

Abstract
Repeated stress releases dynorphins and causes subsequent activation of κ-opioid receptors (KORs) in limbic brain regions. The serotonergic dorsal raphe nucleus (DRN) has previously been found to be an important site of action for the dysphoric effects of dynorphin-κ-opioid receptor system activation during stress-evoked behaviors, and KOR-induced activation of p38α mitogen-activated protein kinase (MAPK) in serotonergic neurons was found to be a critical mediator of the aversive properties of stress. Yet, how dynorphins and KORs functionally regulate the excitability of serotonergic DRN neurons both in adaptive and pathological stress states is poorly understood. Here we report that acute KOR activation by the selective agonist U69,593 [(+)-(5α,7α,8β)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl]benzeneacetamide] inhibits serotonergic neuronal excitability within the DRN through both presynaptic inhibition of excitatory synaptic transmission and postsynaptic activation of G-protein-gated inwardly rectifying potassium channels (GIRKs) electrophysiologically recorded in brain slices. C57BL/6 mice subjected to repeated swim, stress sessions had significantly reduced KOR-mediated GIRK currents recorded in serotonergic neurons in DRN postsynaptically, without significantly affecting presynaptic KOR-mediated regulation of excitatory transmission. This effect was blocked by genetic excision of p38α MAPK selectively from serotonergic neurons. An increase in phospho-immunoreactivity suggests that this functional dysregulation may be a consequence of tyrosine phosphorylation of GIRK (KIR3.1) channels. These data elucidate a mechanism for stress-induced dysregulation of the excitability of neurons in the DRN and identify a functional target of stress-induced p38α MAPK activation that may underlie some of the negative effects of pathological stress exposure.
Introduction
Adaptive responses to stress exposure are critical to an organism's survival and rely upon a precisely tuned constellation of neurotransmitters and neurohormones acting both centrally and peripherally (Korte et al., 2005). Moreover, the etiology of stress-induced mood disorders is thought to involve a combination of pathological neuroadaptations that include alterations in both stress-related neurocircuitry and signaling (Bale, 2006). Dynorphins, the endogenous ligands for the κ-opioid receptor (KOR) (Chavkin et al., 1982), are released and upregulated during stress exposure (Panerai et al., 1987; Shirayama et al., 2004; Bruchas et al., 2007), and these endogenous opioid neuropeptides have emerged as critical mediators of several stress-related behaviors (Knoll and Carlezon, 2010; Lemos and Chavkin, 2010; Wee and Koob, 2010). For example, genetic deletion of either prodynorphin or KOR or pharmacological blockade by KOR antagonists reduce stress-induced anxiety-like, depression-like, and proaddictive behaviors (Bruchas et al., 2010; Knoll and Carlezon, 2010). However, the cellular and molecular mechanisms by which the dynorphin-KOR system regulates neuronal excitability in limbic regions and ultimately produces its behavioral effects remain unclear.
Activation of KOR either pharmacologically or following release of endogenous dynorphins results in both membrane-delimited Gβγ regulation of ion channel conductance and mitogen- activated protein kinase (MAPK) signaling (Bruchas et al., 2010). Prolonged or repeated activation of KOR also stimulates the G-protein kinase 3/β-arrestin signaling cascade that subsequently recruits and activates p38 MAPK (Bruchas et al., 2006, 2007; Xu et al., 2007), and p38 MAPK activation has been shown to be required for the aversive qualities of stress as well as promoting passive coping strategies (Bruchas et al., 2007, 2011). Recently, we demonstrated that KOR-dependent activation of p38α MAPK in 5-HT neurons in the dorsal raphe nucleus (DRN) is both necessary and sufficient to induce a negative affective state (Land et al., 2009; Bruchas et al., 2011).
The importance of the DRN for stress-related behaviors is well established (Maier and Watkins, 2005). The DRN is a principal serotonergic nucleus in the brain that sends afferents to many forebrain limbic regions (O'Hearn and Molliver, 1984; Azmitia and Gannon, 1986; Kosofsky and Molliver, 1987; Vasudeva et al., 2011) and is specifically activated during exposure to uncontrollable stress (Adell et al., 1997; Roche et al., 2003; Maier and Watkins, 2005; Kirby et al., 2007; Land et al., 2009). Therefore, elucidating the regulation of DRN activity by stress-related neuropeptides in naive and stress-exposed individuals is important to understand how the pathological effects of stress impact brain functioning.
In the present study, we measured the effects of KOR activation in DRN using whole-cell voltage-clamp electrophysiological techniques to isolate glutamatergic and GABAergic fast synaptic activity as well as isolating postsynaptic G-protein-gated inwardly rectifying potassium (GIRKs, Kir3) currents. We then investigated how these processes were modified by repeated exposure to stress and specifically sought to identify p38 MAPK-dependent defects in this regulation caused by repeated stress. We hypothesized that repeated stress would result in sustained dynorphin release that would in turn disrupt KOR regulation of 5-HT neuronal excitability in the DRN via a p38α MAPK-dependent process.
Materials and Methods
Subjects
Male C57BL/6 mice, age >50 d, were maintained under a 12 h light/dark cycle (7:00 A.M. to 7:00 P.M. light) with access to standard food and water ad libitum. All procedures on animal subjects were approved by the University of Washington IACUC committee. Mice that were housed together (2–4 per cage) were subjected to the same behavioral treatment. Prodynorphin (Dyn−/−) knock-out mice and p38αCKOSERT mice were derived as described by Sharifi et al. (2001) and Bruchas et al. (2011), respectively, and have been backcrossed to C57BL/6 background >10 generations.
Swim stress
Mice were subjected to a 2 d swim stress procedure in which they were exposed to a 15 min swim session on day 1, then were exposed to four 6 min swim sessions in 29.0–31.0°C water, separated by 6 min and conducted under bright light (690–700 lux) conditions on day 2 as previously described (McLaughlin et al., 2003a).
Tail flick
Stress-induced analgesia was assessed using the warm water (52.5°C) tail withdrawal assay as described previously (Vaught and Takemori, 1979; Melief et al., 2010).
Immunohistochemistry
Tryptophan hydroxylase immunohistochemistry staining and intracellular labeling.
During electrophysiological recordings, cells were filled with biocytin (0.1%; Sigma-Aldrich) present in the recording electrode. After recording, slices were fixed by submersion in 4% paraformaldehyde prepared in 0.1 m phosphate buffer (PB; pH 7.4). Sections were incubated with mouse anti-TPH (tryptophan hydroxylase) antibody (1:500, Sigma-Aldrich) for 12–16 h at room temperature (RT). Subsequently, immunohistochemical labeling was visualized using Alexa Fluor 488 (1:500; Invitrogen)-conjugated goat anti-mouse secondary antibody for 90–120 min at RT. Biocytin was visualized using streptavidin-conjugated Alexa Fluor 647 (1:500, Invitrogen) contained in the same secondary antibody mixture. Between incubations, slices were rinsed with PBS (3 × 10 min) and all incubations were done with mild agitation on an orbital shaker.
Phospho-antibody immunohistochemistry.
Methods for immunohistochemistry experiments were similar to those reported previously (Bruchas et al., 2007). Mice were intracardially perfused with 4% paraformaldehyde in 0.1 m PB. It has been our experience that phospho-protein immunoreactivity (ir) is of higher quality if PBS perfusion before 4% paraformaldehyde is omitted. Brains were dissected and cryoprotected with 30% sucrose in 0.1 m PB at 4°C overnight (or until the brains submerged), frozen and cut into 30 μm sections using a Leica SM200R microtome, and placed in 0.1 m PB with 0.1% sodium azide until processing. We had previously generated a rabbit polyclonal antibody against the phosphorylated Ser369 residue of KOR (McLaughlin et al., 2003b); an equivalent antisera is now available commercially at Abcam. We also generated a rabbit polyclonal antibody against the phosphorylated-Tyr 12 residue of KIR 3.1 (GIRKp) as described by Ippolito et al. (2005). Standard immunohistochemical procedures were performed to obtain phospho-KOR (KORp) (Abcam, ab63511), phospho-p38 MAPK (mouse monoclonal sc-7973) and p38α (rabbit polyclonal sc-535; Santa Cruz Biotechnology) and pY12-KIR 3.1staining. Briefly, floating sections were washed 3 × 10 min in PBS and then blocked for 60 min in 5% normal goat serum, 0.3% Triton-X in PBS. Importantly, we have found that for these phospho-antibodies use of floating sections produces higher quality staining than thaw mounted sections. Sections were incubated in primary antibody at different concentrations, temperatures and incubations for each antibody: KORp (1:200, 72 h at 4°C); mouse anti-phospho-p38 MAPK/rabbit anti-p38α (Santa Cruz Biotechnology; 1:50, 36 h RT or 72 h at 4°C); affinity-purified GIRKp [1:100 for 72 h at 4°C; the concentration was based on a 750 μg protein/ml yield as described previously (Ippolito et al., 2005)]. Often slices were colabeled with mouse anti-TPH (Sigma) at 1:1000; guinea pig anti-Dynorphin B (Peninsula Laboratories) was used at 1:200. Slices were then washed in PBS 10–15 min × 3 and incubated in goat Alexa Fluor antibody (mouse or rabbit, respectively) at 1:500–1:750 for 2 h at RT. Following secondary antibody incubation, slices were washed 3 × 10 min in PBS, then 2 × 10 min in 0.1 m PB. Sections were then mounted on Superfrost plus slides and coverslipped with Vectashield (Vector Laboratories).
Standard epifluorescent and confocal microscopy techniques (Nikon Eclipse E600 and Leica SL, respectively) were used to image sections for both TPH confirmation and phospho-antibody staining. For comparisons of phospho-ir from different behavioral treatment groups, three animals for each group were perfused, sectioned and processed in parallel. Imaging of sections from animals exposed to different treatments were done at the same time with the same camera settings. As it is typical for different rounds of staining to yield different immunoreactive intensities, images from each treatment group displayed in this study came from the same round of staining. The pattern of KORp-ir and GIRKp-ir across treatment groups seen in the dorsal raphe was similar to that previously reported both in vitro and in vitro (Ippolito et al., 2005; Bruchas et al., 2007; Xu et al., 2007; Land et al., 2008; Clayton et al., 2009).
Phospho-p38-ir quantification.
The selectivities of both the p38α and phospho-p38 MAPK antibodies were previously established using both viral vector expression and conditional gene knock-out approaches (Bruchas et al., 2011). We quantified phospho-p38 MAPK-ir using MetaMorph software as a ratio of phospho-p38 MAPK-positive cells to p38α-positive cells (i.e., colocalized). Similar quantification of GIRKp-ir and KORp-ir could not be performed since the antibodies against the unphosphorylated forms of GIRK and KOR were also raised in rabbits. We have previously confirmed that p38α is the isoform that is phosphorylated following KOR activation (Bruchas et al., 2011). Cells are considered positive for the protein if the intensity of the immunoreactivity is one-SD above the average background pixel intensity.
Electrophysiology
Procedures were similar to those previously reported (Lemos et al., 2006, 2011). Mice were decapitated, head placed in ice-cold sucrose buffer during the dissection, and the brain rapidly removed and blocked. The blocked tissue was mounted on the stage of a Leica VT1000S vibratome and surrounded in oxygenated sucrose buffer. Sections (200 μm thick) were cut through the raphe nuclei in sucrose and then placed in oxygenated 95%O2/5%CO2 artificial CSF (ACSF) incubated in a 35−37°C bath for 1 h. The slices were then removed from the bath and kept in oxygenated ACSF at RT. ACSF was composed of the following (in mm): 124 NaCl, 2.5 KCl, 2 NaH2PO4, 2.5 CaCl2, 10 Dextrose, and 26 NaHCO3. The sucrose buffer was ACSF in which NaCl was replaced by 248 mm sucrose.
Slices were placed in a recording chamber (Warner Instruments) and continuously perfused with oxygenated ACSF at ∼1.5 ml/min, maintained at 30−32°C by an inline solution heater (TC-324, Warner Instruments). For recording EPSCs or GIRK currents, the resistance of the electrodes was 5–10 MΩ when filled with an internal solution of (in mm) 130 K-gluconate, 5 NaCl, 10 Na phosphocreatine, 1 MgCl2, 0.02 EGTA, 10 HEPES, 2 MgATP, 0.5 Na2GTP, 0.1% Biocytin, pH 7.3. For measuring IPSCs, the resistance of the electrodes was 4–8 MΩ when filled with an intracellular solution of (in mm) 70 K-gluconate, 70 KCl, 2 NaCl, 10 Na phosphocreatine, 4 EGTA, 10 HEPES, 2 MgATP, 0.3 Na2GTP, 0.1% Biocytin, pH 7.3. Whole-cell recordings in voltage-clamp were performed on cells in the ventromedial and dorsomedial aspect of the DRN. The cell was voltage-clamped at −70 mV using an Axopatch 200B amplifier (Molecular Devices). Signals were digitized by a Digidata 1440 A/D converter (Molecular Devices) and stored using pClamp 10.2 software (Molecular Devices). EPSCs were isolated using picrotoxin (10 μm; Sigma-Aldrich) and CGP 55845 (1 μm; Tocris Bioscience). IPSCs were isolated using DNQX (20 μm) and APV (50 μm; Tocris Bioscience). For evoked recordings, the stimulating electrode was placed 150–200 μm dorsolateral of the recording site. Paired pulse stimulation (1 ms pulse width, 50 ms interstimulus interval, 100–1000 μA) was delivered every 10 s. A mid-range output response was used as baseline for each cell (e.g., approximately the middle of an input–output curve) to allow for observation of either positive or negative regulation by U69,593 [(+)-(5α,7α,8β)-N-methyl-N-[7-(1-pyrrolidinyl)-1-oxaspiro[4.5]dec-8-yl]benzeneacetamide (National Institute of Drug Abuse Drug Supply Program]. For mPSCs, TTX (1 μm; Calbiochem) was added to the bath. For GIRK current experiments, once a recording had been established, the slice was bathed in 5.5 mm K+ ACSF (from 2.5 mm K+) to enhance the currents in the cells at hyperpolarized potentials. As expected, when the slice was incubated in high [K+], the cells became more depolarized and inward rectification was more apparent. This increased current was blocked by 100 μm BaCl2 (Williams et al., 1988). The cells were subjected to a voltage-ramp protocol in which they were brought from −120 mV to −50 mV over 10 s. After three stable ramp measurements (the average of three sweeps) were obtained, U69,593 (1000 nm) was bath applied and the slice was allowed to equilibrate for 3 min. Subsequently three ramp measurements were taken every 3 min for 12 additional minutes and the last three ramp measurements (after 15 min of U69,593 application) were averaged. Following U69,593 application and stable current responses, BaCl2 (100 μm) was added to distinguish Ba2+-sensitive GIRK currents.
Data analysis and statistics
Electrophysiological data were analyzed using Clampfit 10.2 (Molecular Devices) and MiniAnalysis 6.0.7 software (Synaptosoft). One-way repeated- measures ANOVAs or paired t tests were used for within-cell experiments (predrug baseline vs postdrug effect). We used unpaired t tests as well as one-way and two-way ANOVAs as specified in the Results, for comparisons made across behavioral treatment groups and genotypes. The responses of individual cells were considered as a separate N, as is standard for electrophysiology experiments; however, when comparing differences between different behavioral treatment groups (i.e., naive vs stress), a minimum of 3 animals from at least two different cages were used per group.
Results
The dorsal raphe is a heterogeneous nucleus containing ∼70% 5-HT and 30% non-5-HT containing neurons (Kirby et al., 2003). Recordings were confined to the dorso- and ventromedial aspect of the dorsal raphe (see representative low-power image; Fig. 1A, left), which has the highest density of the 5-HT-containing neurons. Additionally, during electrophysiological recordings, cells were filled with biocytin and subsequently slices were postfixed and processed using standard immunohistochemical procedures to distinguish serotonergic (TPH-ir-positive) and nonserotonergic cells (Fig. 1A). Only 12% of cells were not positively identified as serotonergic neurons, and these were not included in the subsequent analysis.
Figure 1.

KOR activation by U69,593 depresses evoked glutamatergic EPSCs recorded in 5-HT neurons of the DRN. A, Confocal fluorescent images [20× and 40× (inset)] of a representative recording site within the DRN. Cells were filled with biocytin and postfixed following the conclusion of recording to assess for the presence of TPH. Cells were considered 5-HT-positive if the biocytin-filled cell (green) colocalized with TPH (red). Recordings were confined to the dorsal and ventromedial aspect of the mid to caudal DRN. Scale bar, 20 μm. B, Representative traces (inset; calibration, 25 ms, 100 pA) and time course of normalized evoked EPSC amplitude before and after U69,593 (500 nm, 1000 nm) application. Electrical stimulation (100–1000 μA) was delivered in a pairwise fashion to the slice with an interstimulus interval of 50 ms (arrows). C, Four responses from baseline. U69,593 values at 500 and 1000 nm were averaged to get mean drug responses relative to baseline (**p < 0.01). Both concentrations of significantly decreased the normalized eEPSC amplitude compared with baseline. D, There was a significant correlation between the percentage inhibition of eEPSC amplitude and the percentage increase in paired pulse ratio (r2 = 0.3912, p < 0.05) N = 12 for all graphs.
κ-opioid activation in serotonergic dorsal raphe neurons has a net inhibitory effect
Following 5–10 min of stable baseline evoked EPSC (eEPSC) amplitude responses, the κ-opioid receptor agonist, U69,593 (500 nm, then 1000 nm) was bath applied to the slice; both eEPSC amplitude and paired paired-pulse ratio were measured. Representative traces are provided demonstrating that KOR activation by U69,593 decreased eEPSC amplitude of the first evoked EPSC (red line) compared with baseline responses (black line) (Fig. 1B, inset). U69,593 at 500 and 1000 nm significantly inhibited eEPSC amplitudes by 31 ± 6% and 37 ± 8% respectively (F(2,29) = 13.53, p < 0.0001, one-way repeated-measures ANOVA with Dunnett's post hoc t test, Fig. 1B,C). We verified that cumulative dosing did not result in KOR desensitization by showing that 5 μm U69,593 added to a previously untreated slice did not produce a greater inhibition than 500 or 1000 nm added cumulatively (data not shown). Inhibition of eEPSC amplitude significantly correlated with the increase in paired pulse ratio (r2 = 0.39, p < 0.05) (Fig. 1D), indicating that KOR activation reduced probability of glutamate release via a presynaptic mechanism.
To further assess the site of KOR action, we also measured the effect of U69,593 (1000 nm) on miniature EPSC (mEPSC) frequency and amplitude, and representative traces are shown (Fig. 2A). Cumulative histograms of both the interevent interval (Fig. 2B) and amplitude (Fig. 2C) corresponding to these traces are also shown. U69,593 significantly decreased mEPSC frequency from baseline (from 7.1 ± 1.8 Hz to 4.1 ± 1.1 Hz, p < 0.05), which was blocked by pretreatment with the KOR antagonist norbinaltorphimine (norBNI) (1 μm; from 7.6 ± 1.3 Hz to 6.1 ± 1 Hz, p > 0.05; norBNI treatment by time, two-way repeated-measures ANOVA, Bonferroni post hoc, *p < 0.05, Fig. 2D). U69,593 did not significantly affect mEPSC amplitude in either the presence or absence of norBNI (norBNI treatment by time, two-way repeated-measures ANOVA, Bonferroni post hoc, p > 0.05, Fig. 2E). These data confirm that acute activation of KOR presynaptically depresses excitatory synaptic transmission.
Figure 2.

U69,593 produces a norBNI-sensitive decrease in mEPSC frequency. A, Representative traces of pharmacologically isolated mEPSCs recorded from 5-HT DRN neurons before and following U69,593 (1000 nm) application. Calibration, 40 pA, 1 s. B, Cumulative histogram of the interevent interval of baseline mEPSC and that after U69,593 application corresponding to representative traces in A. C, Cumulative histogram of mEPSC amplitudes before and following drug application corresponding to the representative traces in A. D, U69,593 significantly decreased mEPSC frequency by 43% on average that was blunted in slices incubated with norBNI following a stable baseline Bonferroni post hoc, *p < 0.05, N = 7 for both groups). E, U69,593 had no effect on mEPSC amplitude in slices with or without norBNI (*p < 0.05, N = 7 for both groups).
Because KORs regulate both glutamatergic and GABAergic transmission with similar efficacies in other brain regions (Wagner et al., 1992, 1993; Halasy et al., 2000; Hjelmstad and Fields, 2001, 2003), we next assessed the effects of U69,593 on both evoked IPSC (eIPSC) amplitude and miniature IPSC frequency and amplitude. As in the eEPSC experiments, a paired pulse stimulation was delivered to the slice and baseline recordings of pharmacologically isolated eIPSCs (e.g., in the presence of DNQX and APV) were obtained. KOR activation by U69,593 had no significant effect on eIPSC amplitude (representative traces, Fig. 3A; time course, Fig. 3B; p > 0.05) nor did it significantly affect paired pulse ratio (p > 0.05). Similarly, U69,593 had no effect on miniature IPSC (mIPSC) (representative traces, Fig. 3C), frequency (paired t test, p > 0.05, Fig. 3D), or amplitude (paired t test, p > 0.05, Fig. 3E). These data demonstrate that KORs presynaptically inhibit excitatory synaptic input without affecting GABAergic transmission in DRN 5-HT neurons, producing a net reduction in excitability.
Figure 3.

KOR activation by U69,593 has no effect of evoked GABAergic IPSCs or mIPSCs. A, Representative traces of eIPSCs before and following U69,593 (1000 nm) application to the slice. Calibration, 50 ms, 200 pA. B, Time course of normalized evoked IPSC amplitude responses to U69,593 (1000 nm) relative to baseline demonstrating no effect of KOR activation on GABAergic synaptic transmission (N = 9). C, Representative traces of mIPSCs before and after U69,593 (1000 nm) bath application (Calibration, 40 pA, 1 s). D, E, U69,593 had no effect of mIPSC frequency or amplitude (N = 6).
KOR activation has also been shown to have direct, postsynaptic effects in the cells of the spinal trigeminal nucleus (Grudt and Williams, 1993). However, this has not previously been examined in DRN. Both the conductance (slope) and peak inward currents were determined for predrug and postdrug voltage-ramp measurements (see representative traces, Fig. 4A). U69,593 significantly increased both the conductance (3.7 ± 0.4 nS to 4.5 ± 0.4 nS, paired t test, p < 0.01, Fig. 4B) and the peak Ba2+-sensitive inward current (87.8 ± 24.1 pA to 145 ± 24.0 pA, paired t test, p < 0.01, Fig. 4C). U69,593 increased the GIRK current 48.7 pA above baseline, and this effect was blocked by 1 μm norBNI pretreatment (unpaired t test, p < 0.001, Fig. 4D). These data suggest that in stress-naive animals, KOR activation has an inhibitory effect on serotonergic neuronal excitability through both presynaptic and postsynaptic sites of action.
Figure 4.

KOR activation increases GIRK currents postsynaptically. A, Representative traces of baseline inward rectifying currents and KOR-activated (U69,593-induced) currents in which the Ba2+-insensitive current has been subtracted. B, C, U69,593 increases significantly increases the conductance (slope) and the peak inward current (measured at −120 mV) compared with baseline measurements (**p < 0.01, N = 7). D, For both untreated slices and slices preincubated with norBNI (1000 nm), the baseline current was subtracted from the current following U69,593 application to get an absolute change in peak inward current (denoted as GIRK current). U69,593 produced a 48.7 pA GIRK current that was absent in cells that had been preincubated with norBNI (**p < 0.01, N = 7 for both groups).
Repeated stress exposure reduces KOR inhibitory regulation on 5-HT neuronal excitability postsynaptically
We found evidence for dynorphin B-ir within the DRN, but in contrast to a previous study by Fu et al. (2010), this staining was present in both TPH-ir-positive and TPH-ir-negative cells (Fig. 5A, yellow and white arrows, respectively), suggesting that dynorphin may be locally released from a heterogeneous population of cells that includes serotonergic neurons. To evoke endogenous dynorphin release in DRN, we used the previously validated 2 d repeated forced swim stress (FSS) paradigm (Mague et al., 2003; McLaughlin et al., 2003a; Fig. 5B). This modified Porsolt procedure produced escalating immobility within the first 15 min session as well as enhanced immobility over the second day of swim sessions (F(6,54) = 40.59, p < 0.0001, one-way repeated-measures ANOVA, Fig. 6A), indicative of increased passive coping behavior (Cryan et al., 2005). In agreement with our previous findings (McLaughlin et al., 2003a), this two-day swim paradigm produced significant stress-induced analgesia (as measured with the warm water tail immersion assay) that could be prevented by norBNI pretreatment (time by behavioral treatment, F(2,36) = 9.969, p < 0.001, two-way repeated-measures ANOVA, Fig. 5D), indicating that this stress exposure evoked dynorphin release. We previously showed that dynorphin release can be detected by an increase in KOR phosphorylation (KORp-ir) (McLaughlin et al., 2003b); agonist activated KOR is a substrate for G-protein Receptor Kinase (GRK3) which phosphorylates ser369 in the C-terminal tail of KOR. Endogenous dynorphin released by neuropathic pain stress or administration of the stress-related peptide corticotropin releasing factor intracerebroventricularly increased KORp-ir in wild-type mice but not in mice lacking the gene for preprodynorphin (Dyn−/−; Xu et al., 2004; Land et al., 2008). Using increases in KORp-ir to detect sites of endogenous dynorphin action in the present study, we found that KORp-ir was significantly increased in the dorsal raphe nucleus evident 30 min following the cessation of the last swim (Fig. 5E). This elevation in KORp-ir returned to basal levels in animals that were allowed to recover for 24 h following the last swim session (Fig. 5E). At higher magnification (60 and 100×), we observed that KORp-ir puncta were robustly localized to the somata of DRN cells and to a lesser extent outside of the somata, interdigitated with synaptophysin- or vGlut1-positive puncta (data not shown). However, even at this resolution we could not distinguish KORp-ir in presynaptic from postsynaptic subcellular profiles. These results confirmed that the two-day swim stress paradigm produced escalating depression-like behaviors, robust and transient activation of KORs through release of endogenous dynorphins, and dynorphin-KOR-dependent stress-induced analgesia.
Figure 5.
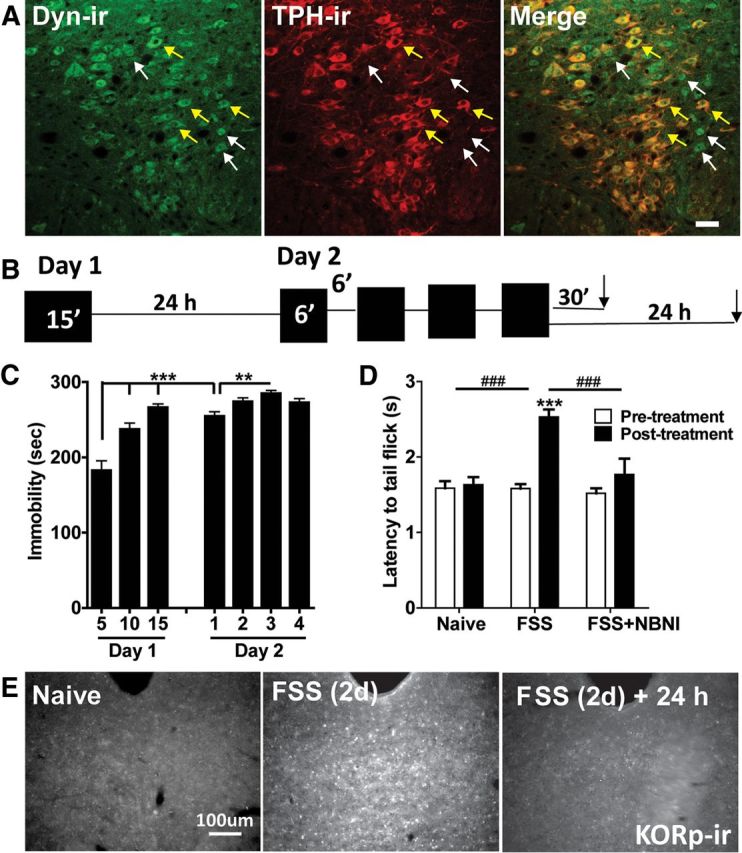
Repeated forced swim stress causes the release of dynorphin and KOR activation. A, 20× fluorescent images of dynorphin and TPH colocalization with the DRN. There was evidence of dynorphin-positive cells in both TPH-negative (white arrows) and TPH-positive cells (yellow arrows) within the DRN. Scale bar, 40 μm. B, Schematic of the 2 d repeated forced swim paradigm used in the study. C, Mice displayed significantly escalating immobility within the first 15 min session on day 1, from day 1 to day 2, and also across sessions on day 2 (**p < 0.01, ***p < 0.001, N = 10). D, Mice showed a significant stress-induced increase in tail-withdrawal latency following exposure to the 2 d swim paradigm that was blocked by norBNI (10 mg/kg) pretreatment (###p < 0.001 interaction; ***p < 0.001 post hoc Bonferroni, N = 7–21). E, Two day repeated swim stress produced an increase in phospho-KOR-ir compared with naive animals that returned to basal levels 24 h after the last swim session. Scale bar, 100 μm.
Figure 6.
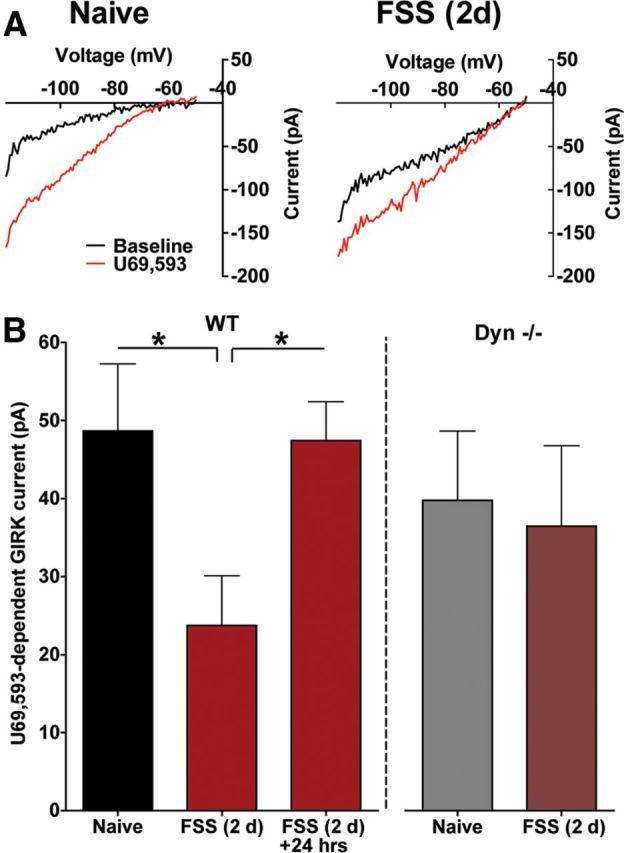
Repeated forced swim stress causes a reduction of KOR-activated GIRK current. A, Representative traces of baseline and KOR-induced GIRK currents in 5-HTcells recorded from the DRN of a naive or stress-exposed mouse. B, In WT animals, repeated swim stress causes a significant decrease in KOR-activated GIRK current that recovered 24 h following the final swim session (*p < 0.05, N = 7–8). In animals lacking the preprodynorphin gene (Dyn−/−), stress exposure did not significantly decrease KOR-mediated GIRK current relative to naive animals (N = 7–9).
Repeated swim stress exposure significantly reduced subsequent in vitro postsynaptic responses to U69,593 in slices prepared 30 min after the final swim session (representative traces, Fig. 6A). There was no significant difference in basal GIRK current between naive and swim-stressed groups [Naive: 76.4 ± 23 pA; FSS (2 d): 66 ± 19.5 pA, unpaired t test, p > 0.05]. The increase in GIRK current caused by U69,593 (1 μm) was 51% smaller in serotonergic neurons recorded in slices from FSS-treated compared with those from naive mice (Fig. 6B). The reduction in KOR stimulated GIRK currents was not evident in slices from mice allowed to recover for 24 h after the final swim session (F(2,19) = 4.432, p < 0.05, one-way ANOVA, Fig. 6B). Mice exposed to additional 5 min daily swim sessions for 5 more days (FSS 7 d) did not further decrease subsequent in vitro KOR responses [Naive: 48.7 ± 8.6 pA; FSS (2 d): 23.7 ± 6.4 pA; FSS (7 d): 25.7 ± 3.6 pA; (F(2,18) = 4.217, p < 0.05, one-way ANOVA]. The dysregulation of the KOR response caused by forced swim was not evident in Dyn−/− mice (p > 0.05, unpaired t test, Fig. 6B). Thus, although multiple neurotransmitters were likely released during swim stress exposure (Cryan et al., 2005), the most parsimonious explanation for stress-induced dysregulation of KOR responsivity is that dynorphins were being repeatedly released to activate KORs. While this does not preclude the involvement of other stress-related neurotransmitters, these data demonstrate that the reduction in KOR regulation of inward current required stress-induced endogenous dynorphin release.
Interestingly, repeated stress exposure had no significant effect on KOR regulation of excitatory presynaptic transmission recorded in serotonergic neurons (Fig. 7). U69,593 had a similar inhibitory effects on evoked EPSC amplitude in slices from naive and FSS-exposed mice (Naive: 38% inhibition for 1000 nm; FSS: 43% inhibition for 1000 nm; p > 0.05, two-way repeated-measures ANOVA, Fig. 7A). Cells recorded from stress-exposed mice had a significantly larger mEPSC amplitudes compared with naive animals (two-way repeated-measures ANOVA, main effect of behavioral treatment, F(1,10) = 16.08, p < 0.01) suggesting that these cells were more excitable following FSS. The inhibition of both mEPSC frequency (behavioral treatment by time, F(1,10) = 0.3210, p > 0.05, two-way repeated-measures ANOVA) and amplitude (behavioral by time, F(1,10) = 3.024, p > 0.05, two-way repeated-measures ANOVA) by U69,593 was not significantly different in 5-HT neurons from naive compared with stress-exposed animals (Fig. 7B,C). The trend for U69,593 to slightly decrease mEPSC amplitude observed in naive animals was significant in stress-exposed animals (post hoc t test, p < 0.01). The lack of a sustained effect of dynorphin release on the excitatory inputs suggests that either the presynaptic KORs were not desensitized or that their desensitization quickly recovered. Together, these data demonstrate that repeated stress exposure selectively disrupts the inhibitory actions of KOR on excitability postsynaptically while keeping the presynaptic inhibition of glutamatergic synaptic drive intact (represented schematically in Fig. 7D).
Figure 7.
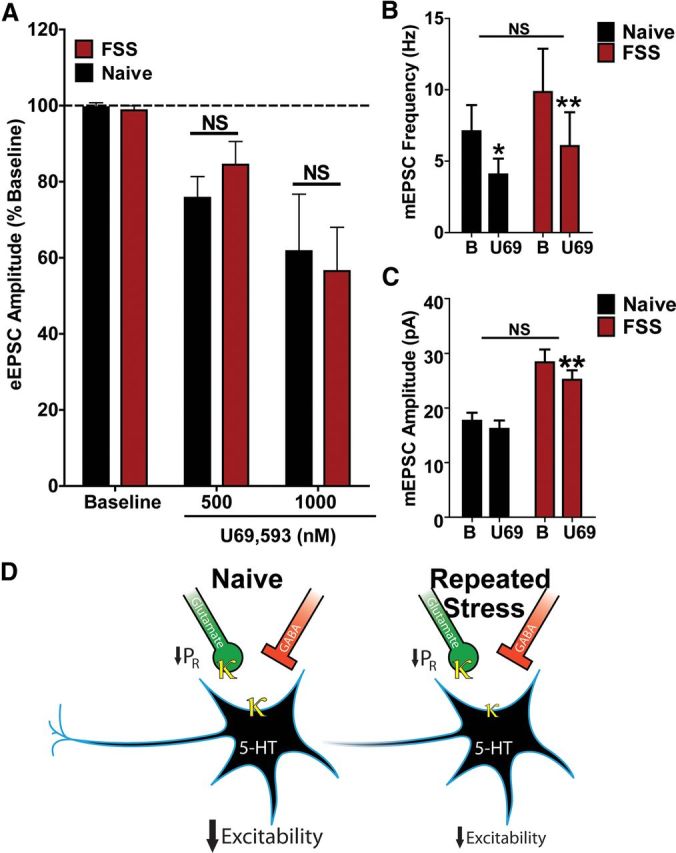
Repeated stress exposure does not alter KOR-mediated depression of glutamatergic synaptic transmission. A, Repeated swim stress did not alter KOR inhibition of normalized eEPSC amplitude at either 500 or 1000 nm relative to measurements obtained in stress-naive animals (N = 10–12). B, U69,593 (1000 nm) caused a similar inhibition of mEPSC frequency in 5-HT cells recorded from naive versus stress-exposed mice (behavioral treatment by time, F(1,10) = 0.3210, p > 0.05, two-way repeated-measures ANOVA; Naive: 62 ± 7.0% of baseline; FSS: 58 ± 7.8% of baseline, Bonferroni post hoc tests, *p < 0.05, **p < 0.01, N = 5–7). C, 5-HT cells recorded from stress-exposed animals had significantly larger mEPSC amplitudes (two-way repeated-measures ANOVA, main effect of behavioral treatment, F(1,10) = 16.08, p < 0.01). There was not a significant difference in U69,593-induced inhibition of mEPSC amplitude between naive and stress-exposed mice (behavioral by time, F(1,10) = 3.024, p > 0.05, two-way repeated-measures ANOVA; Naive: 92 ± 3.3% of baseline; FSS: 89 ± 2.4% of baseline). There was a trend for a small (8%) decrease in mEPSC amplitude by U69,593 in naive animals, but this was not significant (Bonferroni post hoc test, p > 0.05). U69,593 did produce a small (10%), but significant decrease in mEPSC amplitude in 5-HT cells of stress-exposed animals (Bonferroni post hoc t test, **p < 0.01), N = 5–7. D, Summary of KOR regulation of DRN 5-HT neuronal excitability presynaptically and postsynaptically in stress-naive and stress-exposed mice.
Stress-induced reduction in KOR signaling in 5-HT neurons of the DRN is p38α MAPK dependent
Animals perfused 30 min following the final repeated stress exposure had significantly increased phospho-p38-ir compared with naive animals in DRN (Fig. 8A,B). The increased phospo-p38-ir recovered to baseline levels 24 h following the swim stress, and the increase was blocked by norBNI administered before the swim stress (representative images, Fig. 8A; F(3,12) = 4.630, p < 0.05, one-way ANOVA, Fig. 8B). Stress exposure did not change the number of p38α-positive cells within the DRN (p > 0.05, Fig. 8C).
Figure 8.
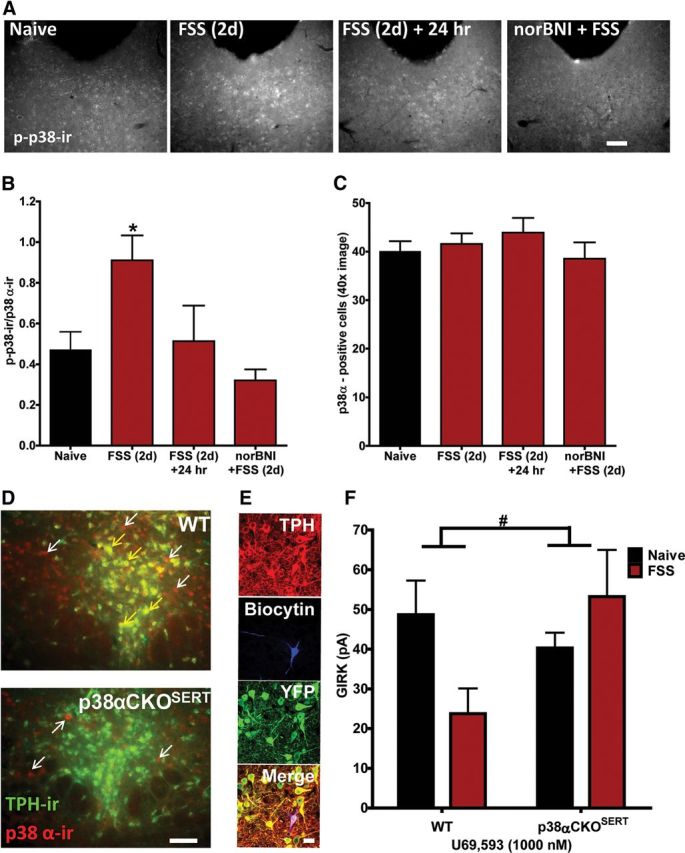
p38α MAPK mediates the stress-induced reduction in KOR-activated GIRK current. A, Representative grayscale fluorescent images of phospho-p38 MAPK-ir in the DRN in naive animals, stress-exposed animals, stress-exposed animals allowed to recover for 24 h, and stress-exposed animals pretreated with norBNI. Scale bar, 200 μm. B, Two day swim stress significantly increased the ratio of phospho-p38 MAPK-ir: p38α-ir cells compared with naive animals sampled from a 40× image of the medial DRN. Following 24 h of recovery, this ratio returned to baseline levels. norBNI pretreatment significantly blocked stress-induced activation of phospho-p38MAPK-ir (*p < 0.05, N = 3–5 animals, triplicate sampling per N). C, There was no significant difference in total number of p38α-positive cells across groups (N = 3–5 animals, triplicate sampling per N). D, Representative fluorescent images of p38α-ir (red fluorescence) and TPH-ir (green fluorescence) colocalization in the DRN of WT and p38αCKOSERT animals. Colocalization of both proteins is indicated by the presence of yellow fluorescence above background. p38α-ir was present in both TPH-positive (yellow fluorescence, yellow arrows) and TPH-negative (red fluorescence only, white arrows) cells within the DRN of WT animals. In contrast, p38α-ir was only present above background staining in TPH-negative cells in p38αCKOSERT (red fluorescence only, white arrows). Scale bar, 100 μm. E, Slices obtained from p38αCKOSERT animals used for electrophysiological recording were postfixed following recordings and triple-labeled for biocytin (blue), TPH (red), and YFP (green) to confirm that the recorded cells were both TPH positive and YFP positive, indicating p38α had been excised. Scale bar, 50 μm. F, Stress-induced reduction in KOR-activated GIRK current in 5-HT cells was absent in 5-HT neurons from stress-exposed p38αCKOSERT animals compared with their naive counterparts (#p < 0.05 interaction, N = 7–12).
Using a conditional knock-out animal (p38αCKOSERT) in which p38α is specifically deleted from serotonin transporter (SERT)-containing neurons (Bruchas et al., 2011), we assessed whether p38α MAPK activation following swim stress mediated the stress-induced reduction in KOR activation of GIRK current. Compared with wild-type (WT) littermates that have p38α expressed in both TPH-ir-positive and-negative neurons (Fig. 8D, top), in p38αCKOSERT animals, p38α-ir was only detected in TPH-ir-negative neurons. These mice also ubiquitously express a ROSA-eYFP stop-floxed reporter, where Cre recombinase expression induces a YFP signal; we also used this signal as an indirect proxy of the floxed p38α gene excision. Thus, to ensure that the analyzed cells from CKO animals were both TPH-ir-positive and had p38α MAPK excised, we only included biocytin filled cells that were both TPH-ir-positive and YFP-ir-positive (Fig. 8E).
Prior studies using in vitro transfected cell culture systems had shown that opioid receptor activation of p38 MAPK caused Src phosphorylation of tyrosine-12 (Y12) in the N-terminal domain of Kir3.1 that enhances GIRK channel deactivation (Ippolito et al., 2002; Clayton et al., 2009). GIRK channel phosphorylation by this mechanism might have been responsible for the reduction in response to U69,593 evident in slices from FSS-exposed mice. Consistent with this hypothesis, p38α MAPK excision from serotonergic neurons prevented the stress-induced reduction in KOR-activated GIRK current (stress exposure by genotype, F(1,34) = 4.344, p < 0.05, two-way ANOVA, Fig. 8F). To further test this hypothesis, we used a previously characterized antibody, GIRKp, able to detect phosphorylation of the Y12 residue of KIR3.1 (Clayton et al., 2009) after either κ agonist treatment with U50,488 (20 mg/kg, i.p.) or repeated swim stress. Both of these manipulations produced robust increases in GIRKp-ir in TPH-ir-positive neurons compared with basal levels (Fig. 9A). The increase in GIRKp-ir caused by FSS was blocked by pretreatment with 10 mg/kg norBNI (Fig. 9A). Stress-induced phosphorylation of p38 MAPK and KIR3.1 colocalized in individual cells within the DRN (Fig. 9B).
Figure 9.
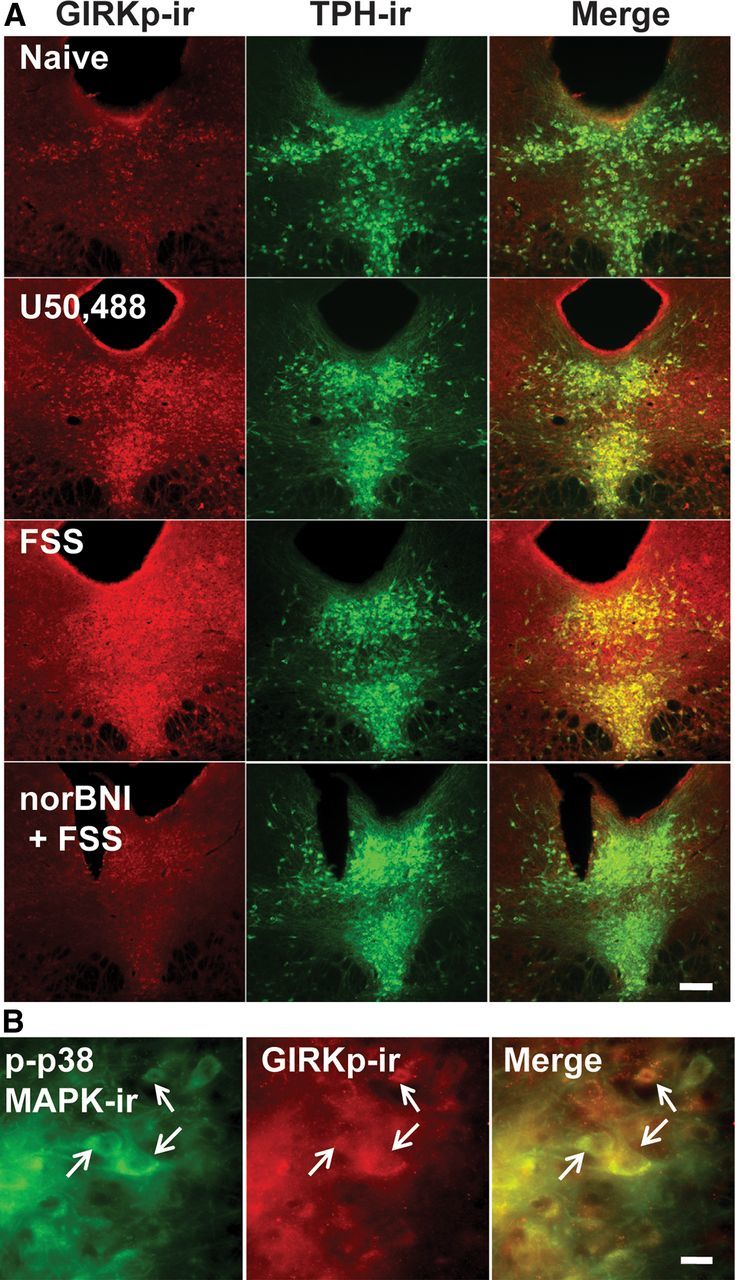
Repeated stressor exposure produces a KOR-dependent increase in phosphorylation of the tyrosine 12 residue of KIR 3.1 in the DRN. A, Fluorescent images of phospho-GIRK-ir and TPH labeling in the DRN of a stress-naive, U50,488 (20 mg/kg)-injected, stress-exposed, and norBNI + stress-exposed animal. Relative to basal levels of phospho-GIRK-ir present in naive animals, U50,488 or repeated swim stress robustly increased GIRKp-ir in TPH-ir cells that was blocked by norBNI pretreatment. Scale bar, 200 μm. B, Fluorescent images (40×) demonstrating colocalization of phospho-p38MAPK-ir and phospho-GIRK-ir. Stress exposure increased phospho-p38MAPK-ir and phospho-GIRK-ir within the same cells in the DRN. Scale bar, 50 μm.
To determine whether p38 MAPK activation also promoted phosphorylation at the KOR ser-369 site important for receptor desensitization (McLaughlin et al., 2003b), we conducted IHC experiments in which WT or p38αCKOSERT animals were exposed to repeated swim stress and compared with naive controls. Serial sections of the DRN from these different animals were stained for either KORp-ir or GIRKp-ir. As previously shown, repeated swim stress increased both KORp-ir and GIRKp-ir with the DRN compared with naive WT animals (Fig. 10A, left). Interestingly, in p38αCKOSERT animals, the stress-induced increase in KORp-ir was still apparent compared with naive CKO animals (Fig. 10A, top). In contrast, stress-induced GIRKp-ir was markedly blunted in CKO animals when compared with both naive CKO animals and stress-exposed WT animals (Fig. 10A, bottom). These results are an in vivo demonstration that p38α MAPK activation is required for the phosphorylation of the Y12 residue of KIR3.1, but p38α MAPK deletion does not affect the phosphorylation of the Ser369 residue of KOR (Bruchas et al., 2006, 2007). A schematic summary of these results is shown (Fig. 10B).
Figure 10.

Excision of p38α from 5-HT neurons blocks stress-induced phosphorylation of GIRK, but not KOR. A, Top, Repeated swim stress increases phospho-KOR-ir in the DRN compared with naive animals in both WT and p38αCKOSERT animals. Bottom, Excision of p38α reduced stress-induced phospho-GIRK-ir in the DRN compared with WT animals. Scale bar, 100 μm. B, Schematic depicting indirect tyrosine phosphorylation of the GIRK channel by p38α MAPK following stress-induced dynorphin release and subsequent KOR activation.
Discussion
In this study, we demonstrate that KOR activation produces a net inhibitory regulation of 5-HT neuronal excitability through presynaptic inhibition of glutamatergic synaptic activity onto serotonergic neurons coupled with an increase in postsynaptic GIRK current activation. We found that the postsynaptic, but not the presynaptic inhibitory effects of KOR activation were reduced following repeated stress exposure, demonstrating a stress-induced change in the regulation of serotonergic neuron excitability. We interrogated the mechanism underlying this stress-induced dysregulation of KOR function through use of functional neuroanatomy (i.e., phospho-antibody immunohistochemistry) as well as p38αCKOSERT transgenic animals and found that stress-induced activation of p38α MAPK was responsible for the reduction in acute KOR-activated GIRK current seen in 5-HT cells in the DRN of stress-exposed animals. Consistent with our previous work (Ippolito et al., 2005; Bruchas et al., 2006; Clayton et al., 2009), we found that p38α MAPK-dependent inhibition of KOR function was likely a consequence of the tyrosine phosphorylation of the GIRK channel initiated by p38α MAPK, following p38α MAPK's activation by KOR.
Mechanism of agonist-induced reduction of KOR activated GIRK current ex vivo
There are several possible mechanisms that could underlie our observation that KOR activation of GIRK current was reduced in stress-exposed animals. First, stress may desensitize KOR signaling through a β-arrestin-dependent mechanism. KOR desensitization and internalization caused by phosphorylation of Ser369 by GRK3 can result in sustained analgesic tolerance that can take weeks to recover (McLaughlin et al., 2003b). However, we found that within the DRN, stress-induced KORp-ir as well as reduction of KOR-mediated GIRK current recovered to initial baseline levels in 24 h, suggesting that phosphorylation of KOR itself was not responsible for the observed dysregulation of response.
Alternatively, there is evidence suggesting that tyrosine phosphorylation at the N-terminal cytoplasmic domain of the GIRK channel is a potential mechanism for the p38-dependent reduction of GIRK current. In previous in vitro work, we showed that phosphorylation of two obligatory/cooperative tyrosine residues (demonstrated through BDNF exposure and activation of TrkB receptors) on KIR3.1 (Y12 and Y67) or KIR3.4 (Y32 and Y53) inhibit U69,693-evoked GIRK current through accelerated deactivation (Rogalski et al., 2000; Ippolito et al., 2002). Phosphorylation of these residues does not cause basal reduction in current, consistent with our observations that basal GIRK currents were not affected by stress (Rogalski et al., 2000; Ippolito et al., 2002). Moreover, suppression of U69,593-induced GIRK current was dependent on p38 MAPK activation of Src kinase (Clayton et al., 2009). In vivo, neuropathic pain or swim stress caused an increase in phospho-GIRK-ir in the spinal cord that was absent in mice lacking GRK3 (Ippolito et al., 2005; Clayton et al., 2009), which would in turn prevent p38 MAPK recruitment. Here we show p38α MAPK excision from serotonergic neurons was sufficient to block both stress-induced reduction of KOR-induced GIRK as well as stress-induced phosphorylation of KIR 3.1, while having no appreciable effect of phosphorylation of KOR. Since p38 is a ser/thr-selective kinase, tyrosine phosphorylation of the channel likely indirectly follows from the MAPK activation of a src-like kinase in serotonergic neurons, as previously demonstrated in transfected AtT20 cells (Clayton et al., 2009). Thus, our findings provide a functional consequence of repeated stress exposure in vivo that is consistent with prior work in heterologous systems.
Disparate actions of stress on presynaptic versus postsynaptic KOR function
The disparate effect of stress exposure on presynaptic versus postsynaptic KOR actions points to a fundamental difference in the molecular machinery regulating these two subcellular compartments. Recently, agonist sensitivity and differences in regulatory mechanisms of opioid receptors based on subcellular localization has been examined in pro-opiomelanocortin (POMC) neurons. Pennock and Hentges (2011) found that agonist-induced desensitization occurs postsynaptically, but not presynaptically. Moreover, these investigators found that there was greater efficacy of opioid agonism at presynaptic receptors compared with postsynaptic receptors, indicating that there may be more spare receptors available presynaptically. While clearly there are temporal differences between these findings on acute desensitization and the current study, the results of Pennock and Hentges underscores differences in agonist-induced regulation of a receptor that are dependent on subcellular localization.
Furthermore, in the current work, it is likely that the differences in pre- versus postsynaptic regulation by stress are related to subcellular differences in expression of GRK3, p38α MAPK and Src kinase. For example, we only observe p38 MAPK-ir in the soma and dendrites of serotonin-containing neurons. Moreover, it is likely that KOR-dependent GIRK current activation did not regulate glutamatergic release probability. It has been shown that KOR inhibition of excitatory transmission in the hippocampus is dependent on dendrotoxin-sensitive Shaker-type voltage-gated potassium channels (Simmons and Chavkin, 1996). There has been one prior report of KOR regulation of EPSPs in the DRN in putative rat 5-HT neurons using current-clamp techniques with the selective KOR agonist enadoline (Pinnock, 1992). However, it was important to further characterize the presynaptic and postsynaptic regulation by KOR using voltage-clamp techniques and a combination of evoked and miniature PSCs to advance our understanding of stress-induced alterations of KOR function. Here we found that behaviorally evoked endogenous ligand release can have different effects on pre- versus postsynaptic receptors within the same cell type of a brain region.
Serotonin, stress coping, and mood disorder
While the role of serotonin in cognition and emotion is generally well appreciated, the mechanisms of serotonin action have been difficult to define because of its wide regional distribution and large number of cloned receptors (Graeff et al., 1996; Lucki, 1998; Cools et al., 2008). There have been two prevailing theories of serotonin function that have been posited and tested. First, is the hypothesis that reduction in serotonin tone/release below basal levels enhances the perception of threat or punishment and promotes a negative affective bias or aversion (Deakin and Graeff, 1991; Cools et al., 2008). This hypothesis has been supported experimentally through serotonin depletion experiments in rats, monkeys, and humans and is also supported by clinical evidence that enhancement of serotonin through selective serotonin reuptake inhibitors (SSRIs) ameliorates depression (Graeff et al., 1996; Cools et al., 2008). The second theory, also supported by preclinical evidence, posits that there is a direct relationship between serotonin levels and behavioral or response inhibition (i.e., increase in serotonin would cause an increase in behavioral inhibition; Lucki, 1998). Furthermore, it is plausible that both decreases or increases in behavioral inhibition in stressful situations translate to active versus passive coping strategies (Waselus et al., 2011). It is likely that serotonin has a role in both these processes; however, it also possible that they are supported by serotonin acting in disparate regions. For example, it has been shown that an acute stress exposure causes elevation in serotonin in the dorsolateral striatum (behavioral inhibition) and simultaneous decreases in serotonin in the lateral septum and the amygdala (aversive learning; Kirby et al., 1995). Importantly, dysregulation of either or both of these behavioral processes can lead to stress vulnerability by producing behavioral hyper-responsivity/enhanced passive coping and enhanced perception of punishment or aversion. Understanding the regulation of dorsal raphe firing is critical to disentangle the role of serotonin in both adaptive behaviors and psychiatric disease.
The current work demonstrated that in naive animals, KOR activation in the dorsal raphe caused a net reduction in 5-HT neuronal excitability, which may act to return firing back to homeostatic levels after heightened periods of activation. KOR regulation of excitatory synaptic input in DRN is consistent with similar inhibitory effects on glutamatergic transmission in the hippocampus (Bausch et al., 1998; Stögmann et al., 2002). Importantly, KOR activation in DRN of naive animals may simultaneously encode the negative affective properties of a stressor through acute reduction in serotonin tone in limbic regions as a transient teaching signal. Stress-induced dynorphin release is likely to produce a net reduction in serotonergic transmission by both presynaptic inhibition of excitatory drive and postsynaptic hyperpolarization of TPH-ir neurons. Sustained dynorphin action caused a p38α MAPK-dependent reduction in the postsynaptic inhibitory regulation by dynorphin/KOR; however, presynaptic inhibition of excitatory drive was not attenuated. Although 5-HT neurons in stress-exposed rats show enhanced excitability (Kirby et al., 2007), the net effect on DRN circuit excitability was not defined in the present study. However, p38-mediated dysregulation of the serotonergic output has been previously shown to contribute to the dysphoric effects of stress (Land et al., 2008; Bruchas et al., 2011) and is likely to involve local circuit adaptations in DRN.
We have shown that in addition to mediating the dysphoric properties of stress, p38 MAPK activation within the DRN contributes to the increased immobility or passive coping behaviors evident in the swim paradigm following prior stress exposure (Bruchas et al., 2011). Our current data are consistent with the notion that prior stress experience promotes a shift to passive coping/behavioral inactivation to subsequent stress exposures caused by an enhanced stress-induced release of 5-HT into the forebrain. This shift resulting from pathogenic stress exposure may lead to an enhanced aversive state in the presence of mild stressors or even non-noxious stimuli. In conclusion, these data offer a mechanism for stress-induced dysregulation of the excitability of neurons in the DRN, a critical locus of stress neurocircuitry and identify a functional target of stress-induced p38MAPK activation that may mediate some of the-negative aspects of pathological stress.
Footnotes
This work was supported by National Institutes of Health Grants F31-MH086269 (J.C.L.), R01-DA030074 and KO5-DA29570 (C.C.), and R01-MH079292 (P.E.M.P.). We thank Peter Groblewski for helpful discussion. The floxed p38α (p38αlox) transgenic mice were provided by Dr. K. Otsu (Osaka University) though the RIKEN Bioresearch Center. The SERT-Cre mice were provided by Dr. Xiaoxi Zhuang (University of Chicago). Dr. Ute Hochgeschwender (Duke University) provided the prodynorphin knock-out mice.
The authors declare no competing financial interests.
References
- Adell A, Casanovas JM, Artigas F. Comparative study in the rat of the actions of different types of stress on the release of 5-HT in raphe nuclei and forebrain areas. Neuropharmacology. 1997;36:735–741. doi: 10.1016/s0028-3908(97)00048-8. [DOI] [PubMed] [Google Scholar]
- Azmitia EC, Gannon PJ. The primate serotonergic system: a review of human and animal studies and a report on Macaca fascicularis. Adv Neurol. 1986;43:407–468. [PubMed] [Google Scholar]
- Bale TL. Stress sensitivity and the development of affective disorders. Horm Behav. 2006;50:529–533. doi: 10.1016/j.yhbeh.2006.06.033. [DOI] [PubMed] [Google Scholar]
- Bausch SB, Esteb TM, Terman GW, Chavkin C. Administered and endogenously released kappa opioids decrease pilocarpine-induced seizures and seizure-induced histopathology. J Pharmacol Exp Ther. 1998;284:1147–1155. [PubMed] [Google Scholar]
- Bruchas MR, Macey TA, Lowe JD, Chavkin C. Kappa opioid receptor activation of p38 MAPK is GRK3- and arrestin-dependent in neurons and astrocytes. J Biol Chem. 2006;281:18081–18089. doi: 10.1074/jbc.M513640200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Aita M, Xu M, Barot SK, Li S, Chavkin C. Stress-induced p38 mitogen-activated protein kinase activation mediates kappa-opioid-dependent dysphoria. J Neurosci. 2007;27:11614–11623. doi: 10.1523/JNEUROSCI.3769-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Land BB, Chavkin C. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Res. 2010;1314:44–55. doi: 10.1016/j.brainres.2009.08.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruchas MR, Schindler AG, Shankar H, Messinger DI, Miyatake M, Land BB, Lemos JC, Hagan CE, Neumaier JF, Quintana A, Palmiter RD, Chavkin C. Selective p38alpha MAPK deletion in serotonergic neurons produces stress resilience in models of depression and addiction. Neuron. 2011;71:498–511. doi: 10.1016/j.neuron.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavkin C, James IF, Goldstein A. Dynorphin is a specific endogenous ligand of the kappa opioid receptor. Science. 1982;215:413–415. doi: 10.1126/science.6120570. [DOI] [PubMed] [Google Scholar]
- Clayton CC, Xu M, Chavkin C. Tyrosine phosphorylation of Kir3 following kappa-opioid receptor activation of p38 MAPK causes heterologous desensitization. J Biol Chem. 2009;284:31872–31881. doi: 10.1074/jbc.M109.053793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cools R, Roberts AC, Robbins TW. Serotoninergic regulation of emotional and behavioural control processes. Trends Cogn Sci. 2008;12:31–40. doi: 10.1016/j.tics.2007.10.011. [DOI] [PubMed] [Google Scholar]
- Cryan JF, Valentino RJ, Lucki I. Assessing substrates underlying the behavioral effects of antidepressants using the modified rat forced swimming test. Neurosci Biobehav Rev. 2005;29:547–569. doi: 10.1016/j.neubiorev.2005.03.008. [DOI] [PubMed] [Google Scholar]
- Deakin JF, Graeff FG. 5-HT and mechanisms of defence. J Psychopharmacol. 1991;5:305–315. doi: 10.1177/026988119100500414. [DOI] [PubMed] [Google Scholar]
- Fu W, Le Maître E, Fabre V, Bernard JF, David Xu ZQ, Hökfelt T. Chemical neuroanatomy of the dorsal raphe nucleus and adjacent structures of the mouse brain. J Comp Neurol. 2010;518:3464–3494. doi: 10.1002/cne.22407. [DOI] [PubMed] [Google Scholar]
- Graeff FG, Guimarães FS, De Andrade TG, Deakin JF. Role of 5-HT in stress, anxiety, and depression. Pharmacol Biochem Behav. 1996;54:129–141. doi: 10.1016/0091-3057(95)02135-3. [DOI] [PubMed] [Google Scholar]
- Grudt TJ, Williams JT. kappa-Opioid receptors also increase potassium conductance. Proc Natl Acad Sci U S A. 1993;90:11429–11432. doi: 10.1073/pnas.90.23.11429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halasy K, Rácz B, Maderspach K. Kappa opioid receptors are expressed by interneurons in the CA1 area of the rat hippocampus: a correlated light and electron microscopic immunocytochemical study. J Chem Neuroanat. 2000;19:233–241. doi: 10.1016/s0891-0618(00)00068-5. [DOI] [PubMed] [Google Scholar]
- Hjelmstad GO, Fields HL. Kappa opioid receptor inhibition of glutamatergic transmission in the nucleus accumbens shell. J Neurophysiol. 2001;85:1153–1158. doi: 10.1152/jn.2001.85.3.1153. [DOI] [PubMed] [Google Scholar]
- Hjelmstad GO, Fields HL. Kappa opioid receptor activation in the nucleus accumbens inhibits glutamate and GABA release through different mechanisms. J Neurophysiol. 2003;89:2389–2395. doi: 10.1152/jn.01115.2002. [DOI] [PubMed] [Google Scholar]
- Ippolito DL, Temkin PA, Rogalski SL, Chavkin C. N-terminal tyrosine residues within the potassium channel Kir3 modulate GTPase activity of Galphai. J Biol Chem. 2002;277:32692–32696. doi: 10.1074/jbc.M204407200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ippolito DL, Xu M, Bruchas MR, Wickman K, Chavkin C. Tyrosine phosphorylation of K(ir)3.1 in spinal cord is induced by acute inflammation, chronic neuropathic pain, and behavioral stress. J Biol Chem. 2005;280:41683–41693. doi: 10.1074/jbc.M507069200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby LG, Allen AR, Lucki I. Regional differences in the effects of forced swimming on extracellular levels of 5-hydroxytryptamine and 5-hydroxyindoleacetic acid. Brain Res. 1995;682:189–196. doi: 10.1016/0006-8993(95)00349-u. [DOI] [PubMed] [Google Scholar]
- Kirby LG, Pernar L, Valentino RJ, Beck SG. Distinguishing characteristics of serotonin and non-serotonin-containing cells in the dorsal raphe nucleus: electrophysiological and immunohistochemical studies. Neuroscience. 2003;116:669–683. doi: 10.1016/s0306-4522(02)00584-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirby LG, Pan YZ, Freeman-Daniels E, Rani S, Nunan JD, Akanwa A, Beck SG. Cellular effects of swim stress in the dorsal raphe nucleus. Psychoneuroendocrinology. 2007;32:712–723. doi: 10.1016/j.psyneuen.2007.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knoll AT, Carlezon WA., Jr Dynorphin, stress, and depression. Brain Res. 2010;1314:56–73. doi: 10.1016/j.brainres.2009.09.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korte SM, Koolhaas JM, Wingfield JC, McEwen BS. The Darwinian concept of stress: benefits of allostasis and costs of allostatic load and the trade-offs in health and disease. Neurosci Biobehav Rev. 2005;29:3–38. doi: 10.1016/j.neubiorev.2004.08.009. [DOI] [PubMed] [Google Scholar]
- Kosofsky BE, Molliver ME. The serotoninergic innervation of cerebral cortex: different classes of axon terminals arise from dorsal and median raphe nuclei. Synapse. 1987;1:153–168. doi: 10.1002/syn.890010204. [DOI] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C. The dysphoric component of stress is encoded by activation of the dynorphin {kappa}-opioid system. J Neurosci. 2008;28:407–414. doi: 10.1523/JNEUROSCI.4458-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Land BB, Bruchas MR, Schattauer S, Giardino WJ, Aita M, Messinger D, Hnasko TS, Palmiter RD, Chavkin C. Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. Proc Natl Acad Sci U S A. 2009;106:19168–19173. doi: 10.1073/pnas.0910705106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos JC, Chavkin C. Kappa opioid receptor function. In: Pasternak G, editor. The opiate receptors. Ed 2. New York: Humana; 2010. pp. 265–305. [Google Scholar]
- Lemos JC, Pan YZ, Ma X, Lamy C, Akanwa AC, Beck SG. Selective 5-HT receptor inhibition of glutamatergic and GABAergic synaptic activity in the rat dorsal and median raphe. Eur J Neurosci. 2006;24:3415–3430. doi: 10.1111/j.1460-9568.2006.05222.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemos JC, Zhang G, Walsh T, Kirby LG, Akanwa A, Brooks-Kayal A, Beck SG. Stress-hyperresponsive WKY rats demonstrate depressed dorsal raphe neuronal excitability and dysregulated CRF-mediated responses. Neuropsychopharmacology. 2011;36:721–734. doi: 10.1038/npp.2010.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucki I. The spectrum of behaviors influenced by serotonin. Biol Psychiatry. 1998;44:151–162. doi: 10.1016/s0006-3223(98)00139-5. [DOI] [PubMed] [Google Scholar]
- Mague SD, Pliakas AM, Todtenkopf MS, Tomasiewicz HC, Zhang Y, Stevens WC, Jr, Jones RM, Portoghese PS, Carlezon WA., Jr Antidepressant-like effects of kappa-opioid receptor antagonists in the forced swim test in rats. J Pharmacol Exp Ther. 2003;305:323–330. doi: 10.1124/jpet.102.046433. [DOI] [PubMed] [Google Scholar]
- Maier SF, Watkins LR. Stressor controllability and learned helplessness: the roles of the dorsal raphe nucleus, serotonin, and corticotropin-releasing factor. Neurosci Biobehav Rev. 2005;29:829–841. doi: 10.1016/j.neubiorev.2005.03.021. [DOI] [PubMed] [Google Scholar]
- McLaughlin JP, Marton-Popovici M, Chavkin C. Kappa opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. J Neurosci. 2003a;23:5674–5683. doi: 10.1523/JNEUROSCI.23-13-05674.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin JP, Xu M, Mackie K, Chavkin C. Phosphorylation of a carboxyl-terminal serine within the kappa-opioid receptor produces desensitization and internalization. J Biol Chem. 2003b;278:34631–34640. doi: 10.1074/jbc.M304022200. [DOI] [PubMed] [Google Scholar]
- Melief EJ, Miyatake M, Bruchas MR, Chavkin C. Ligand-directed c-Jun N-terminal kinase activation disrupts opioid receptor signaling. Proc Natl Acad Sci U S A. 2010;107:11608–11613. doi: 10.1073/pnas.1000751107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hearn E, Molliver ME. Organization of raphe-cortical projections in rat: a quantitative retrograde study. Brain Res Bull. 1984;13:709–726. doi: 10.1016/0361-9230(84)90232-6. [DOI] [PubMed] [Google Scholar]
- Panerai AE, Bianchi M, Brini A, Sacerdote P. Endogenous opioids and their receptors in stress-induced analgesia. Pol J Pharmacol Pharm. 1987;39:597–607. [PubMed] [Google Scholar]
- Pennock RL, Hentges ST. Differential expression and sensitivity of presynaptic and postsynaptic opioid receptors regulating hypothalamic proopiomelanocortin neurons. J Neurosci. 2011;31:281–288. doi: 10.1523/JNEUROSCI.4654-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinnock RD. Activation of kappa-opioid receptors depresses electrically evoked excitatory postsynaptic potentials on 5-HT-sensitive neurones in the rat dorsal raphe nucleus in vitro. Brain Res. 1992;583:237–246. doi: 10.1016/s0006-8993(10)80029-0. [DOI] [PubMed] [Google Scholar]
- Roche M, Commons KG, Peoples A, Valentino RJ. Circuitry underlying regulation of the serotonergic system by swim stress. J Neurosci. 2003;23:970–977. doi: 10.1523/JNEUROSCI.23-03-00970.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogalski SL, Appleyard SM, Pattillo A, Terman GW, Chavkin C. TrkB activation by brain-derived neurotrophic factor inhibits the G protein-gated inward rectifier Kir3 by tyrosine phosphorylation of the channel. J Biol Chem. 2000;275:25082–25088. doi: 10.1074/jbc.M000183200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifi N, Diehl N, Yaswen L, Brennan MB, Hochgeschwender U. Generation of dynorphin knockout mice. Brain Res Mol Brain Res. 2001;86:70–75. doi: 10.1016/s0169-328x(00)00264-3. [DOI] [PubMed] [Google Scholar]
- Shirayama Y, Ishida H, Iwata M, Hazama GI, Kawahara R, Duman RS. Stress increases dynorphin immunoreactivity in limbic brain regions and dynorphin antagonism produces antidepressant-like effects. J Neurochem. 2004;90:1258–1268. doi: 10.1111/j.1471-4159.2004.02589.x. [DOI] [PubMed] [Google Scholar]
- Simmons ML, Chavkin C. k-Opioid receptor activation of a dendrotoxin-sensitive potassium channel mediates presynaptic inhibition of mossy fiber neurotransmitter release. Mol Pharmacol. 1996;50:80–85. [PubMed] [Google Scholar]
- Stögmann E, Zimprich A, Baumgartner C, Aull-Watschinger S, Höllt V, Zimprich F. A functional polymorphism in the prodynorphin gene promotor is associated with temporal lobe epilepsy. Ann Neurol. 2002;51:260–263. doi: 10.1002/ana.10108. [DOI] [PubMed] [Google Scholar]
- Vasudeva RK, Lin RC, Simpson KL, Waterhouse BD. Functional organization of the dorsal raphe efferent system with special consideration of nitrergic cell groups. J Chem Neuroanat. 2011;41:281–293. doi: 10.1016/j.jchemneu.2011.05.008. [DOI] [PubMed] [Google Scholar]
- Vaught JL, Takemori AE. Differential effects of leucine and methionine enkephalin on morphine-induced analgesia, acute tolerance and dependence. J Pharmacol Exp Ther. 1979;208:86–90. [PubMed] [Google Scholar]
- Wagner JJ, Caudle RM, Chavkin C. Kappa-opioids decrease excitatory transmission in the dentate gyrus of the guinea pig hippocampus. J Neurosci. 1992;12:132–141. doi: 10.1523/JNEUROSCI.12-01-00132.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner JJ, Terman GW, Chavkin C. Endogenous dynorphins inhibit excitatory neurotransmission and block LTP induction in the hippocampus. Nature. 1993;363:451–454. doi: 10.1038/363451a0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waselus M, Valentino RJ, Van Bockstaele EJ. Collateralized dorsal raphe nucleus projections: a mechanism for the integration of diverse functions during stress. J Chem Neuroanat. 2011;41:266–280. doi: 10.1016/j.jchemneu.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wee S, Koob GF. The role of the dynorphin-kappa opioid system in the reinforcing effects of drugs of abuse. Psychopharmacology (Berl) 2010;210:121–135. doi: 10.1007/s00213-010-1825-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JT, Colmers WF, Pan ZZ. Voltage- and ligand-activated inwardly rectifying currents in dorsal raphe neurons in vitro. J Neurosci. 1988;8:3499–3506. doi: 10.1523/JNEUROSCI.08-09-03499.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Petraschka M, McLaughlin JP, Westenbroek RE, Caron MG, Lefkowitz RJ, Czyzyk TA, Pintar JE, Terman GW, Chavkin C. Neuropathic pain activates the endogenous kappa opioid system in mouse spinal cord and induces opioid receptor tolerance. J Neurosci. 2004;24:4576–4584. doi: 10.1523/JNEUROSCI.5552-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Bruchas MR, Ippolito DL, Gendron L, Chavkin C. Sciatic nerve ligation-induced proliferation of spinal cord astrocytes is mediated by kappa opioid activation of p38 mitogen-activated protein kinase. J Neurosci. 2007;27:2570–2581. doi: 10.1523/JNEUROSCI.3728-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]