

SUMMARY
The integrity of the epidermis and mucosal epithelia is highly dependent on resident self-renewing stem cells, which makes them vulnerable to physical and chemical insults compromising the repopulating capacity of the epithelial stem cell compartment. This is frequently the case in cancer patients receiving radiation or chemotherapy, many of whom develop mucositis, a debilitating condition involving painful and deep mucosal ulcerations. Here, we show that inhibiting the mammalian target of rapamycin (mTOR) with rapamycin increases the clonogenic capacity of primary human oral keratinocytes and their resident self-renewing cells by preventing stem cell senescence. This protective effect of rapamycin is mediated by the increase expression of mitochondrial superoxide dismutase (MnSOD), and the consequent inhibition of ROS formation and oxidative stress. mTOR inhibition also protects from the loss of proliferative basal epithelial stem cells upon ionizing radiation in vivo, thereby preserving the integrity of the oral mucosa and protecting from radiation-induced mucositis.
INTRODUCTION
Both epidermis and mucosal epithelia are highly dependent on resident self-renewing stem cells, making them particularly vulnerable to physical and chemical insults compromising the repopulating capacity of the epithelial stem cell compartment. This is often the case in cancer patients receiving radiation or chemotherapy, many of whom develop mucositis, a debilitating condition involving painful and deep mucosal ulcerations as a result of damage to the normal tissue (Sonis, 2009, 2010). Mucositis causes distress to the patients and results also in substantial increase in per-patient care cost (Nonzee et al., 2008). Radiotherapy is one of the most widely used cancer treatments (Begg et al., 2011), and although technical advances now allow a more targeted delivery of radiation to the cancer cells, indirect damage to the normal surrounding tissues still remains a common and frequently weakening side effect (Citrin et al., 2010). Hence, therapeutic interventions that can reduce the deleterious effects of radiation on the normal epithelial stem cells will have a great impact on the quality of life of cancer patients and treatment outcome.
The mammalian target of rapamycin (mTOR) plays a central role in the regulation of cell growth and cancer progression, but paradoxically, increased mTOR activity can also cause certain cells to undergo differentiation or senescence, thereby exiting the proliferative cell pool (Castilho et al., 2009; Chen et al., 2008; Lee et al., 2010; Liu et al., 2007; Zhang et al., 2006; reviewed in Iglesias-Bartolome and Gutkind, 2011). In particular, mTOR activation is one of the most frequent events in human malignancies arising from the epithelial cells lining the oral mucosa, tongue and pharynx, collectively referred to as head and neck squamous cell carcinomas (HNSCC) (Molinolo et al., 2007). Inhibition of mTOR by the use of rapamycin and its derivatives has shown promising antitumoral activity in a variety of experimental models of HNSCC (Amornphimoltham et al., 2008; Amornphimoltham et al., 2005; Czerninski et al., 2009; Nathan et al., 2007), and in recent clinical trials. While the clinical benefits of blocking mTOR in HNSCC as a single agent is under current clinical evaluation, we have asked whether mTOR inhibition with rapamycin could synergize with the antitumoral effects of radiation, one of the most frequent approaches for HNSCC treatment (Delaney et al., 2005). We observed only a subtle increase in the cytotoxic effect of ionizing radiation in a panel of HNSCC-derived cells lines. Surprisingly, however, we found that mTOR inhibition protects normal oral epithelial cells from ionizing radiation-induced epithelial stem cell depletion. Furthermore, rapamycin also protected epithelial progenitor cells from replicative senescence, extending dramatically their life span in vitro. Emerging evidence suggest that this protective effect of mTOR inhibition is mediated by the increased expression of mitochondrial superoxide dismutase (MnSOD), and the consequent suppression of oxidative stress caused by the accumulation of reactive oxygen species (ROS) in normal but not in cancer cells. These observations prompted us to ask whether this differential effect of rapamycin in normal and tumor epithelial cells could be exploited to protect the oral mucosa against the deleterious effects of radiation in vivo. We found that inhibition of mTOR prevents the loss of proliferative epithelial progenitor stem cells upon radiation and enhances their tissue repopulating capacity, thereby preserving the integrity of the oral mucosa and protecting from radiation-induced mucositis.
RESULTS
mTOR inhibition protects primary human keratinocytes from loss of clonogenic capacity
Sensitization of cancer cells to the cytotoxic effect of radiation by adjuvant therapy targeting oncogenic signaling circuitries may represent a promising approach to improve radiation outcome (Begg et al., 2011). In this regard, activation of the mTOR pathway has been recently recognized as a frequent event driving neoplastic transformation, including squamous cell carcinomas of the head and neck region (HNSCC) (Molinolo et al., 2007), the 6th most prevalent cancer among men worldwide (Parkin et al., 2005). Given that mTOR inhibitors can sensitize other tumor types to radiation (Eshleman et al., 2002; Konstantinidou et al., 2009; Murphy et al., 2009), we tested whether mTOR blockade by rapamycin increases radiation-induced cancer cell death in representative HNSCC cells lines. Using HN12 and Cal27 cells as typical examples of HNSCC cells that are highly sensitive to the antitumoral activity of rapamycin (Amornphimoltham et al., 2005), however, we detected only a limited synergistic effect on radiation-induced cell death in vitro, as judged by the surviving fraction of cells exposed to clinically-relevant, sub-lethal doses of radiation in the presence of rapamycin (Figures 1A and 1B; see scheme in Figure S1A). The size of the colonies growing after radiation was also not significantly affected by rapamycin, which completely inhibited mTOR activity as assessed by the phosphorylation of S6 (Figures 1A and 1B). Similar results were observed in a large collection of HNSCC cells (not shown). Hence, these in vitro studies suggest that inhibition of mTOR in HNSCC cells may only sensitize slightly to radiation-induced cancer cell death.
Figure 1. mTOR inhibition slightly sensitizes HNSCC to radiation-induced death but protects primary human keratinocytes from loss of clonogenic capacity.
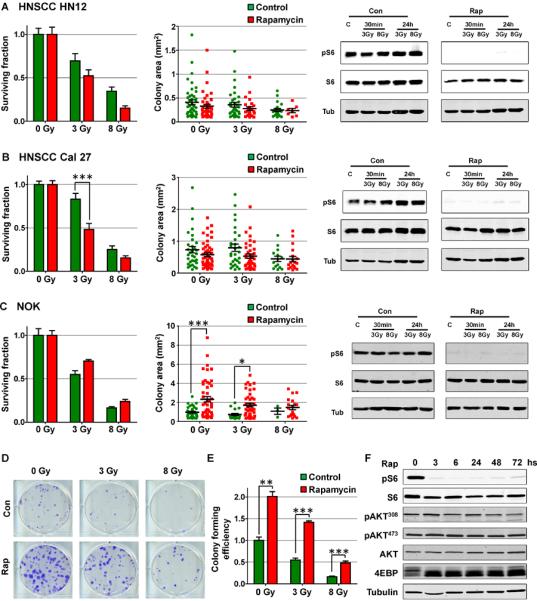
(A and B), Surviving fractions and individual colony areas from clonogenic assays of the HNSCC cell lines HN12 and Cal 27. mTOR activity is shown in control (Con) or rapamycin (Rap) treated cells by Western blot of pS6 at 30 min and 24 h after the indicated dose of radiation in grays (Gy). (C) Surviving fractions and colony areas from clonogenic assays of human primary NOK. mTOR inhibition is also shown by the levels of pS6 in Western blot. (D) Representative dishes from clonogenic assays of NOK. (E) Colony forming efficiency from clonogenic assays of human primary NOK. (F) Western blot analysis of NOK treated with rapamycin for 72 h. mTOR inhibition is shown by the levels of pS6 and the molecular weight shift of 4EBP. No differences are seen in Akt phosphorylation at positions 308 (activating phosphorylation) and 473 (mTORC2-dependent). See Methods section for experimental details. See also Figure S1.
Of interest, however, the impact of rapamycin treatment on normal primary oral epithelial cells was quite remarkable. As a control for the experiments using HNSCC cells, we evaluated the effect of rapamycin on normal oral keratinocytes (NOK). Simple procedures are currently available to isolate primary human NOK that include cells with long-term self-renewal capacity, as judged by their ability to support the prolonged survival of corneal implants in humans (Chen et al., 2009; Nakamura et al., 2007). Using similar procedures, we isolated NOK from gingival biopsies from healthy volunteers, and confirmed that these cultures lack fibroblast contamination (Figures S1B and S1C), and include epithelial stem cells exhibiting tissue regenerative capacity, as judged by organotypic co-cultures (Figure S1D) and by their ability to regenerate stratified epithelium when grafted into nude mice (Figure S1E). Using the same radiation treatment scheme as for HNSCC cells (Figure S1A), we did not observe a decrease in the surviving fraction of NOK undergoing rapamycin treatment (Figure 1C). On the contrary, rapamycin significantly enhanced the colony size of NOK compared with vehicle treated cells, and this increase was maintained after radiation (Figures 1C and 1D). Along with the increase in colony size, rapamycin also increased the colony forming efficiency of control and irradiated human NOK (Figure 1E). Since each large colony is expected to grow from a single surviving self-renewing epithelial stem cell (Jensen et al., 2010), these findings indicate that rapamycin increases the survival and repopulating capacity of epithelial progenitors, hence protecting from radiation-induced loss of this tissue regenerative cell population. Inhibition of mTOR can result in Akt activation due to the disruption of a negative feedback loop in certain cellular systems (Zoncu et al., 2011). Of interest, however, while mTOR activity was completely inhibited by rapamycin in these cells, we did not observe any effect on the phosphorylation levels of Akt after rapamycin treatment (Figures 1C and 1F). This suggests that the increased proliferative capacity of radiated NOK cells upon rapamycin exposure is unlikely related to an elevated Akt activity, and hence to its pro-survival and growth promoting functions (Manning and Cantley, 2007).
Rapamycin decreases senescence in primary human keratinocytes
To begin investigating the mechanisms underlying the higher repopulating capacity of rapamycin treated NOK we evaluated different molecular events involved in apoptosis, senescence and differentiation. We could not observe significant differences on the levels of p53 in NOK after radiation exposure (Figure 2A), nor in the number of apoptotic cells as judged by TdT-mediated dUTP nick end labeling (TUNEL) assays (Figure 2B), indicating that the activation of p53 and the elimination of severely damaged cells by apoptotic death occurs normally after irradiation in control and rapamycin treated cells. An alternative explanation for the enhanced clonogenic capacity of rapamycin-treated human primary NOK after radiation is that rapamycin may induce cell quiescence, as radiation sensitivity can be dependent on cell proliferation (Gudkov and Komarova, 2003). However, rapamycin treatment reduced only slightly the proliferation of NOK as judged by labeling of individual proliferating cells by EdU staining (Figure S2). This is aligned with the observation that rapamycin did not prevent the apoptotic response to radiation, which reflects a similar sensitivity to radiation-induced cell death in control and rapamycin treated cells. Thus, the differences in basal cell proliferation may contribute to, but are unlikely to account for the increased clonogenic capacity of NOK after radiation upon rapamycin treatment.
Figure 2. Rapamycin decreases senescence in primary human keratinocytes.

(A) Western blot analysis of p53 expression in irradiated control (Con) or rapamycin (Rap) treated human primary NOK at 30 min and 24 hs after the indicated dose of radiation in grays (Gy). (B) TdT-mediated dUTP nick end labeling (TUNEL) assay of NOK 24 hs after radiation exposure (8 Gy). Bar: 100μm. (C) Western blot analysis of γH2AX levels in irradiated control (Con) or rapamycin (Rap) treated human primary NOK. Quantification of 3 independent experiments is also shown. (D) Representative pictures of γH2AX (red) levels in irradiated control and rapamycin treated human primary NOK, counterstained to show nuclei (blue) and actin (green). Bar: 20μm. (E and F) Western blot analysis of p16 levels in irradiated control (Con) or rapamycin (Rap) treated human primary NOK. Quantification of 3 independent experiments is also shown. (G) Proportion with respect to control cells and representative pictures of NOK positive for senescence-associated β-galactosidase (SA-βgal) 3 days after rapamycin treatment. (H) Colony forming efficiency from clonogenic assays of human primary NOK transfected with p16 siRNA. Insert shows western blot analysis of p16 levels in NOK transfected with control siRNA (siRNA Con) or siRNA for p16. See also Figure S2.
Interestingly, we found that rapamycin treated NOK exposed to radiation show reduced levels of the DNA damage response marker γH2AX at early and late time points (Figures 2C and 2D). This suggests that rapamycin treatment may reduce the extent of DNA damage in NOK (Bonner et al., 2008). The activation of the DNA damage response is one of the main mediators of cell senescence, which results in irreversible growth arrest (Kuilman et al., 2010; Rodier and Campisi, 2011). Accordingly, we observed reduced levels of the senescence marker p16 in rapamycin-treated NOK after radiation (Figure 2E and 2F), and also observed that rapamycin treated cells show slightly reduced levels of the differentiation marker involucrin (Figure 2E). These findings suggested the possibility that while rapamycin may not protect from radiation-induced cell death, it might instead prevent cells from entering senescence and terminal differentiation by reducing DNA damage and the consequent induction of senescence, therefore increasing NOK replicative capacity. To test this hypothesis, we measured the percentage of cells positive for senescence-associated beta-galactosidase (SA-βgal), a known marker of senescent cells (Debacq-Chainiaux et al., 2009). Compared with control cells, rapamycin treatment reduced the accumulation of senescent cells in both control and radiated cells (Figure 2G). The activation of the RB tumor suppressive pathway by the accumulation of p16 is often considered a key mechanism triggering cell senescence in several systems, including skin keratinocytes (Kuilman et al., 2010; Luis et al., 2011). Indeed, knockdown of p16 in NOK (Figure 2H, insert) resulted in a significant increase in the colony forming efficiency of NOK under control conditions as well as after ionizing radiation (Figure 2H). Together, these findings suggest that mTOR inhibition may increase the clonal proliferative capacity of oral epithelial stem cells by preventing the activation of specific cell senescence mechanisms.
Rapamycin prolongs the lifespan of NOK cultures by limiting epithelial stem cell exhaustion due to replicative senescence
Based on these results, we next asked if mTOR inhibition would also protect NOK from replicative senescence, a process that involves the progressive depletion of progenitor cells due to senescence and differentiation, and often serves as an in vitro surrogate for aging (Zeng, 2007). Surprisingly, continuous treatment of human primary NOK with rapamycin extended dramatically the lifespan of the cells in culture (Figure 3A). Specifically, rapamycin treated cells almost tripled their lifespan and total cumulative population doublings when compared to control cells (Figures 3B, 3C and 3D). We can estimate that while in the absence of rapamycin a single cell will give rise approximately to 106 cells before undergoing senescence, while in the presence of rapamycin a single cell will give rise to around 1017 cells. Similar results were observed in multiple independent NOK isolates. This effect was mediated by the continuous inhibition of mTOR, as cells quickly undergo replicative senescence after rapamycin removal (Figure 3E).
Figure 3. Rapamycin prolongs the lifespan of NOK cultures.

(A) Cumulative population doubling from a representative culture of primary NOK in control conditions or continuous presence of rapamycin. Each dot represents a passage and dotted line indicates that cells could not be further expanded. (B and C) Average of the days before replicative senescence and the Maximum cumulative population doublings from three different cultures of NOK cultured in in control conditions (Con) or continuous presence of rapamycin (Rap). (D) Bright field images of NOK treated or not with rapamycin for 15 or 45 days. (E) Cumulative population doubling from a representative culture of primary NOK in which rapamycin treatment was withdrawn (Rap removal) or kept (Rap) after 42 days. Each dot represents a passage and dotted line indicates that cells could not be further expanded. (F) Western blot analysis of NOK treated (Rap) or not (Con) with rapamycin for 15 or 35 days. mTOR inhibition is reflected by the levels of pS6. No consistent differences were seen in Akt phosphorylation at positions 308 and 473. p16 and p63 levels were reduced or preserved, respectively, by rapamycin treatment. (G) Staining for senescence-associated β-galactosidase (SA-βgal) 35 days after rapamycin treatment. (F) Accumulation of different cytokines associated with the senescence secretory phenotype in conditioned media from human primary NOK treated or not (control) with rapamycin expressed as fold change. See methods sections for cytokines concentration levels.
Western blot analysis revealed that cells continued to respond to rapamycin along the entire treatment, as assessed by the inhibition of phosphorylation of S6 by rapamycin (Figure 3F). While we did not observed significant changes in the phosphorylation status of Akt (Figure 3F), there was a clear reduction in the levels of p16 in rapamycin-treated NOK when compared to the increasing p16 expression levels with time in control cells (Figure 3F), concomitant with a massive increase in SA-βgal in control cells but not in rapamycin treated cells (Figure 3G). As replicative senescence involves the progressive depletion of progenitor cells in primary cultures, we analyzed the effects of rapamycin treatment on the levels of the epithelial stem cell marker p63 (Blanpain and Fuchs, 2009). As shown if figure 3F, we observed that rapamycin prevented the disappearance of the epithelial progenitor marker p63, indicating that mTOR inhibition reduces the depletion of stem cells from the culture. These results highlight the possibility that mTOR inhibition may increase the proliferative capacity of human primary NOK by preventing the senescence of their repopulating stem cells.
Senescent cells acquire the ability to secrete a variety of cytokines, often referred to as the senescence-associated secretory phenotype (SASP). These secreted cytokines have a myriad of effects on neighboring cells, including the promotion of growth arrest and senescence (Acosta et al., 2008; Coppé et al., 2008; Rodier and Campisi, 2011). This pro-senescence autocrine/paracrine positive feedback loop can aid in the accelerated depletion of repopulating cells from tissues and primary cultures. As shown in figure 4H, rapamycin significantly reduced the secretion of most senescence-associated cytokines, an effect that in turn may contribute to the retention of the proliferative capacity of rapamycin-treated primary epithelial cells.
Figure 4. Rapamycin protects from oxidative stress through increased MnSOD expression.

(A) Analysis of reactive oxygen species (ROS) levels by Dihydroethidium hydroethidine (DHE) staining 24 hours after radiation in NOK and HN12 cells pretreated or not (control) with rapamycin (upper panel) and FACS analysis of ROS levels by DHE staining in NOK after 10 or 40 days of continuous rapamycin treatment (lower panel). (B) Western blot of senescence (p16) and DNA damage (γH2AX) marker levels after 24 hs of H2O2 treatment in NOK pretreated or not (control) with rapamycin. (C) Western blot of DNA damage (γH2AX) marker levels after 24 hs of H2O2 treatment in the HNSCC HN12 cells pretreated or not (control) with rapamycin. (D) Western blot analysis of protein levels of ROS scavenging enzymes (MnSOD, Cu-ZnSOD and catalase) in NOK and HN12 cells after 72 hs of rapamycin treatment. mTOR inhibition by rapamycin is shown by the levels of pS6. (E) Quantification of MnSOD protein expression levels by western blot in NOK and HN12 cells after 72 hs of rapamycin treatment. (F) Western blot analysis of p16 and γH2AX levels in control (Con) or rapamycin (Rap) treated human primary NOK transfected with control siRNA (siCon) or siRNA targeted against MnSOD (siMnSOD). (G) Western blot analysis of MnSOD expression and colony forming efficiency from clonogenic assays of human primary NOK transfected with control siRNA (siCon) or siRNA targeted against MnSOD (siMnSOD). Cells were pretreated or not (control) with rapamycin for 72 hs after siRNA transfection, irradiated with 0 or 3 grays (Gy) and 24 hs later subjected to clonogenic assays. See also Figure S3.
Rapamycin protects from oxidative stress through increased MnSOD expression
In search for the potential mechanisms mediating the protective effect of mTOR inhibition in epithelial stem cell senescence, we explored whether rapamycin treatment of NOK results in the activation of molecular events often associated with delayed aging. After rapamycin treatment, however, we did not observe an increase in the expression, nuclear localization, or transcriptional activation of FoxO proteins, nor changes in the expression of SIRT family members, all of which have been shown to influence lifespan and stem cell response to stress (Calnan and Brunet, 2008; Finkel et al., 2009) (not shown). On the other hand, reactive oxygen species (ROS) can mediate the deleterious effects of radiation and contribute to DNA damage and the activation of senescence pathways in cells (Diehn et al., 2009; Kuilman et al., 2010; Sonis, 2009; Spitz et al., 2004). By analyzing the levels of ROS by DHE staining, we found that rapamycin treatment prevented the induction of ROS after radiation in primary NOK, while it failed to protect from ROS accumulation in HNSCC cells (Figure 4A). Furthermore, rapamycin prevented the remarkable increase in ROS preceding NOK cell growth arrest (Figure 4A, 10 days) as well as in cells undergoing replicative senescence (Figure 4A, 40 days). These observations suggested that mTOR inhibition might limit the accumulation of ROS in NOK by decreasing ROS formation or by accelerating their inactivation. Regarding the latter, we hypothesized that if the protective effects of rapamycin is mediated by an increase in the cellular ROS scavenging capacity, mTOR inhibition should also protect from hydrogen peroxide (H2O2) induced senescence. Indeed, pretreatment of primary NOK with rapamycin prevented the appearance of the DNA damage marker γH2AX and the expression of the senescence marker p16 following H2O2 treatment (Figure 4B), while it did not prevent the increase in DNA damage in HNSCC cells (Figure 4C). These results further supported the possibility that rapamycin may prevent normal cells from entering senescence and increase their replicative capacity by suppressing ROS accumulation, and hence oxidative stress.
Rapamycin treatment did not induce significant changes in the ratio of reduced/oxidized glutathione (GSH) (Figure S3A), indicating that this might not be the mechanism by which mTOR inhibition scavenges ROS. We next measured the levels of different enzymes involved in the detoxification of ROS. Interestingly, among these, rapamycin treatment did not affect the expression of catalase or cytosolic superoxide dismutase (Cu-ZnSOD), but specifically increased the protein levels of MnSOD, the mitochondrial-localized superoxide dismutase, in NOK but not in HN12 cells (Figure 4D and 4E). Of direct relevance to our studies, MnSOD has been linked to radiation resistance, and its deficiency can promote the appearance of accelerated aging phenotypes (Hosoki et al., 2012; Velarde et al., 2012; Weyemi et al., 2012). Accordingly, MnSOD knockdown in NOK increased the basal levels of p16 expression and DNA damage as measured by γH2AX, and this effect could only be partially rescued by rapamycin (Figure 4F). Furthermore, decreased expression of MnSOD abrogated the increased clonogenic capacity of rapamycin-treated NOK under normal conditions and after radiation (Figure 4G). Interestingly, mTOR inhibition did not increase the mRNA levels of MnSOD (Figure S3B), indicating that translational or postranslational mechanisms rather than transcriptional events may account for the increased MnSOD protein expression caused by rapamycin in NOK. In particular, MnSOD activity and stability are highly regulated by post-translational modifications, including protein acetylation in the mitochondria (Takahashi et al., 2011; Tao et al., 2010). Indeed, both total and acetylated MnSOD increased upon rapamycin treatment in human NOK (Figure S3C).
mTOR inhibition prevents the deleterious effects of radiation in normal oral epithelia in vivo
Considering the remarkable effects of mTOR inhibition in vitro, we next asked if rapamycin could rescue the deleterious effects of ionizing radiation in normal oral epithelia. For these experiments, 8 weeks old C3H mice received fractionated radiation of a 6 grays (Gy) dose/day/mouse for 5 days and, before each radiation dose, mice were injected with vehicle (control) or rapamycin (Figure 5A). Mice were euthanized at day 5, one hour after the last dose of radiation, and at day 10, 5 days after the last radiation dose. Tongues were removed and processed for analysis to examine the immediate impact of radiation and its long term effects, respectively. In control animals, mTOR activity is restricted to the upper, more differentiated and non-proliferative layers of the oral mucosa (Figure 5B), as we have recently reported in the skin (Squarize et al., 2010). Five days of radiation (day 5) did not result in an increase in mTOR activity in vehicle-treated animals as judged by phosphorylation of S6 (Figure 5B). However, we could observe aberrant activation of mTOR throughout the basal layer of the tongue epithelium 5 days after the last dose of radiation (day 10), which was prevented by rapamycin treatment during the radiation period (Figure 5B). Surprisingly, mTOR inactivation by rapamycin concomitant with radiation almost completely prevented the appearance of mucositis/ulcers in irradiated mice (Figure 5C). Histological analysis of the tongues revealed the preservation of the epithelial layer in irradiated rapamycin-treated mice, although radiation-associated tissue changes were observed (Figure 5D). No differences were observed in response to rapamycin innon-irradiated mice (Figure S4A). In contrast, extensive ulceration, up to 50% of the surface of the tongue, was evident in the animals treated with vehicle, which was absent in all rapamycin treated mice (Figure 5D). Interestingly, rapamycin did not protect from mucositis when mice were irradiated with high intensity in a single dose (15 Gy, Figures S4B and S4C), indicating that the protection conferred by rapamycin treatment might only be effective at clinically-relevant fractionated radiation doses.
Figure 5. Rapamycin prevents the deleterious effects of radiation in normal oral epithelia in vivo.

(A) Fractionated radiation and drug treatment scheme for mice. (B) Representative pictures of pS6 staining (red) in control (non-irradiated) animals and irradiated animals at the end of the radiation period (day 5) or 5 days after the last radiation dose (day 10), receiving vehicle (radiation) or rapamycin (radiation+rapamycin). Tissues were counterstained to label actin (green) and nuclei (blue). Right panels show details at higher magnification. Location of the basal membrane is indicated with a white dotted line. Bar: 100μm. (C). Representative pictures and quantification of tongues stained with toluidine blue from irradiated animals on day 5 following the final radiation dose, receiving vehicle (Radiation) or rapamycin (Radiation+Rapamycin). Lack of protective epithelial barrier and therefore ulcer formation is indicated by deep, royal blue staining in epithelium defects. Quantification represents at least 6 animals per group in 3 independent experiments. (D) Histological analysis of the tongues from control (non-irradiated) animals and irradiated animals 5 days after the last radiation dose (day 10), receiving vehicle (radiation) or rapamycin (radiation+rapamycin). Small panels show details at higher magnification. Control mouse tongue shows normal dorsal and ventral epithelial morphology. Epithelial layer is preserved in irradiated rapamycin-treated mice (radiation+rapamycin), although radiation-associated cellular changes are evident. Ulcerations, up to 50% of the surface of the tongue and radiation-associated cellular changes are evident in irradiated animals treated with vehicle. An ulcerated and non-ulcerated area is magnified. See also Figure S4.
Rapamycin treatment prevents the loss of proliferative capacity of the epithelial stem cell compartment after radiation in vivo
Aligned with our in vitro results, we did not observe substantial differences between control and rapamycin-treated mice in the visible accumulation of p53 in basal epithelial cells after radiation (Figure 6A). However, we observed a remarkable reduction in the levels of γH2AX and increased levels of MnSOD (Figure 6A), indicating that rapamycin may protect normal epithelial cells from DNA damage and oxidative stress in vivo. Indeed, aligned with this possibility, we have recently shown that the persistent activation of mTOR by Wnt1 leads to the accumulation of DNA damage and accelerated epithelial stem cell senescence, which can be prevented by rapamycin treatment (Castilho et al., 2009; Liu et al., 2007). Hence, our in vitro and in vivo observations suggest the possibility that rapamycin might protect from radiation-induced depletion of epithelial progenitors due to senescence or terminal differentiation, thereby increasing their survival and repopulating capacity. This prompted us to analyze the presence of the basal stem cell marker p63 (Pellegrini et al., 2001) in mice exposed to radiation and treated with rapamycin or vehicle. As shown in Figure 6B, we found that rapamycin prevented the radiation-induced disappearance of the epithelial progenitor marker p63 and the basal cell marker cytokeratin 5 (Blanpain and Fuchs, 2009). Furthermore, by using the proliferation marker Ki-67, we found that rapamycin prevented also the radiation-induced block in proliferation of the basal progenitor layer of the oral epithelia at the end of the radiation regime, and preserved the proliferative and tissue repopulating function of the basal epithelial cells 5 days after the last radiation dose (Figure 6B). Rapamycin treatment in non-irradiated mice did not affect the number and distribution pattern of p63 positive cells (not shown); we only observed a slight and yet significant increase in the levels of p63 expression in basal progenitor cells upon rapamycin treatment (Figure S5). Taken together, our observations indicate that rapamycin treatment prevents the loss of proliferative capacity of the epithelial progenitor and stem cell compartment after radiation, which in turn, protects from epithelial stem cell depletion and the consequent appearance of widespread ulcers and mucositis.
Figure 6. Rapamycin treatment prevents the loss of proliferative capacity of the epithelial stem cell compartment after radiation in vivo.

(A) p53, γH2AX and MnSOD expression (red) in representative pictures in tongues of control (non-irradiated) animals and irradiated animals at the end of the radiation period (day 5), receiving vehicle (radiation: Rad) or rapamycin (Rad+Rap). Tissues were counterstained to label actin (green) and/or nuclei (blue). Right panels show details at higher magnification. Location of the basal membrane is indicated with a white dotted line. Bar: 100μm. Graphs show the average fluorescence value per nucleus (p53 and γH2AX) or in the basal layer (MnSOD), expressed in arbitrary units and quantified as described in the methods section. (B) Representative pictures and quantification of the stem cell marker p63 (red) and the basal progenitor marker cytokeratin 5 (K5, green) (first panel) and the proliferation marker Ki67 (red, second panel). Staining was performed in tongues from unirradiated animals and irradiated animals at the end of the radiation period (day 5) or 5 days after the last radiation dose (day 10), receiving vehicle (radiation) or rapamycin (radiation+rapamycin). Tissues were counterstained to label nuclei (blue) and/or actin (green). Right side of each panels show details at higher magnification. Location of the basal membrane is indicated with a white dotted line. Bar: first panel 50μm, second panel 100μm. Graphs show the percentage of basal p63+ cells and the Ki-67 index (percentage of Ki67+ cells/total epithelial cells) in non-irradiated (No IRR) or irradiated (IRR) mice at day 5 and 10, quantified as described in the methods section. See also Figures S5 and S6.
DISCUSSION
While emerging evidence supports the central role of the mTOR pathway in cell growth and cancer progression, increased mTOR activity paradoxically can also cause cells to undergo differentiation or senescence, thereby exiting the proliferative cell pool (reviewed in Iglesias-Bartolome and Gutkind, 2011). In this context, we have recently shown that the persistent activation of mTOR by Wnt1 leads to accelerated epithelial stem cell senescence and premature aging in mice (Castilho et al., 2009; Liu et al., 2007). Here, we show that mTOR inhibition decreases epithelial stem cell depletion. Our results indicate that rapamycin prevents cells from entering senescence and terminal differentiation programs during replicative senescence or when exposed to radiation-induced stress conditions, without protecting HNSCC cells from radiation-induced cell death.
Senescence can be triggered by multiple mechanisms, including those resulting in ROS production and oxidative stress (Kuilman et al., 2010; Rodier and Campisi, 2011). The generation of ROS is also an important mediator of the cellular alterations caused by radiation exposure (Diehn et al., 2009; Sonis, 2009; Spitz et al., 2004), including mucositis (Citrin et al., 2010; Cotrim et al., 2007; Cotrim et al., 2005). Our results show that mTOR inhibition causes a remarkable reduction in ROS accumulation in irradiated NOK as well as in NOK undergoing replicative senescence, thus suggesting that inhibition of ROS formation may represent one the of mechanisms by which rapamycin protects from cell senescence and stem cell depletion. Consistent with this possibility, mTOR inhibition by rapamycin prevented the appearance of DNA damage and the expression of the senescence marker p16 after the direct exposure of human primary NOK to hydrogen peroxide-induced oxidative stress.
In this regard, a reduction in ROS formation after mTOR blockade has been documented in drosophila cells and human T cells, albeit by a yet to be determined molecular mechanism (Lee et al., 2010; Schieke et al., 2006). Here, we observed that ROS inhibition by rapamycin correlated with increased levels of MnSOD. MnSOD is the main ROS scavenger in the inner mitochondrial matrix, and acts as the first line of defense against mitochondrial oxidative damage (Holley et al., 2011). Furthermore, we obtained evidence that increase MnSOD mediates the beneficial effects of mTOR inhibition in primary human NOK. Indeed, MnSOD knock down reverted the decrease in p16 expression and the protective effects of rapamycin in NOK clonogenic capacity under basal conditions and after radiation exposure. Consistent with a protective role of MnSOD, its targeted deletion in connective tissue results in reduced lifespan that correlates with an increase in cell senescence due to the expression of p16 (Treiber et al., 2011). Moreover, mitochondrial oxidative stress resulting from MnSOD deficiency results in cellular senescence in mouse skin, a process associated with increased nuclear DNA damage (Velarde et al., 2012). The precise mechanism by which mTOR blockade results in increased MnSOD protein expression is at the present not known. Rapamycin treatment did not increase mRNA levels of MnSOD, indicating that its enhanced protein expression may result from increased translation or yet to be identified postranslational events, such as enhanced protein stability. We observed that rapamycin promotes an increase in both total and acetylated form of MnSOD, indicating that rapamycin may affect the stability of MnSOD through changes in acetylation of the enzyme. These, as well as other possibilities warrant further investigation.
Rapamycin treatment on mice was effective at limiting the loss of proliferative basal oral epithelial stem cells upon radiation, thereby enhancing their tissue repopulating capacity and preventing the appearance of ulcers. Aligned with our observations supporting that increased MnSOD levels prevent cell senescence in human primary NOK, this protective effect of mTOR inhibition involved increased expression of MnSOD in the basal progenitor epithelial cell layer. Thus, these results raise the possibility that by increasing the expression of MnSOD, mTOR inhibitors may reduce ROS-induced DNA damage, oxidative stress, and ultimately epithelial stem cell senescence, protecting from radiation-induced depletion of tissue-repopulating stem cells and the concomitant appearance of ulcers and mucositis (Figure S6). On the other hand, rapamycin decreased the release of multiple senescence-associated cytokines, suggesting that mTOR inhibition might also ameliorative mucositis by preventing the release of a pro-senescent and pro-inflammatory secretome from normal and irradiated cells (Sonis, 2009).
While mTOR inhibition by rapamycin protects normal cells from initiating senescence programs, intriguingly rapamycin does not increase the viability or survival of HNSCC cells. This is aligned with the inability of rapamycin to increase MnSOD in these cells, which highlights the fact that signaling pathways and stress responses are often differentially wired in normal and cancer cells. HNSCC arises from multiple genetic and epigenetic alterations, and among them, loss of heterozygosity in the p16 locus concomitant with the inability to express the remaining allele due to p16 promoter methylation is one of the earliest events in HNSCC progression (Rosas et al., 2001). Approximately 20% of all HNSCC cases are also now known to be associated with HPV16 infection (zur Hausen, 2009). While the E6 HPV oncogene promotes p53 degradation, the HPV E7 oncogene binds and inhibits RB, thereby rendering p16 unable to stimulate its tumor suppressive functions (Moody and Laimins, 2010). Thus, p16 inactivation by genetic and epigenetic events or by viral oncogenes is a shared feature in most HNSCC lesions, which therefore renders mTOR inhibitors unable to affect p16 expression or function in these cells. This could in turn help explain why rapamycin exposure does not have any impact on the activation of p16-dependent cell senescence pathways in HNSCC.
The demise of stem cells by senescence or differentiation in response to extrinsic or intrinsic cellular stress pathways may represent a key mechanism driving stem cell depletion, thereby contributing to reduced tissue regenerative capacity. In this regard, our results highlight the importance of tissue renewal mediated by adult stem cells in organs that are continuously exposed to environmental assaults, and demonstrate that the aberrant activation of the mTOR pathway plays a predominant role mediating the depletion of the epithelial stem cell compartment. Furthermore, our results support the likely contribution of mitochondrial oxidative damage to mTOR-dependent cellular senescence in adult tissue-regenerating cells. On the other hand, loss of proliferative and repopulating activity of normal epithelial stem cells is an undesired secondary effect of most antineoplastic treatments. In the oral mucosa, this process, in conjunction with other factors, can lead to mucositis, a mucosal injury characterized by disruption of epithelial integrity, ulceration and pain (Sonis, 2009, 2010). As radiotherapy is one of the most widely used antineoplastic treatments, including almost 80% of patients with head and neck cancer (Delaney et al., 2005), interventional strategies reducing the deleterious effect of radiation on normal epithelia and hence preserving the integrity of the oral mucosa will have a great impact in the quality of life of cancer patients (Citrin et al., 2010). Ultimately, our findings indicate that by preserving the tissue repopulating capacity of the epithelial stem cells, mTOR inhibition with rapamycin and newly developed agents (Wander et al., 2011) may prevent the appearance of radiation-induced mucositis and its consequent negative impact on anticancer treatment outcome and cost
EXPERIMENTAL PROCEDURES
Cell Culture
NOK isolation and culture was performed as described (Leelahavanichkul and Gutkind, 2012). NOK were maintained in defined keratinocyte serum free media (KSFM) (Invitrogen) supplemented with antibiotics at 37°C in the presence of 5% CO2 and passed every 3–4 days. NOK between passages 2 to 4 were used for the experiments. HN12 and Cal27 have been described previously (Amornphimoltham et al., 2005). Detailed methods for NOK isolation and cell culture are described in Supplemental Experimental Procedures.
Irradiation and clonogenic assays
Surviving fraction was calculated as previously described (Franken et al., 2006) relative to the respective non-irradiated cells. Colonies were counted and measured with calibrated images in ImageJ. Colony forming efficiency was expressed as the proportion of plated cells that formed colonies relative to the number of colonies formed by non-irradiated control cells.
Immunoblot Analysis and Cytokine measurements
Cells were lysed in lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40) supplemented with protease (protease inhibitor cocktail, Sigma) and phosphatase inhibitors (1mM Na3VO4 and 1mM NaF). Equal amounts of total cell lysate proteins were subjected to SDS-polyacrylamide gel electrophoresis. Bands were detected by using near-infrared fluorescence (Odyssey LI-COR Biotechnology). Cytokines were analyzed in conditioned media by the Cytokine Core Laboratory, University of Maryland, using the Luminex Multianalyte System. Values of cytokines were corrected to the protein and expressed as percentage of the control.
Cell proliferation, apoptosis and senescence
Cell proliferation was evaluated using incorporation of 5-ethynyl-2′-deoxyuridine (EdU) with the Click-iT; apoptosis was detected using the Click-iT TUNEL Alexa Fluor 594 Imaging Assay (Invitrogen); and SA-β-gal activity was measured using the SA-β-gal kit (Cell Signaling Technology) according to the manufacturer's instructions. The proportion of positive cells was determined by fluorescence or transmission microscopy and quantified using ImageJ. Population doublings were calculated as previously described (Cristofalo et al., 1998).
ROS measurement and Immunofluorescence
ROS measurement was performed by FACS in cells incubated with Dihydroethidium (hydroethidine) (Molecular Probes) at a final concentration of 1.5 μM for 10 min at 37 °C. For immunofluorescence, cells and tissues (cryosections) were fixed with 3.2% paraformaldehyde in PBS and permeabilized with Triton×100 0.1% in glycine 200mM in PBS. Nonspecific binding was blocked with 3% of bovine serum albumin (BSA) or 10% FBS in PBS. Cells and tissues were incubated with the primary antibodies overnight at 4°C, followed by 1.5 hs incubation with the secondary antibodies Nuclei were stained with Hoechst 33342 (1:2000 Invitrogen) and actin with Alexa 546-phalloidin (Invitrogen) according to the manufacturers' instructions. Images were taken using Zeiss Axio Imager Z1 microscope equipped with an Apotome device (Carl Zeiss) and a motorized stage. Detailed protocols and image quantification procedures are described in Supplemental Experimental Procedures
Animal irradiation
Female C3H mice were used for this study. The head and neck area was irradiated by placing each animal in a specially built Lucite jig. For fractionated radiation, mice were injected intraperitoneally every day for 5 days with rapamycin (5mg/kg) and irradiated at 6 Gy/day. Ionizing radiation was delivered with a Therapax DXT300 X-ray irradiator (Precision X-ray) at a dose rate of 1.9 Gy/min. Result corresponds to 6 animals and is representative of 3 independent experiments. Detailed methods are described in Supplemental Experimental Procedures
Supplementary Material
Highlights
-
-
Rapamycin increases the repopulating capacity of human epithelial stem cells.
-
-
mTOR inhibition prevents epithelial stem cell senescence.
-
-
mTOR inhibition protects from oxidative stress by increasing MnSOD expression.
-
-
Rapamycin protects from radiation-induced oral ulcers and mucositis.
ACKNOWLEDGMENTS
We thank Bruce J. Baum (NIH, NIDCR) and Thomas Bugge (NIH, NIDCR) for comments and discussions. We thank Mina Konigsberg (Universidad Autonoma, Mexico) for insightful suggestions regarding oxidative stress and senescence. This work was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Dental and Craniofacial Research, and by the Human Frontier Science Program Grant # RGP0041-2011.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Acosta JC, O'Loghlen A, Banito A, Guijarro MV, Augert A, Raguz S, Fumagalli M, Da Costa M, Brown C, Popov N, et al. Chemokine Signaling via the CXCR2 Receptor Reinforces Senescence. Cell. 2008;133:1006–1018. doi: 10.1016/j.cell.2008.03.038. [DOI] [PubMed] [Google Scholar]
- Amornphimoltham P, Leelahavanichkul K, Molinolo A, Patel V, Gutkind JS. Inhibition of Mammalian target of rapamycin by rapamycin causes the regression of carcinogen-induced skin tumor lesions. Clin Cancer Res. 2008;14:8094–8101. doi: 10.1158/1078-0432.CCR-08-0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amornphimoltham P, Patel V, Sodhi A, Nikitakis NG, Sauk JJ, Sausville EA, Molinolo AA, Gutkind JS. Mammalian Target of Rapamycin, a Molecular Target in Squamous Cell Carcinomas of the Head and Neck. Cancer Research. 2005;65:9953–9961. doi: 10.1158/0008-5472.CAN-05-0921. [DOI] [PubMed] [Google Scholar]
- Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nat Rev Cancer. 2011;11:239–253. doi: 10.1038/nrc3007. [DOI] [PubMed] [Google Scholar]
- Blanpain C, Fuchs E. Epidermal homeostasis: a balancing act of stem cells in the skin. Nat Rev Mol Cell Biol. 2009;10:207–217. doi: 10.1038/nrm2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calnan DR, Brunet A. The FoxO code. Oncogene. 2008;27:2276–2288. doi: 10.1038/onc.2008.21. [DOI] [PubMed] [Google Scholar]
- Castilho RM, Squarize CH, Chodosh LA, Williams BO, Gutkind JS. mTOR mediates Wnt-induced epidermal stem cell exhaustion and aging. Cell Stem Cell. 2009;5:279–289. doi: 10.1016/j.stem.2009.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Liu Y, Liu R, Ikenoue T, Guan KL, Zheng P. TSC-mTOR maintains quiescence and function of hematopoietic stem cells by repressing mitochondrial biogenesis and reactive oxygen species. The Journal of experimental medicine. 2008;205:2397–2408. doi: 10.1084/jem.20081297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HC, Chen HL, Lai JY, Chen CC, Tsai YJ, Kuo MT, Chu PH, Sun CC, Chen JK, Ma DH. Persistence of transplanted oral mucosal epithelial cells in human cornea. Invest Ophthalmol Vis Sci. 2009;50:4660–4668. doi: 10.1167/iovs.09-3377. [DOI] [PubMed] [Google Scholar]
- Citrin D, Cotrim AP, Hyodo F, Baum BJ, Krishna MC, Mitchell JB. Radioprotectors and Mitigators of Radiation-Induced Normal Tissue Injury. The Oncologist. 2010;15:360–371. doi: 10.1634/theoncologist.2009-S104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé J-P, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez P-Y, Campisi J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008;6:e301. doi: 10.1371/journal.pbio.0060301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cotrim AP, Hyodo F, Matsumoto K-I, Sowers AL, Cook JA, Baum BJ, Krishna MC, Mitchell JB. Differential Radiation Protection of Salivary Glands versus Tumor by Tempol with Accompanying Tissue Assessment of Tempol by Magnetic Resonance Imaging. Clinical Cancer Research. 2007;13:4928–4933. doi: 10.1158/1078-0432.CCR-07-0662. [DOI] [PubMed] [Google Scholar]
- Cotrim AP, Sowers AL, Lodde BM, Vitolo JM, Kingman A, Russo A, Mitchell JB, Baum BJ. Kinetics of Tempol for Prevention of Xerostomia Following Head and Neck Irradiation in a Mouse Model. Clinical Cancer Research. 2005;11:7564–7568. doi: 10.1158/1078-0432.CCR-05-0958. [DOI] [PubMed] [Google Scholar]
- Cristofalo VJ, Allen RG, Pignolo RJ, Martin BG, Beck JC. Relationship between donor age and the replicative lifespan of human cells in culture: A reevaluation. Proceedings of the National Academy of Sciences. 1998;95:10614–10619. doi: 10.1073/pnas.95.18.10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czerninski R, Amornphimoltham P, Patel V, Molinolo AA, Gutkind JS. Targeting mammalian target of rapamycin by rapamycin prevents tumor progression in an oral-specific chemical carcinogenesis model. Cancer Prev Res (Phila) 2009;2:27–36. doi: 10.1158/1940-6207.CAPR-08-0147. [DOI] [PubMed] [Google Scholar]
- Debacq-Chainiaux F, Erusalimsky JD, Campisi J, Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-betagal) activity, a biomarker of senescent cells in culture and in vivo. Nat Protoc. 2009;4:1798–1806. doi: 10.1038/nprot.2009.191. [DOI] [PubMed] [Google Scholar]
- Delaney G, Jacob S, Barton M. Estimation of an optimal external beam radiotherapy utilization rate for head and neck carcinoma. Cancer. 2005;103:2216–2227. doi: 10.1002/cncr.21084. [DOI] [PubMed] [Google Scholar]
- Diehn M, Cho RW, Lobo NA, Kalisky T, Dorie MJ, Kulp AN, Qian D, Lam JS, Ailles LE, Wong M, et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature. 2009;458:780–783. doi: 10.1038/nature07733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshleman JS, Carlson BL, Mladek AC, Kastner BD, Shide KL, Sarkaria JN. Inhibition of the mammalian target of rapamycin sensitizes U87 xenografts to fractionated radiation therapy. Cancer Res. 2002;62:7291–7297. [PubMed] [Google Scholar]
- Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature. 2009;460:587–591. doi: 10.1038/nature08197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franken NA, Rodermond HM, Stap J, Haveman J, van Bree C. Clonogenic assay of cells in vitro. Nat Protoc. 2006;1:2315–2319. doi: 10.1038/nprot.2006.339. [DOI] [PubMed] [Google Scholar]
- Gudkov AV, Komarova EA. The role of p53 in determining sensitivity to radiotherapy. Nat Rev Cancer. 2003;3:117–129. doi: 10.1038/nrc992. [DOI] [PubMed] [Google Scholar]
- Holley AK, Bakthavatchalu V, Velez-Roman JM, St Clair DK. Manganese superoxide dismutase: guardian of the powerhouse. International journal of molecular sciences. 2011;12:7114–7162. doi: 10.3390/ijms12107114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosoki A, Yonekura S, Zhao QL, Wei ZL, Takasaki I, Tabuchi Y, Wang LL, Hasuike S, Nomura T, Tachibana A, et al. Mitochondria-Targeted Superoxide Dismutase (SOD2) Regulates Radiation Resistance and Radiation Stress Response in HeLa Cells. Journal of radiation research. 2012;53:58–71. doi: 10.1269/jrr.11034. [DOI] [PubMed] [Google Scholar]
- Iglesias-Bartolome R, Gutkind JS. Signaling circuitries controlling stem cell fate: to be or not to be. Curr Opin Cell Biol. 2011;23:716–723. doi: 10.1016/j.ceb.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen KB, Driskell RR, Watt FM. Assaying proliferation and differentiation capacity of stem cells using disaggregated adult mouse epidermis. Nat Protoc. 2010;5:898–911. doi: 10.1038/nprot.2010.39. [DOI] [PubMed] [Google Scholar]
- Konstantinidou G, Bey EA, Rabellino A, Schuster K, Maira MS, Gazdar AF, Amici A, Boothman DA, Scaglioni PP. Dual phosphoinositide 3-kinase/mammalian target of rapamycin blockade is an effective radiosensitizing strategy for the treatment of non-small cell lung cancer harboring K-RAS mutations. Cancer Res. 2009;69:7644–7652. doi: 10.1158/0008-5472.CAN-09-0823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes & development. 2010;24:2463–2479. doi: 10.1101/gad.1971610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Budanov AV, Park EJ, Birse R, Kim TE, Perkins GA, Ocorr K, Ellisman MH, Bodmer R, Bier E, et al. Sestrin as a Feedback Inhibitor of TOR That Prevents Age-Related Pathologies. Science. 2010;327:1223–1228. doi: 10.1126/science.1182228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leelahavanichkul K, Gutkind JS. In Method in Molecular Biology: Epithelial Cell Culture Protocols (Humana Press) 2012. Oral and Pharyngeal Epithelial Keratinocyte Culture. [DOI] [PubMed] [Google Scholar]
- Liu H, Fergusson MM, Castilho RM, Liu J, Cao L, Chen J, Malide D, Rovira, Schimel D, Kuo CJ, et al. Augmented Wnt signaling in a mammalian model of accelerated aging. Science. 2007;317:803–806. doi: 10.1126/science.1143578. [DOI] [PubMed] [Google Scholar]
- Luis, Nuno M, Morey L, Mejetta S, Pascual G, Janich P, Kuebler B, Roma G, Nascimento E, Frye M, Di Croce L, et al. Regulation of Human Epidermal Stem Cell Proliferation and Senescence Requires Polycomb- Dependent and -Independent Functions of Cbx4. Cell Stem Cell. 2011;9:233–246. doi: 10.1016/j.stem.2011.07.013. [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB Signaling: Navigating Downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinolo AA, Hewitt SM, Amornphimoltham P, Keelawat S, Rangdaeng S, Meneses Garcia A, Raimondi AR, Jufe R, Itoiz M, Gao Y, et al. Dissecting the Akt/mammalian target of rapamycin signaling network: emerging results from the head and neck cancer tissue array initiative. Clin Cancer Res. 2007;13:4964–4973. doi: 10.1158/1078-0432.CCR-07-1041. [DOI] [PubMed] [Google Scholar]
- Moody CA, Laimins LA. Human papillomavirus oncoproteins: pathways to transformation. Nat Rev Cancer. 2010;10:550–560. doi: 10.1038/nrc2886. [DOI] [PubMed] [Google Scholar]
- Murphy JD, Spalding AC, Somnay YR, Markwart S, Ray ME, Hamstra DA. Inhibition of mTOR radiosensitizes soft tissue sarcoma and tumor vasculature. Clin Cancer Res. 2009;15:589–596. doi: 10.1158/1078-0432.CCR-08-1019. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Inatomi T, Cooper LJ, Rigby H, Fullwood NJ, Kinoshita S. Phenotypic investigation of human eyes with transplanted autologous cultivated oral mucosal epithelial sheets for severe ocular surface diseases. Ophthalmology. 2007;114:1080–1088. doi: 10.1016/j.ophtha.2006.09.034. [DOI] [PubMed] [Google Scholar]
- Nathan C-AO, Amirghahari N, Rong X, Giordano T, Sibley D, Nordberg M, Glass J, Agarwal A, Caldito G. Mammalian Target of Rapamycin Inhibitors as Possible Adjuvant Therapy for Microscopic Residual Disease in Head and Neck Squamous Cell Cancer. Cancer Research. 2007;67:2160–2168. doi: 10.1158/0008-5472.CAN-06-2449. [DOI] [PubMed] [Google Scholar]
- Nonzee NJ, Dandade NA, Patel U, Markossian T, Agulnik M, Argiris A, Patel JD, Kern RC, Munshi HG, Calhoun EA, et al. Evaluating the supportive care costs of severe radiochemotherapy-induced mucositis and pharyngitis : results from a Northwestern University Costs of Cancer Program pilot study with head and neck and nonsmall cell lung cancer patients who received care at a county hospital, a Veterans Administration hospital, or a comprehensive cancer care center. Cancer. 2008;113:1446–1452. doi: 10.1002/cncr.23714. [DOI] [PubMed] [Google Scholar]
- Parkin DM, Bray F, Ferlay J, Pisani P. Global cancer statistics, 2002. CA: a cancer journal for clinicians. 2005;55:74–108. doi: 10.3322/canjclin.55.2.74. [DOI] [PubMed] [Google Scholar]
- Pellegrini G, Dellambra E, Golisano O, Martinelli E, Fantozzi I, Bondanza S, Ponzin D, McKeon F, De Luca M. p63 identifies keratinocyte stem cells. Proc Natl Acad Sci U S A. 2001;98:3156–3161. doi: 10.1073/pnas.061032098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol. 2011;192:547–556. doi: 10.1083/jcb.201009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas SLB, Koch W, Carvalho M.d.G.d.C., Wu L, Califano J, Westra W, Jen J, Sidransky D. Promoter Hypermethylation Patterns of p16, O6-Methylguanine-DNA-methyltransferase, and Death-associated Protein Kinase in Tumors and Saliva of Head and Neck Cancer Patients. Cancer Research. 2001;61:939–942. [PubMed] [Google Scholar]
- Schieke SM, Phillips D, McCoy JP, Aponte AM, Shen R-F, Balaban RS, Finkel T. The Mammalian Target of Rapamycin (mTOR) Pathway Regulates Mitochondrial Oxygen Consumption and Oxidative Capacity. Journal of Biological Chemistry. 2006;281:27643–27652. doi: 10.1074/jbc.M603536200. [DOI] [PubMed] [Google Scholar]
- Sonis ST. Mucositis: The impact, biology and therapeutic opportunities of oral mucositis. Oral Oncol. 2009;45:1015–1020. doi: 10.1016/j.oraloncology.2009.08.006. [DOI] [PubMed] [Google Scholar]
- Sonis ST. New thoughts on the initiation of mucositis. Oral Diseases. 2010;16:597–600. doi: 10.1111/j.1601-0825.2010.01681.x. [DOI] [PubMed] [Google Scholar]
- Spitz DR, Azzam EI, Li JJ, Gius D. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: a unifying concept in stress response biology. Cancer Metastasis Rev. 2004;23:311–322. doi: 10.1023/B:CANC.0000031769.14728.bc. [DOI] [PubMed] [Google Scholar]
- Squarize CH, Castilho RM, Bugge TH, Gutkind JS. Accelerated Wound Healing by mTOR Activation in Genetically Defined Mouse Models. PLoS One. 2010;5:e10643. doi: 10.1371/journal.pone.0010643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Suzuki T, Shirai A, Matsuyama A, Dohmae N, Yoshida M. Mitochondrial localization of fission yeast manganese superoxide dismutase is required for its lysine acetylation and for cellular stress resistance and respiratory growth. Biochemical and biophysical research communications. 2011;406:42–46. doi: 10.1016/j.bbrc.2011.01.103. [DOI] [PubMed] [Google Scholar]
- Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W, et al. Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Molecular cell. 2010;40:893–904. doi: 10.1016/j.molcel.2010.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treiber N, Maity P, Singh K, Kohn M, Keist AF, Ferchiu F, Sante L, Frese S, Bloch W, Kreppel F, et al. Accelerated aging phenotype in mice with conditional deficiency for mitochondrial superoxide dismutase in the connective tissue. Aging Cell. 2011;10:239–254. doi: 10.1111/j.1474-9726.2010.00658.x. [DOI] [PubMed] [Google Scholar]
- Velarde MC, Flynn JM, Day NU, Melov S, Campisi J. Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging. 2012;4:3–12. doi: 10.18632/aging.100423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wander SA, Hennessy BT, Slingerland JM. Next-generation mTOR inhibitors in clinical oncology: how pathway complexity informs therapeutic strategy. The Journal of Clinical Investigation. 2011;121:1231–1241. doi: 10.1172/JCI44145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyemi U, Parekh PR, Redon CE, Bonner WM. SOD2 deficiency promotes aging phenotypes in mouse skin. Aging. 2012 doi: 10.18632/aging.100433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X. Human Embryonic Stem Cells: Mechanisms to Escape Replicative Senescence? Stem Cell Reviews and Reports. 2007;3:270–279. doi: 10.1007/s12015-007-9005-x. [DOI] [PubMed] [Google Scholar]
- Zhang J, Grindley JC, Yin T, Jayasinghe S, He XC, Ross JT, Haug JS, Rupp D, Porter-Westpfahl KS, Wiedemann LM, et al. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature. 2006;441:518–522. doi: 10.1038/nature04747. [DOI] [PubMed] [Google Scholar]
- Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- zur Hausen H. Papillomaviruses in the causation of human cancers — a brief historical account. Virology. 2009;384:260–265. doi: 10.1016/j.virol.2008.11.046. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.