

Abstract
Drug discovery programs increasingly are focusing on allosteric modulators as a means to modify the activity of G protein-coupled receptor (GPCR) targets. Allosteric binding sites are topographically distinct from the endogenous ligand (orthosteric) binding site, which allows for co-occupation of a single receptor with the endogenous ligand and an allosteric modulator that can alter receptor pharmacological characteristics. Negative allosteric modulators (NAMs) inhibit and positive allosteric modulators (PAMs) enhance the affinity and/or efficacy of orthosteric agonists. Established approaches for estimation of affinity and efficacy values for orthosteric ligands are not appropriate for allosteric modulators, and this presents challenges for fully understanding the actions of novel modulators of GPCRs. Metabotropic glutamate receptor 5 (mGlu5) is a family C GPCR for which a large array of allosteric modulators have been identified. We took advantage of the many tools for probing allosteric sites on mGlu5 to validate an operational model of allosterism that allows quantitative estimation of modulator affinity and cooperativity values. Affinity estimates derived from functional assays fit well with affinities measured in radioligand binding experiments for both PAMs and NAMs with diverse chemical scaffolds and varying degrees of cooperativity. We observed modulation bias for PAMs when we compared mGlu5-mediated Ca2+ mobilization and extracellular signal-regulated kinase 1/2 phosphorylation data. Furthermore, we used this model to quantify the effects of mutations that reduce binding or potentiation by PAMs. This model can be applied to PAM and NAM potency curves in combination with maximal fold-shift data to derive reliable estimates of modulator affinities.
Introduction
The metabotropic glutamate receptors (mGlus) are G protein-coupled receptors for the neurotransmitter glutamate that play important roles in regulating a range of major circuits in the central nervous system. The mGlus include eight subtypes (Niswender and Conn, 2010). Historically, it has been difficult to develop ligands with strong subtype selectivity among the mGlus because of the high level of sequence conservation of the orthosteric (i.e., glutamate) binding site; this has led to the search for compounds that interact with these receptors at “allosteric” sites that are topographically distinct from the orthosteric glutamate binding site. Such compounds, which are referred to as allosteric modulators, can affect the affinity and/or efficacy of orthosteric ligands (a property referred to as cooperativity), which allows them to modulate endogenous agonist activity. Modulators that inhibit orthosteric ligand binding and/or activity are negative allosteric modulators (NAMs), whereas those that enhance binding and/or activity are positive allosteric modulators (PAMs). A third category, i.e., silent (or neutral) allosteric modulators, includes compounds that bind but do not modulate responses to orthosteric agonists.
Allosteric modulators offer a number of theoretical advantages over their competitive counterparts in addition to improvements in receptor selectivity (Melancon et al., 2012). For modulators that possess no intrinsic efficacy, there is the potential for spatial and temporal modulation of receptor activity. This is an especially important consideration for potential therapeutic agents for the central nervous system, where “fine-tuning” of neurotransmission is likely to yield better therapeutic outcomes than sustained blockade or activation by an orthosteric ligand. Furthermore, the cooperativity between the two sites is saturable, such that allosteric modulators have a “ceiling level” to their effects and therefore may have greater therapeutic indices.
Efforts to develop allosteric modulators for one mGlu subtype, mGlu5, have been especially successful, and a broad range of allosteric modulators and radioligands for allosteric sites have been developed for this mGlu subtype. Since the first identification of 6-methyl-2-(phenylazo)-3-pyridinol (SIB-1757) and (E)-2-methyl-6-(2-phenylethenyl)pyridine (SIB-1893) and the structural analogs 2-methyl-6-(phenylethynyl)pyridine (MPEP) and 3-[(2-methyl-1,3-thiazol-4-yl) ethynyl]pyridine (MTEP) as selective mGlu5 NAMs (Gasparini et al., 1999; Varney et al., 1999; Cosford et al., 2003b), a diverse array of allosteric modulators have been identified, including pure PAMs, PAMs with agonist activity, weak and full NAMs, and silent allosteric modulators (O'Brien et al., 2004; Kinney et al., 2005; Rodriguez et al., 2005, 2009, 2010; Chen et al., 2007, 2008; Liu et al., 2008; Noetzel et al., 2012). mGlu5 PAMs have potential utility for treatment of cognitive disorders and schizophrenia, whereas NAMs are being pursued for treatment of fragile X syndrome, depression, anxiety, l-DOPA-induced dyskinesia, and gastroesophogeal reflux disorder (Niswender and Conn, 2010).
In allosteric modulator drug discovery programs, potency and maximal effects are routinely used to drive iterative medicinal chemistry efforts and to select compounds for further characterization. Commonly, NAMs are assessed for inhibition of a submaximal (EC80) concentration of orthosteric agonist, whereas PAMs are assayed for potentiation of a low agonist concentration (EC20) (Melancon et al., 2012). However, PAM or NAM potencies represent the combined contributions of modulator affinity and cooperativity with agonist and are dependent on the agonist concentration present (Gregory et al., 2010). Furthermore, allosteric modulator structure-activity relationships (SARs) are often steep, and small changes in a molecule may result in complete loss of activity, which may be related to changes in modulator cooperativity and/or affinity (Wood et al., 2011). Finally, allosteric ligands are prone to “molecular switches,” in which subtle changes to a NAM scaffold yield a PAM (or vice versa), an effect that is related to cooperativity changes (Wood et al., 2011). Validated approaches for quantitative analyses of the pharmacological characteristics of allosteric modulators are needed to delineate cooperativity versus affinity. We have taken advantage of the large array of tools to study allosteric sites on mGlu5 to validate the use of the operational model of allosterism (Leach et al., 2007). Our data suggest that this quantitative model provides a robust method to determine cooperativity and affinity estimates from modulator potency curves. Derivation of affinity estimates from functional assays should be especially useful for assessment of the affinities of novel allosteric modulators that act at sites for which radioligands have yet to be developed.
Materials and Methods
Materials.
Dulbecco's modified Eagle's medium (DMEM), fetal bovine serum, and antibiotics were purchased from Invitrogen (Carlsbad, CA). 3H-labeled 3-methoxy-5-(pyridin-2-ylethynyl)pyridine (methoxy-PEPy) (76.3 Ci/mmol) was custom-synthesized by PerkinElmer Life and Analytical Sciences (Waltham, MA). 3-Cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl) benzamide (CDPPB), 4-nitro-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide (VU29), N-{4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl}-2-hydroxybenzamide (CPPHA), 4-butoxy-N-(2,4-difluorophenyl)benzamide (VU0357121), 2-{4-[2-(benzyloxy)acetyl]piperazin-1-yl}benzonitrile (VU0364289), 1-{[4-(2-phenylethynyl)phenyl]carbonyl}piperidin-4-ol (VU0092273), N-cyclobutyl-6-[(3-fluorophenyl)ethynyl]nicotinamide hydrochloride (VU0360172), 3-fluoro-5-[3-(pyridin-2-yl)-1,2,4-oxadiazol-5-yl]benzonitrile (VU0285683), 2-(1,3-benzoxazol-2-ylamino)-4-(4-fluorophenyl)pyrimidine-5-carbonitrile (VU0366058), 2-[2-(3-methoxyphenyl) ethynyl]-5-methylpyridine (M-5MPEP), N-(3-chloro-2-fluorophenyl)-3-cyano-5-fluorobenzamide (VU0366248), and N-(3-chloro-4-fluorophenyl)-3-cyano-5-fluorobenzamide (VU0366249) were all synthesized in-house by using previously reported methods (Kinney et al., 2005; Rodriguez et al., 2005, 2010; Chen et al., 2007, 2008; Felts et al., 2010; Hammond et al., 2010; Zhou et al., 2010; Mueller et al., 2012). [5-[(3-Fluorophenyl)ethynyl]pyridin-2-yl](3-hydroxyazetidin-1-yl)methanone (VU0405398), N-tert-butyl-5-[(3-fluorophenyl)ethynyl]picolinamide (VU0405386), and N-tert-butyl-6-[2-(3-fluorophenyl)ethynyl]pyridine-3-carboxamide (VU0415051) were synthesized in-house by using the methods described in the supplemental materials. Unless stated otherwise, all other reagents were purchased from Sigma-Aldrich (St. Louis, MO) and were of analytical grade.
Cell Culture and Mutagenesis.
Mutations were introduced into the wild-type rat mGlu5 gene in pCI:Neo by using site-directed mutagenesis (Quikchange II; Agilent Technologies, Santa Clara, CA) and were verified through sequencing. Wild-type and mutant rat mGlu5 receptor constructs were transfected into HEK293A cells by using Fugene6 (Promega, Madison, WI) as the transfection reagent. Stable polyclonal cell lines were derived for rat mGlu5 mutant constructs by maintaining the cells at subconfluence in the presence of 1 mg/ml G418 (Geneticin; Mediatech, Herndon, VA) for at least four passages. Stably transfected cell lines were subsequently maintained at 37°C in complete DMEM supplemented with 10% fetal bovine serum, 2 mM l-glutamine, 20 mM HEPES, 0.1 mM nonessential amino acids, 1 mM sodium pyruvate, antibiotic/antimycotic solution (Invitrogen), and 500 μg/ml G418, in a humidified incubator containing 5% CO2/95% O2.
Intracellular Ca2+ Mobilization.
The day before assays, mGlu5-expressing HEK293A cells were seeded in poly-d-lysine-coated, black-wall, clear-bottom, 96-well plates at 50,000 cells/well, in assay medium (DMEM supplemented with 10% dialyzed fetal bovine serum, 20 mM HEPES, and 1 mM sodium pyruvate). On the day of the assays, the cell-permeant Ca2+ indicator dye Fluo-4 (Invitrogen) was used to assay receptor-mediated Ca2+ mobilization as described previously (Hammond et al., 2010), with a Flexstation II microplate reader (Molecular Devices, Sunnyvale, CA). A five-point smoothing function was applied to the raw fluorescence traces, and basal fluorescence values for individual wells were determined during the first 20 s. The peak increase in fluorescence over basal levels was determined before normalization to the maximal peak response elicited by glutamate.
ERK1/2 Phosphorylation.
Receptor-mediated ERK1/2 phosphorylation was determined by using an AlphaScreen-based ERK SureFire kit (PerkinElmer Life and Analytical Sciences; TGR Biosciences, Thebarton, Australia). mGlu5-expressing HEK293A cells were plated in poly-d-lysine-coated, clear, 96-well plates at a density of 40,000 cells/well, in assay medium, 16 to 24 h before assays. The medium was aspirated, cells were washed once with serum-free medium (DMEM supplemented with 16 mM HEPES), and then cells were serum-starved for a minimum of 6 h before assays. Serum-free medium was exchanged for fresh medium 20 min before exposure of the cells to modulators and/or glutamate. At room temperature, the time course for mGlu5-mediated ERK phosphorylation was characterized by an initial peak at 7 to 8 min and a return to baseline levels by 15 min (data not shown). For interaction experiments with allosteric modulators, cells were exposed to allosteric modulator or vehicle for 1 min before stimulation with glutamate for 7 min. Assays were terminated through the aspiration of ligand-containing medium and the addition of 50 μl of lysis buffer per well. After agitation for 10 min, 4 μl of lysate was transferred to a white 384-well plate (Costar; Corning Life Sciences, Tewksbury, MA). Under low-light conditions, 7 μl of reaction buffer mixture [containing activation buffer/reaction buffer at 1:6 and donor/acceptor beads at 1:250 (v/v)] were added to each well. After a 90-min incubation at 37°C, the AlphaScreen signal was measured by using a H4 synergy reader (BioTek Instruments, Winooski, VT), with standard AlphaScreen settings. Data are expressed as fold increases over basal levels of phosphorylated ERK.
Radioligand Binding.
Membranes were prepared from HEK293A cells expressing rat mGlu5 and mutants as follows. Cells were harvested through trypsinization and were pelleted through centrifugation for 3 min at 300g. Cell pellets were resuspended in ice-cold homogenization buffer (50 mM Tris-HCl, 10 mM EDTA, 0.9% NaCl, pH 7.4) and were homogenized with three 10-s bursts with a Tekmar TP-18/10S1 homogenizer (Teledyne Tekmar, Cincinnati, OH), which were separated by 30-s periods on ice. Cell fractions were separated through centrifugation for 10 min at 1000g. The supernatant was centrifuged for 1 h at 30,000g, and the resulting pellet was resuspended in ice-cold Ca2+ assay buffer. For saturation binding experiments, membranes (20–50 μg/well) were incubated for 1 h at room temperature with a range of [3H]methoxy-PEPy concentrations (0.5–60 nM) in binding buffer (50 mM Tris-HCl, 0.9% NaCl, pH 7.4), with shaking. MPEP (10 μM) was used to determine nonspecific binding. For inhibition binding experiments, membranes were incubated for 1 h at room temperature with ∼2 nM [3H]methoxy-PEPy and a range of concentrations of test ligands (100 pM to 100 μM), in the absence or presence of 1 mM glutamate (added simultaneously), in Ca2+ assay buffer with 1% dimethylsulfoxide (final concentration), with shaking. Binding assays were terminated with rapid filtration through GF/B Unifilter plates (PerkinElmer Life and Analytical Sciences), by using a 96-well plate harvester (Brandel Inc., Gaithersburg, MD), and three washes with ice-cold binding buffer to separate bound from free radioligand. Plates were allowed to dry overnight before the addition of MicroScint 20 scintillation cocktail (40 μl/well; PerkinElmer Life and Analytical Sciences). Radioactivity was counted after at least 2 h of incubation, by using a TopCount scintillation counter (PerkinElmer Life and Analytical Sciences).
Data Analyses.
All computerized nonlinear regression analyses were performed by using Prism 5.01 (GraphPad Software Inc., San Diego, CA). Data sets on the inhibition of [3H]methoxy-PEPy binding were fitted to a one-site inhibition binding model, and estimates of inhibitor dissociation constants (KI) were derived by using the Cheng-Prusoff equation for competitive ligands (Cheng and Prusoff, 1973). For ligands that did not fully displace the radioligand, the following version of the allosteric ternary complex model (Lazareno and Birdsall, 1995) was fitted to the inhibition binding data:
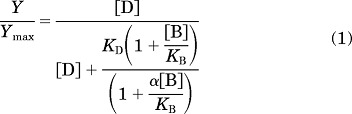 |
where Y/Ymax is the fractional specific binding, [D] is the radioligand concentration, [B] is the molar concentration of the allosteric modulator, KD is the radioligand equilibrium dissociation constant, KB is the allosteric modulator equilibrium dissociation constant, and α denotes the cooperativity factor; α values of >1 describe positive cooperativity, α values of <1 (but >0) denote negative cooperativity, and α values of 1 denote neutral cooperativity.
Shifts of glutamate concentration-response curves with allosteric modulators were globally fitted to an operational model of allosterism (Leach et al., 2007)
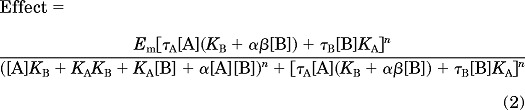 |
where [A] is the molar concentration of the orthosteric agonist glutamate, KA is the equilibrium dissociation constant of the orthosteric agonist glutamate, and KB and [B] are as described above. Affinity modulation is governed by the cooperativity factor α, and efficacy modulation is governed by β. The parameters τA and τB are related to the ability of orthosteric and allosteric ligands, respectively, to yield receptor activation. Em and n denote the maximal possible system response and the transducer function that links occupancy to response, respectively. Unless otherwise stated, all parameters were derived from global fitting of glutamate concentration-response curves in the absence and presence of allosteric modulators.
In the absence of discernible allosteric agonism, it was assumed that τB was equal to 0, such that eq. 2 could be simplified to
 |
Theoretical PAM or NAM concentration-response curves in the presence of different concentrations of agonist were derived from progressive fold shifts of an agonist concentration-response curve simulated by using eq. 3. For these simulations, the following parameters were held constant for both NAMs and PAMs: pKA = 6, pKB = 7, τA = 10, logα = 0, n = 2, Em = 100, and basal = 0. Modulator concentrations ranged from 100 pM to 30 μM. For PAMs, cooperativity was set to logβ = 1; for NAMs, β was assumed to approach 0, such that logβ = −100.
A simplified version of this operational model was applied to estimate a composite cooperativity parameter (αβ) for PAMs (Leach et al., 2007),
where basal denotes the baseline (ligand-independent) level of the system response and all other parameters are as described above.
Allosteric modulator and agonist concentration-response curves were fitted to a four-parameter logistic equation to determine potency estimates,
where bottom and top are the lower and upper plateau levels, respectively, of the concentration-response curve, nH is the Hill coefficient that describes the steepness of the curve, [A] is the molar concentration of the orthosteric agonist glutamate, and EC50 is the molar concentration of modulator required to generate a response halfway between the top and bottom values.
Allosteric modulator concentration-response curves also were fitted to the following version of the operational model of allosterism (eq. 7) in concert with a control glutamate concentration-response curve (eq. 6), to estimate modulator affinity and cooperativity values.
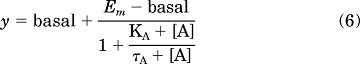 |
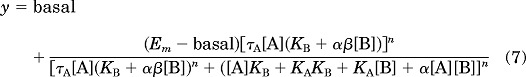 |
where all parameters are as described above. KA, τA, Em, and basal values were shared across analyses; for modulator curves, [A] was held constant at the molar agonist concentration (EC20 for PAMs or EC80 for NAMs) present in the assay.
All affinity, cooperativity, and potency parameters were estimated as logarithms and are expressed as mean ± S.E.M. Statistical analyses were performed as indicated, by using one-way analysis of variance with Dunnett's post hoc test for comparisons with control values or Tukey's post hoc test for multiple comparisons.
Results
Operational Model of Allosterism.
Allosteric modulators routinely are screened for their potencies in either inhibiting the response to a submaximal concentration of orthosteric agonist or potentiating the response to a low concentration of agonist. However, allosteric modulator potency is dependent on the concentration of orthosteric agonist used (Fig. 1, A and B). Analysis of the available literature findings for mGlu5 NAMs revealed that potencies and affinities were well correlated and potencies for the majority of mGlu5 NAMs were within 10-fold of their affinity estimates (Fig. 1C). In contrast, the potencies of mGlu5 PAMs were often higher than their estimated affinities at the prototypical allosteric site labeled with [3H]MPEP and [3H]methoxy-PEPy (de Paulis et al., 2006; Liu et al., 2008; Vanejevs et al., 2008; Rodriguez et al., 2010; Sams et al., 2011; Zou et al., 2011). Only 15 of 61 reported mGlu5 PAMs had potency values within 10-fold of their affinity estimates (Fig. 1D). These discrepancies are likely attributable to the influence of cooperativity with glutamate, because modulator potency reflects the combined contributions of modulator affinity and cooperativity. In addition, some of these investigated PAMs do not bind at the labeled site in a completely competitive manner, and affinity estimates from equations that assume competitive interactions in equilibrium competition binding analyses might not reflect the actual affinities (Chen et al., 2008; Hammond et al., 2010). The vast majority of NAMs that have been investigated are structurally related to the NAM radioligands ([3H]MPEP and [3H]methoxy-PEPy) used to measure affinity, whereas the PAMs belong to a broader range of structural classes.
Fig. 1.

Influences of agonist concentration, modulator affinity, and cooperativity on allosteric modulator potency. A and B, simulations of the effects of different agonist concentrations on the potencies of a negative allosteric modulator (A) and a positive allosteric modulator (B). EC100**, agonist concentration 30-fold in excess of that required to elicit a maximal response. C and D, literature findings on mGlu5 NAM potency and affinity estimates (C) and mGlu5 PAM potency and affinity estimates (D) (Huang et al., 2004; Poon et al., 2004; Roppe et al., 2004; Chua et al., 2005; Tehrani et al., 2005; de Paulis et al., 2006; Kulkarni et al., 2006, 2009; Jaeschke et al., 2007; Milbank et al., 2007; Liu et al., 2008; Vanejevs et al., 2008; Felts et al., 2009, 2010; Galambos et al., 2010; Rodriguez et al., 2010; Wágner et al., 2010; Zhang et al., 2010; Alagille et al., 2011; Gilbert et al., 2011; Lindemann et al., 2011; Sams et al., 2011; Weiss et al., 2011; Zou et al., 2011; Mueller et al., 2012). Dashed lines, unity; dotted lines, potency and affinity within 3-fold of each other; solid lines, 10-fold difference.
Given the discrepancies in measures of potencies versus affinity estimates, we were interested in using mGlu5 as a model system to validate the use of the operational model of allosterism originally developed by Leach et al. (2007) to quantify allosteric interactions. In this model, KA is the equilibrium dissociation constant of the orthosteric agonist and KB is the equilibrium dissociation constant of the allosteric modulator. The coupling efficiencies of the orthosteric agonist and an allosteric modulator are described by τA and τB, respectively. Modulation of affinity when the receptor is simultaneously bound is represented by the cooperativity factor α, whereas efficacy modulation is governed by a second cooperativity factor, β.
Estimation of Allosteric Modulator Affinities for mGlu5 with Radioligand Binding Assays.
A total of 16 mGlu5 allosteric modulators were chosen for validation of affinity and cooperativity estimates (Fig. 2). These compounds represent 11 different chemical scaffolds and a range of allosteric modulator activities, including pure PAMs, PAMs with agonist activity, full NAMs, and weak NAMs (also referred to as partial antagonists or NAMs with low negative cooperativity) (Gasparini et al., 1999; Kinney et al., 2005; Rodriguez et al., 2005, 2010; Chen et al., 2007, 2008; Felts et al., 2010; Hammond et al., 2010; Zhou et al., 2010; Mueller et al., 2012). Inhibition of [3H]methoxy-PEPy binding to HEK293A cell membranes stably expressing wild-type rat mGlu5 showed that affinity estimates for these allosteric modulators spanned >3 orders of magnitude (Fig. 3; Table 1). A number of modulators did not fully displace [3H]methoxy-PEPy binding. VU0357121 was reported previously not to displace [3H]methoxy-PEPy binding significantly (Hammond et al., 2010). However, we used a different cell background and assay conditions (low mGlu5 expression levels, 1% dimethylsulfoxide, and Ca2+ assay buffer, compared with high mGlu5 expression levels and a Tris-based buffer) and observed ∼35% displacement at 30 μM. This is consistent with an allosteric interaction between [3H]methoxy-PEPy and VU0357121. Therefore, the inhibition curve for VU0357121 was fitted to the allosteric ternary complex model (eq. 1) to estimate affinity and cooperativity between these two allosteric sites. CPPHA, VU29, and VU0360172 also were fitted to this model. In the case of VU0364289 and VU0366249, inhibition was consistent with competitive binding limited by solubility. Inhibition of binding for a representative compound from each chemical scaffold was also assessed in the presence of a saturating concentration of glutamate (1 mM); consistent with previous reports (Cosford et al., 2003a; Bradley et al., 2011), glutamate had no effect on specific [3H]methoxy-PEPy binding. The presence of glutamate had no effect on the apparent affinity of these modulators or on the cooperativity (logα) between the apparently noncompetitive PAMs and [3H]methoxy-PEPy (Table 1).
Fig. 2.

Structures of mGlu5 allosteric modulators included in this study. VU0366248 and VU0366249 were originally reported as compounds 41 and 42, respectively (Felts et al., 2010).
Fig. 3.

Inhibition of [3H]methoxy-PEPy binding to wild-type mGlu5-expressing HEK293A cell membranes. Data represent the mean ± S.E.M. from at least three independent determinations.
TABLE 1.
Summary of affinity and cooperativity estimates for mGlu5 allosteric modulators determined from [3H]methoxy-PEPy inhibition binding assays
Data represent the mean ± S.E.M. from at least three independent determinations.
| Modulator | pKI (logα)a |
|
|---|---|---|
| Without Glutamate | With 1 mM Glutamateb | |
| CDPPB | 6.65 ± 0.11 | N.D. |
| VU29 | 6.69 ± 0.10 (−0.95 ± 0.01) | 7.20 ± 0.21 (−0.96 ± 0.05) |
| CPPHA | 5.52 ± 0.06 (−0.64 ± 0.02) | 5.92 ± 0.21 (−0.62 ± 0.01) |
| VU0357121 | 5.65 ± 0.19 (−0.24 ± 0.01) | 5.78 ± 0.20 (−0.35 ± 0.04) |
| VU0364289 | 4.82 ± 0.15 | 5.26 ± 0.21 |
| VU0092273 | 5.97 ± 0.09 | N.D. |
| VU0360172 | 6.55 ± 0.03 (−1.21 ± 0.17) | 6.75 ± 0.07 (−1.39 ± 0.09) |
| VU0405398 | 6.60 ± 0.14 | N.D. |
| VU0415051 | 6.88 ± 0.04 | N.D. |
| VU0405386 | 7.98 ± 0.05 | 8.31 ± 0.14 |
| MPEP | 8.00 ± 0.04 | 8.26 ± 0.10 |
| M-5MPEP | 6.89 ± 0.16 | N.D. |
| VU0285683 | 7.68 ± 0.04 | 7.60 ± 0.07 |
| VU0366248 | 6.18 ± 0.06 | 6.39 ± 0.10 |
| VU0366249 | 5.55 ± 0.08 | N.D. |
| VU0366058 | 6.92 ± 0.06 | 6.83 ± 0.13 |
pKI, negative logarithm of the equilibrium dissociation constant determined through nonlinear regression analysis of [3H]methoxy-PEPy binding data; logα, logarithm of the cooperativity factor for the interaction between the indicated allosteric modulator and [3H]methoxy-PEPy; N.D., not determined.
Modulators that did not fully displace [3H]methoxy-PEPy were fitted with an allosteric model to derive affinity and cooperativity estimates (eq. 1).
Allosteric modulator affinity estimates were not significantly different (P < 0.05) in the presence of 1 mM glutamate, with one-way analysis of variance and Tukey's post hoc test.
Estimation of Allosteric Modulator Affinities for mGlu5 with Receptor-Mediated Ca2+ Mobilization Assays.
Shifts in the glutamate concentration-response curves for intracellular Ca2+ mobilization were assessed for all 16 modulators (Supplemental Fig. 1) (Noetzel et al., 2012), and data for a representative pure PAM, i.e., CPPHA (Fig. 4A), a PAM with agonist activity, i.e., CDPPB (Fig. 4B), a full NAM, i.e., MPEP (Fig. 4C), and two weak NAMs, i.e., M-5MPEP and VU0366249 (Fig. 4, D and E), are shown. To derive estimates of allosteric modulator affinity and cooperativity values, data sets were globally fitted to an operational model of allosterism (eq. 2) in which the affinity of glutamate (pKA) was held constant, on the basis of the value from a previous study in which glutamate affinity was determined by using the orthosteric radioligand [3H]quisqualate (Mutel et al., 2000). For analysis of interactions between glutamate and PAMs, a composite cooperativity parameter (logαβ) that incorporated both affinity and efficacy modulation was derived (Table 2). However, to allow for changes in the maximal response to glutamate, an effect driven by β, it was necessary to consider these two aspects of cooperativity independently. Constraining α to be neutral between glutamate and each of the PAMs yielded similar estimates of PAM affinity (pKB), compared with determinations of affinity on the basis of composite cooperativity logαβ (Fig. 4F; Table 2). Therefore, the interactions between glutamate and PAMs at mGlu5 in this assay could be accommodated solely with efficacy cooperativity (logβ) (Fig. 4G). A composite cooperativity parameter (logαβ) could not be derived for NAMs, because changes in agonist Emax were mediated solely by β. Instead, α either was derived with β or was constrained to equal 1; NAMs showed either neutral α cooperativity or low positive α values (Table 3). The assumption that α = 1 yielded similar estimates of pKB for all NAMs and logβ values for weak NAMs (Fig. 4, F and G). Strong correlations between modulator affinity estimates derived from these functional interaction assays and pKI values derived from experiments measuring inhibition of [3H]methoxy-PEPy binding were observed (Fig. 4H). In general, the functional estimates of modulator affinity were within 3-fold of values derived from binding data. Likewise, allosteric modulator affinity estimates for both PAMs and NAMs determined in a cell line expressing high levels of mGlu5 showed good agreement with values determined in a cell line expressing low levels (Fig. 5F).
Fig. 4.

Allosteric modulation of glutamate concentration-response curves for Ca2+ mobilization in the low-level mGlu5-expressing HEK293A cell line. A and B, in the low-level, wild-type mGlu5-expressing HEK293A cell line, CPPHA (A) and CDPPB (B) induced leftward shifts in the glutamate concentration-response curve for intracellular Ca2+ mobilization, with no change in the maximal response. C to E, MPEP (C), VU0366249 (D), and M-5MPEP (E) inhibited glutamate-stimulated intracellular Ca2+ mobilization. F, modulator affinity estimates that were calculated by determining the composite cooperativity parameter αβ for PAMs (●) and allowing α to float for NAMs (○) (x-axis) or assuming α = 1 and calculating logβ (y-axis) were compared. G, for calculated cooperativity estimates, the assumption of α = 1 had no effect on the apparent cooperativity between glutamate and PAMs (●) and weak NAMs (○). H, affinity estimates for PAMs (●) and NAMs (○) determined from radioligand binding assays (x-axis) and Ca2+ mobilization assays (y-axis) in low-level mGlu5-expressing HEK293A cells were compared. Dashed line, unity. Data represent the mean ± S.E.M. from at least three independent determinations. Error bars not shown lie within the dimensions of the symbols.
TABLE 2.
Summary of operational model parameters for positive allosteric modulation of glutamate-mediated intracellular Ca2+ mobilization in HEK cells expressing either low or high levels of mGlu5
Interactions between glutamate and allosteric modulators were quantified by using eq. 2 (for α = 1), eq. 3 (for τB = 0), or eq. 4 (when αβ was determined); glutamate affinity was held constant at a previously reported value (logKA = −6.155) (Mutel et al., 2000). The presence of allosteric modulators did not affect estimates of glutamate coupling efficiency (logτA), transduction coefficient (n), maximal system response (Em), or basal response levels; the assumption of α = 1 also had no effect on these estimates (one-way analysis of variance) (Supplemental Table 1). In the low-level mGlu5-expressing cell line, with αβ being determined, logτA = 0.75 ± 0.03, n = 2.54 ± 0.12, Em = 102.5 ± 1.0, and basal = 1.0 ± 0.17; with α = 1, logτA = 0.80 ± 0.02, n = 2.66 ± 0.13, Em = 103.5 ± 1.07, and basal = 1.15 ± 0.19. In the high-level mGlu5-expressing cell line, logτA = 0.37 ± 0.02, n = 2.84 ± 0.16, Em = 118.3 ± 2.3, and basal = 0.81 ± 0.23. Data represent the mean ± S.E.M. from at least three independent determinations.
| CDPPB | VU29 | CPPHA | VU0357121 | VU0364289 | VU0092273 | VU0360172 | VU0405398 | VU0415051 | VU0405386 | |
|---|---|---|---|---|---|---|---|---|---|---|
| Low-level mGlu5, determining αβ | ||||||||||
| pKB | 6.31 ± 0.10 | 6.20 ± 0.09 | 6.03 ± 0.10 | 6.46 ± 0.09 | 5.18 ± 0.13 | 6.45 ± 0.11 | 6.98 ± 0.05 | 7.30 ± 0.24 | 7.45 ± 0.24 | 8.17 ± 0.24 |
| logαβ | 0.78 ± 0.16 | 0.81 ± 0.08 | 0.65 ± 0.09 | 0.42 ± 0.05 | 0.97 ± 0.08 | 0.49 ± 0.04 | 0.51 ± 0.02 | 0.33 ± 0.04 | 0.36 ± 0.06 | 0.60 ± 0.11 |
| logτB | −0.42 ± 0.23 | −0.67 ± 0.06 | N.A. | N.A. | N.A. | N.A. | N.A. | N.A. | N.A. | N.A. |
| Low-level mGlu5, assuming α = 1 | ||||||||||
| pKB | 6.38 ± 0.10 | 6.23 ± 0.09 | 6.06 ± 0.10 | 6.48 ± 0.17 | 5.22 ± 0.16 | 6.46 ± 0.12 | 7.00 ± 0.05 | 7.29 ± 0.26 | 7.46 ± 0.20 | 8.18 ± 0.28 |
| logβ | 0.66 ± 0.15 | 0.71 ± 0.08 | 0.56 ± 0.07 | 0.37 ± 0.05 | 0.90 ± 0.09 | 0.44 ± 0.05 | 0.47 ± 0.02 | 0.30 ± 0.04 | 0.30 ± 0.05 | 0.54 ± 0.07 |
| logτB | −0.32 ± 0.15 | −0.64 ± 0.07 | N.A. | N.A. | N.A. | N.A. | N.A. | N.A. | N.A. | N.A. |
| High-level mGlu5, assuming α = 1 | ||||||||||
| pKB | 6.99 ± 0.04 | 6.59 ± 0.13 | 5.55 ± 0.14 | 6.00 ± 0.15 | 5.83 ± 0.20 | 6.68 ± 0.06 | 7.07 ± 0.14 | 7.29 ± 0.20 | 8.02 ± 0.25 | 8.00 ± 0.29 |
| logβ | 0.29 ± 0.05 | 0.54 ± 0.13 | 0.58 ± 0.07 | 0.92 ± 0.01 | 0.78 ± 0.05 | 0.77 ± 0.11 | 0.88 ± 0.15 | 0.87 ± 0.06* | 0.74 ± 0.19 | 1.10 ± 0.16* |
| logτB | 0.00 ± 0.01 | −0.05 ± 0.06 | −0.14 ± 0.15 | N.A. | −0.38 ± 0.03 | −0.23 ± 0.04 | −0.13 ± 0.07 | −0.69 ± 0.15 | −0.19 ± 0.11 | −0.14 ± 0.03 |
pKB, negative logarithm of the allosteric modulator equilibrium dissociation constant; logαβ, logarithm of the composite cooperativity factor, αβ, which reflects both affinity and efficacy modulation, as quantified with eq. 4; logτB, logarithm of the coupling efficiency of the allosteric modulator; logβ, logarithm of the efficacy cooperativity factor, β, as quantified with either eq. 2 or 3 as appropriate; N.A., not applicable because of lack of appreciable agonism by the allosteric modulator.
Significantly different (P < 0.05) from the value for the modulator determined in low-level mGlu5-expressing HEK293A cells for Ca2+ mobilization, with one-way analysis of variance and Tukey's post hoc test.
TABLE 3.
Summary of operational model parameters for negative allosteric modulation of glutamate-mediated intracellular Ca2+ mobilization in HEK cells expressing low and high mGlu5 levels
Parameters are defined and quantification was performed as for Table 2. Estimates of glutamate coupling efficiency (logτA), transduction coefficient (n), maximal system response (Em), and basal response levels are presented in Supplemental Table 2. Data represent the mean ± S.E.M. from at least three independent determinations.
| Cell Line/Parameter | MPEP | M-5MPEP | VU0285683 | VU0366248 | VU0366249 | VU0366058 |
|---|---|---|---|---|---|---|
| Low-level mGlu5 α unconstrained | ||||||
| pKB | 8.44 ± 0.12 | 6.99 ± 0.14 | 7.40 ± 0.17 | 6.62 ± 0.13 | 6.36 ± 0.08 | 6.71 ± 0.20 |
| logβ | −100a | −0.75 ± 0.05 | −100a | −0.83 ± 0.10 | −0.59 ± 0.08 | −100a |
| logα | 0.12 ± 0.06 | 0.25 ± 0.07 | 0.20 ± 0.11 | −0.04 ± 0.14 | 0.25 ± 0.12 | 0.41 ± 0.14 |
| Low-level mGlu5 α = 1 | ||||||
| pKB | 8.55 ± 0.09 | 7.04 ± 0.15 | 7.59 ± 0.10 | 6.63 ± 0.08 | 6.35 ± 0.08 | 7.09 ± 0.10 |
| logβ | −100a | −0.72 ± 0.05 | −100a | −0.90 ± 0.09 | −0.52 ± 0.10 | −100a |
| High-level mGlu5 | ||||||
| pKB | 8.53 ± 0.03 | 7.14 ± 0.08 | 7.55 ± 0.06 | 6.72 ± 0.07 | 6.58 ± 0.04 | 6.69 ± 0.07 |
| logβ | −100a | −100a | −100a | −100a | −0.48 ± 0.04 | −100a |
For NAMs that abolished the response to glutamate, β = 0 was assumed and therefore logβ was constrained to −100.
Fig. 5.

Allosteric modulation of glutamate concentration-response curves for Ca2+ mobilization in the high-level mGlu5-expressing HEK293A cell line. A and B, in the high-level mGlu5-expressing HEK293A cell line, increased agonist activity was seen for CPPHA (A) and CDPPB (B), as well as induction of a leftward shift in the glutamate concentration-response curve for Ca2+ mobilization. C to E, MPEP (C), VU0366249 (D), and M-5MPEP (E) inhibited glutamate stimulation of Ca2+ mobilization. F, affinity estimates for NAMs (○) and PAMs (●) determined from Ca2+ mobilization assays in low-level (x-axis) versus high-level (y-axis) mGlu5-expressing cell lines were compared. Dashed line, unity. Data represent the mean ± S.E.M. from at least three independent determinations. Error bars not shown lie within the dimensions of the symbols.
Quantification of Allosteric Modulator Agonist Activities and Cooperativity with Glutamate in Receptor-Mediated Intracellular Ca2+ Mobilization Assays.
High levels of mGlu5 expression in HEK293A cells result in a greater propensity for the exhibition of agonist activity by PAMs (Noetzel et al., 2012). The high-level mGlu5-expressing HEK293A cell line exhibited a 3-fold greater mGlu5 density than did the low-level mGlu5-expressing cell line (2.3 ± 0.04 versus 0.6 ± 0.1 pmol/mg; data not shown). Glutamate potency was lower in the high-level mGlu5-expressing cell line than in the low-level mGlu5-expressing cell line (541 ± 0.31 versus 149 ± 0.08 nM), which corresponded to a 2.4-fold decrease in the glutamate coupling efficiency logτA (0.37 ± 0.02 versus 0.80 ± 0.02). In the operational model of allosterism, the capacity for intrinsic activity of an allosteric modulator is described by logτB. Phenotypic differences in modulator pharmacological characteristics were observed between the high-level and low-level mGlu5-expressing HEK293A cell lines (Figs. 4, A–E, and 5, A–E). With the exception of VU0357121, all PAMs showed increases in agonist activity or logτB (Table 2). Cooperativity (logβ) values for PAMs were similar in the low-level and high-level mGlu5-expressing cell lines with the exception of VU0405386 and VU0405398, for which cooperativity values were significantly increased by 3-fold (0.54 ± 0.07 versus 1.10 ± 0.16 for VU0405386 and 0.30 ± 0.04 versus 0.87 ± 0.06 for VU0405398 in the low-level versus high-level mGlu5-expressing cell lines). For the three modulators classified as weak NAMs in the low-level mGlu5-expressing cell line (M-5MPEP, VU0366248, and VU0366249), phenotypic changes in pharmacological characteristics were observed, with greater decreases in glutamate Emax being observed in the high-level mGlu5-expressing cell line (Fig. 5, D and E; Supplemental Fig. 2). VU0366249 decreased the Emax of glutamate by ∼40% in the high-level mGlu5-expressing cell line, compared with ∼15% in the low-level mGlu5-expressing line; however, the logβ values were similar in the two cell lines. M-5MPEP and VU0366248 fully abolished the response to glutamate in the high-level mGlu5-expressing cell line. Complete abrogation of the glutamate Emax may be indicative of increased negative cooperativity for these two modulators, with β = 0; however, with the low coupling efficiency of glutamate in the high-level mGlu5-expressing cell line, NAMs with β values of <0.1 are indistinguishable from those with β values of 0. Given this potential for phenotypic differences in allosteric modulator pharmacological features, a second measure of receptor function was used to examine compound activity.
Quantification of Allosteric Modulator Pharmacological Characteristics in Assays of mGlu5-Mediated ERK1/2 Phosphorylation.
Allosteric modulator pharmacological features in our low-level mGlu5-expressing HEK293A cell line most closely resembled those observed in astrocytes (Noetzel et al., 2012). Therefore, translocation of the glutamate-mediated ERK1/2 phosphorylation concentration-response curve was assessed in the presence of each of the 16 allosteric modulators (Fig. 6, A–E; Supplemental Fig. 3) in the low-level mGlu5-expressing cell line alone. Glutamate had ∼10-fold lower coupling efficiency for ERK1/2 phosphorylation, as evidenced by its decreased potency (149 ± 0.08 versus 8671 ± 5071 nM) and logτA (0.80 ± 0.02 versus −0.36 ± 0.13), relative to values for Ca2+ mobilization in the same cell line. Modulator affinity estimates from these ERK1/2 phosphorylation assays showed significant correlations with those derived from Ca2+ mobilization assays in the same cell background (Fig. 6F). Overall, logβ values from ERK1/2 phosphorylation assays were lower than those from Ca2+ mobilization assays for all PAMs. However, all of the PAMs showed intrinsic activity for ERK1/2 phosphorylation (Fig. 6, A and B; Table 4). VU0357121, VU0415051, and VU0405398, i.e., compounds that showed weaker cooperativity in the Ca2+ mobilization assays, generally had lower logτB values, whereas the remaining PAMs, which showed more-robust potentiation and/or agonist activity, had higher logτB values. For NAMs, there was no evidence of inverse agonist activity; MPEP, VU0285683, VU0366058, and VU0366249 exhibited the same pharmacological profiles as in the Ca2+ mobilization assays (Fig. 6, C–E; Table 4; Supplemental Fig. 3). M-5MPEP (Fig. 6E) and VU0366248 (Supplemental Fig. 3) fully abolished ERK1/2 phosphorylation in response to glutamate, which may be indicative of greater negative cooperativity or may reflect the decreased efficacy of glutamate in this assay.
Fig. 6.

Allosteric modulation of glutamate concentration-response curves for ERK1/2 phosphorylation in the low-level mGlu5-expressing HEK293A cell line. A and B, in the low-level mGlu5-expressing cell line, CPPHA (A) and CDPPB (B) displayed agonist activity and potentiated the glutamate concentration-response curve for phosphorylation of ERK1/2. C to E, MPEP (C), VU0366249 (D), and M-5MPEP (E) inhibited the glutamate-stimulated phosphorylation of ERK1/2. F, affinity estimates for PAMs (●) and NAMs (○) determined from ERK1/2 phosphorylation assays (y-axis) and Ca2+ mobilization assays (x-axis) in the low-level mGlu5-expressing cell line were compared. Dashed line, unity. Data represent the mean ± S.E.M. from at least three independent determinations. Error bars not shown lie within the dimensions of the symbols.
TABLE 4.
Summary of operational model parameters for allosteric modulation of glutamate-mediated ERK1/2 phosphorylation in HEK cells expressing low mGlu5 levels
For ERK1/2 phosphorylation in the low-level mGlu5-expressing cell line, data were expressed as fold increases over basal values, with the Em defined as the response to 10% fetal bovine serum (9.4-fold). In the presence of allosteric modulators, logτA (−0.36 ± 0.13) and n (4.43 ± 0.50) were not significantly different (one-way analysis of variance) (Supplemental Table 3). Parameters are as defined for Table 2. Data represent the mean ± S.E.M. from at least three independent determinations.
| Modulator | pKB | logβ | logτB |
|---|---|---|---|
| Positive allosteric modulators | |||
| CDPPB | 7.39 ± 0.18 | 0.13 ± 0.05 | −0.05 ± 0.11 |
| VU29 | 7.63 ± 0.25* | 0.25 ± 0.10 | −0.07 ± 0.08 |
| CPPHA | 6.88 ± 0.14 | 0.05 ± 0.02 | −0.13 ± 0.05 |
| VU0357121 | 7.56 ± 0.30 | 0.03 ± 0.02 | −0.17 ± 0.08 |
| VU0364289 | 6.29 ± 0.11* | 0.16 ± 0.02 | 0.04 ± 0.05 |
| VU0092273 | 7.16 ± 0.08 | 0.11 ± 0.02 | 0.01 ± 0.02 |
| VU0360172 | 7.16 ± 0.26 | 0.12 ± 0.04 | 0.00 ± 0.03 |
| VU0405398 | 7.62 ± 0.32 | 0.05 ± 0.05 | −0.39 ± 0.20 |
| VU0415051 | 8.32 ± 0.21 | 0.03 ± 0.02 | −0.27 ± 0.08 |
| VU0405386 | 8.45 ± 0.19 | 0.19 ± 0.07 | 0.00 ± 0.11 |
| Negative allosteric modulators | |||
| MPEP | 8.20 ± 0.32 | −100 | |
| M-5MPEP | 6.60 ± 0.07 | −100 | |
| VU0285683 | 7.74 ± 0.14 | −100 | |
| VU0366248 | 6.37 ± 0.23 | −100 | |
| VU0366249 | 6.40 ± 0.19 | −0.31 ± 0.12 | |
| VU0366058 | 7.23 ± 0.25 | −100 |
Significantly different (P < 0.05) from the value for the modulator determined in low-level mGlu5-expressing HEK293A cells for Ca2+ mobilization, with one-way analysis of variance and Tukey's post hoc test.
Quantification of Effects of Single Point Mutations of mGlu5 on Allosteric Modulator Affinities and Cooperativity.
In addition to quantifying the affinities of different modulators in various assays, we were interested in using the model to quantify the effects of mGlu5 mutations on the pharmacological characteristics of allosteric modulators. Val substitutions of Tyr658 and Ala809 were reported previously to result in loss of appreciable [3H]MPEP binding and decreased potency for inhibition of quisqualate activity by MPEP (Pagano et al., 2000; Malherbe et al., 2003, 2006; Mühlemann et al., 2006). However, quantification of effects on affinity and/or cooperativity has not been described. MPEP affinity was assessed from concentration-response curves for the inhibition of glutamate-induced Ca2+ mobilization in stable polyclonal HEK293A cell lines expressing Y658V and A809V mGlu5 mutants (Fig. 7). The pKB of MPEP was reduced ∼100-fold with A809V and Y658V, compared with the estimate determined in the polyclonal wild-type mGlu5-expressing HEK293A cell line (Table 5). L743V is known to reduce the affinity of [3H]MPEP by ∼3-fold (Malherbe et al., 2003). In this study, the MPEP pKB in a functional assay was reduced 3-fold. With all three mutations, MPEP retained very high negative cooperativity with glutamate and was able to abolish the response fully. A809V was reported to reduce potentiation by VU29 (Chen et al., 2008); analysis of glutamate potentiation by VU29 with this construct (Fig. 8) showed significantly reduced affinity (30-fold) (Table 5), compared with wild-type values. Cooperativity between glutamate and VU29 was not affected by this mutation. L743V had no effect on the affinity of VU29 but did increase its cooperativity with glutamate (∼3-fold). The non-MPEP site PAM, i.e., CPPHA, was reported previously to show a loss of potentiation with the F585I mutation, at a single concentration (1 μM) (Chen et al., 2008). Compared with the wild-type receptor, the affinity of CPPHA with the F585I construct was reduced ∼3-fold; however, this finding did not reach significance (Fig. 8; Table 5).
Fig. 7.

Effects of single point mutations of mGlu5 on MPEP inhibition of mGlu5-mediated Ca2+ mobilization in response to glutamate. Translocation of glutamate concentration-response curves in the presence of the indicated concentrations of MPEP in polyclonal HEK293A cells expressing wild-type (A), Y658V (B), L743V (C), or A809V (D) mGlu5 was determined. Data represent the mean ± S.E.M. from at least three independent determinations. Error bars not shown lie within the dimensions of the symbols.
TABLE 5.
Quantification of effects of single point mGlu5 mutations on pharmacological characteristics of allosteric modulators
Data represent the mean ± S.E.M. from at least three independent determinations. Parameters are as defined for Table 2.
| Cell Line | MPEP, pKB | VU29 |
CPPHA |
||
|---|---|---|---|---|---|
| pKB | logβ | pKB | logβ | ||
| Wild-type | 8.58 ± 0.17 | 6.87 ± 0.19 | 0.40 ± 0.03 | 5.62 ± 0.16 | 0.78 ± 0.14 |
| F585I | 8.28 ± 0.15 | N.D. | N.D. | 5.14 ± 0.15 | 0.61 ± 0.07 |
| Y658V | 6.57 ± 0.13* | N.D. | N.D. | N.D. | N.D. |
| L743V | 8.04 ± 0.10* | 6.52 ± 0.17 | 1.04 ± 0.09* | N.D. | N.D. |
| A809V | 6.52 ± 0.12* | 5.31 ± 0.26* | 0.58 ± 0.10 | N.D. | N.D. |
N.D., not determined.
Significantly different (P < 0.05) from wild-type value, with one-way analysis of variance and Dunnett's post hoc test.
Fig. 8.

Effects of single point mutations of mGlu5 on potentiation by VU29 and CPPHA of mGlu5-mediated Ca2+ mobilization in response to glutamate. A and B, translocation of glutamate concentration-response curves in the presence of the indicated concentrations of VU29 in polyclonal HEK293A cells expressing wild-type (A) or A809V (B) mGlu5 was determined. C and D, potentiation of glutamate concentration-response curves for Ca2+ mobilization by the indicated concentrations of CPPHA in polyclonal HEK293A cells expressing wild-type (C) or F585I (D) mGlu5 was determined. Data represent the mean ± S.E.M. from at least three independent determinations. Error bars not shown lie within the dimensions of the symbols.
Estimation of Allosteric Modulator Affinities from Modulator Concentration-Response Curves in the Presence of a Single Concentration of Agonist.
The majority of drug discovery programs use concentration-response curves for allosteric modulators in the presence of a single concentration of agonist (a low dose for potentiators and a submaximal dose for inhibitors) to determine structure-activity relationships. Therefore, we were interested in investigating whether valid estimates of affinity and/or cooperativity values could be derived from such data sets. Simulations of the interaction between an agonist and a PAM showed that the PAM concentration-response curve would be translocated to the left in the presence of increasing concentrations of agonist (Fig. 1B). For a NAM, the concentration-response curve would be translocated to the right (Fig. 1A). For an allosteric modulator that potentiates the response to a level less than the maximal response to agonist or does not fully inhibit the response to agonist, modulator cooperativity and affinity values can be determined directly from the modulator potency curve assessed in parallel with the agonist concentration-response curve (Fig. 9, A–C; Table 6). For a PAM (CPPHA or VU0364289) that potentiates the response to agonist to a level equal to or greater than the maximal response to agonist alone, however, cooperativity and affinity values cannot be extrapolated from such potency curves, because similar or identical potency and Emax estimates can be achieved with vastly different cooperativity or affinity values. This is a consequence of the fact that the top plateau of the modulator concentration-response curve might reflect either achievement of the maximal system response or the limit of positive cooperativity. To determine CPPHA and VU0364289 pKB estimates from potency curves, β was constrained such that the values for the apparent cooperativity between these PAMs and glutamate were equal to the maximal leftward shift of the glutamate concentration-response curve in the presence of a high concentration of potentiator (30 μM). In particular, the logarithms of the fold shifts caused by 30 μM VU0364289 (0.86 ± 0.06) and CPPHA (0.76 ± 0.13) were used to constrain logβ. For NAMs that fully inhibited the response to glutamate, the cooperativity factor β was assumed to approach 0 (Fig. 9C; Table 6). With this approach, affinity estimates from modulator potency curves showed good correlation with those from more-rigorous and time-consuming progressive fold-shift analyses (Fig. 9D). Cooperativity factors for weaker PAMs and NAMs determined from potency analyses agreed well with those determined from progressive fold-shift analyses (Fig. 9E; Table 6).
Fig. 9.

Estimation of allosteric modulator cooperativity and affinity values from potency curves. A and B, allosteric modulator potency curves were determined for the indicated positive allosteric modulators in the presence of an EC20 concentration of glutamate. CPPHA and VU0364289 both achieved maximal responses to glutamate; therefore, logβ values were constrained to equal the average maximal leftward shift in the glutamate concentration-response curves (Table 6) for estimations of pKB. For all other PAMs, both logβ and pKB values were determined through nonlinear regression analyses. C, negative allosteric modulators were assessed for their ability to inhibit a submaximal glutamate response. D, affinity estimates from potency curves (y-axis) and from progressive fold-shift analyses (x-axis) for PAMs (●) and NAM (○) showed strong correlation. E, strong correlation was observed between logβ values estimated through nonlinear regression from modulator potency curves (y-axis) and from progressive fold-shift analyses (x-axis). Data represent the mean ± S.E.M. from at least three independent determinations. Error bars not shown lie within the dimensions of the symbols. Dashed line, unity.
TABLE 6.
Estimates of allosteric modulator affinities from modulator concentration-response curves in the presence of an EC20 (PAMs) or EC80 (NAMs) concentration of agonist
Operational analysis of potency curves was performed by simultaneously applying eqs. 6 and 7, with pKA constrained to 6.155, to derive pKB and logβ estimates for modulators. Data represent the mean ± S.E.M. from at least three independent determinations.
| Modulator | PAM pEC50/NAM pIC50 | Operational Analysis of Potency Curves |
Logβ′ (Fold Shift) | |
|---|---|---|---|---|
| pKB | logβ | |||
| CDPPB | 7.16 ± 0.08 | 6.85 ± 0.08 | 0.56 ± 0.05 | 0.78 ± 0.14 (7.1) |
| VU29 | 7.25 ± 0.14 | 6.65 ± 0.20 | 0.88 ± 0.13 | 0.85 ± 0.12 (8.8) |
| CPPHA | 6.35 ± 0.19 | 5.51 ± 0.09 | 0.76a | 0.76 ± 0.13 (7.8) |
| VU0357121 | 6.72 ± 0.12 | 6.46 ± 0.10 | 0.48 ± 0.05 | 0.32 ± 0.03 (2.1) |
| VU0364289 | 6.50 ± 0.11 | 5.50 ± 0.19 | 0.86a | 0.86 ± 0.06 (7.6) |
| VU0092273b | 7.05 ± 0.08b | 6.77 ± 0.10 | 0.51 ± 0.05 | 0.51 ± 0.05 (3.3) |
| VU0360172b | 7.45 ± 0.07b | 7.13 ± 0.05 | 0.53 ± 0.04 | 0.44 ± 0.06 (2.8) |
| VU0405398 | 7.79 ± 0.12 | 7.52 ± 0.09 | 0.34 ± 0.05 | 0.29 ± 0.03 (2.0) |
| VU0415051 | 7.91 ± 0.09 | 7.82 ± 0.05 | 0.41 ± 0.04 | 0.34 ± 0.04 (2.3) |
| VU0405386 | 8.65 ± 0.13 | 8.30 ± 0.13 | 0.50 ± 0.05 | 0.70 ± 0.07 (5.3) |
| MPEP | 8.07 ± 0.07 | 8.10 ± 0.17 | −100c | N.A. |
| M-5MPEP | 6.66 ± 0.09 | 6.85 ± 0.13 | −0.41 ± 0.03 | N.A. |
| VU0285683 | 6.93 ± 0.15 | 7.06 ± 0.15 | −100c | N.A. |
| VU0366248 | 5.85 ± 0.13 | 6.24 ± 0.13 | −0.70 ± 0.13 | N.A. |
| VU0366249 | 5.74 ± 0.20 | 5.73 ± 0.19 | −0.22 ± 0.01 | N.A. |
| VU0366058 | 6.52 ± 0.27 | 6.64 ± 0.11 | −100c | N.A. |
pEC50, negative logarithm of the concentration of modulator that caused half-maximal potentiation of a low concentration (EC20) of glutamate (eq. 5); pIC50, negative logarithm of the concentration of modulator that caused half-maximal inhibition of a submaximal concentration (EC80) of glutamate (eq. 5); pKB, negative logarithm of the equilibrium dissociation constant of an allosteric modulator; logβ, logarithm of the efficacy cooperativity factor β; logβ′, logarithm of the efficacy cooperativity factor β estimated from the maximal leftward shift of the glutamate curve caused by an allosteric modulator; N.A., not applicable.
For PAMs that potentiated up to the maximal response of glutamate, logβ was constrained to this value for estimations of affinities.
Potentiator concentration-response curves were reported previously by Noetzel et al. (2012).
For NAMs that abolished the response to glutamate, β = 0 was assumed and therefore logβ was constrained to −100.
Discussion
Drug discovery programs for GPCRs rely heavily on functional assays for primary screening, lead identification, and optimization. Routinely, compounds are selected and advanced on the basis of their potency, a measure that reflects both affinity and cooperativity. Affinity is generally a secondary measure, estimated either from inhibition of a radiolabeled allosteric modulator or from interactions with an orthosteric radioligand. For the vast majority of GPCRs, orthosteric and allosteric radioligands have yet to be developed, and an easily used framework for quantification of allosteric behaviors is necessary for the estimation of allosteric modulator affinity and cooperativity values from functional assays. Here we validated the use of the operational model of allosterism (Leach et al., 2007) to derive estimates of allosteric modulator affinity and cooperativity values for a representative class C GPCR, mGlu5. Eleven chemical scaffolds were assessed for their interactions with glutamate; the compounds studied ranged from low to high affinity, varied in their degrees of positive and negative cooperativity, and included allosteric agonists. The utility of the model for quantification of the effects of single amino acid substitutions on modulator cooperativity and affinity values was exemplified. Moreover, we present a strategy to determine affinity values from modulator concentration-response curves by incorporating the fold shift at a set concentration as an estimate of cooperativity.
Allosteric modulators of mGlu5 primarily influenced receptor function through modulation of efficacy (β). In particular, there was no change in the affinity of representative PAMs or NAMs when the inhibition of [3H]methoxy-PEPy binding was assessed in the presence of 1 mM glutamate. Furthermore, we observed that PAM cooperativity estimates were unaffected when the assumption of α = 1 was made. Conceptually, efficacy modulation may arise from increased affinity of intracellular signaling partners (such as G proteins and arrestins) for the conformations engendered by the ternary receptor-agonist-PAM complex, compared with the binary receptor-agonist complex. Alternatively, the presence of PAMs may prevent receptor desensitization or other negative signaling outcomes, yielding enhancement of an agonist response. The lack of affinity modulation contrasts with findings from a study by Bradley et al. (2011), in which 30 μM quisqualate increased apparent PAM affinity values in assays of the inhibition of [3H]MPEP binding to cortical astrocytes and rat cortex preparations. Both 30 μM quisqualate and 1 mM glutamate would be expected to occupy the available binding sites maximally (∼1000 × KA). The absence of affinity modulation observed with glutamate may be attributable to the probe-dependent nature of allosteric interactions or may reflect context-dependent pharmacological differences between native tissue and HEK293A cells.
Strong correlation was observed between functional allosteric modulator affinity estimates and estimates from inhibition of [3H]methoxy-PEPy binding. Functional affinity estimates also showed good correlation, which indicated that affinity values were independent of receptor expression levels or the measures of receptor activation. Phenotypic differences in the pharmacological characteristics of allosteric modulators were observed. Agonism by PAMs and inhibition of the maximal response to glutamate by certain NAMs differed depending on receptor expression levels and the assays of receptor function. Glutamate showed lower efficacy for Ca2+ mobilization and ERK1/2 phosphorylation in the high-level mGlu5-expressing cell line. The agonist PAMs VU29 and CDPPB showed the opposite profile, which indicated biased agonism. Increased receptor expression levels ordinarily would be expected to increase agonist efficacy and/or potency (as observed for agonist PAMs). Clearly this is not the case for glutamate acting at mGlu5 to mobilize intracellular Ca2+. To interpret this discrepancy for glutamate, it is important to consider the overall potential impact of receptor overexpression on cellular responses, beyond the established effects of increased receptor reserves on the potency of full agonists for GPCRs. The frequency of Ca2+ oscillations arising from mGlu5 activation was demonstrated to be receptor density dependent, through a “dynamic uncoupling” mechanism whereby mGlu5 undergoes cycles of rapid phosphorylation and dephosphorylation (Kawabata et al., 1996; Nash et al., 2002). It is conceivable that high levels of mGlu5 expression in HEK cells result in changes that ultimately negatively regulate Ca2+ mobilization, such as saturation of rate-limiting signaling partners, coupling to alternative pathways, altered phosphorylation, dimerization or other protein-protein interactions, desensitization, or increased numbers of uncoupled receptors at the cell surface. Although multiple mechanisms are possible, additional studies would be needed to evaluate this phenomenon and to determine whether this is likely to be physiologically relevant under normal physiological or pathological conditions.
Although the affinities tended to be 3-fold higher, cooperativity (logβ) values for all PAMs were lower for ERK1/2 phosphorylation, compared with Ca2+ mobilization. Furthermore, there was a lack of consensus between logβ values for positive allosteric modulation of Ca2+ mobilization in the low-level versus high-level mGlu5-expressing cell lines. Some PAMs showed greater agonist efficacy than glutamate in inducing ERK1/2 phosphorylation, which suggests that glutamate behaves as a partial agonist in stimulating this response. This suggests that the active receptor conformations, and therefore the downstream signaling events, induced by PAMs are different from those induced by glutamate alone. The presence of mGlu5 PAMs may bias mGlu5 signaling toward increased ERK1/2 phosphorylation, relative to calcium mobilization. Although the detailed mechanisms underlying this effect are not fully understood, the possibility that mGlu5 PAMs can induce changes in mGlu5 signaling that differ from those observed with maximal glutamate concentrations is consistent with the report that mGlu5 PAMs promote increases in the frequency of Ca2+ oscillations to a greater extent than does glutamate alone (Bradley et al., 2009). Mechanistically, mGlu5-mediated Ca2+ mobilization and ERK1/2 phosphorylation can arise from independent signaling cascades in both neurons and recombinant cell lines (Thandi et al., 2002; Yang et al., 2006). Stimulus bias produced by allosteric and orthosteric ligands of other family C GPCR family members was reported recently (Davey et al., 2012; Emery et al., 2012). If activation or inhibition of one pathway over another could be attributed to a specific disease state or therapeutic outcome, then compound development might eventually be optimized for biased modulation (Kenakin and Miller, 2010).
Traditionally, affinity determinations and confirmation of an allosteric mechanism of action have used radioligand binding techniques, such as incomplete displacement and changes in the dissociation kinetics of orthosteric radioligands (Ehlert, 1988). However, these techniques cannot be used for detection if an allosteric interaction is driven exclusively by efficacy modulation, as found here. Quantification of affinity and cooperativity without the need for radioligand binding assays presents a number of advantages. For GPCRs and binding sites for which radioligands are not available, including the majority of mGlu family members, the framework of the operational model of allosterism allows for optimization of affinity. Drug discovery programs increasingly are incorporating a need for verification of target engagement, largely through the use of positron emission tomography to assess receptor occupancy by novel compounds at the desired site of action. We also described an approach to analyze PAM potency data in combination with maximal fold-shift data to estimate modulator affinity values. Because optimal radioligands and positron emission tomography tracers exhibit high affinity and specificity, such a method could easily be incorporated into established screening protocols to guide chemistry-related efforts with respect to modulator affinity, which would allow parallel identification of a lead compound and the needed tools to establish target engagement. Furthermore, affinity estimation would allow for correlation of in vivo parameters, such as minimal effective doses and unbound brain concentrations, with receptor occupancy and cooperativity data.
In addition to guiding SAR and lead optimization efforts, the ability to estimate affinity values from functional assays would enable delineation of effects on affinity and cooperativity in structure-function studies. Four previously identified point mutations were studied here for quantification of their effects on allosteric modulator interactions with mGlu5. Val substitution of Ala809 and Tyr658 were reported previously to result in loss of appreciable [3H]MPEP binding and decreased MPEP potency (Pagano et al., 2000; Malherbe et al., 2003, 2006; Mühlemann et al., 2006), effects attributed to 100-fold reductions in MPEP affinity for the mutant receptors. L743V, which was reported to cause a 3-fold reduction in [3H]MPEP affinity, also was assessed (Malherbe et al., 2003). In confirmation of the utility of the model for detection of mutational effects on affinity, L743V was found to decrease the MPEP functional affinity estimate by 3-fold. A809V and F585I also resulted in loss of potentiation by the PAMs VU29 and CPPHA, respectively (Chen et al., 2008), which might be attributable to decreased affinity and/or cooperativity. VU29 affinity was reduced 30-fold with A809V, and CPPHA affinity was reduced 3-fold with F585I; neither mutation affected the cooperativity of PAMs with glutamate. L743V enhanced VU29 cooperativity but had no effect on affinity. Differential interactions with amino acids within a common binding site are likely to underscore potentiation versus inhibition, as well as to contribute to pharmacological mode switches within distinct allosteric modulator scaffolds. Studies are ongoing to probe the molecular determinants of allosteric interactions at mGlu5 and the interactions that govern affinity and cooperativity.
In validating the operational model of allosterism for quantification of allosteric interactions at mGlu5, we discovered evidence for signal bias for both “pure” and agonist PAMs of mGlu5, compared with glutamate. Furthermore, we describe a strategy to estimate affinity values from PAM potency curves. Quantification of allosteric interactions provides the means to better interpret SAR and structure-function assay findings and to identify signal bias.
Supplementary Material
Acknowledgments
We thank Julie R. Field for fruitful discussions, and we acknowledge the invaluable technical assistance of Kiran Gogi and Daryl Venable.
 The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
The online version of this article (available at http://molpharm.aspetjournals.org) contains supplemental material.
This work was supported by the National Institutes of Health National Institute of Mental Health [Grant 2R01-MH062646-13]; the National Institutes of Health National Institute of Neurological Disorders and Stroke [Grant 2R01-NS031373-16A2]; the National Institutes of Health National Institute of Drug Abuse [Grant 1R01-DA023947]; the Molecular Libraries Probe Production Centers Network [Grants 5U54-MH84659-03, 5U54-MH84659-03S1]; a National Institutes of Health National Institute of Neurological Disorders and Stroke National Research Service Award [Grant F32-NS071746] (to M.J.N.); a National Institutes of Health National Institute of Mental Health National Research Service Award [Grant F32-MH088234-02] (to J.M.R.); a National Alliance for Research on Schizophrenia and Depression Maltz Investigator Award (to K.J.G.); an American Australian Association Merck Foundation fellowship (to K.J.G.); and a National Health and Medical Research Council (Australia) overseas biomedical postdoctoral training fellowship (to K.J.G.). P.J.C. is a consultant for Seaside Therapeutics and receives research support from Seaside Therapeutics and Johnson and Johnson/Janssen Pharmaceutica.
This work was previously presented in part as follows: Gregory KJ, Dong EN, Reiff SD, Rook JM, Noetzel MJ, Plumley HC, Kaufmann KW, Manka JT, Zhou YS, Vinson PN, et al. (2011) Application of an operational model of allosterism to investigate the structural determinants of metabotropic glutamate receptor 5 allosteric modulation. Proceedings of the 7th International Meeting on Metabotropic Glutamate Receptors; Insituto Neurologico Mediterraneo, Pozzilli, 2–7 Oct 2011; Taormina, Italy.
Article, publication date, and citation information can be found at http://molpharm.aspetjournals.org.
- mGlu
- metabotropic glutamate receptor
- methoxy-PEPy
- 3-methoxy-5-(pyridin-2-ylethynyl)pyridine
- CDPPB
- 3-cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide
- CPPHA
- N-{4-chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl}-2-hydroxybenzamide
- DMEM
- Dulbecco's modified Eagle's medium
- ERK
- extracellular signal-regulated kinase
- GPCR
- G protein-coupled receptor
- HEK
- human embryonic kidney
- M-5MPEP
- 2-[2-(3-methoxyphenyl)ethynyl]-5-methylpyridine
- MPEP
- 2-methyl-6-(phenylethynyl)pyridine
- MTEP
- 3-[(2-methyl-1,3-thiazol-4-yl)ethynyl]pyridine
- NAM
- negative allosteric modulator
- PAM
- positive allosteric modulator
- SAR
- structure-activity relationship
- SIB-1757
- 6-methyl-2-(phenylazo)-3-pyridinol
- SIB-1893
- (E)-2-methyl-6-(2-phenylethenyl)pyridine
- VU0092273
- 1-{[4-(2-phenylethynyl)phenyl]carbonyl}piperidin-4-ol
- VU0285683
- 3-fluoro-5-[3-(pyridin-2-yl)-1,2,4-oxadiazol-5-yl]benzonitrile
- VU0357121
- 4-butoxy-N-(2,4-difluorophenyl)benzamide
- VU0360172
- N-cyclobutyl-6-[(3-fluorophenyl)ethynyl]nicotinamide hydrochloride
- VU0364289
- 2-{4-[2-(benzyloxy)acetyl]piperazin-1-yl}benzonitrile
- VU0366248
- N-(3-chloro-2-fluorophenyl)-3-cyano-5-fluorobenzamide
- VU0366249
- N-(3-chloro-4-fluorophenyl)-3-cyano-5-fluorobenzamide
- VU0405386
- N-tert-butyl-5-[(3-fluorophenyl)ethynyl]picolinamide
- VU0405398
- [5-[(3-fluorophenyl)ethynyl]pyridin-2-yl](3-hydroxyazetidin-1-yl)methanone
- VU0415051
- N-tert-butyl-6-[2-(3-fluorophenyl)ethynyl]pyridine-3-carboxamide
- VU0366058
- 2-(1,3-benzoxazol-2-ylamino)-4-(4-fluorophenyl)pyrimidine-5-carbonitrile
- VU29
- 4-nitro-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide.
Authorship Contributions
Participated in research design: Gregory, Noetzel, Rook, Vinson, Rodriguez, Niswender, and Conn.
Conducted experiments: Gregory, Noetzel, Rook, and Vinson.
Contributed new reagents or analytic tools: Stauffer, Emmitte, Zhou, Chun, Felts, Chauder, and Lindsley.
Performed data analysis: Gregory, Noetzel, Rook, and Vinson.
Wrote or contributed to the writing of the manuscript: Gregory, Niswender, and Conn.
References
- Alagille D, DaCosta H, Chen Y, Hemstapat K, Rodriguez A, Baldwin RM, Conn PJ, Conn JP, Tamagnan GD. (2011) Potent mGlu5 antagonists: pyridyl and thiazolyl-ethynyl-3,5-disubstituted-phenyl series. Bioorg Med Chem Lett 21:3243–3247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley SJ, Watson JM, Challiss RA. (2009) Effects of positive allosteric modulators on single-cell oscillatory Ca2+ signaling initiated by the type 5 metabotropic glutamate receptor. Mol Pharmacol 76:1302–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley SJ, Langmead CJ, Watson JM, Challiss RA. (2011) Quantitative analysis reveals multiple mechanisms of allosteric modulation of the mGlu5 receptor in rat astroglia. Mol Pharmacol 79:874–885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Goudet C, Pin JP, Conn PJ. (2008) N-{4-Chloro-2-[(1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)methyl]phenyl}-2-hydroxybenzamide (CPPHA) acts through a novel site as a positive allosteric modulator of group 1 metabotropic glutamate receptors. Mol Pharmacol 73:909–918 [DOI] [PubMed] [Google Scholar]
- Chen Y, Nong Y, Goudet C, Hemstapat K, de Paulis T, Pin JP, Conn PJ. (2007) Interaction of novel positive allosteric modulators of metabotropic glutamate receptor 5 with the negative allosteric antagonist site is required for potentiation of receptor responses. Mol Pharmacol 71:1389–1398 [DOI] [PubMed] [Google Scholar]
- Cheng Y, Prusoff WH. (1973) Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 22:3099–3108 [DOI] [PubMed] [Google Scholar]
- Chua PC, Nagasawa JY, Bleicher LS, Munoz B, Schweiger EJ, Tehrani L, Anderson JJ, Cramer M, Chung J, Green MD, et al. (2005) Cyclohexenyl- and dehydropiperidinyl-alkynyl pyridines as potent metabotropic glutamate subtype 5 (mGlu5) receptor antagonists. Bioorg Med Chem Lett 15:4589–4593 [DOI] [PubMed] [Google Scholar]
- Cosford ND, Roppe J, Tehrani L, Schweiger EJ, Seiders TJ, Chaudary A, Rao S, Varney MA. (2003a) [3H]-Methoxymethyl-MTEP and [3H]-methoxy-PEPy: potent and selective radioligands for the metabotropic glutamate subtype 5 (mGlu5) receptor. Bioorg Med Chem Lett 13:351–354 [DOI] [PubMed] [Google Scholar]
- Cosford ND, Tehrani L, Roppe J, Schweiger E, Smith ND, Anderson J, Bristow L, Brodkin J, Jiang X, McDonald I, et al. (2003b) 3-[(2-Methyl-1,3-thiazol-4-yl)ethynyl]-pyridine: a potent and highly selective metabotropic glutamate subtype 5 receptor antagonist with anxiolytic activity. J Med Chem 46:204–206 [DOI] [PubMed] [Google Scholar]
- Davey AE, Leach K, Valant C, Conigrave AD, Sexton PM, Christopoulos A. (2012) Positive and negative allosteric modulators promote biased signaling at the calcium-sensing receptor. Endocrinology 153:1232–1241 [DOI] [PubMed] [Google Scholar]
- de Paulis T, Hemstapat K, Chen Y, Zhang Y, Saleh S, Alagille D, Baldwin RM, Tamagnan GD, Conn PJ. (2006) Substituent effects of N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamides on positive allosteric modulation of the metabotropic glutamate-5 receptor in rat cortical astrocytes. J Med Chem 49:3332–3344 [DOI] [PubMed] [Google Scholar]
- Ehlert FJ. (1988) Estimation of the affinities of allosteric ligands using radioligand binding and pharmacological null methods. Mol Pharmacol 33:187–194 [PubMed] [Google Scholar]
- Emery AC, DiRaddo JO, Miller E, Hathaway HA, Pshenichkin S, Takoudjou GR, Grajkowska E, Yasuda RP, Wolfe BB, Wroblewski JT. (2012) Ligand bias at metabotropic glutamate 1a receptors: molecular determinants that distinguish β-arrestin-mediated from G protein-mediated signaling. Mol Pharmacol 82:291–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felts AS, Lindsley SR, Lamb JP, Rodriguez AL, Menon UN, Jadhav S, Jones CK, Conn PJ, Lindsley CW, Emmitte KA. (2010) 3-Cyano-5-fluoro-N-arylbenzamides as negative allosteric modulators of mGlu5: identification of easily prepared tool compounds with CNS exposure in rats. Bioorg Med Chem Lett 20:4390–4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felts AS, Saleh SA, Le U, Rodriguez AL, Weaver CD, Conn PJ, Lindsley CW, Emmitte KA. (2009) Discovery and SAR of 6-substituted-4-anilinoquinazolines as non-competitive antagonists of mGlu5. Bioorg Med Chem Lett 19:6623–6626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galambos J, Wágner G, Nógrádi K, Bielik A, Molnár L, Bobok A, Horváth A, Kiss B, Kolok S, Nagy J, et al. (2010) Carbamoyloximes as novel non-competitive mGlu5 receptor antagonists. Bioorg Med Chem Lett 20:4371–4375 [DOI] [PubMed] [Google Scholar]
- Gasparini F, Lingenhöhl K, Stoehr N, Flor PJ, Heinrich M, Vranesic I, Biollaz M, Allgeier H, Heckendorn R, Urwyler S, et al. (1999) 2-Methyl-6-(phenylethynyl)-pyridine (MPEP), a potent, selective and systemically active mGlu5 receptor antagonist. Neuropharmacology 38:1493–1503 [DOI] [PubMed] [Google Scholar]
- Gilbert AM, Bursavich MG, Lombardi S, Adedoyin A, Dwyer JM, Hughes Z, Kern JC, Khawaja X, Rosenzweig-Lipson S, Moore WJ, et al. (2011) 3-(Pyridin-2-yl-ethynyl)benzamide metabotropic glutamate receptor 5 negative allosteric modulators: hit to lead studies. Bioorg Med Chem Lett 21:195–199 [DOI] [PubMed] [Google Scholar]
- Gregory KJ, Sexton PM, Christopoulos A. (2010) Overview of receptor allosterism. Curr Protoc Pharmacol Unit 1.21 [DOI] [PubMed] [Google Scholar]
- Hammond AS, Rodriguez AL, Townsend SD, Niswender CM, Gregory KJ, Lindsley CW, Conn PJ. (2010) Discovery of a novel chemical class of mGlu5 allosteric ligands with distinct modes of pharmacology. ACS Chem Neurosci 1:702–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Poon SF, Chapman DF, Chung J, Cramer M, Reger TS, Roppe JR, Tehrani L, Cosford ND, Smith ND. (2004) 2-(2-[3-(Pyridin-3-yloxy)phenyl]-2H-tetrazol-5-yl)pyridine: a highly potent, orally active, metabotropic glutamate subtype 5 (mGlu5) receptor antagonist. Bioorg Med Chem Lett 14:5473–5476 [DOI] [PubMed] [Google Scholar]
- Jaeschke G, Porter R, Büttelmann B, Ceccarelli SM, Guba W, Kuhn B, Kolczewski S, Huwyler J, Mutel V, Peters JU, et al. (2007) Synthesis and biological evaluation of fenobam analogs as mGlu5 receptor antagonists. Bioorg Med Chem Lett 17:1307–1311 [DOI] [PubMed] [Google Scholar]
- Kawabata S, Tsutsumi R, Kohara A, Yamaguchi T, Nakanishi S, Okada M. (1996) Control of calcium oscillations by phosphorylation of metabotropic glutamate receptors. Nature 383:89–92 [DOI] [PubMed] [Google Scholar]
- Kenakin T, Miller LJ. (2010) Seven transmembrane receptors as shapeshifting proteins: the impact of allosteric modulation and functional selectivity on new drug discovery. Pharmacol Rev 62:265–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney GG, O'Brien JA, Lemaire W, Burno M, Bickel DJ, Clements MK, Chen TB, Wisnoski DD, Lindsley CW, Tiller PR, et al. (2005) A novel selective positive allosteric modulator of metabotropic glutamate receptor subtype 5 has in vivo activity and antipsychotic-like effects in rat behavioral models. J Pharmacol Exp Ther 313:199–206 [DOI] [PubMed] [Google Scholar]
- Kulkarni SS, Nightingale B, Dersch CM, Rothman RB, Newman AH. (2006) Design and synthesis of noncompetitive metabotropic glutamate receptor subtype 5 antagonists. Bioorg Med Chem Lett 16:3371–3375 [DOI] [PubMed] [Google Scholar]
- Kulkarni SS, Zou MF, Cao J, Deschamps JR, Rodriguez AL, Conn PJ, Newman AH. (2009) Structure-activity relationships comparing N-(6-methylpyridin-yl)-substituted aryl amides to 2-methyl-6-(substituted-arylethynyl)pyridines or 2-methyl-4-(substituted-arylethynyl)thiazoles as novel metabotropic glutamate receptor subtype 5 antagonists. J Med Chem 52:3563–3575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazareno S, Birdsall NJ. (1995) Detection, quantitation, and verification of allosteric interactions of agents with labeled and unlabeled ligands at G protein-coupled receptors: interactions of strychnine and acetylcholine at muscarinic receptors. Mol Pharmacol 48:362–378 [PubMed] [Google Scholar]
- Leach K, Sexton PM, Christopoulos A. (2007) Allosteric GPCR modulators: taking advantage of permissive receptor pharmacology. Trends Pharmacol Sci 28:382–389 [DOI] [PubMed] [Google Scholar]
- Lindemann L, Jaeschke G, Michalon A, Vieira E, Honer M, Spooren W, Porter R, Hartung T, Kolczewski S, Büttelmann B, et al. (2011) CTEP: a novel, potent, long-acting, and orally bioavailable metabotropic glutamate receptor 5 inhibitor. J Pharmacol Exp Ther 339:474–486 [DOI] [PubMed] [Google Scholar]
- Liu F, Grauer S, Kelley C, Navarra R, Graf R, Zhang G, Atkinson PJ, Popiolek M, Wantuch C, Khawaja X, et al. (2008) ADX47273 [S-(4-fluoro-phenyl)-{3-[3-(4-fluoro-phenyl)-[1,2,4]-oxadiazol-5-yl]-piperidin-1-yl}-methanone]: a novel metabotropic glutamate receptor 5-selective positive allosteric modulator with preclinical antipsychotic-like and procognitive activities. J Pharmacol Exp Ther 327:827–839 [DOI] [PubMed] [Google Scholar]
- Malherbe P, Kratochwil N, Mühlemann A, Zenner MT, Fischer C, Stahl M, Gerber PR, Jaeschke G, Porter RH. (2006) Comparison of the binding pockets of two chemically unrelated allosteric antagonists of the mGlu5 receptor and identification of crucial residues involved in the inverse agonism of MPEP. J Neurochem 98:601–615 [DOI] [PubMed] [Google Scholar]
- Malherbe P, Kratochwil N, Zenner MT, Piussi J, Diener C, Kratzeisen C, Fischer C, Porter RH. (2003) Mutational analysis and molecular modeling of the binding pocket of the metabotropic glutamate 5 receptor negative modulator 2-methyl-6-(phenylethynyl)-pyridine. Mol Pharmacol 64:823–832 [DOI] [PubMed] [Google Scholar]
- Melancon BJ, Hopkins CR, Wood MR, Emmitte KA, Niswender CM, Christopoulos A, Conn PJ, Lindsley CW. (2012) Allosteric modulation of seven transmembrane spanning receptors: theory, practice, and opportunities for central nervous system drug discovery. J Med Chem 55:1445–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milbank JB, Knauer CS, Augelli-Szafran CE, Sakkab-Tan AT, Lin KK, Yamagata K, Hoffman JK, Zhuang N, Thomas J, Galatsis P, et al. (2007) Rational design of 7-arylquinolines as non-competitive metabotropic glutamate receptor subtype 5 antagonists. Bioorg Med Chem Lett 17:4415–4418 [DOI] [PubMed] [Google Scholar]
- Mueller R, Dawson ES, Meiler J, Rodriguez AL, Chauder BA, Bates BS, Felts AS, Lamb JP, Menon UN, Jadhav SB, et al. (2012) Discovery of 2-(2-benzoxazoyl amino)-4-aryl-5-cyanopyrimidine as negative allosteric modulators (NAMs) of metabotropic glutamate receptor 5 (mGlu5): from an artificial neural network virtual screen to an in vivo tool compound. ChemMedChem 7:406–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mühlemann A, Ward NA, Kratochwil N, Diener C, Fischer C, Stucki A, Jaeschke G, Malherbe P, Porter RH. (2006) Determination of key amino acids implicated in the actions of allosteric modulation by 3,3′-difluorobenzaldazine on rat mGlu5 receptors. Eur J Pharmacol 529:95–104 [DOI] [PubMed] [Google Scholar]
- Mutel V, Ellis GJ, Adam G, Chaboz S, Nilly A, Messer J, Bleuel Z, Metzler V, Malherbe P, Schlaeger EJ, et al. (2000) Characterization of [3H]quisqualate binding to recombinant rat metabotropic glutamate 1a and 5a receptors and to rat and human brain sections. J Neurochem 75:2590–2601 [DOI] [PubMed] [Google Scholar]
- Nash MS, Schell MJ, Atkinson PJ, Johnston NR, Nahorski SR, Challiss RA. (2002) Determinants of metabotropic glutamate receptor-5-mediated Ca2+ and inositol 1,4,5-trisphosphate oscillation frequency: receptor density versus agonist concentration. J Biol Chem 277:35947–35960 [DOI] [PubMed] [Google Scholar]
- Niswender CM, Conn PJ. (2010) Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol 50:295–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noetzel MJ, Rook JM, Vinson PN, Cho HP, Days E, Zhou Y, Rodriguez AL, Lavreysen H, Stauffer SR, Niswender CM, et al. (2012) Functional impact of allosteric agonist activity of selective positive allosteric modulators of metabotropic glutamate receptor subtype 5 in regulating central nervous system function. Mol Pharmacol 81:120–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien JA, Lemaire W, Wittmann M, Jacobson MA, Ha SN, Wisnoski DD, Lindsley CW, Schaffhauser HJ, Rowe B, Sur C, et al. (2004) A novel selective allosteric modulator potentiates the activity of native metabotropic glutamate receptor subtype 5 in rat forebrain. J Pharmacol Exp Ther 309:568–577 [DOI] [PubMed] [Google Scholar]
- Pagano A, Ruegg D, Litschig S, Stoehr N, Stierlin C, Heinrich M, Floersheim P, Prezèau L, Carroll F, Pin JP, et al. (2000) The non-competitive antagonists 2-methyl-6-(phenylethynyl)pyridine and 7-hydroxyiminocyclopropan[b]chromen-1α-carboxylic acid ethyl ester interact with overlapping binding pockets in the transmembrane region of group I metabotropic glutamate receptors. J Biol Chem 275:33750–33758 [DOI] [PubMed] [Google Scholar]
- Poon SF, Eastman BW, Chapman DF, Chung J, Cramer M, Holtz G, Cosford ND, Smith ND. (2004) 3-[3-Fluoro-5-(5-pyridin-2-yl-2H-tetrazol-2-yl)phenyl]-4-methylpyridine: a highly potent and orally bioavailable metabotropic glutamate subtype 5 (mGlu5) receptor antagonist. Bioorg Med Chem Lett 14:5477–5480 [DOI] [PubMed] [Google Scholar]
- Rodriguez AL, Grier MD, Jones CK, Herman EJ, Kane AS, Smith RL, Williams R, Zhou Y, Marlo JE, Days EL, et al. (2010) Discovery of novel allosteric modulators of metabotropic glutamate receptor subtype 5 reveals chemical and functional diversity and in vivo activity in rat behavioral models of anxiolytic and antipsychotic activity. Mol Pharmacol 78:1105–1123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez AL, Nong Y, Sekaran NK, Alagille D, Tamagnan GD, Conn PJ. (2005) A close structural analog of 2-methyl-6-(phenylethynyl)-pyridine acts as a neutral allosteric site ligand on metabotropic glutamate receptor subtype 5 and blocks the effects of multiple allosteric modulators. Mol Pharmacol 68:1793–1802 [DOI] [PubMed] [Google Scholar]
- Rodriguez AL, Williams R, Zhou Y, Lindsley SR, Le U, Grier MD, Weaver CD, Conn PJ, Lindsley CW. (2009) Discovery and SAR of novel mGlu5 non-competitive antagonists not based on an MPEP chemotype. Bioorg Med Chem Lett 19:3209–3213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roppe J, Smith ND, Huang D, Tehrani L, Wang B, Anderson J, Brodkin J, Chung J, Jiang X, King C, et al. (2004) Discovery of novel heteroarylazoles that are metabotropic glutamate subtype 5 receptor antagonists with anxiolytic activity. J Med Chem 47:4645–4648 [DOI] [PubMed] [Google Scholar]
- Sams AG, Mikkelsen GK, Brodbeck RM, Pu X, Ritzén A. (2011) Efficacy switching SAR of mGlu5 allosteric modulators: highly potent positive and negative modulators from one chemotype. Bioorg Med Chem Lett 21:3407–3410 [DOI] [PubMed] [Google Scholar]
- Tehrani LR, Smith ND, Huang D, Poon SF, Roppe JR, Seiders TJ, Chapman DF, Chung J, Cramer M, Cosford ND. (2005) 3-[Substituted]-5-(5-pyridin-2-yl-2H-tetrazol-2-yl)benzonitriles: identification of highly potent and selective metabotropic glutamate subtype 5 receptor antagonists. Bioorg Med Chem Lett 15:5061–5064 [DOI] [PubMed] [Google Scholar]
- Thandi S, Blank JL, Challiss RA. (2002) Group-I metabotropic glutamate receptors, mGlu1a and mGlu5a, couple to extracellular signal-regulated kinase (ERK) activation via distinct, but overlapping, signalling pathways. J Neurochem 83:1139–1153 [DOI] [PubMed] [Google Scholar]
- Vanejevs M, Jatzke C, Renner S, Müller S, Hechenberger M, Bauer T, Klochkova A, Pyatkin I, Kazyulkin D, Aksenova E, et al. (2008) Positive and negative modulation of group I metabotropic glutamate receptors. J Med Chem 51:634–647 [DOI] [PubMed] [Google Scholar]
- Varney MA, Cosford ND, Jachec C, Rao SP, Sacaan A, Lin FF, Bleicher L, Santori EM, Flor PJ, Allgeier H, et al. (1999) SIB-1757 and SIB-1893: selective, noncompetitive antagonists of metabotropic glutamate receptor type 5. J Pharmacol Exp Ther 290:170–181 [PubMed] [Google Scholar]
- Wágner G, Wéber C, Nyéki O, Nógrádi K, Bielik A, Molnár L, Bobok A, Horváth A, Kiss B, Kolok S, et al. (2010) Hit-to-lead optimization of disubstituted oxadiazoles and tetrazoles as mGlu5 NAMs. Bioorg Med Chem Lett 20:3737–3741 [DOI] [PubMed] [Google Scholar]
- Weiss JM, Jimenez HN, Li G, April M, Uberti MA, Bacolod MD, Brodbeck RM, Doller D. (2011) 6-Aryl-3-pyrrolidinylpyridines as mGlu5 receptor negative allosteric modulators. Bioorg Med Chem Lett 21:4891–4899 [DOI] [PubMed] [Google Scholar]
- Wood MR, Hopkins CR, Brogan JT, Conn PJ, Lindsley CW. (2011) “Molecular switches” on mGlu allosteric ligands that modulate modes of pharmacology. Biochemistry 50:2403–2410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Mao L, Chen H, Catavsan M, Kozinn J, Arora A, Liu X, Wang JQ. (2006) A signaling mechanism from Gαq-protein-coupled metabotropic glutamate receptors to gene expression: role of the c-Jun N-terminal kinase pathway. J Neurosci 26:971–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Zou MF, Rodriguez AL, Conn PJ, Newman AH. (2010) Structure-activity relationships in a novel series of 7-substituted-aryl quinolines and 5-substituted-aryl benzothiazoles at the metabotropic glutamate receptor subtype 5. Bioorg Med Chem 18:3026–3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Manka JT, Rodriguez AL, Weaver CD, Days EL, Vinson PN, Jadhav S, Hermann EJ, Jones CK, Conn PJ, et al. (2010) Discovery of N-aryl piperazines as selective mGlu5 potentiators with improved in vivo utility. ACS Med Chem Lett 1:433–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou MF, Cao J, Rodriguez AL, Conn PJ, Newman AH. (2011) Design and synthesis of substituted N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamides as positive allosteric modulators of the metabotropic glutamate receptor subtype 5. Bioorg Med Chem Lett 21:2650–2654 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.