapine administration differentially affects the activity of two subpopulations of midbrain dopamine neurons. J Neurosci 5:2539–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creese I, Burt DR, Snyder SH. (1976) Dopamine receptors and average clinical doses. Science 194:546. [DOI] [PubMed] [Google Scholar]
- Grace AA. (1992) The depolarization block hypothesis of neuroleptic action: implications for the etiology and treatment of schizophrenia. J Neural Transm Suppl 36:91–131 [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. (1983) Intracellular and extracellular electrophysiology of nigral dopaminergic neurons. 1. Identification and characterization. Neuroscience 10:301–315 [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. (1986) Induction of depolarization block in midbrain dopamine neurons by repeated administration of haloperidol: analysis using in vivo intracellular recording. J Pharmacol Exp Ther 238:1092–1100 [PubMed] [Google Scholar]
- Grace AA, Bunney BS, Moore H, Todd CL. (1997) Dopamine-cell depolarization block as a model for the therapeutic actions of antipsychotic drugs. Trends Neurosci 20:31–37 [DOI] [PubMed] [Google Scholar]
- Gurevich EV, Bordelon Y, Shapiro RM, Arnold SE, Gur RE, Joyce JN. (1997) Mesolimbic dopamine D3 receptors and use of antipsychotics in patients with schizophrenia: A postmortem study. Arch Gen Psychiatry 54:225–232 [DOI] [PubMed] [Google Scholar]
- Gyertyán I, Sághy K, Laszy J, Elekes O, Kedves R, Gémesi LI, Pásztor G, Zájer-Balázs M, Kapás M, Agai Csongor E, et al. (2008) Subnanomolar dopamine D3 receptor antagonism coupled to moderate D2 affinity results in favourable antipsychotic-like activity in rodent models: II. behavioural characterisation of RG-15. Naunyn Schmiedebergs Arch Pharmacol 378:529–539 [DOI] [PubMed] [Google Scholar]
- Hwang R, Zai C, Tiwari A, Müller DJ, Arranz MJ, Morris AG, McKenna PJ, Munro J, Potkin SG, Lieberman JA, et al. (2010) Effect of dopamine D3 receptor gene polymorphisms and clozapine treatment response: exploratory analysis of nine polymorphisms and meta-analysis of the Ser9Gly variant. Pharmacogenomics J 10:200–218 [DOI] [PubMed] [Google Scholar]
- Institute of Laboratory Animal Resources (1996) Guide for the Care and Use of Laboratory Animals 7th ed Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council, Washington, DC [Google Scholar]
- Joyce JN, Gurevich EV. (1999) D3 receptors and the actions of neuroleptics in the ventral striatopallidal system of schizophrenics. Ann NY Acad Sci 877:595–613 [DOI] [PubMed] [Google Scholar]
- Kapur S, Mamo D. (2003) Half a century of antipsychotics and still a central role for dopamine D2 receptors. Prog Neuropsychopharmacol Biol Psychiatry 27:1081–1090 [DOI] [PubMed] [Google Scholar]
- Kiss B, Horváth A, Némethy Z, Schmidt E, Laszlovszky I, Bugovics G, Fazekas K, Hornok K, Orosz S, Gyertyán I, et al. (2010) Cariprazine (RGH-188), a dopamine D3 receptor-preferring, D3/D2 dopamine receptor antagonist-partial agonist antipsychotic candidate: in vitro and neurochemical profile. J Pharmacol Exp Ther 333:328–340 [DOI] [PubMed] [Google Scholar]
- Kuzhikandathil EV, Yu W, Oxford GS. (1998) Human dopamine D3 and D2L receptors couple to inward rectifier potassium channels in mammalian cell lines. Mol Cell Neurosci 12:390–402 [DOI] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA. (1987) Dopamine acts on D2 receptors to increase potassium conductance in neurones of the rat substantia nigra zona compacta. J Physiol 392:397–416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li SM, Collins GT, Paul NM, Grundt P, Newman AH, Xu M, Grandy DK, Woods JH, Katz JL. (2010) Yawning and locomotor behavior induced by dopamine receptor agonists in mice and rats. Behav Pharmacol 21:171–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. (2007) Aberrant hippocampal activity underlies the dopamine dysregulation in an animal model of schizophrenia. J Neurosci 27:11424–11430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. (2009) Gestational methylazoxymethanol acetate administration: a developmental disruption model of schizophrenia. Behav Brain Res 204:306–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodge DJ, Grace AA. (2011) Hippocampal dysregulation of dopamine system function and the pathophysiology of schizophrenia. Trends Pharmacol Sci 32:507–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lomazzi M, Slesinger PA, Lüscher C. (2008) Addictive drugs modulate GIRK-channel signaling by regulating RGS proteins. Trends Pharmacol Sci 29:544–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli M, Rudick CN, Hu XT, White FJ. (2006) Exitability of dopamine neurons: modulation and physiological consequences. CNS Neurol Disord Drug Targets 5:79–97 [DOI] [PubMed] [Google Scholar]
- Melis M, Mereu G, Lilliu V, Quartu M, Diana M, Gessa GL. (1998) Haloperidol does not produce dopamine cell depolarization-block in paralyzed, unanesthetized rats. Brain Res 783:127–132 [DOI] [PubMed] [Google Scholar]
- Merchant KM, Gill GS, Harris DW, Huff RM, Eaton MJ, Lookingland K, Lutzke BS, Mccall RB, Piercey MF, Schreur PJ, et al. (1996) Pharmacological characterization of U-101387, a dopamine D4 receptor selective antagonist. J Pharmacol Exp Ther 279:1392–1403 [PubMed] [Google Scholar]
- Mercuri NB, Saiardi A, Bonci A, Picetti R, Calabresi P, Bernardi G, Borrelli E. (1997) Loss of autoreceptor function in dopaminergic neurons from dopamine D2 receptor deficient mice. Neuroscience 79:323–327 [DOI] [PubMed] [Google Scholar]
- Millan MJ, Svenningsson P, Ashby CR, Jr, Hill M, Egeland M, Dekeyne A, Brocco M, Di Cara B, Lejeune F, Thomasson N, et al. (2008) S33138 [N-4–2-[(3aS,9bR)-8-cyano-1,3a,4,9b-tetrahydro[1]-benzopyrano[3,4-c] pyrrol-2(3H)-yl-ethyl]phenylacetamide], a preferential dopamine D3 versus D2 receptor antagonist and potential antipsychotic agent. II. A neurochemical, electrophysiological and behavioral characterization in vivo. J Pharmacol Exp Ther 324:600–611 [DOI] [PubMed] [Google Scholar]
- Miyamoto S, Duncan GE, Marx CE, Lieberman JA. (2005) Treatments for schizophrenia: a critical review of pharmacology and mechanisms of action of antipsychotic drugs. Mol Psychiatry 10:79–104 [DOI] [PubMed] [Google Scholar]
- Moore H, Jentsch JD, Ghajarnia M, Geyer MA, Grace AA. (2006) A neurobehavioral systems analysis of adult rats exposed to methylazoxymethanol acetate on E17: implications for the neuropathology of schizophrenia. Biol Psychiatry 60:253–264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neve KA, Seamans JK, Trantham-Davidson H. (2004) Dopamine receptor signaling. J Recept Signal Transduct Res 24:165–205 [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. (1986) The Rat Brain in Stereotaxic Coordinates. Academic Press, Sydney, Australia [Google Scholar]
- Pucak ML, Grace AA. (1991) Partial dopamine depletions result in an enhanced sensitivity of residual dopamine neurons to apomorphine. Synapse 9:144–155 [DOI] [PubMed] [Google Scholar]
- Rahman Z, Schwarz J, Gold SJ, Zachariou V, Wein MN, Choi KH, Kovoor A, Chen CK, DiLeone RJ, Schwarz SC, et al. (2003) RGS9 modulates dopamine signaling in the basal ganglia. Neuron 38:941–952 [DOI] [PubMed] [Google Scholar]
- Schotte A, Janssen PF, Gommeren W, Luyten WH, Van Gompel P, Lesage AS, De Loore K, Leysen JE. (1996) Risperidone compared with new and reference antipsychotic drugs: in vitro and in vivo receptor binding. Psychopharmacology (Berl) 124:57–73 [DOI] [PubMed] [Google Scholar]
- Seeman P. (1987) Dopamine receptors and the dopamine hypothesis of schizophrenia. Synapse 1:133–152 [DOI] [PubMed] [Google Scholar]
- Seeman P. (2002) Atypical antipsychotics: mechanism of action. Can J Psychiatry 47:27–38 [PubMed] [Google Scholar]
- Seeman P, Chau-Wong M, Tedesco J, Wong K. (1975) Brain receptors for antipsychotic drugs and dopamine: direct binding assays. Proc Natl Acad Sci U S A 72:4376–4380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stilo SA, Murray RM. (2010) The epidemiology of schizophrenia: replacing dogma with knowledge. Dialogues Clin Neurosci 12:305–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svensson K, Carlsson A, Huff RM, Kling-Petersen T, Waters N. (1994) Behavioral and neurochemical data suggest functional differences between dopamine D2 and D3 receptors. Eur J Pharmacol 263:235–243 [DOI] [PubMed] [Google Scholar]
- Szechtman H, Dai H, Mustafa S, Einat H, Sullivan RM. (1994) Effects of dose and interdose interval on locomotor sensitization to the dopamine agonist quinpirole. Pharmacol Biochem Behav 48:921–928 [DOI] [PubMed] [Google Scholar]
- Urraca N, Camarena B, Aguilar A, Fresán A, Apiquián R, Orozco L, Carnevale A, Nicolini H. (2011) Association study of DRD3 gene in schizophrenia in Mexican sib-pairs. Psychiatry Res 190:367–368 [DOI] [PubMed] [Google Scholar]
- Valenti O, Cifelli P, Gill KM, Grace AA. (2011) Antipsychotic drugs rapidly induce dopamine neuron depolarization block in a developmental rat model of schizophrenia. J Neurosci 31:12330–12338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vehof J, Burger H, Wilffert B, Al Hadithy A, Alizadeh BZ, Snieder H, and GROUP investigators (2012) Clinical response to antipsychotic drug treatment: Association study of polymorphisms in six candidate genes. Eur Neuropsychopharmacol 22:625–631 [DOI] [PubMed] [Google Scholar]
- Vrana SL, Kluttz BW, Vrana KE. (1995) Application of quantitative RT-PCR to the analysis of dopamine receptor mRNA levels in rat striatum. Mol Brain Res 34:127–134 [DOI] [PubMed] [Google Scholar]
- Zhang F, Fan H, Xu Y, Zhang K, Huang X, Zhu Y, Sui M, Sun G, Feng K, Xu B, et al. (2011) Converging evidence implicates the dopamine D3 receptor gene in vulnerability to schizophrenia. Am J Med Genet B Neuropsychiatr Genet 156B:613–619 [DOI] [PubMed] [Google Scholar]