

Abstract
The diagnosis and treatment of cancers, which rank among the leading causes of mortality in developed nations, presents substantial clinical challenges. The genetic and epigenetic heterogeneity of tumors can lead to differential response to therapy and gross disparities in patient outcomes, even for tumors originating from similar tissues. High-throughput DNA sequencing technologies hold promise to improve the diagnosis and treatment of cancers through efficient and economical profiling of complete tumor genomes, paving the way for approaches to personalized oncology that consider the unique genetic composition of the patient’s tumor. Here we present a novel method to leverage the information provided by cancer genome sequencing to match an individual tumor genome with commercial cell lines, which might be leveraged as clinical surrogates to inform prognosis or therapeutic strategy. We evaluate the method using a published lung cancer genome and genetic profiles of commercial cancer cell lines. The results support the general plausibility of this matching approach, thereby offering a first step in translational bioinformatics approaches to personalized oncology using established cancer cell lines.
1. Introduction
Despite innovations in relevant diagnostics and therapeutics over the past decades, cancers remain among the leading causes of mortality in developed nations. Although many common molecular drivers of oncogenesis are known to exist, the majority of cancers are heterogeneous in their molecular characteristics, leading to disparities in response to standard cancer therapies. High-throughput sequencing technologies, with promise to offer complete DNA sequence profiling of cancer genomes, present novel opportunities understanding the unique molecular characteristics of tumors profiled in clinical populations. Knowledge of the unique molecular characteristics of a tumor, as detailed by its genomic sequence, could inform diagnosis, prognosis and treatment, thereby establishing a basis for personalized oncology.
In order to gain clinical utility from personal cancer genomes, the molecular characteristics latent in the cancer genomic sequence must be related to a broader biological context. Aberrations in a cancer genome, such as somatic variations in single nucleotides, copy number or novel gene fusions can serve as informative biomarkers that inform diagnosis, prognosis or treatment. For example, mutations in the epidermal growth factor receptor (EGFR) have been associated with response to gefitinib in non-small cell lung cancer (NSCLC)1, and mutations in KRAS are known to be predictive of response to cetuximab in colon cancers2. Such markers have great clinical value when they are well characterized, however a complete genomics sequence of a cancer is likely to present many novel molecular aberrations that have minimal to no precedence in the literature. Furthermore, consideration for only a subset of the markers available in a fully sequenced cancer genome might miss molecular and biological features important for individualized treatment.
In order to assess functional correlates of disease progression or therapeutic susceptibility, approaches to personalized oncology need to consider molecular phenotypes salient in individual tumor biology along with the tumor’s genotype. For example, expression levels of human epidermal growth factor receptor 2 (HER2) are predictive of response to trastuzumab3, and various cellular metabolic features have been associated with tumor progression4. Ideally, it would be possible to functionally investigate these molecular phenotypes towards a personalized course of clinical care (e.g. test the response of several different chemotherapies to determine the best course of treatment), however it is not possible to conduct such clinical experimentation in vivo without placing the patient in danger of serious harm. One solution is to create autologous tumor cell lines from tumor tissue excised from the patient. However, the technical capacity to establish, maintain, and functionally test autologous cell lines is not at all common in most clinical settings, and therefore may not be as viable as a therapeutic option during the course of clinical care for cancer patients.
Here we describe a method to match a personal cancer genome with commonly studied commercially available cancer cell lines based on shared genetic profiles. Commercial cell lines serve as an attractive option for personalized oncology, because they are readily and economically available through commercial suppliers, and the pharmacological and biochemical characteristics of many of the available cancer cell lines are well reported in the literature. Furthermore, it has been shown that large collections of cancer cell lines can serve as “systems” to functionally characterize the pathophysiological properties of individual tumors5. Once a personal cancer genome is matched to a commercial cell line, it is possible that the cell line and the prior knowledge around that cell line could serve as an in vitro surrogate for clinical functional assessment of tumor biology. We offer a profile similarity approach that matches a cancer genome with commercial cell lines based on profiles of shared somatic variability at multiple loci. The method is assessed using data from a recently published genomic sequence of a lung cancer tumor, which was matched to genotyped cell lines found in the GlaxoSmithKline cancer cell line genomic profiling data.
2. Methods
2.1. Data
A set of somatic single nucleotide variants discovered in a NSCLC genome through paired genome sequencing in a lung cancer patient was obtained from the supplementary information provided by Lee et al6. Variant positions were mapped to dbSNP rsId’s by genomic location. SNP genotype profiles for commercial cancer cell lines were downloaded from the Cancer Biomedical Informatics Grid (caBIG) website (https://cabig.nci.nih.gov/caArray_GSKdata/) via FTP. Allele frequency information was downloaded from data provided by the International HapMap Project Phase IIa7. We aggregated in vivo tumor xenograft screening data made available through the National Cancer Institute (NCI) Developmental Therapeutics Program (DTP) website (http://dtp.nci.nih.gov/webdata.html). The DTP screening data provides assessments of the anti-tumor efficacy of a wide range of chemical compounds evaluated across various clinical endpoints in human tumor xenograft models8.
2.2. Profile similarity
A profile similarity metric was computed by comparing common variant loci between the cancer genome and the cancer cell line SNP profiles. The SNP profiles for the commercial cell lines only represent the genotype of various primary cancer cells, and therefore offer no means to distinguish somatic variants from neutral variation. We used allele frequency data from the HapMap project as a proxy for the normal baseline genotype. In this way, a locus was said to be a cancer-associated variation if it was not found to harbor the associated major allele for that locus found in the HapMap data. We then derived a multi-locus identity metric to compute a similarity score between to genomic profiles based on shared genotypes at somatically variant positions. For each locus an identity-by-similarity (IBS) score was computed based on the number of alleles shared between the profiles at that locus. The IBS score = 0 if no alleles are share, 1 if one allele is shared, or 2 if both profiles are homozygous for the same allele. The multi-locus profile identity score (mIS) was computed by summing the IBS scores across all shared loci and dividing by twice the number of common loci:
Where L is the number of common variant loci between two genomic profiles i and j, and is the genotype of the Ith locus in profile i, and is the genotype of the Ith locus in profile j.
2.3. Matching the lung cancer genome to cell lines
To match the NSCLC genome to cell lines we computed the mIS score between the somatic variants and the SNP profiles for all cell lines found in the GSK data set. To estimate a p-value for mIS scores we computed a random distribution of mIS scores by constructing random genotype profiles by sampling randomly from the GSK data, and computing the mIS score between the NSCLC profile and the random genotype for one thousand iterations. The empirical p-value for an mIS score was computed as the proportion of mIS scores from the random distribution greater than the given mIS score.
2.4. Clustering tumors by therapeutic profiles
The DTP inhibition data was averaged by tumor type and compound. For each tumor type defined in the DTP data set, a chemotherapeutic profile was defined as the average inhibition for each compound against which the tumor was evaluated. A distance matrix was computed between tumors using the Pearson’s correlation of compound inhibition response values. Only statistically significant correlations were retained. Hierarchical clustering was performed on the correlation distance matrix (1 -correlation) using the average agglomeration method. The significance of the compound inhibition clustering was assessed by multiscale bootstrap resampling across 1,000 bootstrap replicates using the pvclust package (http://www.is.titech.ac.jp/~shimo/prog/pvclust/). All computations were performed using the R language for statistical computing (http://www.r-project.org).
3. Results
Using genomic location information we mapped 9,754 somatic single nucleotide variants and their genotypes to dbSNP rsId identifiers. Among these loci we found 391 that overlapped with the SNPs measured on the SNP array used to profile the cancer cell lines in the GSK data set. This common set of loci was used to compute the profile similarity between the NSCLC genome and the cancer cell lines. After computing mIS profile similarity scores (see methods) between the NSCLC genome and all cell lines profiled in the GSK data set, we find 16 cell lines to be significantly associated with the personal cancer genome by genetic profile (Table 1). The distribution of mIS scores across the GSK data set is shown in Figure 1. The top match among the GSK cancer cell lines is bladder carcinoma line J82. While other lung carcinomas are found among the top results, we also find non-obvious associations between various leukemias and lymphomas.
Table 1.
Cancer cell lines from the GSK genomic profiling data set with genetic profiles significantly similar to the individual NSCLC genome based on mIS scores.
| Cancer Type | Cell Line | mIS score | P-value |
|---|---|---|---|
| Carcinoma of Bladder | J82 | 0.84 | 2.3×10−2 |
| Acute T Cell Lymphoblastic Leukemia of Hematopoietic and lymphatic system | CCRFCEM | 0.83 | 3.3 ×10−2 |
| Lymphoma of Hematopoietic and lymphatic system | SR | 0.83 | 3.3 ×10−2 |
| Hodgkin Lymphoma of Hematopoietic and lymphatic system | RPMI6666 | 0.83 | 3.3 ×10−2 |
| Lung Adenocarcinoma | NCIH1975 | 0.82 | 4.8×10−2 |
| Lung Adenocarcinoma | NCIH2228 | 0.82 | 4.8×10−2 |
| Atypical Carcinoid Tumor of Lung | NCIH720 | 0.82 | 4.8×10−2 |
| Small Cell Lung Carcinoma of Lung | NCIH524 | 0.82 | 4.8×10−2 |
| Burkitt Lymphoma of Hematopoietic and lymphatic system | MC116 | 0.82 | 4.8×10−2 |
| Burkitt Lymphoma of Hematopoietic and lymphatic system | 1A2 | 0.82 | 4.8×10−2 |
| Carcinoma of Uterus | KLE | 0.82 | 4.8×10−2 |
| Sarcoma of Bone | SW1353 | 0.82 | 4.8×10−2 |
| Carcinoma of Uterus | RL952 | 0.82 | 4.8×10−2 |
| Myeloma of Hematopoietic and lymphatic system | HuNS1 | 0.82 | 4.8×10−2 |
| Carcinoma of Breast | MT3 | 0.82 | 4.8×10−2 |
| Acute T Cell Lymphoblastic Leukemia of | CEMC1 | 0.82 | 4.8×10−2 |
Figure 1.
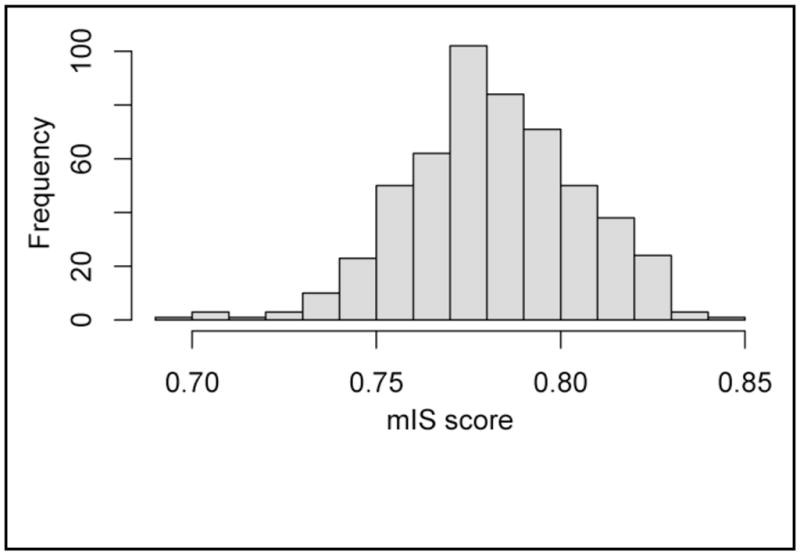
Distribution of genetic profile similarity scores between the lung cancer genome and GSK cancer cell lines.
To explore the plausibility of these cell line associations, we obtained chemotherapeutic screening data from the NCI Developmental Therapeutics Program (DTP) and clustered tumors based on their response to various chemotherapies (Figure 2). Based on chemotherapy response profiles, we find that Lewis lung carcinomas, a model for non-small cell lung cancer, generally cluster with several leukemias and reticular (lymphoid) sarcoma, which is reflective of our cell line match results.
Figure 2.

Hierarchical clustering of tumors profiled by the National Cancer Institute Developmental Therapeutics Program based on their chemotherapeutic inhibition response profiles. Values at the inner nodes represent bootstrap p-values estimated by multiscale bootstrap resampling using 1,000 boostrap replicates.
4. Discussion
In effort to relate a personal cancer genome to cancer cell lines for personalized oncology, we developed a profile similarity method that computes a similarity score between two genetic profiles based on shared alleles at somatically variant sites. We applied this method to a published non-small cell lung cancer genome and a set of SNP profiles from the GSK cancer genomic profiling data set. We found that the personal cancer genome could be significantly matched with 16 cell lines from the GSK data set by genetic profile (Table 1). While we find a number of lung cancer cell lines among these significant matches, we also find equally significant matches for non-lung cancers, including various Hodgkin lymphomas, leukemias and bladder cancer.
It is not immediately apparent why the lung cancer genome would be associated with these seemingly unassociated cancers. One possible explanation is that there are many passenger mutations after the cancer initiation event has started9, and that the similarities are being driven by these mutations. Since passenger mutations are not necessarily causal, and could therefore confound variation based similarity metrics like the one used in this study. In this case, future work might involve inclusion of prior knowledge of cancer causal variants to reduce false positives, or look across multiple cancer genomes to understand patterns of earlier versus later mutations from a data-driven perspective.
Another explanation is that these associations might point towards some shared etiological or pathophysiological characteristics. Smoking is a well-known risk factor for lung cancers, leading to consistent genetic lesions observable in the genomes of lung cancer tumors. Smoking is also a substantial risk factor for bladder cancer10, which is the top match in our results, and is also known to be associated with increased risk of various leukemia’s and lymphomas11. Therefore the computed similarity between the lung cancer genome and these cell lines might have a basis in shared common genetic lesions due to smoking. It is also known that individuals affected by Hodgkin’s lymphoma have an increased risk of lung cancer and non-Hodgkin lymphomas12, suggesting a possible shared molecular pathophysiology among the various forms of cancer. Therefore, despite the fact that many of the matches are not of the same tumor type as the lung cancer genome, it is possible that they still might serve as functional surrogates for personalized clinical investigation.
To gain functional support for the plausibility of these cell line associations, we clustered tumors based on their response to various chemotherapies (Figure 2). Based on chemotherapy response profiles, we find that non-small cell lung cancer model tumors (Lewis lung) cluster significantly with both each other and other non-lung tumor types. A scatterplot of the chemotherapeutic profile similarity between a NSCLC tumor and leukemia is shown in Figure 3. Although the cell lines used in the DTP screening data set are not precise matches for the cell lines in the GSK data set, we can draw support for the notion that unrelated cancers such as lymphomas or leukemias could serve as functionally relevant clinical surrogates for lung cancer tumors.
Figure 3.

Comparison of the chemotherapeutic response profiles between a model of non-small cell lung cancer tumor model and a leukemia characterized in the NCI DTP data. The points represent the inhibition proportion (treatment/control) for a compound.
We find additional support for a plausible functional relationship through investigation of the variants driving the similarity between the lung cancer genome and cell lines. The best match in our data set was a bladder carcinoma cell line (J82). The gene associated variants shared between the lung cancer genome and the J82 cell line are shown in Table 2. Although all of these shared loci are intronic, it’s still possible that they could be disrupting gene function through an effect on alternative splicing, or might serve as surrogate markers for mutational disruption of other loci in the same gene through linkage disequilibrium. Among these genes we find several known to be associated with cancers. PTPRT, a protein tyrosine phosphatase receptor, is a signaling molecule known to be implicated in oncogenic transformation in several different cancers13, including colon cancer14,15, glioma16, and melanoma17. NELL1 and NELL2, growth factor like protein thought to be involved in regulation of cell growth, has also been associated with multiple cancer types, including esophageal adenocarcinoma18, colon cancer and Burkitt’s lymphoma19. TUSC3, a putative tumor suppressor gene, has been associated with pancreatic cancer20, prostate cancer21 and ovarian cancer22. It’s possible that these pleiotropic oncogenes are driving the similarity relationship between the lung cancer genome and J82 based on common patterns of oncogenic mutation. Several other genes underlying this similarity are not known to be oncogenic, however variants in BMP6, COL6A3, C7, GABRG3 and NRG3 are known to be associated with various complex and Mendelian diseases.
Table 2.
Gene-associated variants driving the similarity score between the personal lung cancer genome profile and the top cell-line match bladder carcinoma (J82). Both the lung cancer genome and J82 exhibit somatic variation at these positions and share at least one variant allele.
| dbSNP rsID | Gene region | Gene symbol | Gene description |
|---|---|---|---|
| rs169124 | intronic | BMP6 | bone morphogenetic protein 6 |
| rs13378247 | intronic | ENOX1 | ecto-NOX disulfide-thiol exchanger 1 |
| rs11182675 | intronic | NELL2 | NEL-like 2 (chicken) |
| rs7824149 | intronic | NECAB1 | N-terminal EF-hand calcium binding protein 1 |
| rs938726 | intronic | EIF2C2 | eukaryotic translation initiation factor 2C, 2 |
| rs10983337 | intronic | ASTN2 | astrotactin 2 |
| rs639839 | intronic | NRG3 | neuregulin 3 |
| rs16907794 | intronic | NELL1 | NEL-like 1 (chicken) |
| rs2425562 | intronic | PTPRT | protein tyrosine phosphatase, receptor type, T |
| rs2837583 | intronic | DSCAM | Down syndrome cell adhesion molecule |
| rs10852799 | intronic | DNAH9 | dynein, axonemal, heavy chain 9 |
| rs8024401 | intronic | GABRG3 | gamma-aminobutyric acid (GABA) A receptor, gamma 3 |
| rs9555507 | intronic | MYO16 | myosin XVI |
| rs10483422 | intronic | NPAS3 | neuronal PAS domain protein 3 |
| rs11158839 | intronic | SLC8A3 | solute carrier family 8 (sodium/calcium exchanger), member 3 |
| rs9620769 | intronic | TTC28 | tetratricopeptide repeat domain 28 |
| rs13112477 | intronic | C4orf22 | chromosome 4 open reading frame 22 |
| rs6720773 | intronic | COL6A3 | collagen, type VI, alpha 3 |
| rs10932540 | intronic | VWC2L | von Willebrand factor C domain-containing protein 2-like |
| rs7550703 | intronic | HHAT | hedgehog acyltransferase |
| rs1881410 | intronic | LOC730124 | similar to hCG2041586 |
| rs4730038 | intronic | LHFPL3 | lipoma HMGIC fusion partner-like 3 |
| rs2642484 | intronic | CNTNAP2 | contactin associated protein-like 2 |
| rs7819262 | intronic | TUSC3 | tumor suppressor candidate 3 |
| rs2910639 | intronic | ADAMTS12 | ADAM metallopeptidase with thrombospondin type 1 motif, 12 |
| rs16870537 | intronic | C7 | complement component 7 |
We acknowledge several limitations in our approach. Foremost, we recognize that since the GSK cell lines were profiled by SNP microarray, that the analysis was appreciably constrained to only the loci measured on the array platform. Future work might employ sophisticated imputations algorithms to expand the genotype profiles in the GSK data set, but ideally full genome sequencing data for these cell lines would likely be necessary for clinical application of this approach. We also acknowledge that the DTP chemotherapeutic profiling data can only offer indirect support for functional associations between these cell lines, as many of the cell lines profiled in the GSK data set are not represented in the NCI DTP screening data set. Efforts are needed to comprehensively characterize the chemotherapetuic response profiles of these cell lines and to provide a machine-readable representation of these data in the public domain.
Future work in this area will incorporate improved similarity metrics that give added importance to somatic variations more likely to play a causal role in tumorigenesis or metastasis, such as mutations in evolutionary conserved regions, or in loci known to act as expression quantitative trait loci (eQTLs) for genes associated with oncogenesis. More importantly, future work should incorporate experimental validation of predicted cell line matches to test whether or not the predicted cell line match exhibits clinical characteristics (e.g. chemotherapeutic response) similar to the individual tumor genome to which it was matched. Developments in this area will provide novel directions in personalized oncology that leverage the clinical, economic, and scientific benefits of well studied and characterized commercial cancer cell lines.
Acknowledgments
JTD is supported by the Graduate Training in Biomedical Informatics grant (R01 LM009719) from the National Library of Medicine. AJB is supported by the National Cancer Institute (R01 CA138256), the Lucile Packard Foundation for Children’s Health and the Hewlett Packard Foundation. We thank Alex Skrenchuk and Boris Oskotsky from Stanford University for computer support.
References
- 1.Kobayashi S, et al. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 2.Lievre A, et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res. 2006;66:3992–3995. doi: 10.1158/0008-5472.CAN-06-0191. [DOI] [PubMed] [Google Scholar]
- 3.Hudis CA. Trastuzumab--mechanism of action and use in clinical practice. N Engl J Med. 2007;357:39–51. doi: 10.1056/NEJMra043186. [DOI] [PubMed] [Google Scholar]
- 4.Tennant DA, Duran RV, Gottlieb E. Targeting metabolic transformation for cancer therapy. Nat Rev Cancer. 2010;10:267–277. doi: 10.1038/nrc2817. [DOI] [PubMed] [Google Scholar]
- 5.Neve RM, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee W, et al. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature. 2010;465:473–477. doi: 10.1038/nature09004. [DOI] [PubMed] [Google Scholar]
- 7.International HapMap C. The International HapMap Project. Nature. 2003;426:789–796. doi: 10.1038/nature02168. [DOI] [PubMed] [Google Scholar]
- 8.Teicher BA, Andrews PA. Anticancer drug development guide: preclinical screening, clinical trials, and approval. Humana Press; Totowa, N.J.: 2004. [Google Scholar]
- 9.Greenman C, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–158. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boffetta P. Tobacco smoking and risk of bladder cancer. Scand J Urol Nephrol Suppl. 2008:45–54. doi: 10.1080/03008880802283664. [DOI] [PubMed] [Google Scholar]
- 11.Willett EV, O’Connor S, Smith AG, Roman E. Does smoking or alcohol modify the risk of Epstein-Barr virus-positive or -negative Hodgkin lymphoma? Epidemiology. 2007;18:130–136. doi: 10.1097/01.ede.0000248899.47399.78. [DOI] [PubMed] [Google Scholar]
- 12.van Leeuwen FE, et al. Increased risk of lung cancer, non-Hodgkin’s lymphoma, and leukemia following Hodgkin’s disease. J Clin Oncol. 1989;7:1046–1058. doi: 10.1200/JCO.1989.7.8.1046. [DOI] [PubMed] [Google Scholar]
- 13.Lee JW, et al. Mutational analysis of PTPRT phosphatase domains in common human cancers. APMIS. 2007;115:47–51. doi: 10.1111/j.1600-0463.2007.apm_554.x. [DOI] [PubMed] [Google Scholar]
- 14.Zhao Y, et al. Identification and functional characterization of paxillin as a target of protein tyrosine phosphatase receptor T. Proc Natl Acad Sci U S A. 2010;107:2592–2597. doi: 10.1073/pnas.0914884107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ruivenkamp CA, et al. Ptprj is a candidate for the mouse colon-cancer susceptibility locus Scc1 and is frequently deleted in human cancers. Nat Genet. 2002;31:295–300. doi: 10.1038/ng903. [DOI] [PubMed] [Google Scholar]
- 16.Norman SA, Golfinos JG, Scheck AC. Expression of a receptor protein tyrosine phosphatase in human glial tumors. J Neurooncol. 1998;36:209–217. doi: 10.1023/a:1005840420136. [DOI] [PubMed] [Google Scholar]
- 17.Yu J, et al. Tumor-derived extracellular mutations of PTPRT/PTPrho are defective in cell adhesion. Mol Cancer Res. 2008;6:1106–1113. doi: 10.1158/1541-7786.MCR-07-2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jin Z, et al. Hypermethylation of the nel-like 1 gene is a common and early event and is associated with poor prognosis in early-stage esophageal adenocarcinoma. Oncogene. 2007;26:6332–6340. doi: 10.1038/sj.onc.1210461. [DOI] [PubMed] [Google Scholar]
- 19.Kuroda S, et al. Biochemical characterization and expression analysis of neural thrombospondin-1-like proteins NELL1 and NELL2. Biochem Biophys Res Commun. 1999;265:79–86. doi: 10.1006/bbrc.1999.1638. [DOI] [PubMed] [Google Scholar]
- 20.Bashyam MD, et al. Array-based comparative genomic hybridization identifies localized DNA amplifications and homozygous deletions in pancreatic cancer. Neoplasia. 2005;7:556–562. doi: 10.1593/neo.04586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bova GS, et al. Physical mapping of chromosome 8p22 markers and their homozygous deletion in a metastatic prostate cancer. Genomics. 1996;35:46–54. doi: 10.1006/geno.1996.0321. [DOI] [PubMed] [Google Scholar]
- 22.Pils D, et al. Five genes from chromosomal band 8p22 are significantly down-regulated in ovarian carcinoma: N33 and EFA6R have a potential impact on overall survival. Cancer. 2005;104:2417–2429. doi: 10.1002/cncr.21538. [DOI] [PubMed] [Google Scholar]