

Introduction
The Ras→Raf→MEK(mitogen-activated kinase kinase)→ERK (extracellular-signal-regulated kinase) signalling pathway is one of at least five mitogen-activated protein kinase (MAPK) pathways that control several fundamental cellular processes, driving proliferation, differentiation and cell survival.[1–3] Signal transduction through this particular pathway, which is depicted in Figure 1, is initiated by the binding of a wide variety of ligands, including hormones and growth factors, to receptor tyrosine kinases (RTKs). This leads to the activation of Ras proteins (H-, K- and N-Ras isoforms) previously anchored in the plasma membrane by earlier post-translational reactions, e.g. farnesylation. Subsequently, the Ras proteins are induced to exchange their bound GDP for GTP, which leads to a conformational change in Ras and the initiation of a three-stage phosphorylation cascade that climaxes with the activation of ERK1/2. First, the Raf family of kinases (A-, B- and Raf-1 isoforms), the best studied of which is Raf-1, is recruited to the plasma membrane. Upon its subsequent phosphorylation, Raf-1 then activates (phosphorylates) MAP / ERK kinase 1 and 2 (MEK1/2), which, in turn, activate (phosphorylate) ERK1 and ERK2 (p44 MAPK and p42 MAPK, respectively). ERK1/2 are activated through phosphorylation of both a threonine and a tyrosine residue, namely Thr202 and Tyr204 of ERK1 and Thr183 and Tyr185 of ERK2. MEK1/2 are the only known activators of ERK1/2 and are, thus, dual specificity kinases. Activated ERK1/2 then phosphorylate serine/threonine residues of more than 50 downstream cytosolic and nuclear substrates, leading to alterations in gene expression profiles and an increase in proliferation, differentiation and cell survival.[1–3]
Figure 1.
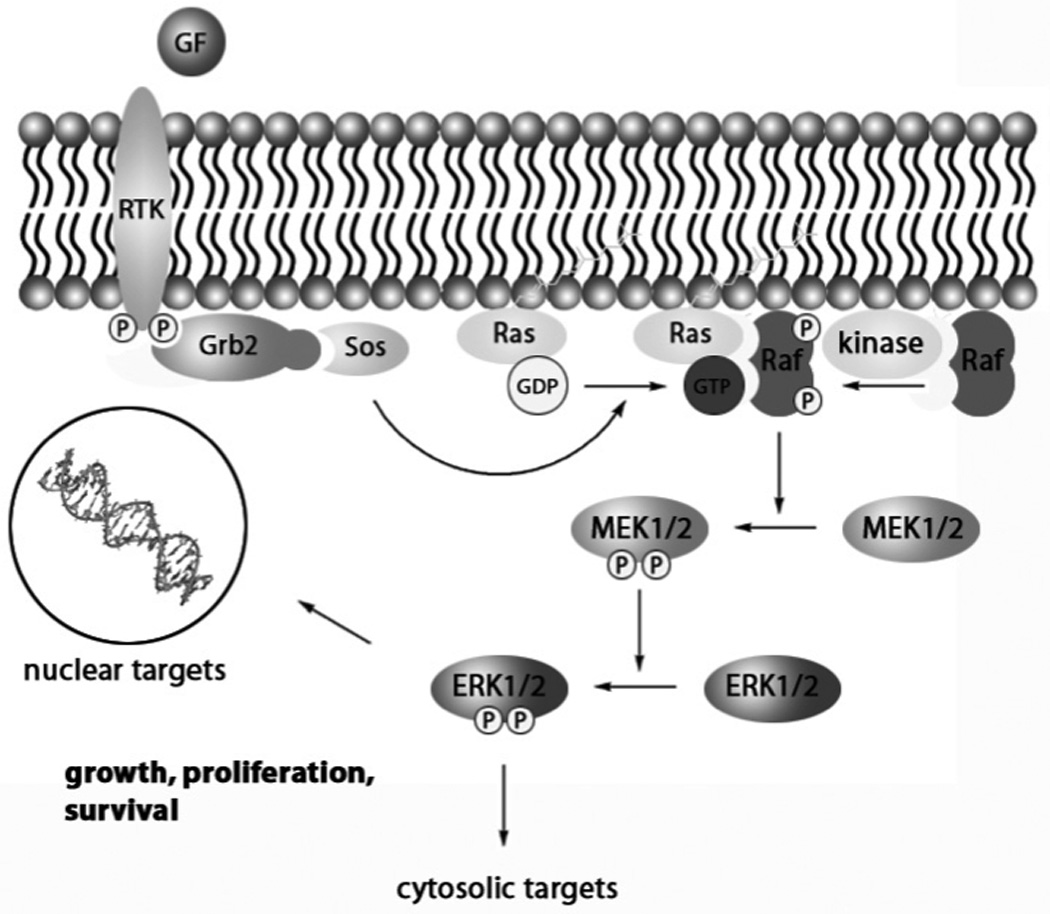
Schematic representation of the Ras→Raf→MEK1/2→ERK1/2 signalling pathway. GF = growth factor, RTK = receptor tyrosine kinase, Grb2 = growth factor receptor-bound protein 2; Sos = son of sevenless; P indicates a phosphorylated serine, threonine or tyrosine residue.
There is now considerable evidence that links the dysregulation of the Ras→Raf→MEK→ERK pathway to the oncogenesis of human cancers. Ras is hyperactivated in around 30% of human cancers, most commonly the K-Ras isoform.[4] More specifically, Ras activating mutations have been reported in about 90% of pancreatic carcinomas, 50% of colon carcinomas, 30% of lung cancers and in around 30% of myeloid leukaemia cases.[4] Activating mutations of Raf have also been reported in around 7% of human cancers.[5,6] In particular, mutations of B-Raf have been observed in over 60% of melanomas, around 30% of ovarian cancer and in approximately 20% of colorectal carcinomas, as well as in several other malignancies at lower frequencies.[5,6] Constitutively activate MEK1/2 and ERK1/2 proteins are present in a relatively high number of human tumours, particularly those from the colon, lung, pancreas, ovary and kidney.[7] Since mutations of the MEK1/2 and ERK1/2 genes have not been observed in human tumours, it seems probable that the hyperactivity of these proteins is a consequence of their constitutive phosphorylation due to hyperactivation of upstream effectors, including receptors, Ras and B-Raf. In summary, the Ras→Raf→MEK1/2→ERK1/2 pathway is an appealing target for the development of potential anti-cancer therapeutics. Moreover, the pathway offers several junctures for signal transduction blockade; due to the converging functions of MEK1/2 and ERK1/2, specific inhibition of these proteins is particularly desirable.
In this mini-review, some of the more prominent small molecule inhibitors of the ERK pathway will be presented, with a particular emphasis on those discovered within the last ten to fifteen years. In the first section, we shall discuss those inhibitors that target proteins upstream of ERK1/2, specifically Raf and MEK1/2. We will then shift to the main focus of this review, which is the direct inhibition of ERK1/2 through targeting either the ATP-binding site (ATP-competitive inhibitors) or the surface of ERK and blocking its protein–protein interactions with its substrates (non-ATP-competitive inhibitors).
Indirect Inhibition of ERK
Raf Inhibitors
Constitutive activation of the ERK pathway, which has been observed in many human cancers, is predominantly due to gain-of-function mutations of Ras or Raf.[4–6] The large number of published Ras inhibitors, in particular the farnesyltransferase inhibitors, along with their varied and mostly disappointing results in clinical trials, is beyond the scope of this review, and the authors direct the reader elsewhere for a discussion on this topic.[8] Of the three Raf isoforms in mammals (A-Raf, B-Raf and Raf-1), it is predominantly the B-Raf isoform that is mutated.[5,6] However, the quest for potent B-Raf inhibitors is very much in its infancy, probably since it was originally considered that Raf-1 was the Raf isoform with the greatest oncogenic potential.[9] In that regard, GW5074 and sorafenib, two potent, ATP-competitive inhibitors of Raf-1, were identified through various screening techniques.
Researchers at GlaxoSmithKline synthesized and screened a series of over 2000 benzylidene oxindole compounds in a Raf1/MEK/ERK2 cascade assay. Optimization of an initial hit led to the discovery of GW5074 (1), which blocks Raf-1 kinase activity in vitro with an IC50 value of 9 nM.[10] The inspiration for the benzylidene oxindole scaffold came from previous research that had shown such compounds inhibited tyrosine-specific protein kinase activity of the epidermal growth factor receptor (EGFR).[11] Moreover, the NH/CO of the oxindole is a hydrogen bond donor/acceptor motif that is found in many kinase inhibitors.[12] It was discovered that the pKa of the phenol component of the benzylidene oxindoles was proportional to the inhibitory activity against Raf-1, such that the more acidic the phenol, the more potent was the compound against Raf-1.10 Furthermore, the exocyclic double bond, which ensures planarity of the phenyl and oxindole cores, was found to be essential. Optimal potency and selectivity were furnished through the incorporation of two groups flanking the phenol OH, with Cl and alkoxy groups preferred. Significantly, GW5074 demonstrated ≥100-fold selectivity for Raf-1 over several other kinases, including CDK1, CDK2, c-Src, ERK2, MEK, p38, Tie2, VEGFR2, and c-Fms. In a further testament to its selectivity, a cell-based mechanistic assay demonstrated that GW5074 down-regulates EGF-stimulated MAP kinase activity without disrupting the upstream target EGF receptor tyrosine kinase.
The bis-aryl urea Raf-1 inhibitor sorafenib (BAY 43–9006, 2) was also identified through a screening approach that was coupled with a subsequent optimization strategy.[13] The syntheses of 2 and several of its analogues are described in the literature.[14] The initial screen used by researchers at Bayer was an in vitro Raf kinase biochemical assay conducted on many thousands of compounds, and then active compounds (IC50 <500 nM) were subjected to a series of three further screens, ending with a tumour cell-based mechanistic assay. Their most active compound was optimized in a structure–activity relationship (SAR) study, leading to the discovery of the potent small molecule sorafenib (2: IC50 (Raf-1) = 12 nM).[13] Sorafenib is a rather promiscuous inhibitor, blocking the activity of B-Raf (IC50 = 22 nM) and a wide range of protein kinases at nanomolar concentrations (e.g. VEGFR2, PDGF and p38 MAPK). However this is not necessarily an unfavourable result. Indeed, it is now recognized that suppressing more than one target may be necessary in order to realize fully effective anti-cancer chemotherapeutics, either through combination therapies or through the development of multi-kinase inhibitors.[15] Sorafenib suppressed tumour growth in several human tumour xenograft models harbouring K-ras mutations (HCT116 colon carcinoma, MiaPaca-2 pancreatic carcinoma and H460 non-small cell lung carcinoma). Owing to the multi-kinase activity of sorafenib, it is probable that this promising preclinical in vivo data is the result of the inhibition of several kinases in addition to the targeted Raf-1 kinase. Sorafenib was recently approved for the treatment of renal cell carcinoma and hepatocellular carcinoma.[16]
It was around the time of the discovery of sorafenib that B-Raf mutations were observed in melanoma.[6] Owing to differences in their regulation, it now seems that B-Raf is more prone to becoming an oncogenic protein through a single point mutation than is Raf-1.[17,18] The most common B-Raf mutation, which accounts for over 90% of all B-Raf mutations, is a valine to glutamic acid substitution at residue 600 (V600E).[5] The mutant glutamic acid residue is believed to mimic the phosphorylation of T598 or S601, allowing the kinase to fold into its active conformation. Accordingly, second-generation, small molecule inhibitors of wild-type B-Raf and B-RafV600E have subsequently been developed that exhibit better selectivity for the Raf kinases than does sorafenib, as well as demonstrating improved in vivo activity and sustained promising pre-clinical activities. These recently reported inhibitors include Novartis’s Raf265[19,20] (3) and Exelsis’s XL281[21] (structure undisclosed), both of which are currently being evaluated in Phase I clinical trials for malignant melanoma, and PLX-4032[22,23] (4) from Plexxikon / Roche (B-RafV600E: IC50 = 44 nM), which is in Phase I clinical trials for various advanced solid tumours. Notably, PLX-4720 (5), a structural analogue of PLX-4032 that was designed to bind to B-RafV600E, demonstrates a 10-fold selectivity for the B-RafV600E mutant over the wild type in vitro and greater than a 100-fold selectivity in cell proliferation assays.[24] Paradoxically, whilst PLX-4032 inhibited signal transduction through the ERK pathway in cultured melanoma cells harbouring the B-RafV600E mutant, compound PLX-4032 actually induced the pathway in B-RafWT melanoma cells through activation of Raf-1.[25] These findings suggest that only patients known to carry the B-RafV600E mutant should be chosen for treatment with this drug.
MEK1/2 Inhibitors
MEK1/2 are activated by several kinases, including Mos, A-Raf, B-Raf, Raf-1 and MEKK. At the same time, ERK1/2 are the only known substrates of MEK1/2.[1–3] Therefore, inhibition of MEK1/2, rather than the Raf kinases, might offer a more effective approach to blocking signal transduction through the ERK pathway. Accordingly, potent and selective inhibitors of MEK1/2 as potential anti-cancer compounds have been identified. These inhibitors operate either in an ATP-competitive fashion, targeting the ATP-binding site, or in a non-ATP-competitive fashion, binding to an alternative site of MEK1/2 that prevents either its activation (phosphorylation) or the activation of ERK1/2 through blocking the docking of its upstream effectors or downstream substrate, respectively.
ATP-Competitive Inhibitors
Several of the naturally occurring resorcylic acid lactones (RALs), which are polyketides with a large, macrocyclic ring fused to resorcylic acid, have also demonstrated inhibition of MEK1/2. The most active of these contain an α,β-unsaturated ketone in the macrocycle, more specifically a cis-enone functionality. These include hypothemycin (6) and L-783,277 (7), which inhibit MEK1 in vitro with IC50 values of 15 and 4 nM, respectively.[26] The correlation of potent activity with the presence of a cis-enone group suggests the RALs act as Michael acceptors, with an irreversible binding mode of action through conjugation to the nucleophilic sulfhydryl groups of cysteine residues. Indeed, NaBH4-mediated reduction of the α,β-unsaturated ketone of L-783,277 led to a dramatic drop in MEK1 inhibition.[26] Furthermore, the potency of the inhibition of MEK1 by L-783,277 was time-dependent, competitive with ATP, and a reversibility assay demonstrated that MEK1 recovered from after its incubation with L-783,277 no longer exhibited catalytic activity. These data suggest that L-783,277 covalently binds to the MEK1 protein in its ATP-binding site. In fact, a co-crystal structure of hypothemycin with the downstream kinase ERK2 revealed a covalent bond between the small molecule and Cys166 in the ATP-binding site.[27] Given this finding, it is not surprising that these natural products are also potent inhibitors of other protein kinases that have an appropriately placed Cys residue in the ATP-binding site; these include VEGFR1, VEGFR2, PDGFR (and ERK).[27] As noted earlier, it is considered by some that poor kinase specificity may actually yield a more fruitful anti-cancer therapeutic, being capable of knocking out several kinases that are linked to the cancer phenotype. However, in this case, given the inherent instability of these compounds towards Michael addition to the cis-enone functionality, it is unlikely the RALs would progress to clinical trials.
Wyeth-Ayerst have also developed MEK inhibitors, in this case based on a 4-anilino-3-cyanoquinoline core that was identified during the evaluation of compounds as inhibitors of other kinases. The most potent compound identified by this team was 8, with an in vitro IC50 value of 2.4 nM against human recombinant MEK1.[28] The cyanoquinoline core was found to be crucial for MEK inhibitory activity, since its replacement with a quinazoline core led to a substantial loss in activity. Moreover, the most potent inhibitors were furnished through substitution of the 6- and 7-positions with alkoxy groups, whilst substitution at the 8-position completely abolished activity. The location of phenoxy substituents on the aniline ring also proved critical, wherein para-substitution afforded the most potent compound and ortho-substitution gave a compound that was completely devoid of MEK inhibitory activity. Importantly, their most active compounds had little effect on ERK2 (IC50 > 10 µM), as well as on several tyrosine kinases, suggesting very good selectivity for MEK. Compound 8 also demonstrated potent activity on several tumour cell lines, inhibiting proliferation of Colo205, Lovo and SW620 human colon carcinoma cells with IC50 values in the range of 190 – 380 nM.[28]
Non-ATP-Competitive Inhibitors
The first reported MEK small molecule inhibitor was the chromone-based compound PD098059 (9), identified by researchers at Parke-Davis in 1995 upon screening a compound library with an in vitro cascade assay.[29] In this assay, ERK1-catalyzed phosphorylation of MBP (myelin basic protein) was measured in the presence of recombinant ERK1 and recombinant partially activated mutant MEK1 (S218E/S222E-MEK1). Order-of-addition experiments confirmed that PD098059 exerted its inhibitory activity by blocking the activity of MEK1 rather than that of ERK1, with an IC50 value of about 10 µM. Similar inhibitory activity of PD098059 was demonstrated in 3T3 whole cells. Despite precedent for the α,β-unsaturated ketone motif of chromones to participate in Michael addition reactions,[30] it was ascertained that the inhibition of MEK1 by PD098059 was, in fact, reversible, indicating no covalent bond was formed between the small molecule and the protein. Furthermore, PD098059 was found to be an allosteric inhibitor, proving to be non-competitive with respect to both ATP- and ERK1/2-binding. PD098059 was not considered suitable for clinical trials due to its weak inhibitory effect in vitro, poor solubility and inadequate bioavailability.
Shortly after the discovery of PD098059, a library of 40,000 compounds was screened for antagonists of activator protein-1 (AP-1) in a cell-based reporter assay.[31] AP-1 is a transcription factor protein whose activity is regulated by ERK. Subsequently, U0126 (10) was identified, and its AP-1 inhibitory activity was found to be manifested through its inhibition of MEK1 and MEK2 (IC50 values of 72 nM and 58 nM, respectively), and exhibited little, if any, activity against several other protein kinases, including ERK1, JNK and p38 MAP kinase). Favata and co-workers then compared the inhibitory activities of PD098059 and U0126 against a constitutively active recombinant MEK1 (ΔN3-S218E/S222D MEK) and found that U0126 was in excess of 100-fold more potent (IC50 values of 0.07 µM for U0126 versus 10 µM for PD98059).[31] On the other hand, both compounds exhibited weaker inhibition of activated wild type MEK, suggesting the subtle conformational differences between the two activated forms of MEK are significant. Since their binding events were found to be mutually exclusive, it follows that the two MEK inhibitors occupy a common or overlapping binding site(s). Importantly, both PD098059 and U0126 were shown to be non-competitive inhibitors of MEK1/2 with respect to both ATP and ERK. More recent studies have shown that these two small molecules inhibit MEK1/2 activation indirectly by binding to the inactive enzyme and thereby blocking the recruitment of Raf-1 kinase.[32,33]
Given the limited success of PD098059, Pfizer continued searching for alternative MEK1/2 inhibitors and a novel N-(iodophenyl)anthranilic acid derivative was identified. Optimization of this hit led to the discovery of the benzhydroxamate ester CI-1040 (PD184352, 11), which boasted an impressive in vitro IC50 value of 17 nM.[34] Kinetics of binding were shown to be non-competitive with respect to ATP, and this was further corroborated by the solution of X-ray crystal structures of ternary CI-1040:ATP:MEK1/2 complexes, revealing the small molecule to be an allosteric inhibitor and binding MEK1/2 adjacent to the ATP-binding site. In addition to its high potency and selectivity in vitro (a large panel of kinases, including ERK, were not substantially inhibited at 10 µM), CI-1040 exhibited excellent whole cell activity, completely suppressing phosphorylation of ERK at 100 nM in PDGF-stimulated serum-starved C26 murine colon carcinoma cells. With improved pharmacological properties, CI-1040 demonstrated impressive attenuation of tumour growth of human colon tumour xenografts in mice upon oral administration. Significantly, this was associated with a suppression of phosphorylated (activated) ERK1/2. CI-1040 was the first small molecule MEK inhibitor to be evaluated in clinical trials, progressing to Phase II; several cancers were examined, including advanced colorectal, pancreatic and breast cancers.[35] However, owing to its low systemic exposure from its poor solubility and rapid metabolism, CI-1040 demonstrated insufficient anti-tumour activity and thus failed clinical trials.
Chemists at Pfizer began optimizing the diphenylamine core and the hydroxylamine side chain of CI-1040 in order to enhance cell potency and solubility whilst retaining oral bioavailability; their efforts ultimately led to the discovery of PD032591 (12), which inhibited MEK1 and MEK2 with Ki values of 1.1 and 0.79 nM, respectively.[36] In addition to being about 100 times as potent at inhibiting the growth of C26 murine colon cancer cells (CI-1040: IC50 = 35 nM; PD0325901: IC50 = 0.33 nM), second-generation MEK inhibitor PD-0325901 exhibited a greater than 190-fold improvement in solubility. Moreover, in subcutaneously implanted murine C26 tumours in mice, after a single oral administration of 25 mg/kg, PD-0325901 demonstrated improved in vivo activity, as measured by the suppression of activation (phosphorylation) of ERK (pERK) by more than 50% at 24 hours post-dosing. PD-0325901 was undergoing clinical trials for the treatment of breast, colon and non-small cell lung cancers as well as for melanoma,[37] although Pfizer has recently reported that these trials have been terminated due to ocular and neurological toxicities.[38]
ARRY-142886 (AZD6244, 13) is another allosteric, non-ATP-competitive inhibitor of MEK1/2, developed by Array Biopharma and now licensed by AstraZeneca, inhibiting MEK1 with an IC50 of 14 nM, and with a similar activity against the MEK2 isoform.[39] Excellent selectivity of ARRY-142886 was demonstrated by the lack of inhibition of more than 40 other protein serine/threonine and tyrosine kinases at 10 µM concentration, consistent with the observation that the binding of ARRY-142886 is non-competitive with ATP. Moreover, impressive whole cell activity of ARRY-142886 against several human tumour cell lines harbouring B-Raf and Ras mutations has been reported. For example, ARRY-142886 inhibited the cell growth of Malme-3M cells, a melanoma cancer cell line, with an IC50 value of 59 nM. Significantly, ARRY-142886 exhibited minimal effect on other cell lines, including Malme-3, a normal skin fibroblast cell line isolated from the same patient as the Malme-3M cell line, suggesting the activity of ARRY-142886 was not due simply to general cytotoxicity. Upon oral administration, ARRY-142886 inhibited both ERK1/2 phosphorylation and the growth of HT-29 xenograft tumours in mice. ARRY-142886 has completed several Phase I and Phase II clinical trials for the treatment of a number of cancers, including multiple melanoma, hepatocellular carcinoma and advanced solid tumours, with 77% reduction in pERK being documented.[40] Several additional Phase II clinical trials are currently ongoing.[41] However, as of writing this review, it appears that the use of MEK inhibitors as part of an anti-cancer chemotherapy regimen may only be appropriate for patients showing an upregulation of the Ras→Raf→MEK→ERK pathway, since it has been suggested that MEK inhibitors are cytostatic and not cytotoxic.[42]
Ardrea Biosciences has recently disclosed a nanomolar inhibitor of MEK1/2; RDEA119 (BAY869766, 14) is a close analogue of PD0325901 (12) with the most significant difference being the replacement of the hydroxamate group with a sulfonamide group. RDEA119 inhibits MEK1 with an IC50 value of 21 nM and MEK2 with an IC50 value of 50 nM.[43] Furthermore, this compound is highly selective for MEK, demonstrating >100-fold selectivities across a panel of 205 kinases, likely due to its non-ATP-competitive mode of action. RDEA119 is currently undergoing Phase I/II clinical trials for advanced tumours.[44]
Direct Inhibition of ERK
As has been discussed, pharmacological inhibitors of Ras, Raf and MEK1/2 have been shown to block signalling through the ERK pathway, and several of these compounds are currently undergoing clinical trials for suppressing proliferation of cancer cells. However, since the proteins of the ERK pathway are involved in many cellular functions, it may prove more advantageous to inhibit the ERK protein directly given its unique role in the Ras→Raf→MEK/1/2→ERK1/2 pathway, regulating the distribution of upstream signals to its cytosolic and nuclear effectors. Accordingly, over recent years, several groups have met with varying degrees of success in the development of ERK inhibitors, through either targeting the ERK kinase domain to furnish ATP-competitive inhibitors, or through targeting substrate docking domains on the ERK protein surface, yielding non-ATP-competitive inhibitors. Each of these two classes shall be discussed in turn.
ATP-Competitive Inhibitors
The ATP-binding domain is highly conserved across the kinase family, so the development of kinase specificity through targeting this domain is challenging. Nevertheless, the FDA approval of Gleevec demonstrates that achieving kinase specificity is possible, leading to, in this case, a potent anti-cancer compound with limited side effects. Indeed, in recent years, there has been especially promising success in the development of selective ERK inhibitors through targeting its kinase domain.
Ohori et al. conducted a high-throughput phosphorylation assay on an in-house chemical library to identify compounds capable of inhibiting ERK-mediated phosphorylation of MBP. Small molecule FR180204 (15) was subsequently identified as an inhibitor of ERK1 and ERK2 with IC50 values of 0.51 µM and 0.33 µM, respectively.[45] Interestingly, replacement of the 3’-NH2 group of the pyrazolopyridazine ring of FR180204 with the isosteric OH group (compound FR180289 (16)) led to a complete loss in inhibition of ERK1 and ERK2, suggesting the amino group at the 3’ position contributes substantially to the interaction with ERK1/2. As well as exhibiting weaker inhibition for the related kinase p38α with an IC50 value of 10 µM, FR180204 proved very selective for ERK1/2 over a series of other kinases, including MEK1, with no activity at concentrations below 30 µM. This is an impressive result given the fact that a Lineweaver-Burk analysis demonstrated that FR180204 is an ATP-competitive inhibitor. Further biochemical analysis revealed that FR180204 inhibited TGFβ-inhibited AP-1 activation in Mv1Lu cells, suggesting that the small molecule is cell-permeable and that it is capable of blocking transduction through the ERK signalling pathway.
The authors were successful in solving the crystal structure of FR180204 with ERK2; the small molecule was found in the ATP-binding pocket, corroborating the results from the Lineweaver-Burk plot. As shown in Figure 6, FR180204-binding to the ATP domain consists of hydrogen bonding of its pyrazolopyridazine 2’ nitrogen atom to the main chain amide NH of Met108, as well as its 3’ amino group to both the side chain CO of Gln105 and the main chain CO of Asp106, and of the 3-position nitrogen of the pyrazolopyridine ring to the side chain amino group of Lys54. This latter interaction is believed to be particularly significant, since it is missing in crystal structures of ERK2 with weaker inhibitors, and the interaction of Lys54 with the phosphate moiety of ATP is known to be critical to ATP binding. Additionally, this compound was shown to have novel hydrophobic contacts; the domain’s Leu156 and Cys166 residues make CH–π and SH–π interactions with both its pyrazolopyridazine and pyrazolopyridine rings, respectively. It is likely the kinase selectivity that was observed is derived, to some degree, from the hydrogen bond between FR180204 and Gln105, which is referred to as the gatekeeper residue; such residues have a well-established role in determining the selectivity of kinase inhibitors.[46]
Figure 6.
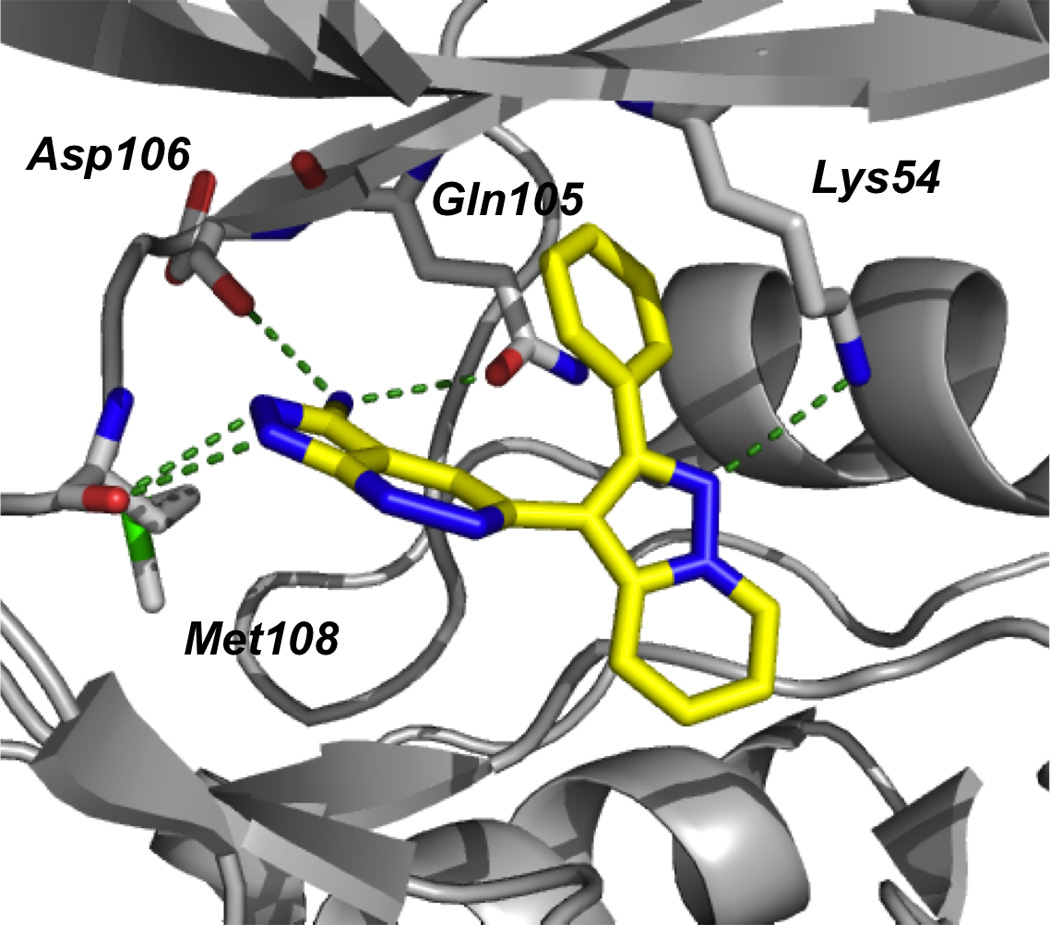
Crystal structure of FR180204 (15) bound in the active site of ERK2 (PDB ID: 1TVO); green dotted lines indicate hydrogen bonds. Image created using the “cartoon” option in PyMOL.[48]
More recently, Ohori and colleagues identified further inhibitors of the ERK2 protein through the use of an enzyme assay to screen compounds isolated from a fermentation culture broth. The most potent ERK2 inhibitor thereby discovered was the resorcylic acid lactone (RAL) FR148083 (17), which suppressed ERK2 activity with an IC50 value of 80 nM.[47] Although FR148083 has been previously reported in the literature for its antibiotic properties under the name LL-Z1640-2, this is the first time its activity against ERK has been documented. TGFβ-induced AP-1-dependent luciferase expression, which proceeds through the ERK and JNK signalling pathways, was also suppressed by compound FR148083 (IC50 = 50 nM). Structure–activity relationship studies of FR148083 revealed that saturation of the double bond at the 1’–2’ positions afforded a 5.5-fold drop in inhibition of ERK2, whilst saturation of both double bonds at the 1’–2’ and 7’–8’ positions led to a complete loss of activity. The fall in activity in both cases is probably due, to some degree, to detrimental changes in the conformation of the 14-membered lactone ring. In the latter case, however, and in light of the fact that hypothemycin forms a covalent bond to ERK2,[27] it is likely that considerable activity has been lost owing to the reduction of the Michael acceptor α,β-unsaturated ketone moiety to a simple ketone, destroying the ability to form covalent bonds and operate as an irreversible inhibitor. Additional SAR information was gained from blockage of the 4’ and 5’ hydroxyl groups as their dimethyl acetonide derivative that led to a 15-fold drop in activity. This suggested either that the hydroxyl groups were engaged in important interactions with the protein, or that the addition of steric bulk in this location was not tolerated.
X-ray crystallography of the ERK2:FR148083 complex revealed the small molecule binds in the ATP pocket by virtue of several hydrogen bonding and hydrophobic interactions. Importantly, and consistent with the findings of Rastelli et al. with the related RAL hypothemycin,[27] a covalent bond was found between the small molecule and the sulfhydryl group of Cys166, confirming that FR148083 operates as an irreversible inhibitor. Indeed, several kinases, including JNK1/2 and p38 isoforms, were not inhibited (IC50 > 10 µM) by FR148083, and it is believed that this is due to a lack of a cysteine residue in the ATP-binding pocket. Instead, these proteins carry leucines at the corresponding position; such residues have no nucleophilic groups to participate in a Michael reaction. Conversely, MEK1 and MKK7, the respective upstream kinases of ERK and JNK, were inhibited by FR148083 with IC50 values of 6 nM and 0.3 µM, respectively, likely owing to the presence of cysteine residues that correspond to Cys166 in ERK2. The absence of cysteine residues appears to assure very good selectivity for ERK2 over several other kinases. However, the authors’ data suggest that the activity of FR148083 does not manifest purely from the covalent bond with the sulfhydryl of Cys166 since the kinase MKK4 also possesses a cysteine residue in the ATP-binding site, yet is not inhibited by this resorcylic acid lactone (IC50 > 3 µM). It is probable that non-covalent interactions assist in positioning the small molecule in the correct orientation to allow the Michael reaction to occur. Nevertheless, small molecule Michael acceptors carry with them a significant caveat in that they can react, rather than more benignly interact, non-specifically with a whole host of proteins and other biomolecules that carry nucleophilic groups, and it is doubtful whether such compounds would reach the clinic.
Researchers at Vertex Pharmaceuticals have also identified potent and selective, ATP-competitive inhibitors of ERK2 (Figure 7).[49] Kinase inhibition by pyrazolylpyrrole-based compounds has previously been reported in the literature, but, upon finding that pyrazolylpyrrole 18 inhibited ERK2 in a high-throughput screen (Ki = 2.3 µM), Aronov et al. began crystallographic studies of 18 complexed to ERK2 in order to optimize potency and selectivity for the ERK2 kinase. Owing to the presence of several, critical hydrogen bonds between the pyrazolylpyrrole amide core of 18 and the ATP-binding site of ERK2, it was decided to keep this part of the small molecule intact for the structure-guided optimization that would follow. In particular, the hydrogen bond between the pyrrole NH and the carboxamide carbonyl of the gatekeeper residue Gln105 was considered especially significant since gatekeeper residues are known to confer selectivity on kinase inhibitors. Varying the phenyl and amide substituents, the latter of which projects into the salt bridge region between Lys54 and Asp167 where there is sufficient room to accommodate such groups, the authors identified a nanomolar inhibitor of ERK2 (19: Ki = 86 nM), although the selectivity profile against the related kinase JNK3 (Ki = 550 nM) was considered inadequate (around six-fold). Thus, crystal structures of 19 complexed with ERK2 and JNK3 were solved in order to assist the authors in optimizing selectivity for the ERK2 kinase. The binding modes of 19 in the two kinases were subsequently revealed to be essentially opposite to each other. This structural data suggested that the addition of a hydrogen bond donor group to the benzylic carbon of 19 would not only be beneficial to ERK2-binding, potentially making hydrogen bonds to Asn154 and Asp167, but would be detrimental to JNK3-binding, since the additional polar group would be result in a steric clash with the main chain atoms of Asp152.
Figure 7.
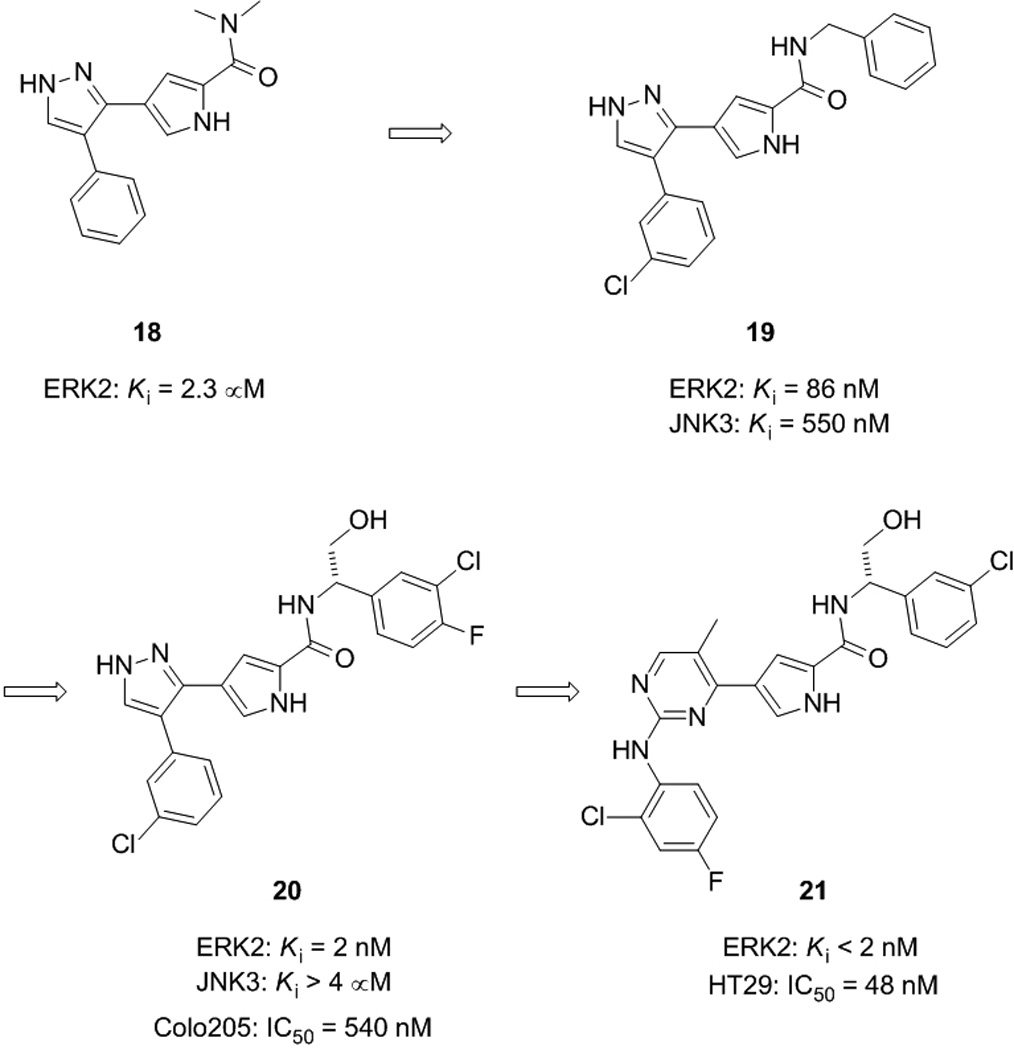
Structural evolution of an especially potent and exquisitely selective ATP-competitive ERK2 inhibitor (20) and its subsequent optimization to enhance whole cell activity, furnishing pyrimidinylpyrrole 21.
In the second round of the SAR program executed by Aronov and colleagues, small molecule 20 was subsequently discovered as an especially potent and selective ERK2 inhibitor.[49] With a significant additional hydroxymethylene moiety at the benzylic position relative to 19, compound 20 inhibited ERK2 with a Ki value of 2 nM with minimal activity against JNK3 (Ki > 4 µM), suggesting a selectivity for ERK2 over JNK3 of at least 2,000-fold. Similar selectivities were seen against MEK1 and p38α, and the smallest selectivity window reported of all the kinases tested was around 200-fold. A final crystal structure of 20 bound to ERK2 revealed all the previous hydrogen bonds had been maintained, with the additional polar hydroxymethylene group forming hydrogen bonds to Asn154 and Asp167, as predicted. These interactions, along with those between the halogens of the small molecule and the glycine-rich loop region of ERK, suggest the rationale behind the potency and selectivity of 20. Undoubtedly, this crystal structure-guided optimization strategy has proven especially successful indeed, and has led, to the best of our knowledge, to the most potent and selective ERK inhibitor currently available. In 2009, the authors reported an extension of their work that resolved the insufficient whole cell activity of pyrazolylpyrrole 20 (IC50 = 540 nM (Colo205 cell proliferation assay) by replacing the pyrazole ring with a pyrimidine. After several rounds of structure-guided optimization, pyrimidinylpyrrole 21, a selective, subnanomolar ERK2 inhibitor with potent cellular activity (IC50 = 48 nM (HT29 cell proliferation assay)), was identified.[50]
Non-ATP-Competitive Inhibitors
Partially due to the expansive and relatively featureless natures of their interfaces, the disruption of protein–protein interactions (PPIs) through binding the docking domain of one protein and physically blocking its interaction with its target protein was, until recently, considered especially daunting.[51] Nevertheless, with the identification of interaction “hot spots” on the protein surface, it is now understood that binding to the entire interfacial region is not necessary to block PPIs effectively, as revealed in a large number of papers over the last decade.[52] As with many signalling cascade proteins, activated ERK phosphorylates its substrates upon initial recruitment through PPIs. In a quest for ERK-specific inhibitors, given the difficulties associated with developing ATP-competitive kinase-specific inhibitors into therapeutic agents, it may prove more beneficial to target the substrate docking region of the ERK protein. In addition, given that ERK has over 50 substrate proteins, such a strategy may yield inhibitors that are specific for selected ERK substrates as opposed to ATP-competitive inhibitors that would inhibit phosphorylation of all substrate proteins. Such inhibitors, as well as being potential therapeutic agents, may also prove useful chemical tools in studying the biological functions of ERK.
In 2005, Shapiro, MacKerell et. al. reported the first small molecular weight non-ATP-competitive ERK inhibitors by use of computer aided drug design (CADD).[53] Previous research had identified the common docking (CD) and ED domains in the C-terminus of the surface of ERK as regions that mediate its PPIs. Interested in developing PPI inhibitors of ERK and, hence, potentially kinase- and substrate-specific inhibitors, Shapiro and colleagues used the program SPHGEN to detect potential binding sites on the surface of the unphosphorylated ERK2 protein. Of the sites identified, those in the vicinity of the CD and ED domains were examined in detail, leading to the discovery of a putative small molecule binding site in between the two domains. Accordingly, given the importance of this region of the protein in ERK2-substrate protein interactions, this cleft was targeted in their CADD in silico screen, after which final compound selection was executed on the basis of Lipinksi’s Rule of 5. Out of 800,000 molecules screened, 80 compounds were thus selected for biological testing, beginning with assays to evaluate levels of ERK specific phosphorylation of its substrate proteins Rsk-1, a serine/threonine kinase, and Elk-1, a transcription factor. Compounds 22 and 23 showed greater than 50% inhibition of Rsk-1 phosphorylation at 100 µM in HeLa (human cervical carcinoma) cells. Moreover, increasing doses of 22 also led to a reduction in ERK-mediated Elk-1 phosphorylation, as measured by immunoblotting analysis. Compound 23 was shown to have little effect on the phosphorylation state of ERK1/2, implying the reductions in ERK substrate phosphorylations was not due to a reduction in active ERK. Furthermore, a cell free kinase assay with the nonspecific substrate MBP demonstrated the catalytic activity of ERK had not been compromised.
Taken together, these data suggest that small molecule 22 is a specific inhibitor of ERK phosphorylation of downstream substrates, whilst exhibiting little effect on its upstream activation by MEK1/2. In fact, fluorescence quenching experiments indicated the biological activity of compound 22 was due to its direct binding to ERK2 (Kd = 5 µM). On the other hand, compound 23 bound poorly to ERK2 (no Kd value given), suggesting its observed biological activity may not be via ERK-specific interactions. Both small molecules 22 and 23 inhibited the proliferation of several cancer cell lines, including HeLa cells with IC50 values of around 15 – 20 µM, and A549 lung carcinoma cells with IC50 values of 25 and 15 µM, respectively. Given the finding that compound 22 binds directly to ERK2, the authors investigated the possible binding mode to the protein. The computational screening applied at the onset of this research predicted that 22 fits into the polar grove located between the CD and ED sites on the ERK2 protein. More specifically, 22 appears to make contacts with several amino acid residues between Asp316 / Asp319 of the CD domain and Thr157 / Thr158 of the ED domain. In particular, as well as a possible cation–π interaction between Arg133 and the phenyl ring of 22, several hydrogen bonds were observed between 22 and Asp316 and Asp319. It is likely that the ammonium group of 22 (protonated at physiological pH) is engaged in salt bridges with Asp316 and Asp319. Indeed, we are currently investigating the significance of the primary amine of 22 by introducing a variety of chemical modifications to reduce the number of polar N-H bonds and to alter the basicity of the nitrogen atom; these results shall be reported in due course.
Zhang and co-workers in 2009 synthesized and biologically evaluated various analogues of compound 22 to identify the pharmacophore responsible for ERK inhibitory activity; their research targeted the phenyl ring, the C=C double bond “spacer” and the ethylamine tail.[54] Again, Rsk1 phosphorylation was employed as a measure of the inhibition of ERK1/2. The primary amino group was found to be critical to activity, since its double reductive amination with formaldehyde to furnish the ethyl(dimethylamine) derivative abolished activity. Replacement of the 4-ethoxyphenyl group with larger, more hydrophobic groups, such as cinnamyl, often enhanced ERK1/2 inhibition but the kinase specificity was reduced, with these compounds exerting varying activities against other kinases, including p38 and MEK1/2. Electron withdrawing groups were not favoured at the 3- or 4- positions of the phenyl ring of 22 since F and NO2 at these locations destroyed inhibition of Rsk1 phosphorylation, whilst a chlorine atom at the 4-position gave similar results to the parent compound 22. The entire ethoxy moiety and its position on the phenyl ring were found to be important factors in influencing the resulting activity, with a shift of the ethoxy group from the 4- to the 2-position furnishing inhibitor 24 that was around three times as active at inhibiting Rsk1 phosphorylation over the parent 22 at 50 µM inhibitor concentration.
Compound 24 also demonstrated improved inhibition of Elk-1 phosphorylation, relative to the parent compound. Moreover, good specificity was maintained since 24 exhibited limited inhibition of p38, MEK and JNK. As in Shapiro’s work with 22, an in vitro cell free kinase assay with active ERK2 and MBP confirmed that the mode of action of 24 was not through blocking the ATP binding site of ERK2 since its catalytic activity was unchanged. Although a whole cell viability assay (MTS) with U937 cells in the presence of 24 indicated no reduction in viability up to a concentration of 30 µM, the small molecule was capable of reducing the cell proliferation rate, as measured by a 60% reduction in the uptake of 3H-thymidine. The authors suggest these findings may be a consequence of the enhanced inhibitory effect of 24 on Rsk1 and Elk1 phosphorylations. In summary, these data advocate the continued investigation into decorating the pharmacophore of 24 with suitable functionalities in order to generate more potent and more specific ERK1/2 inhibitors.
In addition to employing the crystal structure of inactive (non-phosphorylated) ERK, Shapiro, MacKerell and colleagues have also reported the application of CADD to identify PPI inhibitors of active (phosphorylated) ERK, which is the more likely target in rapidly dividing cancer cells.[55] Their initial screen identified many of the same compounds that were identified in their earlier work with inactive (non-phosphorylated) ERK. This finding is consistent with the observation that the 3D crystal structures of the two forms of the protein are similar, including those regions in the vicinity of the CD and ED domains.[56,57] Nevertheless, previously reported differences in deuterium exchange rates between the two forms of the protein in the region containing the CD domain suggests they may harbour subtle differences,[58] and so the authors were hopeful they might identify additional ERK docking domain inhibitors.
Indeed, 45 new compounds were thus identified by conducting a CADD in silico screen with the active structure of ERK2.[55] Of these 45 compounds, 13 were available in the Chembridge chemical library; these were purchased and evaluated further in biological assays. At concentrations of 100 µM, compounds 25 and 26 proved the most effective inhibitors of ERK protein–protein interactions, suppressing phosphorylation of Elk-1 by around 70% or more and that of Rsk-1 by around 50%. Moreover, the entire set of 13 compounds displayed good specificity for ERK since none of the test compounds inhibited p38 MAP kinase, nor was the phosphorylation of ERK1/2 affected, suggesting these compounds do not inhibit MEK1/2 and nor do they block MEK–ERK protein–protein interactions. Small molecules 25 and 26 also inhibited proliferation of HeLa cells with IC50 values of about 5 µM and 10 – 25 µM, respectively. Finally, fluorescence quenching experiments with the ERK2 protein in vitro suggested these compounds mediate their biological effects through binding directly to the ERK2 protein with similar affinities (25: Kd = 13 µM; 26: Kd = 16 µM). While still ongoing, these studies[53,55] indicate the ability of CADD-directed drug discovery to identify non-ATP competitive inhibitors of ERK1/2. Future efforts will build on these successes with the goal of identifying a collection of inhibitors that inhibit different ERK–substrate protein interactions, making them of utility for chemical biology studies of the biological function of ERK as was as possibly being developed into novel therapeutic agents.
Outlook
Efforts to develop inhibitors of ERK1/2 as well as a collection of other kinases have lead to the identification of both ATP-competitive and non-ATP competitive inhibitors. These studies have led to the development of therapeutic agents, most notably Gleevec, that are currently in the clinic. As more knowledge of the biological activities of these inhibitors, including their specificities, and the structures of the inhibitor–kinase complexes is gained, it may be anticipated that more rational approaches to the design of kinase inhibitors with varying degrees of specificity will be developed. Towards this goal, structure-guided optimization and CADD methods in combination with medicinal chemistry and comprehensive biological assays offer great potential.
Figure 2.
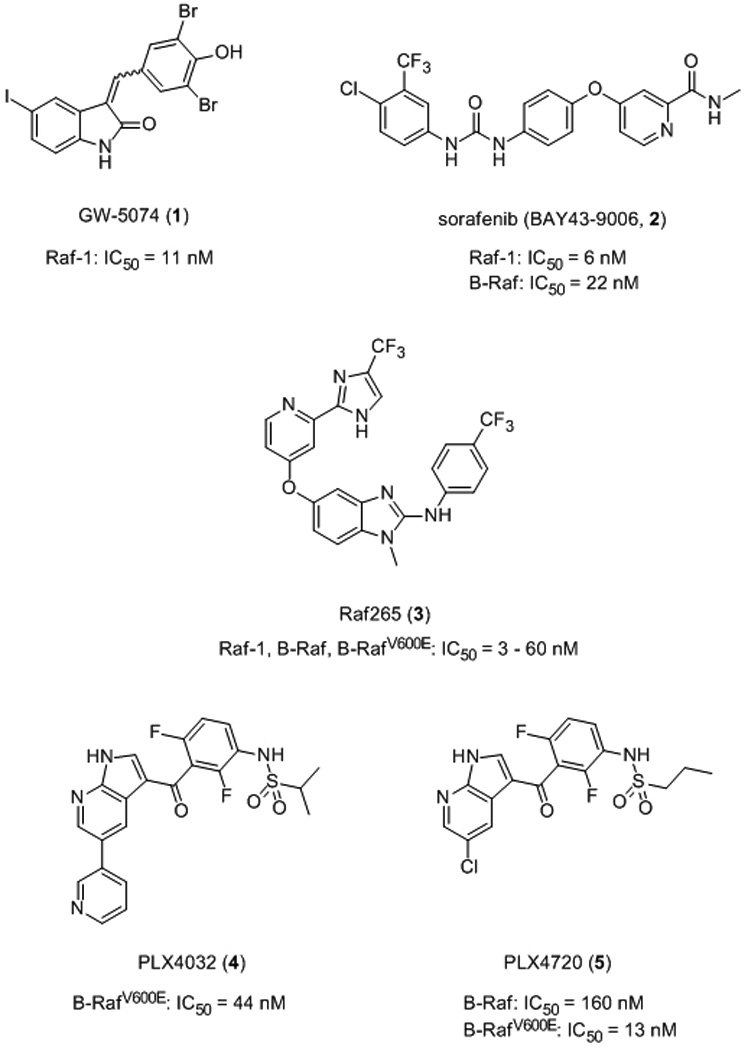
Raf inhibitors.
Figure 3.
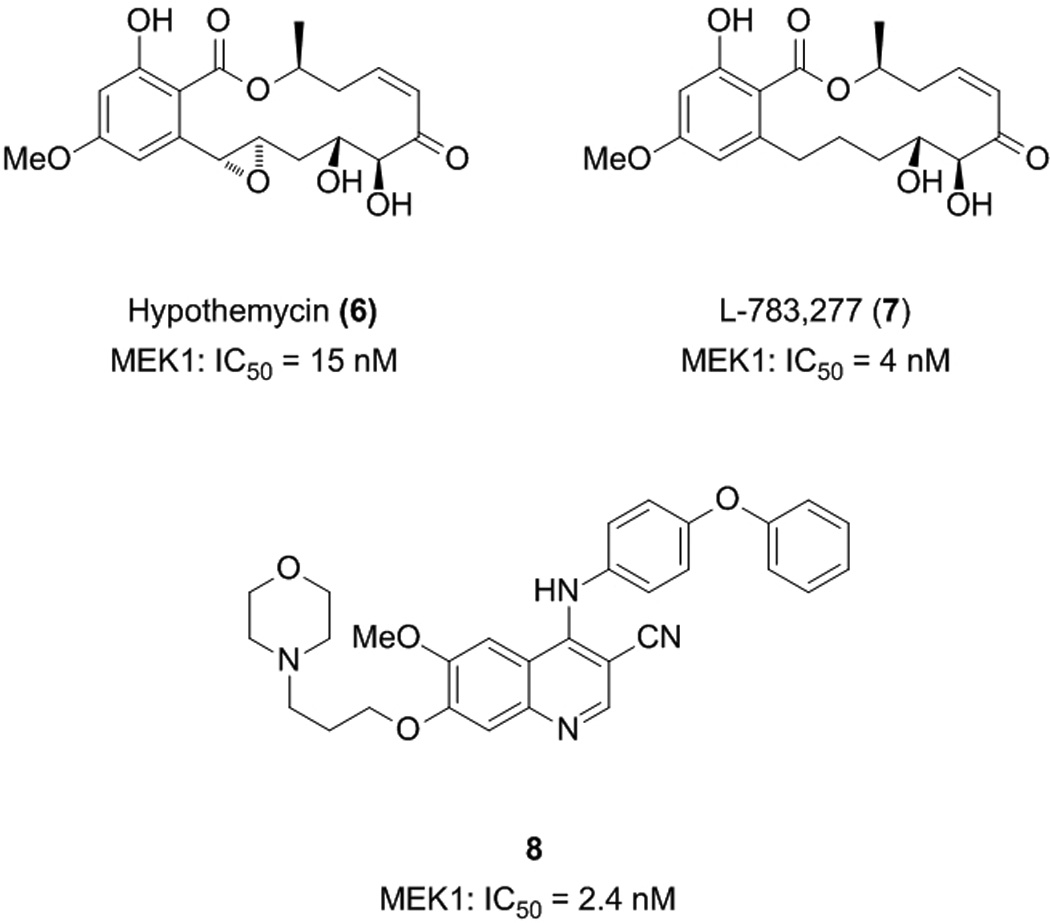
ATP-competitive MEK1/2 inhibitors.
Figure 4.
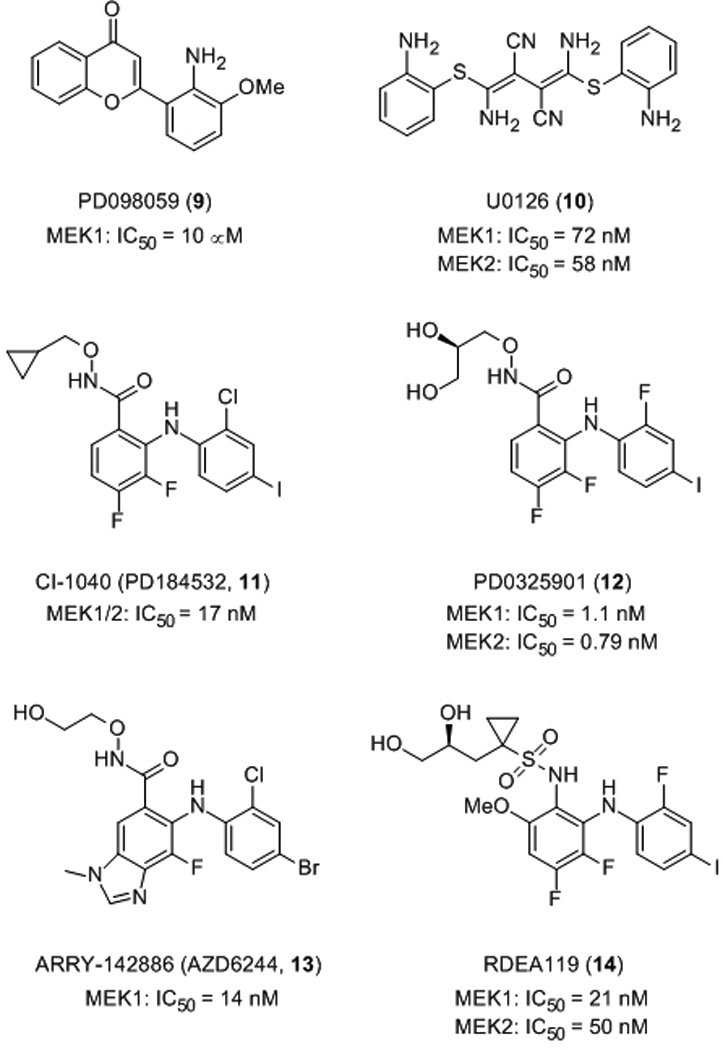
Non-ATP-competitive MEK1/2 inhibitors.
Figure 5.
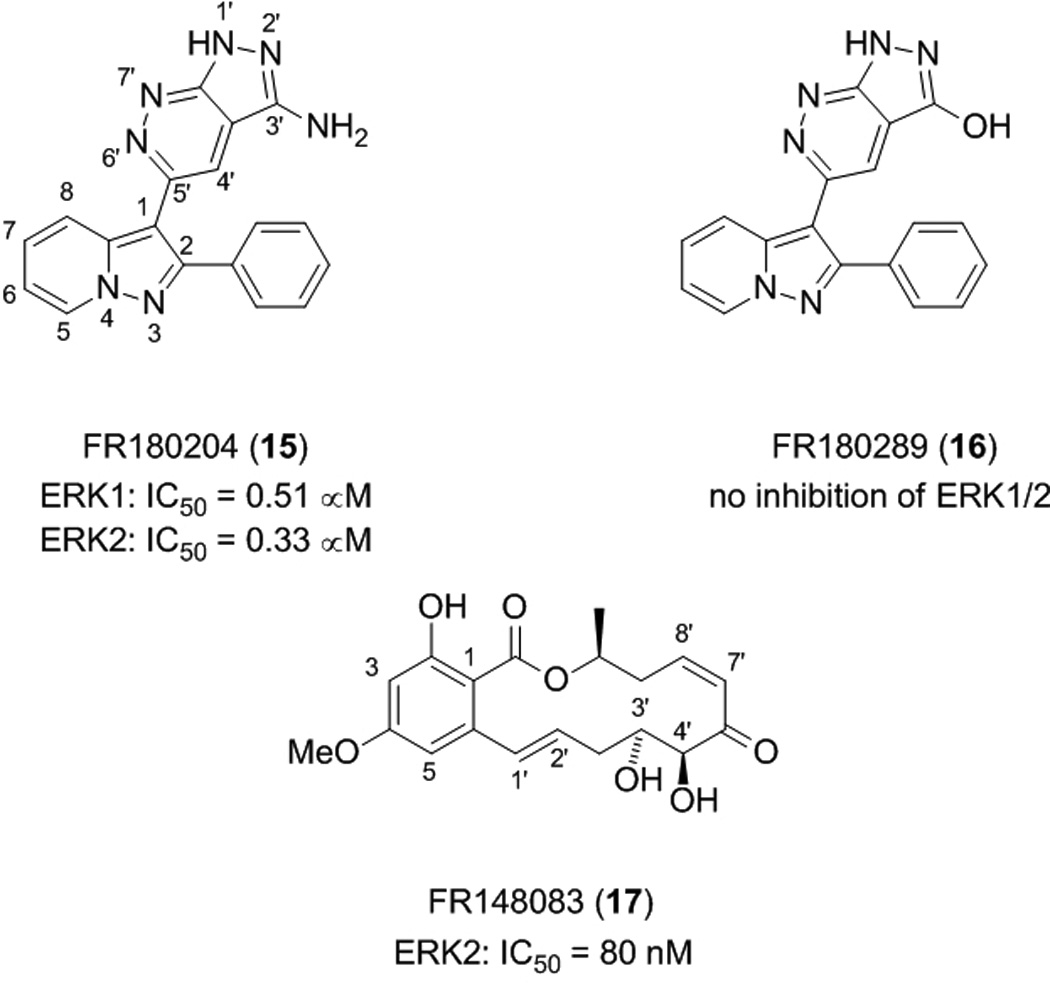
ATP-competitive ERK1/2 inhibitors.
Figure 8.
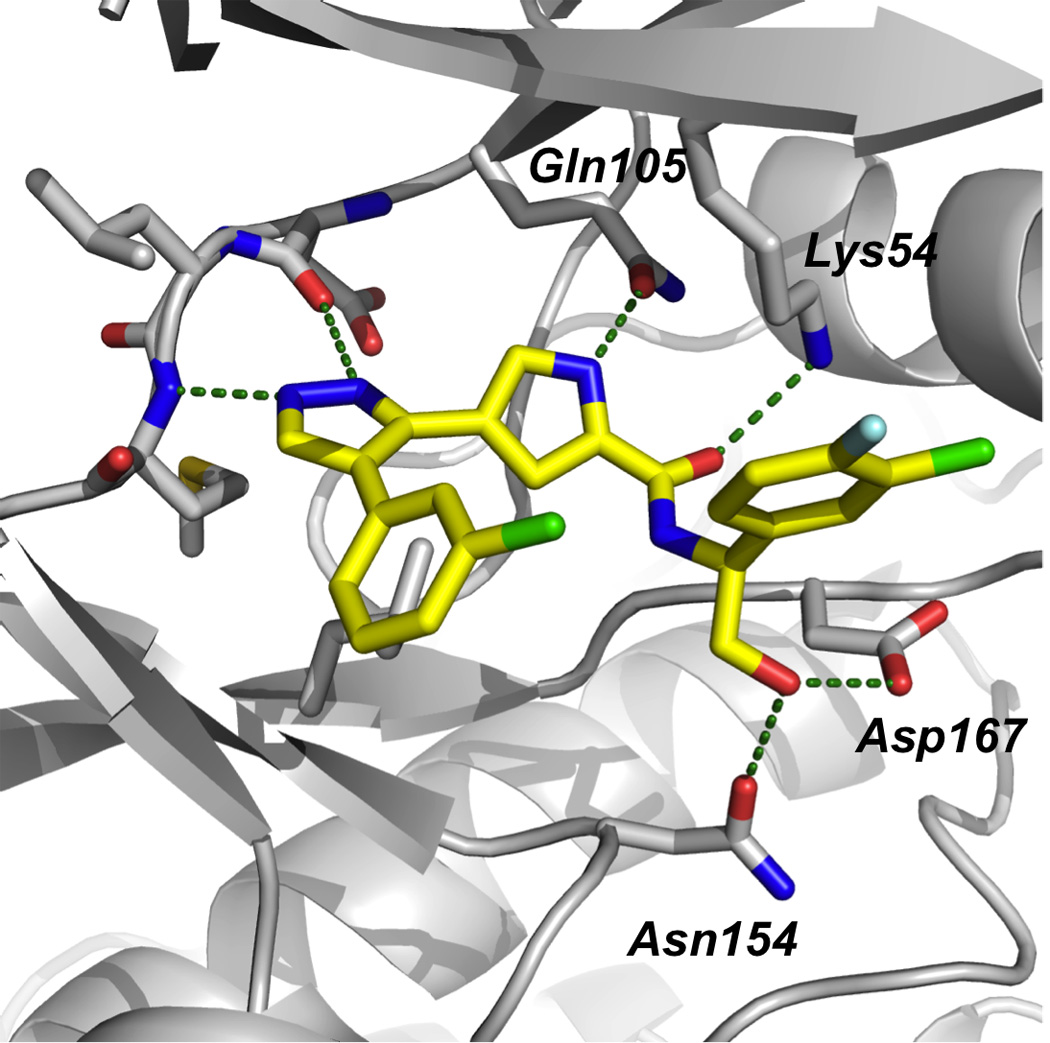
Crystal structure of compound 20 bound in the active site of ERK2 (PDB ID: 2OJJ); green dotted lines indicate hydrogen bonds. Image reproduced with permission from reference 49.
Figure 9.
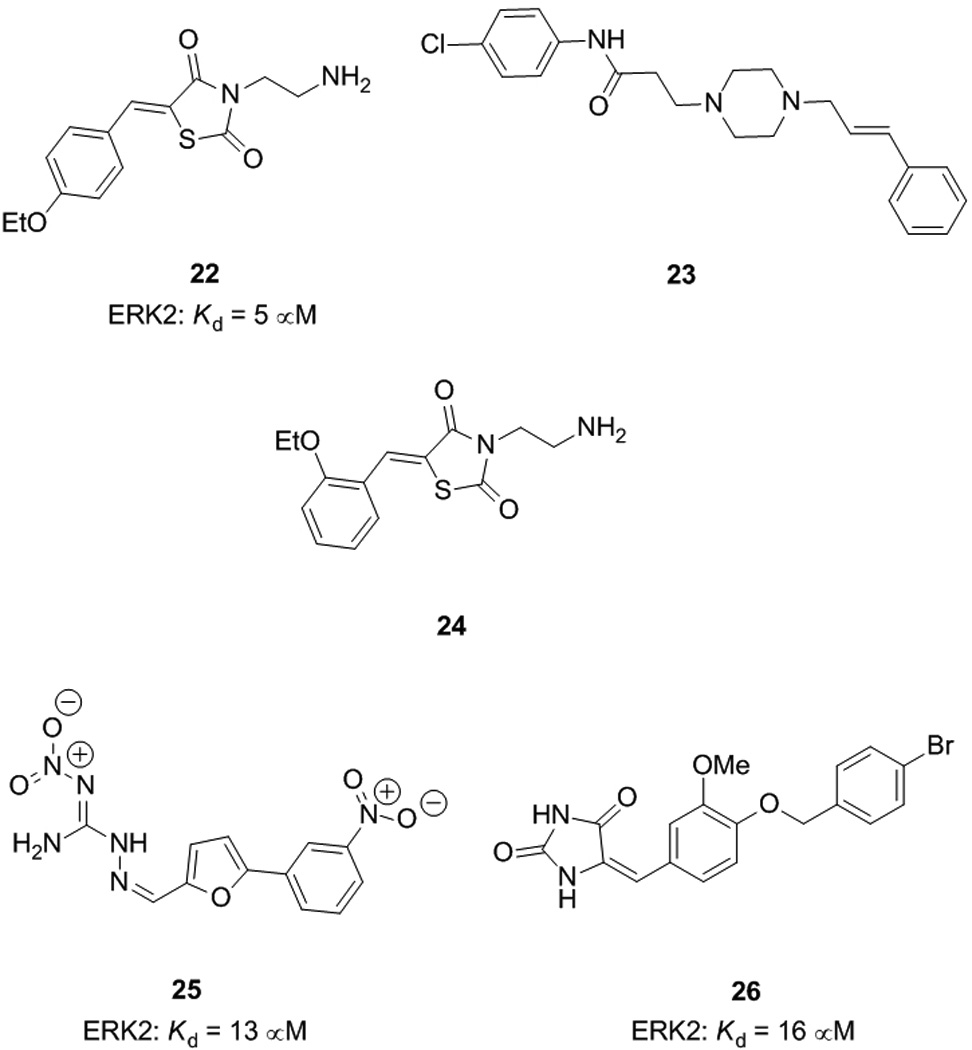
Non-ATP-competitive ERK1/2 inhibitors.
Acknowledgements
We thank the University of Maryland School of Pharmacy (SF), the University of Maryland Computer-Aided Drug Design Center and NIH grant CA120215 (ADM and PS) for financial support of our work in this area.
References
- 1.Seger R, Krebs EG. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- 2.Lewis TS, Shapiro PS, Ahn NG. Adv. Cancer Res. 1998;74:49–139. doi: 10.1016/s0065-230x(08)60765-4. [DOI] [PubMed] [Google Scholar]
- 3.Pearson G, Robinson F, Gibson TB, Xu B-E, Karandikar M, Berman K, Cobb MH. Endocr. Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 4.Bos JL. Cancer Res. 1989;49:4682–4689. [PubMed] [Google Scholar]
- 5.Garnett MJ, Marais R. Cancer Cell. 2004;6:313–319. doi: 10.1016/j.ccr.2004.09.022. [DOI] [PubMed] [Google Scholar]
- 6.Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmier G, Cossu A, Flanagan A, Nicholson A, Ho JWC, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal A. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- 7.Hoshino R, Chatani Y, Yamori T, Tsuruo T, Oka H, Yoshida O, Shimada Y, Ari-i S, Wada H, Fujimoto J, Kohno M. Oncogene. 1999;18:813–822. doi: 10.1038/sj.onc.1202367. [DOI] [PubMed] [Google Scholar]
- 8.Bell I. J. Med. Chem. 2004;47:1869–1878. doi: 10.1021/jm0305467. [DOI] [PubMed] [Google Scholar]
- 9.Wellbrock C, Karasarides M, Marais R. Nat. Rev. Mol. Cell Biol. 2004;5:875–885. doi: 10.1038/nrm1498. [DOI] [PubMed] [Google Scholar]
- 10.Lackey K, Cory M, Davis R, Frye SV, Harris PA, Hunter RN, Jung DK, McDonald OB, McNutt RW, Peel MR, Rutkowske RD, Veal JM, Wood ER. Bioorg. Med. Chem. Lett. 2000;10:223–226. doi: 10.1016/s0960-894x(99)00668-x. [DOI] [PubMed] [Google Scholar]
- 11.Shiraishi T, Domoto T, Imai N, Shimada Y, Watanabe K. Biochem. Biophys. Res. Commun. 1987;147:322–328. doi: 10.1016/s0006-291x(87)80124-9. [DOI] [PubMed] [Google Scholar]
- 12.Kim S-H, Schulze-Gahmen U, Brandsen J, de Azevedo WF. Prog. Cell Cycle Res. 1996;2:137–145. doi: 10.1007/978-1-4615-5873-6_14. [DOI] [PubMed] [Google Scholar]
- 13.Lyons JF, Wilhelm SM, Hibner B, Bollag G. Endocr. Relat. Cancer. 2001;8:219–225. doi: 10.1677/erc.0.0080219. [DOI] [PubMed] [Google Scholar]
- 14.Khire UR, Bankston D, Barbosa J, Brittelli DR, Caringal Y, Carlson R, Dumas J, Gane T, Heald SL, Hibner B, Johnson JS, Katz ME, Kennure N, Kingery-Wood J, Lee W, Liu X-G, Lowinger TB, McAlexander I, Monahan M-K, Natero R, Renick J, Riedl B, Rong H, Sibley RN, Smith RA, Wolanina D. Bioorg. Med. Chem. Lett. 2004;14:783–786. doi: 10.1016/j.bmcl.2003.11.041. [DOI] [PubMed] [Google Scholar]
- 15.Ghoreschi K, Laurence A, O’Shea JJ. Nat. Immunol. 2009;10:356–360. doi: 10.1038/ni.1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. http://www.cancer.gov/cancertopics/druginfo/fda-sorafenib-tosylate.
- 17.Wan PT, Garnett MJ, Roe SM, Lee S, Niculescu-Duvaz D, Good VM, et al. Cell. 2004;116:855–867. doi: 10.1016/s0092-8674(04)00215-6. [DOI] [PubMed] [Google Scholar]
- 18.Emuss V, Garnett M, Mason C, Marais R. Cancer Res. 2005;65:9719–9726. doi: 10.1158/0008-5472.CAN-05-1683. [DOI] [PubMed] [Google Scholar]
- 19. http://www.novartisoncology.com:80/research-i.nnovation/pipeline/raf265.jsp. [Google Scholar]
- 20.Amiri P, Aikawa ME, Dove J, Stuart DD, Poon D, Pick T, Ramurthy S, Subramanian S, Levine B, Costales A, Harris A, Paul R. Proc. Am. Assoc. Cancer Res. 2006;47 Abstract 4855. [Google Scholar]
- 21. http://www.exelixis.com/pipeline/xl281. [Google Scholar]
- 22.Sala E, Mologni L, Truffa S, Gaetano C, Bollag GE, Gambacorti-Passerini C. Mol. Cancer Res. 2008;6:751–759. doi: 10.1158/1541-7786.MCR-07-2001. [DOI] [PubMed] [Google Scholar]
- 23.Flaherty K, Puzanov I, Sosman J, Kim K, Ribas A, McArthur G, Lee RJ, Grippo JF, Nolop K, Chapman P. J. Clin. Oncol. 2009;25:15s. doi: 10.1200/JCO.2011.39.1938. Abstract 9000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, Bremer R, Gillette S, Kong J, Haass NK, Sproesser K, Li L, Smalley KSM, Fong D, Zhu Y-L, Marimuthu A, Nguyen H, Lam B, Liu J, Cheung I, Rice J, Suzuki Y, Luu C, Settachatgul C, Shellooe R, Cantwell J, Kim S-H, Schlessinger J, Zhang KYJ, West BL, Powell B, Habets G, Zhang C, Ibrahim PN, Hirth P, Artis DR, Herlyn M, Bollag G. Proc. Natl. Acad. Sci. USA. 2008;105:3041–3046. doi: 10.1073/pnas.0711741105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Halaban R, Zhang W, Bacchiocchi A, Cheng E, Parisi F, Ariyan S, Krauthammer M, McCusker JP, Kluger Y, Sznol M. Pigment Cell Melanoma Res. 2010;23:190–200. doi: 10.1111/j.1755-148X.2010.00685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao A, Lee SH, Mojena M, Jenkins RG, Patrick DR, Huber HE, Goetz MA, Hensens OD, Zink DL, Vilella D, Dombrowski AW, Lingham RB, Huang L. J. Antibiot. 1999;52:1086–1094. doi: 10.7164/antibiotics.52.1086. [DOI] [PubMed] [Google Scholar]
- 27.Rastelli G, Rosenfeld R, Reid R, Santi DV. J. Struct. Biol. 2008;164:18–23. doi: 10.1016/j.jsb.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Wyeth N, Wu B, Powell D, Wissner A, Floyd MB, Kovacs ED, Toral-Barza L, Kohler C. Bioorg. Med. Chem. Lett. 2000;10:2825–2828. doi: 10.1016/s0960-894x(00)00580-1. [DOI] [PubMed] [Google Scholar]
- 29.Dudley DT, Pang L, Decker SJ, Bridges AJ, Saltiel AR. Proc. Natl. Acad. Sci. USA. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Eicher T, Hauptmann S, Speicher A. The Chemistry of Heterocycles. 2nd edition. Wiley-VCH; 2003. pp. 261–266. [Google Scholar]
- 31.Favata MF, Horiuchi KY, Manos EJ, Daulerio AJ, Stradley DA, Feeser WS, Van Dyk DE, Pitts WJ, Earl RA, Hobbs F, Copeland RA, Magolda RL, Scherle PA, Trzaskos JM. J. Biol. Chem. 1998;273:18623–18632. doi: 10.1074/jbc.273.29.18623. [DOI] [PubMed] [Google Scholar]
- 32.Davies SP, Reddy H, Caivano M, Cohen P. Biochem. J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ahn NG, Nahreini TS, Tolwinski NS, Resing KA. Methods Enzymol. 2001;332:417–431. doi: 10.1016/s0076-6879(01)32219-x. [DOI] [PubMed] [Google Scholar]
- 34.Sebolt-Leoopold JS, Dudley DT, Herrera R, Van Becelaera K, Wiland A, Gowan RC, Tecle H, Barrett SD, Bridges A, Przybranowski S, Leopold WR, Saltiel AR. Nat. Med. 1999;5:810–816. doi: 10.1038/10533. [DOI] [PubMed] [Google Scholar]
- 35.Rinehart J, Adjei AA, LoRusso PM, Waterhouse D, Hecht JR, Natale RB, Hamid O, Varterasian M, Asbury P, Kaldjian EP, Gulyas S, Mitchell DY, Herrera R, Sebolt-Leopold JS, Meyer MB. J. Clin. Oncol. 2004;22:4456–4462. doi: 10.1200/JCO.2004.01.185. [DOI] [PubMed] [Google Scholar]
- 36.Barrett SD, Bridges AJ, Dudley DT, Saltiel AR, Fergus JH, Flamme CM, Delaney AM, Kaufman M, LePage S, Leopold WR, Przybranowski SA, Sebolt-Leopold J, Van Becelaere K, Doherty AM, Kennedy RM, Marston D, Howard WA, Jr, Smith Y, Warmus JS, Tecle H. Bioorg. Med. Chem. Lett. 2008;18:6501–6504. doi: 10.1016/j.bmcl.2008.10.054. [DOI] [PubMed] [Google Scholar]
- 37.Sebolt-Leopold JS. Clin. Cancer Res. 2008;14:3651–3656. doi: 10.1158/1078-0432.CCR-08-0333. [DOI] [PubMed] [Google Scholar]
- 38. http://clinicaltrials.gov/ct2/show/NCT00174369.
- 39.Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, Parry J, Smith D, Brandhuber BJ, Gross S, Marlow A, Hurley B, Lyssikatos J, Lee PA, Winkler JD, Koch K, Wallace E. Clin. Cancer Res. 2007;13:1576–1583. doi: 10.1158/1078-0432.CCR-06-1150. [DOI] [PubMed] [Google Scholar]
- 40. http://clinicaltrials.gov/ct2/show/NCT00372788?term=AZD6244&rank=10; http://clinicaltrials.gov/ct2/show/NCT00710515?term=AZD6244&rank=11; http://clinicaltrials.gov/ct2/show/NCT00514761?term=AZD6244&rank=15.
- 41. http://clinicaltrials.gov/ct2/results?term=AZD6244&pg=1.
- 42.Milella M, Korblau S, Estrov Z, Carter B, Lapillone H, Harris D, Konopleva M, Zhao S, Estey E, Andreeff M. J. Clin. Invest. 2001;108:851–859. doi: 10.1172/JCI12807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iverson C, Larson G, Lai C, Yeh L-T, Dadson C, Weingarten P, Appleby T, Vo T, Maderna A, Vernier J-M, Hamatake R, Miner JN, Quart B. Cancer Res. 2009;69:6839–6847. doi: 10.1158/0008-5472.CAN-09-0679. [DOI] [PubMed] [Google Scholar]
- 44. http://clinicaltrials.gov/ct2/results?term=rdea119.
- 45.Ohori M, Kinoshita T, Okubo M, Sato K, Yamazaki A, Arakawa H, Nishimura S, Inamura N, Nakajima H, Neya M, Miyake H, Fujii T. Biochem. Biophys. Res. Commun. 2005;336:357–363. doi: 10.1016/j.bbrc.2005.08.082. [DOI] [PubMed] [Google Scholar]
- 46.Vieth M, Higgs RE, Robertson DH, Shapiro M, Gragg EA, Hemmerle H. Biochim. Biophys. Acta. 2004;1697:243–257. doi: 10.1016/j.bbapap.2003.11.028. [DOI] [PubMed] [Google Scholar]
- 47.Ohori M, Kinoshita T, Yoshimura S, Warizaya M, Nakajima H, Miyake H. Biochem. Biophys. Res. Commun. 2007;353:633–637. doi: 10.1016/j.bbrc.2006.12.083. [DOI] [PubMed] [Google Scholar]
- 48.DeLano WL. San Carlos, CA, USA: DeLano Scientific; 2002. [Google Scholar]
- 49.Aronov AM, Baker C, Bemis GW, Cao J, Chen G, Ford PJ, Germann UA, Green J, Hale MR, Jacobs M, Janetka JW, Maltais F, Martinez-Botella G, Namchuk MN, Straub J, Tang Q, Xie X. J. Med. Chem. 2007;50:1280–1287. doi: 10.1021/jm061381f. [DOI] [PubMed] [Google Scholar]
- 50.Aronov AM, Tang Q, Martinez-Botella G, Bemis GW, Cao J, Chen G, Ewing NP, Ford PJ, Germann UA, Green J, Hale MR, Jacobs M, Janetka JW, Maltais F, Markland W, Namchuk MN, Nanthakumar S, Poondru S, Straub J, ter Haar E, Xie X. J. Med. Chem. 2009;52:6362–6368. doi: 10.1021/jm900630q. [DOI] [PubMed] [Google Scholar]
- 51.Juliano RL, Astriab-Fisher A, Falke D. Mol. Interv. 2001;1:40–53. [PubMed] [Google Scholar]
- 52.(a) Fletcher S, Hamilton AD. Curr. Opin. Chem. Biol. 2005;9:632–638. doi: 10.1016/j.cbpa.2005.10.006. [DOI] [PubMed] [Google Scholar]; (b) Fletcher S, Hamilton AD. JR. Soc. Interface. 2006;3:215–233. doi: 10.1098/rsif.2006.0115. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Fletcher S, Hamilton AD. Curr. Top. Med. Chem. 2007;7:922–927. doi: 10.2174/156802607780906735. [DOI] [PubMed] [Google Scholar]
- 53.Hancock CN, Macias A, Lee E Kyoung, Yu Su Yeon, MacKerell Alexander D, Jr, Shapiro P. J. Med. Chem. 2005;48:4586–4595. doi: 10.1021/jm0501174. [DOI] [PubMed] [Google Scholar]
- 54.Li Q, Al-Ayoubi A, Guo T, Zheng H, Sarkar A, Nguyen T, Eblen ST, Grant S, Kellogg GE, Zhang S. Bioorg. Med. Chem. Lett. 2009;19:6042–6046. doi: 10.1016/j.bmcl.2009.09.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen F, Hancock CN, Macias AT, Joh J, Still K, Zhong S, Mackerell AD, Jr, Shapiro P. Bioorg. Med. Chem. Lett. 2006;16:6281–6287. doi: 10.1016/j.bmcl.2006.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang F, Strand A, Robbins D, Cobb MH, Goldsmith EJ. Nature. 1994;367:704–711. doi: 10.1038/367704a0. [DOI] [PubMed] [Google Scholar]
- 57.Canagarajah BJ, Khokhlatchev A, Cobb MH, Goldsmith EJ. Cell. 1997;90:859–869. doi: 10.1016/s0092-8674(00)80351-7. [DOI] [PubMed] [Google Scholar]
- 58.Hoofnagle AN, Resing KA, Goldsmith EJ, Ahn NG. Proc. Natl. Acad. Sci. USA. 2001;98:956–961. doi: 10.1073/pnas.98.3.956. [DOI] [PMC free article] [PubMed] [Google Scholar]