

Abstract
The rates of oxidation of fatty acids by CYP119 Compound I were dependent on the pH of the medium. The plot shows log k for reactions of acids as a function of pH, where the slopes indicate mixed third-order and fourth-order dependence on base concentration. For palmitic acid, the rate increased by a factor of 50 over the pH range of 6.8 to 7.3.
Keywords: cytochrome P450, oxidation, fatty acids, kinetics, regioselectivity
Cytochrome P450 enzymes (CYPs or P450s) are ubiquitous heme-containing enzymes with protein thiolate from cysteine serving as the fifth ligand to iron.[1] Most P450s catalyze oxidation reactions, and hepatic P450s oxidize many xenobiotics as well as most of the drugs used by humans.[2] The active oxidants in P450s are widely thought to be oxoiron(IV) porphyrin radical cations analogous to the so-called Compound I transients formed by reactions of peroxidase and catalase enzymes with hydrogen peroxide.[3] P450 Compounds I have not been observed under turnover conditions, but they can be prepared by “shunt reactions” where resting P450s are oxidized by hydroperoxy species. Shunt reactions were implicated early in P450 studies,[4] and spectroscopic evidence that Compound I transients were formed in these reactions accumulated over the past two decades.[5] The best characterized P450 Compound I is that formed by reaction of CYP119 with m-chloroperoxybenzoic acid (MCPBA).[5b, 5d]
Much research has focused on understanding P450-catalyzed hydroxylations,[6] but direct kinetic studies of P450 Compounds I are in their infancy. P450-catalyzed hydroxylations of fatty acids, often at unactivated C-H positions, have a special distinction in that the search for fatty acid hydroxylase enzymes drove research that led to the discovery of one of the first characterized P450 enzymes.[7] Recently, we reported that rate constants for fatty acid oxidations by CYP119 Compound I were dependent on glycol co-solvent concentrations.[8] We now report that both the rates and regioselectivities of fatty acid oxidations by CYP119 Compound I are highly dependent on pH, further implicating a rate-determining conformational changes in the complex of Compound I with substrate for long chain fatty acids.
Kinetic studies of dodecanoic (lauric) acid oxidations by CYP119 Compound I purportedly under the same reaction conditions gave different rate constants. Rittle and Green found that the reaction was so fast that only an apparent second-order rate constant could be obtained,[5d] whereas we found that saturation kinetics could be observed leading to a distinct binding constant and first-order rate constant.[8] Given the obvious environment effect we recently observed and the evidence that the rate-determining step involved a conformational change, we explored the possibility that pH effects influenced the kinetics of fatty acid oxidations.
The kinetics of reactions of octanoic (caprylic) acid, dodecanoic (lauric) acid, and hexadecanoic (palmitic) acid with CYP119 Compound I were studied at varying pH using the stopped-flow kinetic method employed previously.[5d, 8] In a four-syringe stopped-flow kinetic unit, Compound I was produced by oxidation of the ferric enzyme with MCPBA, aged for 100 ms, and mixed with substrate. The rate of formation of ferric enzyme as Compound I reacted was followed. Details of the method have been reported[8–9] and are summarized in the Supporting Information. Results are in Table 1. When pH < 7.4, we observed curvature in the kinetic plots indicating saturation kinetics with reversible formation of a complex and rate-limiting first-order reaction of the complex. At pH 7.4, the rates increased so rapidly with concentration that we were not able to observe curvature in the plots.
Table 1.
Rate constants for reactions of fatty acids with CYP119 Compound I.[a]
| Substrate | pH | Kbind (M−1) | kexp (s−1) | KIE[b] |
|---|---|---|---|---|
| Octanoic acid[c] | 7.0 | (4.3 ± 0.3) × 103 | 6.9 ± 0.2 | 2.3 ± 0.2 |
| 7.2 | (6.7 ± 0.8) × 103 | 33 ± 2 | 3.3 ± 0.4 | |
| 7.3 | (8.9 ± 0.4) × 103 | 69 ± 1 | 4.5 ± 0.2 | |
| Dodecanoic acid[c] | 7.0 | (2.1 ± 0.2) × 104 | 33 ± 2 | 1.00 ± 0.2 |
| 7.1 | (3.1 ± 0.4) × 104 | 45 ± 2 | ||
| 7.2 | (4.1 ± 0.6) × 104 | 199 ± 6 | ||
| 7.3 | (4.5 ± 0.3) × 104 | 269 ± 8 | 0.92 ± 0.10 | |
| Hexadecanoic acid | 6.8 | (3.3 ± 0.3) × 104 | 10.3 ± 0.3 | 1.07 ± 0.08 |
| 6.9 | (4.0 ± 0.4) × 104 | 19.8 ± 0.7 | 1.03 ± 0.08 | |
| 7.0 | (4.4 ± 0.2) × 104 | 52 ± 1 | 1.02 ± 0.03 | |
| 7.1 | (4.6 ± 0.3) × 104 | 117 ± 3 | 1.02 ± 0.12 | |
| 7.2 | (5.0 ± 0.2) × 104 | 241 ± 4 | 1.03 ± 0.04 | |
| 7.3 | (5.8 ± 0.6) × 104 | 514 ± 25 | 1.03 ± 0.16 |
Reactions in 100 mM phosphate buffer at 4 °C with errors at 1σ.
Kinetic isotope effect in kexp. with errors at 2σ.
At pH 7.4, apparent second-order rate constants were kapp = (4.9 ± 0.2) × 105 M−1 s−1 for octanoic acid and kapp = (1.2 ± 0.1) × 107 M−1 s−1 for dodecanoic acid.
Perdeuterated acids were studied for comparison to several of the results with non-deuterated acids, and the kinetic isotope effects (KIEs) in the first-order rate constants are listed in Table 1. Perdeuterated octanoic acid reacted less rapidly than the non-deuterated isotopomer giving modest KIEs. For dodecanoic acid and hexadecanoic acid, the perdeuterated isotopomers reacted with the same rate constants as the non-deuterated substrates. The saturation kinetics data requires that a complex of enzyme and substrate is formed reversibly, but the small or absent KIEs indicate that the first-formed complex is non-reactive and must isomerize to a reactive form (see Supporting Information). Such a model for reactions of CYP119 Compound I was presented previously.[5d, 8]
Because the KIEs for dodecanoic acid and hexadecanoic acid are 1.0 at all pH, the oxidation rate constants for the deuterated acids are greater than the rate-limiting isomerization rate constant. Using a KIE of kH/kD ≈ 7 as found for intramolecular KIEs for dodecanoic and hexadecanoic acids,[8] the oxidation rate constant for hexadecanoic acid at pH 7.3 would be greater than 3500 s−1. Further assuming zero entropy of activation for the first-order process, the activation energy for the reaction at 4 °C would be less than 12 kcal/mol, a remarkably low activation energy for functionalization of unactivated C-H bonds. For comparison, Bell and Groves recently reported a highly reactive model for P450 Compound I,[10] and extrapolation of their Brønsted-Evans-Polanyi relationship predicts that their model would react with an unactivated C-H bond at 10 °C with a rate constant of 0.01 M−1 s−1 or activation energy of about 15 kcal/mol.
The pH effect on the kinetics found here explains diverse results previously reported for reactions of dodecanoic acid with CYP119 Compound I. Rittle and Green found that reaction of dodecanoic acid in 100 mM phosphate buffer at 4 °C was too fast to observe saturation kinetics and reported an apparent second-order rate constant of kapp = 1.2 × 107 M−1 s−1 at a reported pH 7.[5d] We found that the reaction at pH 7.0 displayed saturation kinetics, but the reaction at pH 7.4 studied here has exactly the same rate constant as found by Rittle and Green. In a similar manner, the apparent rate constant for reaction of octanoic acid previously reported was kapp = 4.5 × 105 M−1 s−1 at pH 7,[5d] and we found a similar rate constant at pH 7.4. Thus, careful measurement of pH is necessary in P450 Compound I kinetic studies.
The results clearly demonstrate that the isomerization reactions are strongly accelerated in increasingly basic solutions. Plots of log kexp versus pH demonstrate good linearity (Figure 2), and the slopes, which give the order of dependence on hydroxide concentration, are 3.3 ± 0.1 (octanoic acid), 3.4 ± 0.7 (dodecanoic acid), and 3.5 ± 0.1 (hexadecanoic acid). These values indicate a minimum of two competing reactions in the rate-determining step, one third-order in hydroxide, and the other fourth-order in hydroxide (see Supporting Information). Based on the pH range of the kinetic effect, the rate-determining isomerization reactions appear to involve fast reactions for deprotonated forms of histidine residues. CYP119 contains seven histidines, three of which are within 11 Å of the heme iron in a reported structure,[11] but identification of histidines controlling the rates of the isomerization reactions would seem to require studies with site-directed mutants.
Figure 2.

Plots of log kexp versus pH. The lines are linear regression fits.
The regioselectivities of the oxidation reactions also were found to be dependent on the pH and demonstrate competing reactions with different kinetic order in hydroxide (Table 2). In these experiments, the hydroxylation product mixture was treated with diazomethane to give methyl ester products that were analyzed by GC for quantification and GC-mass spectrometry for identification.[12] An example of the pH effect on regioselectivity is shown in Figure 3 for dodecanoic acid reactions at pH 7.0 and 7.4
Table 2.
Regioselectivity of oxidations of fatty acids by CYP119 Compound I.[a]
| Substrate | pH | ω-3 | ω-2 | ω-1 |
|---|---|---|---|---|
| Dodecanoic acid | 7.0 | 36 | 29 | 35 |
| 7.4 | 3 | 36 | 61 | |
| Hexadecanoic acid | 7.0 | 26 | 50 | 24 |
| 7.4 | 5 | 61 | 34 |
Reactions in 100 mM phosphate buffer at 4 °C. Percentages of alcohol products with errors estimated at ca ± 5% of the reported value.
Figure 3.
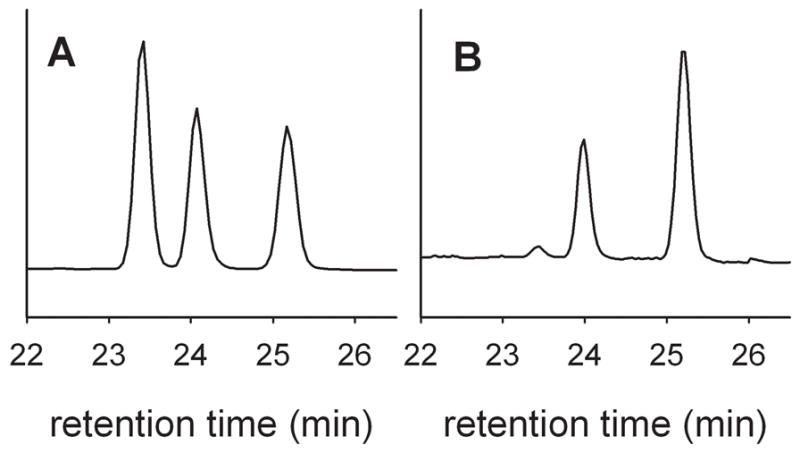
GC traces of methyl esters of products from oxidation of dodecanoic acid at pH 7.0 (A) and pH 7.4 (B). The products elute as follows: methyl 9-hydroxydodecanoate (23.4 min), methyl 10-hydroxydodecanoate (24.1 min), methyl 11-hydroxydodecanoate (25.2 min).
The regioselectivities for oxidations of dodecanoic acid and hexadecanoic acid at pH 7.0 and 7.4 are listed in Table 2. At the lower pH, dodecanoic acid was oxidized at the ω-1, ω-2, and ω-3 positions in comparable amounts, but, at higher pH, it was oxidized almost exclusively at the ω-1 and ω-2 positions. Previously, dodecanoic acid oxidation by CYP119 under turnover conditions was reported to give mainly the ω-1 and ω-2 products in an approximate 3:1 ratio,[12a] and reactions of CYP119 Compound I were reported to give comparable amounts of ω-2 and ω-1 alcohols[5d] or comparable amounts of the three alcohol products found here.[8] The regioselectivity of oxidation of hexadecanoic acid behaved much like that of dodcanoic acid; at pH 7.0, the ω-1, ω-2, and ω-3 products were formed in an approximate 1:2:1 ratio, and at higher pH, the ω-3 product decreased considerably.
In conclusion, we have found that the rates of reactions with CYP119 Compound I with fatty acids are highly dependent on the pH of the medium with rates increasing with greater than third order in base concentration as the pH is raised. The rate-determining steps in the reactions are conformational changes in the complex of activated Compound I plus substrate that, one assumes, bring the substrate into intimate contact with the activated oxygen atom of the oxo-iron(IV) moiety, and the rates of those changes appear to be related to the protonation states of histidines. Combined with the previous demonstration that glycol co-solvents strongly affect rates of fatty acid oxidations by CYP119 Compound I,[8] a high mechanistic complexity for the hydroxylation reactions is apparent. One should exercise caution in predictions of rates of C-H hydroxylations by P450 Compounds I until more detailed information is available.
Experimental Section
Materials
MCPBA (Sigma-Aldrich) was recrystallized before use. Cytochrome P450 119 (CYP119) was expressed in E. coli and purified by the method of Koo et al.[12a] The isolated enzyme had an Rz value (ratio of absorbance at λmax of the Soret band to that at 280 nm) of 1.6 to 1.7 indicating high purity.[12a] Octanoic acid, dodecanoic acid, and hexadecanoic acid (Sigma-Aldrich) and their perdeuterated isotopomers (Cambridge Isotopes) were commercial samples that were used as obtained.
Oxidation products
Bulk oxidations with MCPBA-generated Compound I were conducted by treating CYP119 (1 nmol) and substrate (1 mmol) in 1 mL of 100 mM phosphate buffer (pH 7.0 or 7.4) at 4 °C with MCPBA (10 nmol). After 10 min, the reaction was quenched by addition of dilute HCl to bring the solution to pH 1, and the mixture was extracted with CH2Cl2 (15 mL). The combined CH2Cl2 phases were dried (MgSO4), internal standard was added, and the mixture was treated with excess diazomethane in ether. The resulting mixture of methyl esters was analyzed by GC on a Carbowax 20M bonded phase column for quantitation. For identification purposes, the mixture of methyl esters of the hydroxylated products was treated with excess N-methyl-N-trimethylsilylacetamide to give the O-trimethylsilyl derivatives that were analyzed by GC-mass spectrometry on a DB5 bonded phase column according to their fragmentation patterns as previously described.[12]
Kinetic Studies
The methods for kinetic studies with MCPBA oxidations were the same as previously reported.[5d, 8] In brief, the oxidations were conducted in a four-syringe stopped-flow mixing unit held at 4 °C using photo-multiplier detection with monochromatic light. Equal volumes of buffered solutions of the enzyme (20 μM) and MCPBA (20 μM) were mixed in the first push of the stopped-flow unit, aged for 100 ms, and then mixed with a buffered solution containing substrate at varying concentrations. The growth of signal at λ = 416 (λmax of the ferric protein) was followed. The data analysis is discussed in the Supporting Information.
Supplementary Material
Figure 1.

Rate constants for reactions of CYP119 with fatty acids at 4 °C in 100 mM phosphate buffer solutions at varying pH. The observed pseudo-first-order rate constants are shown as circles. The lines are fits for the kinetic parameters in Table 1. The plots are labeled with the pH of the solutions.
Acknowledgments
This work was supported in part by a grant from the National Institutes of Health (GM-48722).
Footnotes
Supporting information for this article is available on the WWW under http://www.chembiochem.org or from the author.
References
- 1.Ortiz de Montellano PR, editor. Cytochrome P450 Structure, Mechanism, and Biochemistry. 3. Kluwer; New York: 2005. [Google Scholar]
- 2.Guengerich FP. In: Cytochrome P450 Structure, Mechanism, and Biochemistry. 3. Ortiz de Montellano PR, editor. Kluwer; New York: 2005. pp. 377–530. [Google Scholar]
- 3.Sono M, Roach MP, Coulter ED, Dawson JH. Chem Rev. 1996;96:2841–2887. doi: 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- 4.Nordblom GD, White RE, Coon MJ. Arch Biochem Biophys. 1976;175:524–533. doi: 10.1016/0003-9861(76)90541-5. [DOI] [PubMed] [Google Scholar]
- 5.a) Egawa T, Shimada H, Ishimura Y. Biochem Biophys Res Commun. 1994;201:1464–1469. doi: 10.1006/bbrc.1994.1868. [DOI] [PubMed] [Google Scholar]; b) Kellner DG, Hung SC, Weiss KE, Sligar SG. J Biol Chem. 2002;277:9641–9644. doi: 10.1074/jbc.C100745200. [DOI] [PubMed] [Google Scholar]; c) Raner GM, Thompson JI, Haddy A, Tangham V, Bynum N, Reddy GR, Ballou DP, Dawson JH. J Inorg Biochem. 2006;100:2045–2053. doi: 10.1016/j.jinorgbio.2006.09.025. [DOI] [PubMed] [Google Scholar]; d) Rittle J, Green MT. Science. 2010;330:933–937. doi: 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]; e) Jung C, de Vries S, Schünemann V. Arch Biochem Biophys. 2011;507:44–55. doi: 10.1016/j.abb.2010.12.029. [DOI] [PubMed] [Google Scholar]
- 6.Ortiz de Montellano PR. Chem Rev. 2010;110:932–948. doi: 10.1021/cr9002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lu AYH, Coon MJ. J Biol Chem. 1968;243:1331–1332. [PubMed] [Google Scholar]
- 8.Su Z, Chen X, Horner JH, Newcomb M. Chem Eur J. 2012;18:2472–2476. doi: 10.1002/chem.201103170. [DOI] [PubMed] [Google Scholar]
- 9.Chen XH, Su Z, Horner JH, Newcomb M. Org Biomolec Chem. 2011;9:7427–7433. doi: 10.1039/c1ob06035j. [DOI] [PubMed] [Google Scholar]
- 10.Bell SR, Groves JT. J Am Chem Soc. 2009;131:9640–9641. doi: 10.1021/ja903394s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yano JK, Koo LS, Schuller DJ, Li HY, Ortiz de Montellano PR, Poulos TL. J Biol Chem. 2000;275:31086–31092. doi: 10.1074/jbc.M004281200. [DOI] [PubMed] [Google Scholar]
- 12.a) Koo LS, Immoos CE, Cohen MS, Farmer PJ, Ortiz de Montellano PR. J Am Chem Soc. 2002;124:5684–5691. doi: 10.1021/ja017174g. [DOI] [PubMed] [Google Scholar]; b) Nicolaides N, Soukup VG, Ruth EC. Biomed Mass Spec. 1983;10:441–449. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.