

Abstract
Ethanol (EtOH) potentiation of GABAergic neurotransmission in the basolateral amygdala (BLA) may contribute to the acute anxiolytic effects of this drug. Previous studies have shown that BLA pyramidal neurons receive GABAergic input from two distinct sources: local interneurons and a cluster of GABAergic cells termed lateral paracapsular (LPCS) interneurons. It is noteworthy that whereas EtOH enhances local GABAergic synapses via a presynaptic increase in GABA release, EtOH potentiation of LPCS inhibition is mediated via a distinct mechanism that requires adrenoceptor (AR) activation. Here, we sought to further characterize the interaction between the AR system and EtOH enhancement of LPCS GABAergic synapses by using in vitro electrophysiology techniques in male Sprague-Dawley rats. Exogenous norepinephrine (NE) enhanced LPCS-evoked inhibitory postsynaptic currents (eIPSCs) via the activation of β-ARs, because this effect was blocked by propranolol. EtOH potentiation of LPCS eIPSCs was also blocked by propranolol and significantly reduced by NE pretreatment, suggesting that NE and EtOH may enhance LPCS inhibition via a common mechanism. EtOH enhancement of LPCS eIPSCs was significantly reduced by a selective β1-, but not β2- or β3-, AR antagonist, and both EtOH and NE potentiation of LPCS IPSCs was blocked by postsynaptic disruption of cAMP signaling. These data suggest that EtOH enhances LPCS synapses via a postsynaptic β1-AR, cAMP-dependent cascade. Because enhancement of LPCS inhibition can reduce anxiety-like behaviors, these findings shed light on a novel mechanism that may play a role in some of the anxiolytic effects of EtOH that are thought to contribute to the development and progression of alcoholism.
Introduction
Neuronal activity within the basolateral amygdala (BLA) is thought to play an integral role in the regulation of anxiety-like behaviors (LeDoux, 2003). Numerous studies have shown that microinjection of GABAA receptor agonists directly into the BLA can produce anxiolytic effects in a number of behavioral paradigms, whereas microinjection of GABAA receptor antagonists into the BLA can produce increases in some anxiety-like behaviors (for reviews see Menard and Treit, 1999; Engin and Treit, 2008). Moreover, local infusion of a GABAA receptor antagonist into the BLA can block the anxiolytic effects of a systemically administered benzodiazepine (Sanders and Shekhar, 1995), clearly demonstrating the significance of BLA GABAergic tone in the regulation of anxiety behaviors. Ethanol (EtOH) has been shown to enhance GABAergic transmission in many brain regions (Siggins et al., 2005; Breese et al., 2006; Weiner and Valenzuela, 2006), and acute EtOH administration has also been shown to produce anxiolytic effects in a wide range of behavioral paradigms (Tornatzky and Miczek, 1995; Langen et al., 2002; Chandra et al., 2008), suggesting that EtOH potentiation of BLA GABAergic transmission may contribute to the anxiolytic effects of this drug. It is noteworthy that the relief of stress and anxiety brought on by acute EtOH consumption is thought to play an integral role in the development and progression of alcohol use disorders (Koob and Le Moal, 2005; Sinha et al., 2008; Uhart and Wand, 2009; Breese et al., 2011). Therefore, determining the mechanisms through which EtOH enhances GABAergic synapses in the BLA may shed new light on our understanding of the cellular and synaptic events involved in the etiology of alcoholism.
Evidence suggests that the excitability of BLA glutamatergic pyramidal cells, the primary output neurons within this region, is regulated by inhibitory input from at least two distinct GABAergic pathways (Marowsky et al., 2005; Silberman et al., 2008). One pathway arises from the diverse groups of local circuit GABAergic interneurons that provide mainly feedback inhibition within the BLA (Woodruff and Sah, 2007). The second pathway arises from a distinct class of GABAergic cells that reside along the border between the BLA and the external capsule. These lateral paracapsular (LPCS) interneurons are thought to provide cortical feed-forward inhibition to the BLA (Marowsky et al., 2005). Initial studies have demonstrated that EtOH enhances both local and LPCS GABAergic inhibition, albeit via distinct mechanisms. EtOH enhances local GABAergic synapses via a presynaptic facilitation of GABA release, as observed in a number of other brain regions (Weiner and Valenzuela, 2006). In contrast, EtOH enhancement of the LPCS-mediated pathway seems to be mediated via a novel mechanism that may involve postsynaptic adrenergic receptor (AR) activation (Silberman et al., 2008). The BLA receives strong innervation by noradrenergic afferents, which enter the BLA mainly as dense bundles via the external capsule (Fallon et al., 1978). Based on their similar anatomical location, it is likely that norepinephrine (NE) afferents may modulate LPCS inhibition and potentially regulate EtOH potentiation of these synapses. This hypothesis is directly supported by our initial evidence that exogenous NE can enhance LPCS evoked IPSCs (eIPSCs) and antagonism of ARs can block EtOH enhancement of LPCS GABAergic transmission (Silberman et al., 2008). Therefore, the purpose of this study was 3-fold: 1) to further characterize the effect of NE on LPCS GABAergic inhibition in the BLA; 2) to determine how the NE system modulates EtOH potentiation of LPCS synapses; and 3) to identify the AR subtypes responsible for these effects. Our data suggest that NE enhances LPCS GABAergic inhibition in the BLA through a β-AR-dependent mechanism and EtOH potentiation of LPCS GABAergic transmission depends on postsynaptic β1-AR activation.
Materials and Methods
Slice Preparation.
Transverse amygdala slices (400 μm) were prepared from 4- to 6-week-old male Sprague-Dawley rats (Harlan, Indianapolis, IN). Slices were maintained at ambient temperature for at least 1 h in oxygenated artificial cerebrospinal fluid containing 124 mM NaCl, 3.3 mM KCl, 2.4 mM MgCl2, 2.5 mM CaCl2, 1.2 mM KH2PO4, 10 mM d-glucose, and 25 mM NaHCO3, saturated with 95% O2 and 5% CO2.
Electrophysiological Recordings.
Slices were transferred to a recording chamber and superfused with aerated artificial cerebrospinal fluid at 2 ml/min by using a calibrated flowmeter (Gilmont Instruments, Racine, WI). Experiments were performed at ambient temperature because our previous studies have found that this promotes the stability of patch-clamp recordings in brain slices and does not influence EtOH enhancement of GABAA IPSCs (Ariwodola and Weiner, 2004; Silberman et al., 2008). Recording electrodes were prepared from filamented borosilicate glass capillary tubes (inner diameter, 0.86 mm) by using a horizontal micropipette puller (P-97; Sutter Instrument Company, Novato, CA). Whole-cell patch-clamp recordings of eIPSCs were made by using a “standard” filling solution containing 130 mM K-gluconate, 10 mM KCl, 1 mM EGTA, 100 μM CaCl2, 2 mM Mg-ATP, 200 μM Tris-guanosine 5′-triphosphate, and 10 mM HEPES, pH adjusted with KOH, 275 to 280 mOsm. In some experiments, 40 μM Rp-CAMPS or 20 μM PKA inhibitor fragment (6-22) amide (PKA-I) was added to the standard solution to disrupt postsynaptic cAMP signaling. Recordings of spontaneous IPSCs (sIPSCs) were made by using a similar filling solution, exchanging equimolar CsCl for K-gluconate and KCl. In all experiments, 5 mM N-(2,6-dimethyl-phenylcarbamoylmethyl)-triethylammonium chloride (QX-314) was included in the recording solution to block voltage-gated sodium currents and the postsynaptic K+ conductance underlying GABAB IPSCs in the BLA neurons being recorded (Horn et al., 1980; Nathan et al., 1990). Whole-cell patch-clamp recordings were made from BLA pyramidal neurons voltage-clamped at −30 to −40 mV for eIPSCs and −60 to −70 mV for sIPSCs (not corrected for junction potential). The calculated Nernst potential for Cl− using the K-gluconate-based filling solution was −66.3 mV, and the calculated Nernst potential for Cl− using the CsCl-based filling solution was 0.6 mV. Only cells with a stable access resistance of 5 to 20 MΩ were used in these experiments. Whole-cell currents were acquired by using an Axoclamp 2B or Axopatch 200B amplifier, digitized (Digidata1200 or Digidata 1321A; Molecular Devices, Sunnyvale, CA) and analyzed on- and off-line using an IBM (White Plains, NY)-compatible personal computer and pClamp 10.0 software (Molecular Devices).
Pharmacological Isolation of Synaptic Currents.
In all experiments, GABAA IPSCs were pharmacologically isolated by using a mixture of 50 μM 2-amino-5-phosphonovalerate and 20 μM 2,3-dihydroxy-6,7-dinitroquinoxaline to block N-methyl-d-aspartate and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid/kainate receptors, respectively. In most experiments, GABAA IPSCs were evoked every 20 s by electrical stimulation (0.2-ms duration) by using a concentric bipolar stimulating electrode (FHC Inc., Bowdoinham, ME) placed along the external capsule to target LPCS interneurons (see Fig. 1). In these experimental procedures, stimulation intensity was adjusted to evoke responses that were 10 to 20% of maximal currents (typically 80–120 pA). No electrical stimulation was used during sIPSC recordings. sIPSCs were digitized at 5 to 10 kHz in continuous 3-min epochs. Unless otherwise stated, all drugs used were purchased from Sigma (St. Louis, MO). Rp-CAMPS, PKA-I, and the selective AR antagonists 1-[2-((3-carbamoyl-4-hydroxy)phenox y)ethylamino]-3-[4-(1-methyl-4-trifluoromethyl-2-imidazolyl)phenoxy]-2-propanol dihydrochloride (CGP 20712), (±)-erythro-(S*,S*)-1-[2,3-(dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)amino]-2-butanol hydrochloride (ICI 118,551), and 1-(2-ethylphenoxy)-3-[[(1S)-1,2,3,4-tetrahydro-1-naphthalenyl]amino]-(2S)-2-propanol hydrochloride (SR 59230A) were purchased from Tocris Bioscience (Ellisville, MO). Rp-CAMPS and PKA-I were made up as a 100-fold concentrate, and aliquots were added to the pipette recording solution each day before experiments to obtain the final concentration. Other drugs were made up as 100- to 400-fold concentrates and applied to slices via calibrated syringe pumps (Razel Scientific Instruments, Stamford, CT). CGP 20712, ICI 118,551, and SR 59230A were dissolved in water. A 4-M EtOH solution was prepared immediately before each experiment from a 95% stock solution (Aaper Alcohol and Chemical, Shelbyville, KY) kept in a glass storage bottle.
Fig. 1.
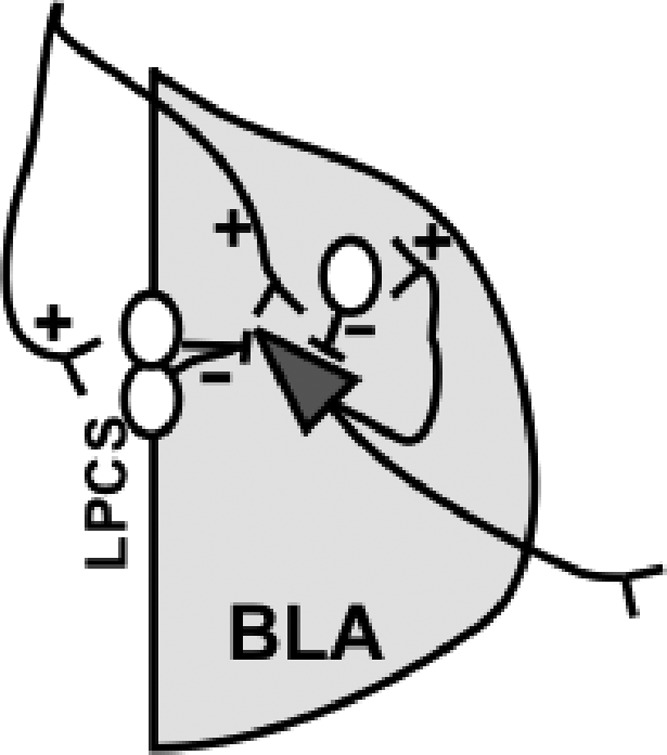
Schematic diagram of the BLA neurocircuitry. Glutamatergic afferents (+) innervate BLA pyramidal neurons (triangle) as well as LPCS GABAergic cells (white circles along the lateral BLA border). LPCS cells provide feed-forward GABAegic inhibitory signaling (dashed line) onto BLA pyramidal neurons. Pyramidal neurons can excite local GABAergic interneurons (white circle within the BLA), which then provide feedback GABAergic inhibition onto BLA pyramidal neurons.
Statistics.
Effects of EtOH and other drugs on evoked IPSCs were quantified as the percentage of change in the area under the curve of synaptic currents relative to the mean of control and washout values. sIPSC events were first identified by using Clampfit event detection software (pClamp 10.0) and then visually inspected to avoid inclusion of spurious responses. sIPSCs in each epoch were then averaged, and the amplitude and decay time of the averaged traces were calculated. Effects of EtOH and other drugs on sIPSCs were quantified as the percentage of change in area, decay, and frequency of spontaneous events relative to the mean of control and washout values. Statistical analyses of drug effects were performed by using the two-tailed Student's paired or unpaired t tests, where applicable, with a minimal level of significance of p < 0.05. Statistical analysis of sIPSC data were performed by using the two-tailed Student's t test for group results, with a minimal level of significance of p < 0.05, and confirmed by the Kolmogorov-Smirnov test on individual cells, with a minimal level of significance of p < 0.01.
Results
NE Pretreatment Significantly Occludes EtOH Potentiation of LPCS Synapses.
Our previous work suggests that exogenous NE can significantly enhance LPCS eIPSCs, an effect that is blocked by a cocktail of α1-, α2-, and β-AR antagonists. Moreover, EtOH potentiation of LPCS, but not local, eIPSCs was also blocked by this same AR antagonist cocktail (Silberman et al., 2008), suggesting that the effect of EtOH at LPCS synapses involves the AR system. To further examine this hypothesis, we first tested the interaction between exogenous NE application and EtOH potentiation of LPCS eIPSCs. Because NE was previously found to have no significant effect on local eIPSCs under our recording conditions, and EtOH potentiation of local synapses was not modulated by the AR antagonist cocktail, we focused mainly on LPCS-mediated GABAergic synaptic transmission in this study.
Confirming our previous reports, bath application of 80 mM EtOH significantly enhanced the area of LPCS GABAA eIPSCs (53.6 ± 10.52% increase from control; n = 10; p < 0.05; Fig. 2, A and D) in a reversible manner. In addition, in accordance with our previous results, bath application of 20 μM NE significantly and reversibly enhanced LPCS eIPSC area (56.0 ± 16.3% increase from control; n = 9; p < 0.05; Fig. 2, B and D Inset). Application of 80 mM EtOH after an 8- to 10-min pretreatment with 20 μM NE resulted in a modest, but significant, further increase in LPCS eIPSC area (75.5 ± 15.0% increase from control; n = 9; p < 0.05; Fig. 2, C and D Inset). It is noteworthy that the potentiating effect of EtOH in the presence of NE was significantly less than that of EtOH alone (15.6 ± 4.1% increase from NE pretreatment levels versus 53.6 ± 10.52% increase from control level when applied alone; p < 0.05; Fig. 2D).
Fig. 2.

Norepinephrine pretreatment significantly occludes EtOH enhancement of LPCS synapses in the BLA. A to C, representative traces and time courses of the potentiating effects of 80 mM EtOH (A), 20 μM NE (B), and EtOH after NE pretreatment (C) on LPCS eIPSCs. Traces are an average of 10 consecutive IPSCs evoked under the given conditions. Lowercase letters (a, b, c) indicate the corresponding time point from which the averaged representative traces were taken. D, bar graph summarizing the mean effect of EtOH alone and EtOH after NE pretreatment on the area under the curve of LPCS evoked GABAA IPSCs. Inset, bar graph summarizing the mean potentiating effects of NE alone and NE+EtOH on the area under the curve of LPCS eIPSCs. Numbers in parentheses indicate the number of cells used in each group. * indicates significant differences from control or pretreatment levels, p < 0.05. # indicates significant differences between groups, p < 0.05.
β-ARs Contribute to Both NE and EtOH Potentiation of LPCS eIPSCs.
Our previous work showed that NE-mediated enhancement of LPCS eIPSCs can be blocked by a cocktail of α1-, α2-, and β-AR antagonists. To better ascertain which AR subtypes were responsible for the effect of NE, we treated BLA slices with modified AR antagonist cocktails, in which individual antagonists were removed, and then examined the effect of NE. We found that treating BLA slices with 10 μM propranolol, a nonselective β-AR antagonist that had no effect on its own (1.7 ± 4.7% increase from control; n = 7), significantly reduced the potentiating effect of 20 μM NE on LPCS eIPSCs by almost 73% (15.3 ± 10.5% increase from control; n = 3; p < 0.05 versus NE alone; Fig. 3). These findings suggest that NE potentiation of LPCS eIPSCs is mediated by β-AR activation.
Fig. 3.

Norepinephrine and EtOH potentiation of LPCS eIPSCs is blocked by propranolol. A, traces from representative cells showing the effects of 20 μM propranolol and 80 mM EtOH on LPCS eIPSCs under control conditions and after pretreatment with 10 μM propranolol. Traces are an average of 10 consecutive IPSCs evoked under the given conditions. B, bar graph summarizing the mean effect of 20 μM norepineprine, 10 μM propranolol, and 80 mM EtOH on the area under the curve of LPCS eIPSCs and the effect of propranolol on norepinephrine and EtOH potentiation of these currents. Note that propranolol has no effect on its own but significantly inhibits both norepinephrine and EtOH enhancement of LPCS eIPSCs both alone and together. * indicates significant differences from control, p < 0.05. # indicates significant differences between compared groups, p < 0.05.
Because NE pretreatment significantly occluded EtOH potentiation of LPCS eIPSCs and NE potentiation of LPCS inhibition was blocked by propranolol, we next tested the hypothesis that β-ARs may also regulate the effects of EtOH on LPCS eIPSCs. To test this hypothesis, BLA slices were treated with 10 μM propranolol for 8 to 10 min, followed by application of 80 mM EtOH for 8 to 10 min. Propranolol treatment significantly reduced EtOH potentiation of LPCS eIPSCs by almost 92% (4.5 ± 6.0% increase from control; n = 7; p < 0.05 versus EtOH alone; Fig. 3), suggesting that β-AR activation is required for EtOH potentiation of LPCS synapses.
β-ARs Do Not Alter the Facilitatory Effects of EtOH on sIPSCs.
We have previously shown that EtOH enhances the frequency of sIPSCs and tetrodotoxin-resistant miniature IPSCs recorded from BLA pyramidal neurons without altering the kinetics of these events, consistent with a presynaptic facilitation of GABA release (Silberman et al., 2009). In addition, the facilitatory effect of EtOH on sIPSC frequency was significantly enhanced by a GABAB receptor antagonist and blocked by pretreatment with a GABAB receptor agonist, consistent with the effects of these modulators on EtOH enhancement of local, but not LPCS, eIPSCs (Silberman et al., 2009). Taken together, these data suggest that, at least under our recording conditions, sIPSCs may only reflect the activity of local interneurons. To provide additional empirical support for this hypothesis, we next tested the effect of propranolol pretreatment on EtOH potentiation of sIPSCs. Because propranolol blocks the effect of EtOH on LPCS, but not local, eIPSCs, and sIPSCs seem to reflect only the activity of local interneurons, we hypothesized that propranolol pretreatment would have no effect on EtOH potentiation of sIPSC frequency. As reported previously, bath application of 80 mM EtOH for 8 to 10 min significantly increased sIPSC frequency (95.1 ± 27.3%; n = 8; p < 0.05; Fig. 4), with no significant change in the amplitude or decay of sIPSC events (17.6 ± 12.8 and 0.5 ± 5.7% increases from control, respectively; n = 8). Bath application of 10 μM propranolol for 8 to 10 min had no effect on the frequency, amplitude, or decay of sIPSCs (14.2 ± 23.7, 9.4 ± 15.7, and 8.5 ± 3.8% increases from control, respectively; n = 5). After propranolol treatment, application of 80 mM EtOH still resulted in a significant increase in sIPSC frequency (84.2 ± 23.3% increase from propranolol levels; n = 6; p < 0.05; Fig. 4) with no significant change in the amplitude or decay of these events (19.3 ± 9.3 and 11.4 ± 6.5% increases, respectively; n = 6; Fig. 4). No significant difference in the magnitude of EtOH enhancement of sIPSC frequency was noted between control and propranolol pretreatment conditions, further suggesting that LPCS synapses do not significantly contribute to sIPSC activity under our recording conditions.
Fig. 4.

EtOH potentiation of spontaneous IPSC frequency is not blocked by propranolol. A, representative traces showing the potentiating effects of 80 mM EtOH on the frequency of sIPSCs under control conditions and after pretreatment with 10 μM propranolol. B, cumulative probability histograms from the recordings illustrated in A demonstrating a significant effect of EtOH on the frequency (p < 0.01), but not amplitude of sIPSCs under control conditions and after propranolol pretreatment, as determined by Kolmogorov-Smirnov tests. C, bar graph summarizing the mean effect of EtOH, propranolol, and EtOH + propranolol on sIPSC frequency, amplitude, and decay. * indicates significant differences from control, p < 0.05.
β1-AR Activation Is Required for EtOH Potentiation of LPCS eIPSCs.
Research has shown that functional β1-, β2-, and β3-ARs may be present in the BLA (Abraham et al., 2008; Silberman et al., 2010). Because propranolol is a nonselective β-AR antagonist that can potentially block all three β-AR subtypes at the concentration used in this study (Hoffmann et al., 2004) we next sought to determine which β-AR subtypes might be responsible for the potentiating effects of EtOH on LPCS eIPSCs. To that end, we tested the effect of various β-AR subtype-selective antagonists on EtOH potentiation of LPCS eIPSCs. Experiments examining the effect of 80 mM EtOH alone were interspersed throughout these studies as positive controls. In these interspersed control experiments, 80 mM EtOH significantly potentiated the area of LPCS eIPSCs (59.4 ± 9.5% increase from baseline; p < 0.05; n = 9; Fig. 5), which was not significantly different from the effect EtOH described earlier (Fig. 1).
Fig. 5.

EtOH potentiation of LPCS eIPSCs is reduced by a β1-, but not a β2- or β3-, adrenergic receptor antagonist. Bar graph summarizes the mean effect of 80 mM EtOH and selective β-AR subtype antagonists [CGP20712A (β1), ICI118,551 (β2), or SR59230A (β3)] on the area under the curve of evoked LPCS GABAA IPSCs both alone and during antagonist pretreatment + EtOH conditions. Between four and nine cells were used in each group. * indicates significant differences from control, p < 0.05. # indicates a significant difference between groups, p < 0.05. NS indicates no significant difference between groups. Dashed line indicates normalized control level.
Bath application of a selective β1-AR antagonist, 1 μM CGP 20712A, for 8 to 10 min had no effect on LPCS eIPSC area (2.9 ± 20.1% increase from control; n = 4; Fig. 5). However, CGP 20712A pretreatment significantly attenuated EtOH potentiation of LPCS eIPSCs (21.1 ± 8.8% increase from CGP 20712A levels; n = 7; Fig. 5), suggesting that a significant portion of the facilitatory effects of EtOH at LPCS GABAergic synapses involves the activation of β1-ARs. Similar experiments were performed with either a selective β2-AR antagonist (1 μM ICI118,551) or a selective β3-AR antagonist (4 μM SR59230A). Bath application of either antagonist alone had no significant effect on LPCS eIPSC area (9.4 ± 6.8 and 4.3 ± 6.1% decreases from baseline, n = 6 and 7, respectively) and also had no significant effect on EtOH potentiation of LPCS eIPSCs (42.8 ± 9.8 and 88.0 ± 22.1% increase from pretreatment levels, p < 0.05 versus pretreatment levels, n = 8 and 6, respectively; Fig. 5).
Synaptic Locus of EtOH Potentiation of LPCS Synapses.
Together, the above findings suggest that β-AR activation is required for NE and EtOH potentiation of LPCS eIPSCs, with the β1-AR subtype playing a predominant role in mediating EtOH facilitation. We next sought to examine the synaptic locus underlying these effects, because prior studies suggested that EtOH enhancement of LPCS IPSCs did not depend on an increase in terminal GABA release (Silberman et al., 2008, 2009). Because sIPSCs, commonly used to determine presynaptic vs. postsynaptic loci of drug effects, reflect only the activity of local GABAergic synapses under our recording conditions, an alternate method was devised to further determine the synaptic locus underlying NE and EtOH facilitation of LPCS synapses. Because β-AR activation is necessary for the potentiating effects of both NE and EtOH, and β-AR activation is typically associated with the initiation of cAMP signaling cascades (Wallukat, 2002), we postulated that if these effects were mediated by a postsynaptic mechanism, then disruption of postsynaptic cAMP signaling may block NE and EtOH enhancement of LPCS eIPSCs. To that end, we added 40 μM Rp-CAMPS to the standard patch pipette recording solution to disrupt cAMP signaling only in the pyramidal neuron being recorded. After a 15-min period to ensure adequate dialysis of Rp-CAMPS, LPCS eIPSCs were evoked as in previous experiments. In comparison with interspersed control experiments using a standard recording solution, in which 20 μM NE significantly potentiated LPCS eIPSC area (50.3 ± 15.1% increase from baseline; n = 5; p < 0.05), inclusion of Rp-CAMPS into the standard recording solution effectively blocked the potentiating effect of NE (10.7 ± 6.0% increase from baseline; n = 8; Rp-CAMPS versus interspersed control; p < 0.05; Fig. 6).
Fig. 6.

Intracellular dialysis of Rp-CAMPS blocks norepinephrine potentiation of LPCS IPSCs. A, time courses from representative cells illustrating the effect of 20 μM NE on LPCS eIPSCs recorded with our standard patch electrode filling solution or one containing 40 μM Rp-CAMPS. B, bar graph summarizing the mean effect of 20 μM NE on LPCS eIPSCs recorded with a standard or Rp-CAMPS solution. Numbers in parentheses indicate number of cells used in each group. * indicates significant differences from baseline levels, p < 0.05. # indicates significant differences between groups, p < 0.05. Dashed line indicates normalized baseline level.
We next conducted a similar series of experiments to assess the effect of cAMP signaling on EtOH potentiation of LPCS eIPSCs. EtOH (80 mM) significantly potentiated the area of LPCS eIPSCs using a standard recording solution (82.3 ± 18.2% increase from baseline; p < 0.05; n = 9). In comparison, inclusion of Rp-CAMPS into the recording solution significantly reduced the potentiating effect of 80 mM EtOH (16.2 ± 7.4% increase from baseline; n = 7; Rp-CAMPS versus interspersed control; p < 0.05; Fig. 7, A and C). There were no significant differences in the effects of 20 μM NE or 80 mM EtOH during the interspersed control experiments compared with the effects of these modulators described in earlier experiments throughout this article.
Fig. 7.

Disruption of postsynaptic cAMP signaling attenuates the EtOH potentiation of LPCS eIPSCs. A, time courses from representative cells showing the effect of 80 mM EtOH on the area of LPCS eIPSCs recorded with our standard intracellular solution or one containing 40 μM Rp-CAMPS. Traces above the graph are an average of 10 consecutive IPSCs evoked under the given conditions. B, time courses from a representative cell showing the effect of 80 mM EtOH on the area of LPCS IPSCs recorded with our standard intracellular solution or one containing the membrane-impermeant PKA inhibitor PKA-I (20 μM). Traces above the graph are an average of 10 consecutive IPSCs evoked under the given conditions. C, bar graph summarizing the mean effect of EtOH on the area of LPCS eIPSCs recorded with the standard, Rp-CAMPS-containing, and PKA-I-containing recording solutions. Numbers in parentheses indicate number of cells used in each group. * indicates significant differences from baseline levels, p < 0.05. # indicates significant differences between groups, p < 0.05. Dashed line indicates normalized baseline level.
As an additional control, we examined the effect of Rp-CAMPS on EtOH potentiation of locally evoked GABAA IPSCs. As predicted from our prior work demonstrating that EtOH enhances local eIPSCs via a presynaptic, AR-independent mechanism, intracellular infusion of Rp-CAMPS had no significant effect on EtOH potentiation of local eIPSCs (standard recording solution: 62.5 ± 12.5%, n = 7; standard recording solution + 40 μM Rp-CAMPS: 82.3 ± 12.1%, n = 7, data not shown).
Finally, although Rp-CAMPS was present only in the patch pipette, this compound can diffuse across cellular membranes. To control for the possibility that this drug may have inhibited EtOH potentiation of LPCS IPSCs by diffusion to presynaptic loci, we repeated the same experimental protocol with a membrane-impermeant peptide inhibitor of PKA (PKA-I). Inclusion of 20 μM PKA-I in the recording solution also blocked EtOH enhancement of LPCS IPSCS (19.0 ± 7.0%; n = 9) (Fig. 7, B and C).
Discussion
The results of this study confirm and expand on our previous findings that EtOH and NE enhance LPCS eIPSCs in the rat BLA (Silberman et al., 2008). NE pretreatment significantly occluded EtOH potentiation of LPCS inhibition, and the potentiating effects of EtOH and NE on LPCS eIPSCs were significantly decreased by the nonselective β-AR antagonist propranolol. Furthermore, EtOH facilitation of LPCS synapses required β1-AR activation, and disruption of postsynaptic cAMP signaling significantly reduced the potentiating effects of both EtOH and NE on LPCS eIPSCs. In contrast, propranolol did not block EtOH-induced increases in sIPSC frequency, a measure of local, and not LPCS, GABAergic interneuron activity under our recording conditions (Silberman et al., 2009), and postsynaptic inhibition of cAMP signaling had no effect on EtOH enhancement of local eIPSCs, suggesting dissociable effects of EtOH at distinct inhibitory synapses onto BLA pyramidal neurons.
The finding that NE pretreatment attenuated EtOH potentiation of LPCS eIPSCs is consistent with the hypothesis that NE and EtOH may facilitate LPCS GABAergic inhibition through a common pathway. This idea is further supported by our data showing that NE and EtOH potentiation of LPCS eIPSCs is significantly reduced by propranolol and postsynaptic disruption of cAMP signaling. These observations are consistent with the hypothesis that acute EtOH exposure may increase NE release in the BLA. The BLA receives dense NE projections from the locus coeruleus and other brain stem structures (Asan, 1998), and at least some of these projections target the external capsule region that was stimulated to evoke LPCS IPSCs (Fuxe et al., 2003). Although no studies, to date, have directly examined the acute effects of EtOH on NE release in brain slice preparations, there is considerable evidence from in vivo studies that plasma NE levels are increased after EtOH exposure in both rats (Patterson-Buckendahl et al., 2005) and humans (Patkar et al., 2004) and acute EtOH treatments stimulate NE circuitry in the central nervous system (Lee et al., 2011). In addition, studies with dopamine (which cannot synthesize NE) β-hydroxylase knockout mice show that NE plays an integral role in EtOH preference and consumption (Weinshenker et al., 2000), and inhibition of β-ARs significantly reduces dependence-induced increases in EtOH self-administration in rats (Gilpin and Koob, 2010). Further studies will be needed to discern how β-AR activation regulates EtOH actions at LPCS synapses; however, our findings suggest that these synapses may represent an important locus that contributes to some of the behaviorally relevant EtOH-NE interactions that have been reported in vivo.
To ascertain which β-AR subtype might be required for EtOH enhancement of LPCS eIPSCs, we tested the effect of β-AR subtype-selective antagonists. Based on prior findings by us and others, demonstrating that β3-AR activation in the BLA selectively enhances LPCS eIPSCs (Silberman et al., 2010) and systemic administration (Stemmelin et al., 2008) or intra-BLA infusion (Silberman et al., 2010) of a β3-AR agonist decreases anxiety-like behaviors, we hypothesized that this AR subtype might mediate EtOH actions at LPCS synapses. We were surprised to find that β1-ARs play the predominant role in mediating EtOH enhancement of LPCS synapses, whereas blockade of β3-ARs had no discernible effect on EtOH enhancement of these responses. Although the density of β-AR subtypes in the BLA has yet to be determined, β2- and β3-ARs probably are expressed at lower levels than β1-ARs in this brain region (Rainbow et al., 1984; Abraham et al., 2008). In addition, β1-ARs are thought to have a higher affinity for NE than β2- or β3-ARs (Molinoff, 1984; Wallukat, 2002). Therefore, if EtOH does indeed act by increasing NE release, it seems plausible that endogenous NE may act predominantly on β1-ARs in the BLA. However, it is worth noting that repeated EtOH exposure as well as stress can profoundly influence GABAergic circuitry and AR function in the BLA (Braga et al., 2004; Buffalari and Grace, 2009; Diaz et al., 2011). Therefore, it will be important in future studies to more thoroughly characterize the contribution of all three β-AR subtypes in regulating in vitro and in vivo effects of EtOH, particularly after chronic exposure and withdrawal from this drug.
To more thoroughly characterize the mechanisms of EtOH action at local and LPCS inhibitory synapses in the BLA, we first tested the effect of propranolol pretreatment on EtOH modulation of sIPSCs. We have previously shown that EtOH significantly increases sIPSC frequency and GABAB receptor modulators can alter this effect in a manner similar to the effect of these modulators on EtOH potentiation of local IPSCs (Silberman et al., 2009). These findings, coupled with the putative distal dendritic origin of LPCS synapses, suggest that sIPSCs largely reflect local interneuron activity in the BLA, at least under our recording conditions. However, if LPCS cells significantly contribute as a source of sIPSCs, then propranolol pretreatment should reduce EtOH enhancement of sIPSC frequency. Our data reveal that EtOH significantly enhanced the frequency of sIPSCs and this effect was not attenuated by propranolol pretreatment. These findings provide further evidence that LPCS interneurons are not a significant source of sIPSCs in brain slice experiments. In addition, we found that selective disruption of postsynaptic cAMP signaling, by inclusion of Rp-CAMPS or a membrane-impermeant peptide inhibitor of PKA (PKA-I) into the patch pipette recording solution, significantly reduced NE and EtOH potentiation of LPCS eIPSCs. In contrast, disruption of cAMP signaling had no effect on EtOH enhancement of local eIPSCs. Together, these findings provide further evidence that EtOH potentiation of LPCS eIPSCs is mediated by a postsynaptic mechanism that requires activation of β1-ARs and the subsequent initiation of cAMP signaling cascades.
It is somewhat surprising that exogenous NE alone could enhance LPCS eIPSCs. NE is thought to play an integral role in the physiological response to stress and anxiety (Morilak et al., 2005). It is generally thought that NE released during times of stress acts to enhance neuronal excitation in target brain regions (Bremner et al., 1996). Because BLA activation plays an integral role in the regulation of anxiety behaviors, and NE is thought to increase central nervous system excitation in response to anxious stimuli, it has been generally accepted that NE increases BLA excitability. Indeed, activation of β1-β2 ARs can increase excitatory synaptic transmission in the BLA (Abraham et al., 2008; Silberman et al., 2010). Such an effect could serve as a cellular mechanism for enhanced anxiety-like behaviors in the face of external stressors. However, in this study we found that NE significantly enhances LPCS-mediated IPSCs, an effect that would be expected to decrease BLA excitability. Indeed, selective enhancement of LPCS GABAergic transmission in vivo is sufficient to decrease experimental measures of anxiety-like behavior (Silberman et al., 2010). It is also worth noting that NE can enhance BLA GABA release via an α1-AR mediated mechanism that increases the firing rate of local interneurons (Braga et al., 2004; Kaneko et al., 2008). In addition, in vivo electrophysiological evidence suggests that NE can both enhance and depress the excitability of BLA neurons (Buffalari and Grace, 2007; Chen and Sara, 2007).
One emerging hypothesis from this collective body of evidence may be that NE modulation of emotional behaviors, like anxiety, arises via an equilibrated balance between the excitatory and inhibitory effects of this neurotransmitter on BLA excitability. It is noteworthy that chronic stressors have been shown to reduce the ability of NE to increase BLA GABAergic tone (Braga et al., 2004) and decrease BLA excitability (Buffalari and Grace, 2009), whereas stress may actually sensitize excitatory effects of NE in this brain region (Buffalari and Grace, 2009). These data suggest that some of the inhibitory effects of NE on BLA excitability may become uncoupled after chronic stress, whereas NE influences on BLA excitatory transmission may remain unchanged or even grow more pronounced. These findings raise the intriguing possibility that an imbalance between the normal excitatory and inhibitory actions of NE within the BLA may contribute to the etiology of anxiety-related disorders by tipping the scales toward increased BLA excitability. Furthermore, numerous reports have shown that withdrawal from chronic intermittent EtOH exposure results in pronounced increases in anxiety-like behaviors (for review see Kliethermes, 2005; McCool et al., 2010). In addition, cycles of chronic stress can substitute for cycles of chronic intermittent EtOH exposure (Knapp et al., 2007). Therefore, it also seems possible that chronic EtOH exposure may disrupt inhibitory effects of NE in the BLA and thus contribute to the progressive increase in anxiety associated with alcohol abuse disorders. In fact, propranolol significantly attenuates dependence-induced increases in EtOH self-administration in rats while having no effect on drinking in nondependent animals (Gilpin and Koob, 2010).
In summary, our findings suggest that EtOH potentiation of LPCS GABAergic transmission may be mediated via a postsynaptic mechanism that requires the activation of β1-ARs and the subsequent initiation of cAMP signaling cascades. Because enhancement of LPCS inhibition is sufficient to decrease at least some measures of anxiety-like behavior, these findings identify a novel mechanism that probably contributes to some of the anxiolytic effects of EtOH that are thought to play an integral role in the etiology of alcohol use disorders.
This work was supported by the National Institutes of Health National Institute on Alcohol Abuse and Alcoholism [Grants AA 17531, AA 17056, AA 17039].
Article, publication date, and citation information can be found at http://jpet.aspetjournals.org.
- BLA
- basolateral amygdala
- EtOH
- ethanol
- AR
- adrenoceptor
- LPCS
- lateral paracapsular
- NE
- norepinephrine
- IPSC
- inhibitory postsynaptic current
- eIPSC
- evoked IPSC
- sIPSC
- spontaneous IPSC
- GABA
- γ-aminobutyric acid
- PKA
- protein kinase A
- PKA-I
- PKA inhibitor fragment (6-22) amide
- CGP 20712
- 1-[2-((3-carbamoyl-4-hydroxy)phenox y)ethylamino]-3-[4-(1-methyl-4-trifluoromethyl-2-imidazolyl)phenoxy]-2-propanol dihydrochloride
- ICI 118,551
- (±)-erythro-(S*,S*)-1-[2,3-(dihydro-7-methyl-1H-inden-4-yl)oxy]-3-[(1-methylethyl)amino]-2-butanol hydrochloride
- Rp-CAMPS
- adenosine-3′,5′-cyclic phosphorothioate
- SR 59230A
- 1-(2-ethylphenoxy)-3-[[(1S)-1,2,3,4-tetrahydro-1-naphthalenyl]amino]-(2S)-2-propanol hydrochloride
- QX-314
- N-(2,6-dimethyl-phenylcarbamoylmethyl)-triethylammonium chloride.
Authorship Contributions
Participated in research design: Silberman and Weiner.
Conducted experiments: Silberman and Ariwodola.
Performed data analysis: Silberman and Weiner.
Wrote or contributed to the writing of the manuscript: Silberman and Weiner.
References
- Abraham PA, Xing G, Zhang L, Yu EZ, Post R, Gamble EH, Li H. (2008) β1- and β2-adrenoceptor induced synaptic facilitation in rat basolateral amygdala. Brain Res 1209:65–73 [DOI] [PubMed] [Google Scholar]
- Ariwodola OJ, Weiner JL. (2004) Ethanol potentiation of GABAergic synaptic transmission may be self-limiting: role of presynaptic GABAB receptors. J Neurosci 24:10679–10686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asan E. (1998) The catecholaminergic innervation of the rat amygdala. Adv Anat Embryol Cell Biol 142:1–118 [DOI] [PubMed] [Google Scholar]
- Braga MF, Aroniadou-Anderjaska V, Manion ST, Hough CJ, Li H. (2004) Stress impairs α1A adrenoceptor-mediated noradrenergic facilitation of GABAergic transmission in the basolateral amygdala. Neuropsychopharmacology 29:45–58 [DOI] [PubMed] [Google Scholar]
- Breese GR, Criswell HE, Carta M, Dodson PD, Hanchar HJ, Khisti RT, Mameli M, Ming Z, Morrow AL, Olsen RW, et al. (2006) Basis of the gabamimetic profile of ethanol. Alcohol Clin Exp Res 30:731–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breese GR, Sinha R, Heilig M. (2011) Chronic alcohol neuroadaptation and stress contribute to susceptibility for alcohol craving and relapse. Pharmacol Ther 129:149–171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremner JD, Krystal JH, Southwick SM, Charney DS. (1996) Noradrenergic mechanisms in stress and anxiety: I. Preclinical studies. Synapse 23:28–38 [DOI] [PubMed] [Google Scholar]
- Buffalari DM, Grace AA. (2007) Noradrenergic modulation of basolateral amygdala neuronal activity: opposing influences of α-2 and β receptor activation. J Neurosci 27:12358–12366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buffalari DM, Grace AA. (2009) Chronic cold stress increases excitatory effects of norepinephrine on spontaneous and evoked activity of basolateral amygdala neurons. Int J Neuropsychopharmacol 12:95–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandra D, Werner DF, Liang J, Suryanarayanan A, Harrison NL, Spigelman I, Olsen RW, Homanics GE. (2008) Normal acute behavioral responses to moderate/high dose ethanol in GABAA receptor α4 subunit knockout mice. Alcohol Clin Exp Res 32:10–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen FJ, Sara SJ. (2007) Locus coeruleus activation by foot shock or electrical stimulation inhibits amygdala neurons. Neuroscience 144:472–481 [DOI] [PubMed] [Google Scholar]
- Diaz MR, Christian DT, Anderson NJ, McCool BA. (2011) Chronic ethanol and withdrawal differentially modulate lateral/basolateral amygdala paracapsular and local GABAergic synapses. J Pharmacol Exp Ther 337:162–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engin E, Treit D. (2008) The effects of intra-cerebral drug infusions on animals' unconditioned fear reactions: a systematic review. Prog Neuropsychopharmacol Biol Psychiatry 32:1399–1419 [DOI] [PubMed] [Google Scholar]
- Fallon JH, Koziell DA, Moore RY. (1978) Catecholamine innervation of the basal forebrain. II. Amygdala, suprarhinal cortex and entorhinal cortex. J Comp Neurol 180:509–532 [DOI] [PubMed] [Google Scholar]
- Fuxe K, Jacobsen KX, Höistad M, Tinner B, Jansson A, Staines WA, Agnati LF. (2003) The dopamine D1 receptor-rich main and paracapsular intercalated nerve cell groups of the rat amygdala: relationship to the dopamine innervation. Neuroscience 119:733–746 [DOI] [PubMed] [Google Scholar]
- Gilpin NW, Koob GF. (2010) Effects of β-adrenoceptor antagonists on alcohol drinking by alcohol-dependent rats. Psychopharmacology (Berl) 212:431–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann C, Leitz MR, Oberdorf-Maass S, Lohse MJ, Klotz KN. (2004) Comparative pharmacology of human β-adrenergic receptor subtypes–characterization of stably transfected receptors in CHO cells. Naunyn Schmiedebergs Arch Pharmacol 369:151–159 [DOI] [PubMed] [Google Scholar]
- Horn R, Brodwick MS, Dickey WD. (1980) Asymmetry of the acetylcholine channel revealed by quaternary anesthetics. Science 210:205–207 [DOI] [PubMed] [Google Scholar]
- Kaneko K, Tamamaki N, Owada H, Kakizaki T, Kume N, Totsuka M, Yamamoto T, Yawo H, Yagi T, Obata K, et al. (2008) Noradrenergic excitation of a subpopulation of GABAergic cells in the basolateral amygdala via both activation of nonselective cationic conductance and suppression of resting K+ conductance: a study using glutamate decarboxylase 67-green fluorescent protein knock-in mice. Neuroscience 157:781–797 [DOI] [PubMed] [Google Scholar]
- Kliethermes CL. (2005) Anxiety-like behaviors following chronic ethanol exposure. Neurosci Biobehav Rev 28:837–850 [DOI] [PubMed] [Google Scholar]
- Knapp DJ, Overstreet DH, Breese GR. (2007) Baclofen blocks expression and sensitization of anxiety-like behavior in an animal model of repeated stress and ethanol withdrawal. Alcohol Clin Exp Res 31:582–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Le Moal M. (2005) Plasticity of reward neurocircuitry and the ‘dark side’ of drug addiction. Nat Neurosci 8:1442–1444 [DOI] [PubMed] [Google Scholar]
- Langen B, Dietze S, Fink H. (2002) Acute effect of ethanol on anxiety and 5-HT in the prefrontal cortex of rats. Alcohol 27:135–141 [DOI] [PubMed] [Google Scholar]
- LeDoux J. (2003) The emotional brain, fear, and the amygdala. Cell Mol Neurobiol 23:727–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Craddock Z, Rivier C. (2011) Brain stem catecholamines circuitry: activation by alcohol and role in the hypothalamic-pituitary-adrenal response to this drug. J Neuroendocrinol 23:531–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marowsky A, Yanagawa Y, Obata K, Vogt KE. (2005) A specialized subclass of interneurons mediates dopaminergic facilitation of amygdala function. Neuron 48:1025–1037 [DOI] [PubMed] [Google Scholar]
- McCool BA, Christian DT, Diaz MR, Läck AK. (2010) Glutamate plasticity in the drunken amygdala: the making of an anxious synapse. Int Rev Neurobiol 91:205–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menard J, Treit D. (1999) Effects of centrally administered anxiolytic compounds in animal models of anxiety. Neurosci Biobehav Rev 23:591–613 [DOI] [PubMed] [Google Scholar]
- Molinoff PB. (1984) α- and β-Adrenergic receptor subtypes properties, distribution and regulation. Drugs 28 (Suppl 2):1–15 [DOI] [PubMed] [Google Scholar]
- Morilak DA, Barrera G, Echevarria DJ, Garcia AS, Hernandez A, Ma S, Petre CO. (2005) Role of brain norepinephrine in the behavioral response to stress. Prog Neuropsychopharmacol Biol Psychiatry 29:1214–1224 [DOI] [PubMed] [Google Scholar]
- Nathan T, Jensen MS, Lambert JD. (1990) The slow inhibitory postsynaptic potential in rat hippocampal CA1 neurones is blocked by intracellular injection of QX-314. Neurosci Lett 110:309–313 [DOI] [PubMed] [Google Scholar]
- Patkar AA, Marsden CA, Naik PC, Kendall DA, Gopalakrishnan R, Vergare MJ, Weinstein SP. (2004) Differences in peripheral noradrenergic function among actively drinking and abstinent alcohol-dependent individuals. Am J Addict 13:225–235 [DOI] [PubMed] [Google Scholar]
- Patterson-Buckendahl P, Kubovcakova L, Krizanova O, Pohorecky LA, Kvetnansky R. (2005) Ethanol consumption increases rat stress hormones and adrenomedullary gene expression. Alcohol 37:157–166 [DOI] [PubMed] [Google Scholar]
- Rainbow TC, Parsons B, Wolfe BB. (1984) Quantitative autoradiography of β1- and β2-adrenergic receptors in rat brain. Proc Natl Acad Sci U S A 81:1585–1589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders SK, Shekhar A. (1995) Anxiolytic effects of chlordiazepoxide blocked by injection of GABAA and benzodiazepine receptor antagonists in the region of the anterior basolateral amygdala of rats. Biol Psychiatry 37:473–476 [DOI] [PubMed] [Google Scholar]
- Siggins GR, Roberto M, Nie Z. (2005) The tipsy terminal: presynaptic effects of ethanol. Pharmacol Ther 107:80–98 [DOI] [PubMed] [Google Scholar]
- Silberman Y, Ariwodola OJ, Chappell AM, Yorgason JT, Weiner JL. (2010) Lateral paracapsular GABAergic synapses in the basolateral amygdala contribute to the anxiolytic effects of β3 adrenoceptor activation. Neuropsychopharmacology 35:1886–1896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberman Y, Ariwodola OJ, Weiner JL. (2009) Differential effects of GABAB autoreceptor activation on ethanol potentiation of local and lateral paracapsular GABAergic synapses in the rat basolateral amygdala. Neuropharmacology 56:886–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberman Y, Shi L, Brunso-Bechtold JK, Weiner JL. (2008) Distinct mechanisms of ethanol potentiation of local and paracapsular GABAergic synapses in the rat basolateral amygdala. J Pharmacol Exp Ther 324:251–260 [DOI] [PubMed] [Google Scholar]
- Sinha R, Fox HC, Hong KA, Bergquist K, Bhagwagar Z, Siedlarz KM. (2008) Enhanced negative emotion and alcohol craving, and altered physiological responses following stress and cue exposure in alcohol dependent individuals. Neuropsychopharmacology 34:1198–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmelin J, Cohen C, Terranova JP, Lopez-Grancha M, Pichat P, Bergis O, Decobert M, Santucci V, Françon D, Alonso R, et al. (2008) Stimulation of the β3-adrenoceptor as a novel treatment strategy for anxiety and depressive disorders. Neuropsychopharmacology 33:574–587 [DOI] [PubMed] [Google Scholar]
- Tornatzky W, Miczek KA. (1995) Alcohol, anxiolytics and social stress in rats. Psychopharmacology (Berl) 121:135–144 [DOI] [PubMed] [Google Scholar]
- Uhart M, Wand GS. (2009) Stress, alcohol and drug interaction: an update of human research. Addict Biol 14:43–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallukat G. (2002) The β-adrenergic receptors. Herz 27:683–690 [DOI] [PubMed] [Google Scholar]
- Weiner JL, Valenzuela CF. (2006) Ethanol modulation of GABAergic transmission: the view from the slice. Pharmacol Ther 111:533–554 [DOI] [PubMed] [Google Scholar]
- Weinshenker D, Rust NC, Miller NS, Palmiter RD. (2000) Ethanol-associated behaviors of mice lacking norepinephrine. J Neurosci 20:3157–3164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodruff AR, Sah P. (2007) Networks of parvalbumin-positive interneurons in the basolateral amygdala. J Neurosci 27:553–563 [DOI] [PMC free article] [PubMed] [Google Scholar]