

Abstract
Polycystic ovary syndrome (PCOS) is a syndrome of variable combinations of menstrual irregularity, hirsutism or acne, and obesity. It can be diagnosed in adolescence and has early childhood antecedents. PCOS is the single most common endocrine cause of anovulatory infertility and a major risk factor for the metabolic syndrome and, in turn, development of type 2 diabetes mellitus (T2DM) in women. Thus, it appears that PCOS increases a woman’s risk of developing cardiovascular disease. Therefore, identifying girls at risk for PCOS and implementing treatment early in the development of PCOS may be an effective means of preventing some of the long-term complications associated with this syndrome. This article reviews the definition, clinical features, diagnosis, and treatment of PCOS.
Definition
Classic polycystic ovary syndrome
The classic syndrome originally was described in 1935 by Stein and Leventhal as the association of amenorrhea with polycystic ovaries in women, of whom about two thirds were hirsute, and one half were obese [1]. The term PCOS was introduced upon recognition of a broader spectrum of clinical symptoms and ovarian histology, including stromal hyperplasia with multiple subcapsular follicles. Approximately two-thirds of patients with classic PCOS have hirsutism (or hirsutism equivalents, acne vulgaris or pattern alopecia), two-thirds have anovulatory symptoms (manifested as amenorrhea, oligomenorrhea, dysfunctional uterine bleeding, or unexplained infertility), and one-half are obese. Thus, only about one-third of classic cases have the full-blown clinical picture (Fig. 1). The laboratory diagnostic criteria for classic PCOS require biochemical evidence of hyperandrogenism with either a polycystic ovary by ultrasound or an increased serum level of luteinizing hormone (LH) or LH to follicle-stimulating hormone (FSH) ratio. These criteria have proven to not necessarily coincide (Fig. 2).
Fig. 1.
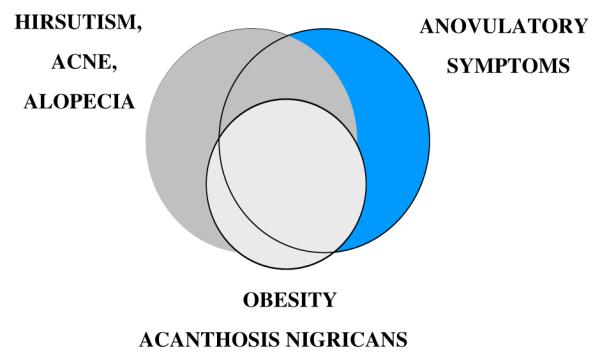
Clinical manifestations of PCOS. The major clinical manifestations of PCOS are shown in approximate proportion to their relative incidence and coincidence. (Modified from Rosenfield RL. Current topics of polycystic ovary syndrome. Bailliere’s Clin Obstet Gynaecol 1997;11:307; with permission.)
Fig. 2.
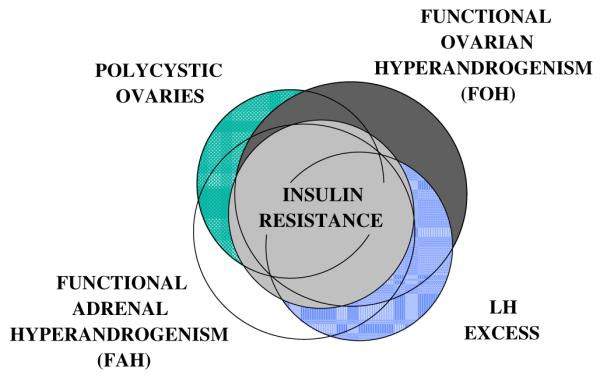
Laboratory manifestations of PCOS. The laboratory manifestations of PCOS are shown in approximate proportion to their relative incidence and coincidence. (Modified from Rosenfield RL. Polycystic ovary syndrome and insulin-resistant hyperinsulinemia. J Am Acad Dermatology 2001;45:5095; with permission.)
Nonclassic and atypical polycystic ovary syndrome
In 1990, the National Institutes of Health (NIH) Conference on PCOS considered the implications of recent research findings for the diagnosis [2]. Fifty percent to 60% of those present concurred that the criteria for PCOS should consist of chronic anovulation with clinical or biochemical signs of hyperandrogenism that was not explained by other etiologies. This recognized the spectrum of the syndrome to include androgen excess in the absence of ultrasonographic and gonadotropic abnormalities (here termed nonclassic PCOS).
The androgen excess of PCOS usually results from characteristic types of functional ovarian hyperandrogenism (FOH) or functional adrenal hyper-androgenism (FAH) [3,4]. FOH is gonadotropin-dependent excessive ovarian androgen production, and it occurs in most women with classic PCOS. It is also found in an equal number of hyperandrogenic women who do not meet the criteria for classic PCOS. It is characterized by 17-hydroxyprogesterone hyper-responsiveness to gonadotropin agonist testing or human chorionic gonadotropin (hCG) testing or by subnormal suppression of plasma-free testosterone after dexamethasone administration [3,5]. It is suppressible by chronic estrogen–progestin or chronic gonadotropin treatment [6,7]. FAH is defined as glucocorticoid-suppressible, ACTH-dependent 17-ketosteroid excess and is found in about one half of women with classic or nonclassic PCOS. Thus, laboratory findings are heterogeneous (Fig. 2) [2]. FOH and FAH are found to be variably associated with polycystic ovaries, LH excess, and one another.
A 2003 Rotterdam international reproductive medicine consensus workshop recommended using polycystic ovaries as an alternative criterion to either hyperandrogenism or anovulation (Box 1) [8]. This proposal indicates the growing acceptance of PCOS being very heterogeneous and including nonclassic forms. On the other hand, it is likely to prove controversial, because hyperandrogenemia is a higher consensus criterion for diagnosis than hirsutism or polycystic ovaries [2], and the combination of anovulation and polycystic ovaries has substantial nonspecificity.
Box 1. Rotterdam diagnostic criteria (2003) for polycystic ovary syndrome.
Two out of the following three after exclusion of other hyperandrogenic disorders:
Oligo- or anovulation
Clinical and/or biochemical signs of hyperandrogenism
Ultrasonographic evidence of polycystic ovaries
From Rotterdam ESHRE/ASRM-sponsored PCOS Consensus Workshop Group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril 2004;81:19; with permission.
Insulin resistance originally was recognized as being related to PCOS after it was found to underlie the common finding of acanthosis nigricans. An abnormal extent of insulin resistance occurs in about half of PCOS cases (see Fig. 2) [9]. Insulin resistance was acknowledged by both the NIH conference and the Rotterdam workshop to be important in the pathogenesis of PCOS. It has not been useful as a diagnostic criterion, however. This is because there is no universally accepted, clinically useful, numeric expression that identifies insulin resistance [10]. Direct measures of it, such as the minimal model derived from the frequently sampled intravenous glucose tolerance test, are clinically impractical, and the fasting insulin level is not reliable because of interassay variability and overlap across the spectrum from normal to abnormal glucose tolerance and insulin sensitivity. Therefore, the metabolic syndrome, a variably expressed cluster of central obesity, hypertension, and dyslipidemia, serves as a surrogate measure for insulin resistance [11,12].
Although the Rotterdam workshop codified the perspective of the reproductive endocrinologist, it recognized that the diagnostic criteria listed do not encompass the entire clinical and endocrinological spectrum of the syndrome. Thus, it accommodates, but does not define, the atypical types of PCOS that are seen in a medical setting rather than a gynecologic or infertility setting. These atypical forms of the syndrome include the hyperandrogenic patients who have normal ovulatory cyclicity and ovarian morphology, yet have obesity with or without acanthosis nigricans or cutaneous signs of hyperandrogenism. These atypical patients may have the typical PCOS-types of ovarian or adrenal dysfunction.
Classic, nonclassic, or atypical PCOS may present in adolescence with typical symptoms. PCOS can be documented in the perimenarcheal stage, as young as 10-years old [13]. Ovarian dysfunction, however, may not become manifest until 3 years after menarche [14]. Adolescent girls with PCOS have higher LH levels and more insulin resistance than nonhyperandrogenic controls [15,16]. The ability to recognize and diagnose the disorder early continues to be enhanced by new research findings. For instance, it is recognized that PCOS may be heralded by congenital virilizing disorders (see Etiology section) intrauterine growth retardation [17], premature adrenarche[18,19], true sexual precocity [20], overgrowth caused by pseudoacromegaly, or intractable obesity in midchildhood [21] (Fig. 3).
Fig. 3.
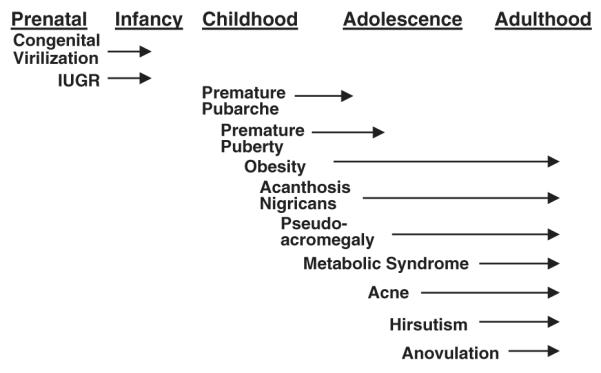
Early clinical manifestations of PCOS. The clinical presentations are listed below the developmental stages in which they first appear, and the arrows indicate duration of the symptoms. (Modified from Baumann EE, Rosenfield RL. Polycystic ovary syndrome in adolescence. The Endocrinologist 2002;12:333; with permission.)
Pathophysiology of polycystic ovary syndrome
Vigorous debate has ensued about whether PCOS is fundamentally a disorder of hypothalamic–pituitary gonadotropin secretion or a disorder of ovarian function. More recently, the question of whether it is basically a metabolic disorder has emerged.
Abnormal pituitary function
Excessive LH secretion once was thought to cause PCOS by a complex cycle of events in which peripheral conversion of adrenal androstenedione to estrone sensitized gonadotropes to gonadotropin-releasing hormone (GnRH) (estrone hypothesis). From this perspective, researchers have argued that LH-induced theca cell hyperplasia accounts for the ovarian secretory abnormalities [22]. This hypothesis, however, does not account for the occurrence of the syndrome in the absence of LH excess or the apparent abnormality of LH-steroid dose–response relationships in the syndrome [5].
The latest research suggests that LH excess may result from abnormal sex steroid feedback at the hypothalamic level. This evidence indicates that androgen excess may cause LH excess by counteracting the LH-suppressive effect of female hormones [23,24]. In women with PCOS, LH pulse frequency was less responsive to luteal phase levels of estradiol and progesterone than normal [25]. Supranormal plasma progesterone concentrations were required to normalize their increased LH pulse frequency. Further examination showed that the antiandrogen flutamide normalized the inhibitory effect of progesterone on LH pulse frequency [24]. These findings indicate that the androgen excess of PCOS may play an important role in desensitizing LH pulse frequency to hypothalamic feedback inhibition by progesterone.
Although in vivo studies have failed to show an effect of insulin on LH secretion in PCOS [26], there is emerging evidence suggesting that insulin nevertheless may enhance LH secretion. Studies in a gonadotrope cell line, LßT2, show that insulin enhances GnRH-activated LHβ gene expression [27]. Another study in these cells observed that insulin alone alters LHβ gene expression independently of GnRH [28]. These studies are compatible with the possibility that hyperinsulinism may contribute to dysregulation of LH at the pituitary level. Hypothalamic GnRH secretion is also dependent on insulin in transgenic mice as discussed below (see Metabolic disturbance).
Abnormal steroidogenesis
Recent studies have suggested the alternative hypothesis that the anovulation of PCOS is attributable to intraovarian androgen excess, which, in turn, arises from FOH [3,5]. The high intraovarian androgen concentrations of FOH seem responsible for the ovarian abnormalities. Substantial intraovarian androgen excess appears to stimulate excessive growth of small follicles while hindering the follicular maturation necessary for the emergence of a dominant follicle [29]. It also causes thecal and stromal hyperplasia. Similar ovarian changes are found in frankly virilizing adrenal disorders, such as poorly controlled classic congenital adrenal hyperplasia, which, thus, cause secondary PCOS. It is not clear whether the available tests are sufficiently sensitive to detect ovarian hyperandrogenism in the exceptional cases of PCOS in which only the mild hyperandrogenemia of FAH can be demonstrated.
Primary FOH and primary FAH appear to be caused by abnormal regulation (dysregulation) of steroidogenesis. Eighty percent or more of cases of PCOS appear to arise from dysregulation of ovarian androgen secretion. The dysregulation is postulated to result from imbalance among various intrinsic and extrinsic factors involved in the modulation of trophic hormone action. A major site of dysregulation affects the function of the thecal enzyme cytochrome P450c17(CYP17), which has 17α-hydroxylase and 17,20-lyase activity, the latter being the rate-limiting step in androgen formation. Theca cells isolated from women with PCOS compared with normal theca cells have elevated gene expression and activity of 17α-hydroxylase in particular, and all the steroidogenic steps [30], with the possible exception of steroidogenic acute regulatory protein and 17β-hydroxysteroid dehydrogenase type 5 [31,32]. This combination of flaws seems to account for the characteristic hyper-responsiveness of excessive 17-hydroxyprogesterone to GnRH agonist or hCG testing.
Granulosa cells also exhibit steroidogenic dysregulation [5]. They are excessively responsive to FSH, particularly at high dosage. In part this is caused by a larger cohort of follicles, in part because of intrinsic granulosa cell dysfunction [33]. This accounts for the tendency of women with PCOS to develop the dangerous ovarian hyperstimulation syndrome during fertility treatment.
The dysregulation seems to result from flaws in the multiple intraovarian systems that modulate steroidogenic responses to gonadotropins and ACTH. Insulin excess appears to play an important role in aggravating the steroidogenic dysregulation [3,5]. The abnormality in LH-steroid dose–response relationships resembles the change in the set point for androgen secretion in response to LH that occurs when normal thecal cells are treated with insulin or insulinlike growth factor.
There is also evidence that the hyperandrogenism seen in women with PCOS may not only result from increased ovarian or adrenal androgen production [5]. The liver and other peripheral target tissues may contribute to the hyperandrogenism observed in PCOS. For example, women with PCOS given oral dehydroepiandrosterone (DHEA) have increased peripheral 5α-reductase activity as shown by increased formation of dihydrotestosterone compared with healthy women without PCOS [34]. These effects may in part be secondary to androgen excess itself and in part to hyperinsulinism. Subcutaneous fat is also a source of testosterone [35].
Metabolic disturbance
The compensatory, insulin-resistant hyperinsulinemia so frequent in PCOS seems to be an important factor contributing to the dysregulation [1,36]. There are many clues to the metabolic effects of insulin excess. The ovaries and adrenal glands function as if responsive to excess insulin in a state of resistance to the glucose–metabolic effects of insulin. Hyperinsulinism seems to account for the epidermal hyperplasia that underlies acanthosis nigricans. It may not only contribute to pituitary LH excess, but potentially may affect hypothalamic GnRH release [37]. Clinically, all forms of insulin resistance, from rare lipodystrophies to common T2DM, are associated with PCOS. Relative peripheral hyperinsulinemia may account for the apparent increase in PCOS prevalence in type 1 diabetes [38]. Notably, insulin-lowering treatments improve ovarian function.
Insulin resistance imposes vascular risk factors upon the syndrome. Although a direct link between PCOS and cardiovascular events has not been established [39], the evidence that PCOS is associated with metabolic syndrome and T2DM is undeniable, with a consequent relative risk of cardio- and cerebrovascular morbidity approximating 1.5 [40].
Etiology of polycystic ovary syndrome
To a great extent, PCOS seems to be a congenital disorder that is first diagnosable at puberty [3,13]. Accumulating evidence suggests that PCOS arises as a complex trait with contributions from heritable and nonheritable factors [41]. A genetic basis for the disorder has been suggested by familial clustering of cases [42,43]. Nearly half of sisters of women with PCOS were found to have an elevated plasma testosterone level. Only half of these sisters, however, had symptoms such as menstrual irregularity. Polycystic ovaries appear to occur as a dominant trait, with a suggestion of a counterpart male phenotype of premature male-pattern baldness [44,45]. Polycystic ovaries from asymptomatic women have an abnormal steroidogenic pattern similar to that of women with PCOS in vitro [46], and ovarian function testing shows them to have a subclinical increase in androgens [47–49].
The underlying cause for the androgen excess is unclear. Various subclinical steroidogenic defects have been reported to be risk factors for the syndrome [50,51]. Recently, theca cells were found to retain their capacity for excessive steroidogenesis through many doublings in culture, supporting an intrinsic mechanism [52]. The excessive androgen secretion is caused by overexpression of several steroidogenic enzymes [30–32]. Thus, some as yet undiscovered, more fundamental disorder, such as a malfunctioning coregulator system, could account for these multiple abnormalities. Another possibility is excessive serine phosphorylation of the steroidogenic enzyme P450c17 and the insulin receptor, which would explain the combination of hyperandrogenism and insulin resistance [53]. Recently, evidence has accrued for a developmental basis for the syndrome. Either prenatal androgen excess or perinatal insults that cause intrauterine growth retardation may predispose to obesity, insulin resistance, and androgen excess in later life [17,54–56].
A subtle increase in insulin resistance sometimes accompanies the subtle increase in androgen levels that occurs in ovulatory women with polycystic ovaries [47]. Thus, insulin resistance may develop in parallel with androgen excess, rather than independently of it. The known association of PCOS with insulin resistance [57] and obesity accounts for the predisposition of these women to metabolic syndrome [58,59] and type 2 diabetes mellitus (T2DM) [60,61]. The authors’ preliminary studies show a stronger relationship of adolescent PCOS to parental metabolic syndrome than to any other parental feature [62]. Insulin resistance, insulin secretion, and all the components of the metabolic syndrome have significant heritable components [63,64]. There are several linkages to genes involved in insulin secretion and action [65]. These data are compatible with PCOS resulting from the interaction among several predisposing and provocative factors, some of which may be environmental (eg, obesity), and some of which may be complex genetic disorders (eg, T2DM) or inherited as incompletely penetrant autosomal dominant traits (such as polycystic ovaries and steroidogenic enzyme variants that predispose to testosterone excess). The extent to which the classic, nonclassic, and atypical forms of PCOS are caused by specific subsyndromes from a genetic perspective is unclear.
Clinical features
Hirsutism is a classic feature of the androgen excess of PCOS and is found in about two thirds of cases (Fig. 1). Hirsutism is excessive sexual hair that appears in a male pattern [66]. It is commonly graded according to the Ferriman-Gallwey system, where a score of less than eight is normal in adults (Fig. 4) [67]. Not all hyperandrogenic women are hirsute, however. The absence of hirsutism in many hyperandrogenic women appears to be caused by individual variability in pilosebaceous unit sensitivity to androgens. Furthermore, some young adolescents whose hyperandrogenism has not evolved fully may not be hirsute.
Fig. 4.

Diagnosis of hirsutism using Ferriman-Gallwey system. Illustrated are the different areas on a woman’s body that constitute a male pattern of excessive sexual hair. The numbers in the diagram provide a scoring system to quantify the presence of excessive hair growth in each individual area, and a total score of less than 8 is normal in adult women. (Reproduced from Hatch R, Rosenfield RL, Kim MH, et al. Hirsutism: implications, etiology, and management. Am J Obstet Gynecol 1981;140:815; with permission.)
Other cutaneous signs of androgen excess are acne, seborrhea, alopecia, or hyperhidrosis [68,69]. Any of these hirsutism equivalents may be the only cutaneous sign of androgen excess. Female-pattern hair loss usually is configured differently than male-pattern baldness: it typically begins in the fronto–parietal area and is fairly diffuse [66].
Menstrual irregularity is found in about two thirds of adolescents with PCOS (see Fig. 1). These girls may present with oligomenorrhea (menstrual bleeding that occurs at intervals over 40 days [ie, fewer than 9 periods yearly]) [70], primary amenorrhea (the absence of menarche by 16 years of age), secondary amenorrhea (the absence of menses for at least 3 months), or dysfunctional uterine bleeding (excessive and irregular vaginal bleeding). Furthermore, monthly menstrual cycles may still be anovulatory, which is suggested when there is a paucity of menstrual cramps, absence of premenstrual molimina (breast tenderness, lower abdominal bloating, or moodiness), or menorrhagia (excessive menstrual bleeding). An abnormal amount of bleeding is suggested by finding anemia.
Obesity is present in approximately half of patients with PCOS (see Fig. 1). It is typically android in type (central adiposity with a waist circumference greater than 88 cm after sexual maturity is reached). It often begins in midchildhood and is accentuated during puberty. Obesity or the accompanying acanthosis nigricans, an indicator of insulin resistance, is often the presenting complaint. Obese patients with the greatest degree of insulin resistance also may have pseudo-Cushing’s syndrome, with marked Cushingoid fat distribution, or pseudoacromegaly, with overgrowth in height and/or prognathism [21,71,72].
Adolescent girls with PCOS are less likely to have demonstrable polycystic ovaries than adults with the syndrome. In a comparison of consecutively studied hyperandrogenic females, polycystic ovaries were found in significantly fewer adolescents (55%) than adults (75%) [13]. This limitation may arise partly from the use of the abdominal route to image the ovaries in adolescents, rather than the vaginal one. The absence of polycystic ovaries in early adolescents may, in part, also relate to the fact that ovarian dysfunction may not be demonstrable until 3 years after menarche [14].
The obesity in PCOS contributes to the metabolic syndrome [73]. It is recommended that obese patients with PCOS be evaluated for the metabolic syndrome and T2DM [8].
Differential diagnosis
Applying the new (2003) diagnostic guidelines for PCOS (see Box 1) to adolescents may be challenging for several reasons. Hirsutism, acne, menstrual irregularity, and obesity are common normal variants during puberty. Three fourths of adolescent girls develop acne, and one quarter have inflammatory acne [74]. Approximately one half of menstrual cycles are anovulatory during the first 2 postmenarchal (gynecologic) years [75]. Therefore, PCOS can be dismissed easily as ordinary physiologic adolescent anovulation with its wide spectrum of menstrual irregularities. Obesity is on the increase, and over one quarter of obese adolescents have metabolic syndrome [76].
Hypertrichosis must be distinguished from hirsutism. Hypertrichosis is distributed in a nonsexual pattern (eg, in a generalized distribution or more prominent on the forehead or shoulders) and is not caused by excess androgen, but excess androgen can exacerbate this condition. It may occur on an ethnic or familial basis. It may also be caused by drugs such as glucocorticoids, phenytoin, or cyclosporine.
Approximately one half of mildly hirsute women and one sixth of moderately hirsute women have idiopathic hirsutism, in the sense of having no biochemical evidence of hyperandrogenism [66]. Usage of this term is evolving, however. Historically it has been used to designate hirsutism associated with normal total testosterone levels and menstrual cyclicity [77], but it now is known that many such women have subtle androgen excess [49].
The differential diagnosis of hyperandrogenism includes virilizing disorders and drugs as diverse as anabolic steroids and valproic acid (Box 2) [78,79]. The most common of these is nonclassic congenital adrenal hyperplasia caused by 21-hydroxylase deficiency [80]. This accounts for less than 5% of cases [13]. Polycystic ovaries occur in about one-third of these, however [80], and the adrenal rests of the ovaries that can occur in this condition may be difficult to distinguish from PCOS [55]. Androgen-secreting tumors of the ovaries or adrenal glands occur in only about 0.2% of cases [81], but many are malignant. Those with only moderate androgen elevations have indolent presentations resembling PCOS [82].
Box 2. Causes of hyperandrogenism.
Functional gonadal hyperandrogenism
Primary (dysregulational) functional ovarian hyperandrogenism (common form of PCOS)
Secondary polycystic ovary syndrome
Poorly controlled classic congenital adrenal hyperplasia
Ovarian steroidogenic blocks
Syndromes of severe insulin resistance
Portohepatic shunting
Epilepsy or valproic acid therapy
Adrenal rests
Hermaphroditism
Chorionic gonadotropin-related
Functional adrenal hyperandrogenism
Primary (dysregulational) functional adrenal hyperandrogenism (uncommon form of PCOS)
Congenital adrenal hyperplasia
Prolactin or growth hormone excess
Dexamethasone-resistant functional adrenal hyperandrogenism
Cushing’s syndrome
Cortisol resistance
Apparent cortisone reductase deficiency
Peripheral androgen overproduction
Obesity
Idiopathic hyperandrogenism
Tumoral hyperandrogenism
Androgenic drugs
From Baumann EE, Rosenfield RL. Polycystic ovary syndrome in adolescence. The Endocrinologist 2002;12:333; with permission.
Hyperprolactinemia [83,84], Cushing’s syndrome [85,86], and acromegaly [87] sometimes mimic PCOS. True hermaphroditism is a rare cause of gonadal androgen excess. On rare occasions, adrenal hyperandrogenism can arise from cortisol resistance [88] or cortisone reductase (type 1 11β-hydroxysteroid dehydrogenase) deficiency [89].
Diagnostic approach
The diagnostic approach for PCOS involves the following steps: first, determining who should be screened; second, demonstrating the presence of androgen excess; third, ruling out disorders that mimic PCOS; and fourth, determining the source of the androgen excess.
Screening for polycystic ovary syndrome
Polycystic ovary syndrome should be suspected in adolescents with hirsutism (or hirsutism equivalents like acne), menstrual irregularity, or obesity. The history and physical examination should be directed toward ascertainment of disorders that mimic PCOS. Premature pubarche or prepubertal acne suggest nonclassic congenital adrenal hyperplasia. Virilizing disorders may be indicated by sudden onset and rapid course, defeminization, or clitoromegaly. Galactorrhea suggests hyperprolactinemia. Cushing’s syndrome should be considered in those with a central fat distribution, especially if violaceous striae or easy bruisability are present. Serious athletes are prone to anabolic steroid abuse.
Although mild hirsutism is often idiopathic, its association with menstrual irregularity or obesity suggests androgen excess. Acne that requires treatment early in adolescence or that is of such severity that it is resistant to standard therapy, such as requiring isotretinoin for management, is an alternative indication for evaluation for PCOS. Because failure to establish a normal adult menstrual pattern by 2 years after menarche or to sustain a normal pattern for 2 years after one has been established carries a greater than two thirds risk of persistent oligo-ovulation [90], the authors recommend that work-up begin at this time, or sooner if other signs of PCOS coexist. Although obesity is common, a history of acceleration during puberty, the presence of acanthosis nigricans, or a family history of metabolic syndrome or T2DM is an indication for work-up for PCOS.
Determining the presence of androgen excess
The second step in diagnosing PCOS is to demonstrate androgen excess. This sometimes is said more easily than done. Some laboratories provide excessively broad normal ranges for total testosterone, because many commercial direct assays are inaccurate [91] and because the general population includes women with asymptomatic mild androgen excess[43,49]. Direct assays of free testosterone are suspect; the most reliable compute free testosterone from total testosterone and sex hormone binding globulin [92]. For these reasons, the current criteria for the diagnosis of PCOS (see Box 1) permit hirsutism to serve as a surrogate for biochemical evidence of hyperandrogenism. Nevertheless, mild hirsutism is a particularly weak criterion.
Testosterone determinations thus are performed best by a specialty laboratory [93]. The authors recommend screening for hyperandrogenism by ordering an androgen panel that includes plasma total testosterone, free testosterone, and another androgen such as DHEA sulfate. The interpretation of androgen levels poses certain problems. Because androgens are produced from ovarian or adrenal secretion or by peripheral metabolism of precursors secreted by these glands, plasma testosterone undergoes pulsatile, diurnal, and cyclic changes, varying about one quarter around the mean [94]. A total testosterone level over about 90 ng/dL, if confirmed, is ordinarily good evidence for the presence of androgen excess.
Plasma-free testosterone is the best single indicator of androgen excess[95,96]. It is high about 50% more often than the total testosterone level in hirsute women because of their relatively low sex hormone binding globulin level. This binding protein is suppressed by the hyperinsulinemia of insulin resistance and by androgen excess itself [97,98]. It determines the fraction of plasma testosterone that is free or bound to albumin [95], and the free is ultimately the bioavailable fraction [98,99]. There is no uniform laboratory standard for free testosterone, so assay-specific normal ranges differ. The authors’ normal early morning range for follicular phase women without hirsutism, acne, or polycystic ovaries is 3 to 10 pg/mL. An adult level is reached at the time of menarche [100].
Initial work-up to rule out disorders that mimic polycystic ovary syndrome
Polycystic ovary syndrome is a diagnosis of exclusion according to gynecologic practice guidelines [8,101,102]. These guidelines were developed because of practical concerns about cost-effectiveness and the inconsistent availability of reliable specialized hormone assays. The diagnosis of PCOS is most solid in hyperandrogenic adolescents who have anovulatory symptoms or a polycystic ovary after ruling out other causes of androgen excess. The combination of anovulatory symptoms and polycystic ovaries for the diagnosis of PCOS (see Box 1) is particularly weak in adolescents because of the normalcy of physiologic anovulation and of multi-follicular ovaries.
The work-up for the other causes of androgen excess should be initiated with a blood sample and an ultrasound examination (Fig. 5). The blood sample is to screen for thyroid dysfunction, hyperprolactinemia, nonclassic congenital adrenal hyperplasia caused by 21-hydroxylase deficiency, and Cushing’s syndrome, which are ordinarily the major considerations. An enlarged or polycystic ovary with androgen excess meet the Rotterdam criteria for PCOS (see Box 1) (although the presence of polycystic ovaries is neither specific nor necessary for this diagnosis), and ultrasound is also valuable to exclude ovarian tumor and screen for adrenal tumor.
Fig. 5.

Initial work-up of androgen excess. This algorithm rules out most disorders that can mimic PCOS.
A polycystic ovary is defined specifically as one with increased volume (over 10 cc using a simplified formula [0.5 × length × width × depth]) or containing 12 or more follicles that are 2 to 9 mm in diameter (microcysts) [8,103]. Alternative criteria are an area of 5.5 cm2 or 10 or more follicles in the maximum plane, respectively. Increased volume is an objective indicator of excessive ovarian stroma. The volume criteria assume the absence of a dominant follicle (over 10 mm diameter) or a corpus luteum. The normal adolescent volume criterion is similar: a volume over 10.8 cc by the formula for a prolate ellipsoid [104]. Ovarian enlargement is the most solid criterion. Multi-follicular ovaries, normal in adolescence and present in other anovulatory states, are distinguishable from polycystic ovaries primarily on the basis of whether volume is normal or abnormal, which can be problematic [104,105].
Detecting polycystic ovaries in adolescents poses diagnostic difficulties. It is important that the ultrasonographer adhere to the guidelines recommended for their ascertainment [103]. To some extent, ascertainment is limited in obese virginal subjects, in whom use of a vaginal ultrasonographic probe is inappropriate.
There are some caveats to a solely exclusionary approach for the diagnosis of PCOS. First, adrenal tumors, while rare, may be indolent and mimic PCOS. Therefore, it is advisable to specifically screen for these by ultrasound imaging of the adrenal glands. In atypical cases, CT scan may be indicated to rule out an adrenal tumor. It is not clear whether ultrasound will distinguish the rare ovarian adrenal rests of nonclassic congenital adrenal hyperplasia from polycystic ovaries [55]. Most importantly, while the remaining disorders are rare, well under 1%, patients must be followed to be sure that they respond to therapy as expected.
Determining the source of androgen excess
To expeditiously determine the source of androgen in PCOS and rule out rare congenital adrenal disorders, the American Association of Clinical Endocrinologists’ guidelines (2001) recommended a comprehensive evaluation of hyperandrogenism, because the prognosis and treatment differ [93]. Although the approach differs among endocrinologists, the authors find that a combination of dexamethasone suppression testing and cosyntropin testing, if necessary, will yield a diagnosis of FOH or FAH in most patients (Fig. 6). This algorithm is a relatively cost-effective one that usually can yield a positive diagnosis of PCOS and rule out other causes of androgen excess.
Fig. 6.

Algorithm to determine the source of androgen excess. This algorithm helps to determine the source of excess androgen production.
Dexamethasone is given in a dosage of 0.5 mg four times daily for 4 days (7 days if weight is over 100 kg), and free testosterone (or alternatively 17-hydroxyprogesterone), DHEA sulfate, and cortisol are measured after a final dose on the following morning. Dexamethasone suppression of ACTH-dependent adrenal function normally causes plasma cortisol to fall below 1.5 μg/dL, DHEA sulfate to fall by 75% to below 80 μg/dL, and total testosterone to fall below 35 ng/dL. A plasma-free testosterone below 8 pg/mL is the most discriminating criterion in the authors’ hands. Because this is a method-dependent criterion, a 17-hydroxyprogesterone level below 50 ng/dL appears to be a comparable alternative [106].
Subnormal androgen suppression with normal adrenocortical suppression indicates a source of androgen other than an ACTH-dependent adrenal one. This is typical of PCOS if tumor has not been found by ultrasound examination. If both cortisol and androgen suppression are subnormal, then the androgen excess may be secondary to noncompliance with taking dexamethasone, Cushing’s syndrome, or defective cortisol metabolism or action.
If androgen suppression is normal, then cosyntropin stimulation testing to assess 17-hydroxyprogesterone and other steroid intermediates is recommended. A clear diagnosis of nonclassic congenital adrenal hyperplasia requires a steroid intermediate peak that is more than five standard deviations above average (eg, 17-hydroxyprogesterone over 1000 to 1500 ng/dL for 21-hydroxylase deficiency or 17-hydroxypregnenolone over 5000 ng/dL for 3β-hydroxysteroid dehydrogenase deficiency) [80,107,108]. Mildly elevated or normal responses to cosyntropin are consistent with FAH or idiopathic hyperandrogenism, respectively. Unexplained mild androgen excess (idiopathic hyperandrogenism), may be caused by abnormalities in the peripheral metabolism of androgen. It is unclear whether this is an atypical manifestation of PCOS. In a very small number of cases, GnRH or hCG testing will be necessary for a positive diagnosis of PCOS.
Management
Management depends on symptoms or the source of androgen excess. Several treatment options are available, which allows for an individualized approach.
Because PCOS patients are at risk for the development of the metabolic syndrome and T2DM, a fasting lipid panel and oral glucose tolerance test are recommended, particularly in obese patients. Likewise, evaluation of primary relatives for these disorders should be recommended.
Treatment of hirsutism, acne, and pattern alopecia
Endocrinologic treatment to interrupt one or more of the steps in the pathway of androgen action is indicated if standard cosmetic or topical dermatologic measures fall short. Cosmetic and dermatologic treatment of hirsutism includes depilation (eg, shaving or chemical depilatories), epilation (eg, plucking or waxing), inhibition of local hair growth (eg, eflornithine hydrochloride cream), or destruction of the dermal papilla (eg, electrolysis or laser therapy). Laser treatment, including equivalents such as diode and flashlamp, recently was approved by the Food and Drug Administration (FDA) for permanent hair reduction [109].
Combination oral contraceptive pills
Oral contraceptive pills (OCPs) are the first line endocrine treatment for women with the dermatologic or menstrual abnormalities of PCOS [66]. They act by suppressing plasma androgens, particularly free testosterone, mainly by inhibiting ovarian function. They also raise sex hormone binding globulin levels and modestly lower DHEA sulfate levels. They normalize androgen levels within the first month of therapy. Treatment with OCPs improves acne within 3 months, arrests progression of hirsutism, and reduces shaving frequency.
All estrogen–progestin combinations are generally satisfactory for women with mild hirsutism, in combination with cosmetic measures. Those with nonandrogenic progestins have generally favorable risk–benefit ratios [110] and optimize lipid profiles [111]. Those with the larger estrogen doses may be necessary in larger women to provide menstrual regularity. Yasmin is a monophasic OCP that contains 30 μg of ethinyl estradiol and 3 mg of drospirenone, a spironolactone-derived progestin. This progestin’s mild antimineralocorticoid and antiandrogenic properties make it attractive for minimizing fluid retention and androgenic adverse effects. Yasmin is equivalent to 25 mg spironolactone and should be used with the same precautions as that drug [112]. Norgestimate is combined with 35 μg of ethinyl estradiol in Ortho-Cyclen or Ortho-Tri-Cyclen, which has received approval from the FDA for the treatment of acne vulgaris in females. Demulen 1/50 contains ethinyl estradiol and ethynodiol diacetate, a progestin of low androgenic potential, and it is useful for obese patients who required a larger dose of estrogen.
It is advisable to recheck patients after 3 months of therapy to assess the efficacy of treatment and normalization of androgen levels. Androgen suppression is supportive of the diagnosis, but not diagnostic [67]. As a general rule, OCP treatment should be continued until the patient is gynecologically mature (5 years postmenarchal) or has lost a substantial amount of excess weight. At that point, withholding treatment for a few months to allow recovery of suppression of pituitary–gonadal function and to ascertain whether the menstrual abnormality persists is usually advisable. In doing so, however, one must keep in mind that the anovulatory cycles of PCOS lead to relative infertility, not absolute infertility. The need for continued use of OCP for contraceptive purposes must be considered.
Glucocorticoids
Glucocorticoid therapy may be of benefit for treating occasional nonobese hirsute PCOS patients who have a prominent component of FAH. Such patients may have androgen excess that cannot be controlled with OCPs. The sequelae of glucocorticoid therapy usually can be minimized by using a modest bedtime dose (eg, 5–7.5 mg prednisone); this reduces secretion of adrenal androgens more than that of cortisol. The authors typically aim to suppress DHEA sulfate to below the adult range without suppressing it completely. Significant glucocorticoid deficiency can be excluded by finding a cortisol level of 10 μg/dL or more at 8 AM or 18 μg/dL 30 minutes after administering a low dose (1.0 μg) of ACTH (cosyntropin) [113].
Antiandrogens
Antiandrogens are required for substantial improvement of hirsutism. Antiandrogens act as competitive antagonists of steroid binding to the androgen receptor and reverse the androgen-induced transformation of vellus to terminal hairs [66]. Thus, the effects of these agents usually are not appreciated for 9 to 12 months because of the long growth cycles of sexual hair follicles. Their use for this purpose is off-label, because all carry the risk of causing pseudohermaphroditism of the male fetus. Therefore, contraception is required. All antiandrogens should be prescribed with a contraceptive. Antiandrogens may have a modest effect on the metabolic abnormalities associated with PCOS [114].
Spironolactone is the safest potent antiandrogen available in the United States [43]. Higher doses are required for its antiandrogenic effect than for its antimineralocorticoid effect; it also is a weak progestin and weak glucocorticoid. It is effective in lowering the hirsutism score by approximately one third, although considerable individual variation exists. The authors recommend starting with 100 mg twice a day until the maximal effect has been achieved and then attempting to reduce the dose to 50 mg twice a day for maintenance therapy. Spironolactone must be administered as long as the patient wishes to maintain her improvement in hirsutism. Administered in these doses, spironolactone usually is well-tolerated, but hyperkalemia may limit its usefulness. It is contraindicated in patients with adrenal, hepatic, or renal insufficiency. Women are at risk of hyperkalemia if on potassium-sparing diuretics, potassium supplements, daily nonsteroidal anti-inflammatory drugs, angiotensin-converting enzyme inhibitors, heparin, or such drugs. Therefore, electrolytes should be monitored. Alone, it tends to cause irregular bleeding. It must be used with an OCP.
Other antiandrogens used to treat hirsutism and hirsutism equivalents include cyproterone acetate, flutamide, and finasteride. Cyproterone acetate is a progestational antiandrogen that has weak antiglucocorticoid effects. It is not available in the United States, but it is available in Europe and Canada as a combination oral contraceptive containing ethinyl estradiol and low-dose cyproterone acetate. Flutamide is a more specific antiandrogen with efficacy similar to that of cyproterone and spironolactone; its use has been limited by expense and the potential for hepatocellular toxicity. Flutamide may enhance ovulation in women with PCOS [114,115]. Its utility for this purpose is precluded by the risk of feminization of the male fetus. It must be used with an OCP. Finasteride, a type 1 5-α-reductase inhibitor, is less effective than other antiandrogens for treating hirsutism [116].
Female-pattern hair loss is less androgen-dependent than is male-pattern baldness. Although topical minoxidil is the only medication approved for its treatment, antiandrogen–OCP therapy appears to be superior in those with PCOS [117,118].
Treatment of menstrual irregularity
Menstrual irregularity should be treated in patients with PCOS, because chronic anovulation is associated with increased risk of developing endometrial hyperplasia and carcinoma. The major treatment options for menstrual irregularity include progestin or OCPs. Insulin-lowering agents, glucocorticoids, and GnRH agonists have a place in some patients.
Progestin
Menstrual irregularities often can be treated effectively with micronized progesterone, 100 to 200 mg daily at bedtime for 7 to 10 days. This induces withdrawal bleeding in most patients, but some do not respond, apparently because of an antiestrogenic effect of androgen excess on the endometrium [119]. Progestin therapy has the appeal of permitting the detection of the emergence of normal menstrual cyclicity. Progestin therapy, however, does not normalize androgen levels and is not an adequate treatment if hirsutism or hirsutism equivalents are a problem. The perimenarcheal girl who responds well to progestin therapy can be maintained at approximately 6-week cycles to permit the detection of spontaneous menses. Adverse effects of progestin include mood symptoms (depression), bloating, and breast soreness. Patients must be informed that oral progestin dosed in this way is not a means of contraception. When progestin is used for dysfunctional uterine bleeding, it may have to be given 2 weeks out of 3.
Combination oral contraceptive pills
Oral contraceptive pills induce regular menstrual periods with a higher degree of reliability than does any other form of treatment. The mechanism of action was discussed in the preceding section.
Dysfunctional uterine bleeding (DUB) usually can be treated with cyclic progestin, but heavy DUB may require estrogen treatment. Estrogen can be given as OCPs, one tablet three to four times daily for 7 days. Any OCP can be used, but one with a relatively large estrogenic component such as Demulen 1/50 is preferred. Treatment then is stopped for 5 days, and the patient is warned that heavy withdrawal bleeding and cramps may occur. Therapy with cyclic OCPs then should be started to prevent recurrence of dysfunctional bleeding.
Oral contraceptive pill therapy decreases excessive menstrual blood loss in anemic patients with menorrhagia by approximately 50%. Gynecologic referral is indicated for patients whose bleeding cannot be controlled medically or who have uncontrollable menstrual abnormalities, because these patients are at risk for developing endometrial carcinoma.
There are, nevertheless, several potential disadvantages to the use of OCPs for managing PCOS in adolescents. They will bring growth to an end in perimenarcheal girls. OCPs, particularly some third-generation OCPs, may be contraindicated in patients who are at risk for venous thrombosis [120]. In patients with migraine headaches, OCPs should be used with caution and in the lowest estrogen dose possible [121]. Patients may use OCPs as an excuse for not losing weight. The patient may believe that the treatment is curative and defer a definitive diagnostic work-up. OCPs do not permit conception if and when it is desired. The long-term consequences of these agents on fertility is unknown; there is the theoretic possibility of post-pill amenorrhea, and high-dose estrogen begun in early adolescence may increase the risk of infertility [122].
Glucocorticoids
Glucocorticoid replacement may be of benefit in the exceptional nonobese PCOS patient with FAH. A 3-month trial of glucocorticoid therapy is sufficient to determine the efficacy of this mode of treatment.
Gonadotropin-releasing hormone agonists
Gonadotropin-releasing hormone agonist therapy (eg, depot leuprolide) may be used to suppress gonadotropin secretion in the exceptional patient who cannot tolerate OCPs. Patients who are treated with GnRH agonists also should be treated with low-dose physiologic estradiol add-back (eg, 50 μg estradiol by transdermal patch) to maintain vaginal lubrication and to ensure bone mineral accrual during adolescence.
Insulin-lowering therapy
Antidiabetic treatments improve ovulation and hyperandrogenemia in PCOS by lowering insulin levels. They are discussed next.
Treatment of obesity and insulin resistance
The treatment of obesity improves ovulation and androgen excess in patients with PCOS [123–125]. Although weight reduction is indicated in obese PCOS patients, it is typically difficult to achieve.
The new insulin-lowering agents, metformin and thiazolidinediones, are promising adjuncts for treating PCOS. Their mechanisms of action differ, metformin primarily inhibiting hepatic glucose output and thiazolidinediones increasing insulin sensitivity primarily by promoting fat mobilization from the bloodstream [126,127]. Both types of drugs promote ovulation and lower androgen levels up to about 20% [128,129], although in current regimens, they have little if any direct effect on steroidogenesis [130,131]. These agents are unlikely to normalize androgen levels that are greater than two times normal, however. They seem to be effective even in PCOS patients who have no indication of being insulin resistant [132].
Metformin appears to have the most utility in managing adolescents, because it suppresses appetite and enhances weight loss, albeit to a modest degree [133]. It is effective only to the extent that weight is lost [134]. The extended-release form of metformin often is tolerated more than the short-acting form. Therapy should start with 500 mg daily before the evening meal, with an increase in the dose by 500 mg per week to a maximal dose of 2000 mg daily as tolerated. The greater doses often are tolerated better when divided into two daily doses. One is advised to obtain a comprehensive metabolic panel at baseline and every 3 to 6 months, or as necessary, because of the rare complication of lactic acidosis.
The prototype thiazolidinedione, troglitazone, has been withdrawn from the market in the United States and the United Kingdom because of the risk of hepatotoxicity. The new generation of more potent thiazolidinediones (eg, pioglitazone and rosiglitazone) appears to be safe and beneficial in women with PCOS [127,135]. Unfortunately, this class of drugs tends to cause weight gain.
Summary
PCOS should be considered in any adolescent girl with hirsutism or hirsutism equivalents, menstrual irregularity, or obesity. Recognizing and treating PCOS in adolescents is important, because it poses a risk for the metabolic syndrome, and chronic anovulation is associated with increased risk of development of endometrial hyperplasia and carcinoma. A plasma free testosterone level above the normal adult range, as determined by a specialty laboratory, is the preferred screening test. PCOS must be distinguished from other hyperandrogenic disorders that require specific therapy (eg, virilizing tumors, nonclassic congenital adrenal hyperplasia, hyperprolactinemia, and Cushing’s syndrome). Ultrasonography is an important testing modality that helps in this differential diagnosis and may demonstrate the polycystic ovaries that recently have been designated one of the diagnostic criteria. The approach to the diagnosis of PCOS varies among specialists. According to practice guidelines, PCOS is a diagnosis of exclusion. A positive diagnosis, however, can be reached most expeditiously by a combination of dexamethasone suppression and cosyntropin testing. Management of PCOS is determined by the symptoms, and several treatment options are available, including OCPs, progestins, glucocorticoids, antiandrogens, and insulin-lowering treatments.
Acknowledgments
These studies were supported in part by the Robert Wood Johnson Foundation (CB) and Unites States Public Health Service grants K12-HD-043387-0 (CB), HD-39267, HD-U54-041859, and RR-00055 (RLR).
References
- [1].Rosenfield RL. Polycystic ovary syndrome and insulin-resistant hyperinsulinemia. J Am Acad Dermatol. 2001;45:S095. doi: 10.1067/mjd.2001.117430. [DOI] [PubMed] [Google Scholar]
- [2].Zawadzki J, Dunaif A. Diagnostic criteria for polycystic ovary syndrome: towards a rational approach. In: Dunaif A, Givens J, Haseltine F, et al., editors. Polycystic ovary syndrome. Blackwell Scientific Publications; Cambridge (MA): 1992. p. 377. [Google Scholar]
- [3].Ehrmann DA, Barnes RB, Rosenfield RL. Polycystic ovary syndrome as a form of functional ovarian hyperandrogenism due to dysregulation of androgen secretion. Endocr Rev. 1995;16:322. doi: 10.1210/edrv-16-3-322. [DOI] [PubMed] [Google Scholar]
- [4].Ehrmann DA, Rosenfield RL, Barnes RB, et al. Detection of functional ovarian hyperandrogenism in women with androgen excess. N Engl J Med. 1992;327:157. doi: 10.1056/NEJM199207163270304. [DOI] [PubMed] [Google Scholar]
- [5].Rosenfield RL. Ovarian and adrenal function in polycystic ovary syndrome. Endocrinol Metab Clin North Am. 1999;28:265. doi: 10.1016/s0889-8529(05)70070-0. [DOI] [PubMed] [Google Scholar]
- [6].Dunaif A, Green G, Futterweit W, et al. Suppression of hyperandrogenism does not improve peripheral or hepatic insulin resistance in the polycystic ovary syndrome. J Clin Endocrinol Metab. 1990;70:699. doi: 10.1210/jcem-70-3-699. [DOI] [PubMed] [Google Scholar]
- [7].Rittmaster R, Thompson D. Effect of leuprolide and dexamethasone on hair growth and hormone levels in hirsute women: the relative importance of the ovary and the adrenal in the pathogenesis of hirsutism. J Clin Endocrinol Metab. 1990;70:1096. doi: 10.1210/jcem-70-4-1096. [DOI] [PubMed] [Google Scholar]
- [8].Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril. 2004;81:19. doi: 10.1016/j.fertnstert.2003.10.004. [DOI] [PubMed] [Google Scholar]
- [9].Kahn SE. Regulation of beta-cell function in vivo: from health to disease. Diabetes Reviews. 1996;4:372. [Google Scholar]
- [10].American Diabetes Association Consensus Development Conference on Insulin Resistance. Diabetes Care. 1998;21:310. doi: 10.2337/diacare.21.2.310. [DOI] [PubMed] [Google Scholar]
- [11].Alberti KG, Zimmet PZ. Report of a WHO consultation. Part 1: diagnosis and classification of diabetes mellitus. World Health Organization; Geneva (Switzerland): 1999. Definition, diagnosis and classification of diabetes mellitus and its complications. [DOI] [PubMed] [Google Scholar]
- [12].Grundy SM, Brewer HB, Jr, Cleeman JI, et al. Definition of metabolic syndrome: report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation. 2004;109:433. doi: 10.1161/01.CIR.0000111245.75752.C6. [DOI] [PubMed] [Google Scholar]
- [13].Rosenfield RL, Ghai K, Ehrmann DA, et al. Diagnosis of polycystic ovary syndrome in adolescence. Comparison of adolescent and adult hyperandrogenism. J Pediatr Endocrinol Metab. 2000;13:1285. [PubMed] [Google Scholar]
- [14].Ibañez L, de Zegher F, Potau N. Anovulation after precocious pubarche: early markers and time course in adolescence. J Clin Endocrinol Metab. 1999;84:2691. doi: 10.1210/jcem.84.8.5883. [DOI] [PubMed] [Google Scholar]
- [15].Apter D, Bützow G, Laughlin A, et al. Metabolic features of polycystic ovary syndrome are found in adolescent girls with hyperandrogenism. J Clin Endocrinol Metab. 1995;80:2966. doi: 10.1210/jcem.80.10.7559882. [DOI] [PubMed] [Google Scholar]
- [16].Apter D, Bützow T, Laughlin G, et al. Accelerated 24-hour luteinizing hormone pulsatile activity in adolescent girls with ovarian hyperandrogenism: relevance to the developmental phase of polycystic ovarian syndrome. J Clin Endocrinol Metab. 1994;79:119. doi: 10.1210/jcem.79.1.8027216. [DOI] [PubMed] [Google Scholar]
- [17].Ibañez L, Potau N, Francois I, et al. Precocious pubarche, hyperinsulinism, and ovarian hyperandrogenism: relation to reduced fetal growth. J Clin Endocrinol Metab. 1998;83:3558. doi: 10.1210/jcem.83.10.5205. [DOI] [PubMed] [Google Scholar]
- [18].Ibañez L, Potau N, Virdis R, et al. Postpubertal outcome in girls diagnosed of premature pubarche during childhood: increased frequency of functional ovarian hyperandrogenism. J Clin Endocrinol Metab. 1993;76:1599. doi: 10.1210/jcem.76.6.8501168. [DOI] [PubMed] [Google Scholar]
- [19].Meas T, Chevenne D, Thibaud E, et al. Endocrine consequences of premature pubarche in postpubertal Caucasian girls. Clin Endocrinol (Oxf) 2002;57:101. doi: 10.1046/j.1365-2265.2002.01579.x. [DOI] [PubMed] [Google Scholar]
- [20].Rosenfield RL. Are adrenal and ovarian function normal in true precocious puberty? Eur J Endocrinol. 1995;133:399. doi: 10.1530/eje.0.1330399. [DOI] [PubMed] [Google Scholar]
- [21].Baumann E, Weiss R, Deplewski D, et al. Pseudo-Cushing syndrome and pseudoacromegalic gigantism associated with insulin resistant hyperinsulinism as childhood precursors of polycystic ovary syndrome. Pediatr Res. 2002;51:117A. [Google Scholar]
- [22].Sahin Y, Kelestimur F. 17-hydroxyprogesterone response to buserelin testing in the polycystic ovary syndrome. Clin Endocrinol. 1993;39:151. doi: 10.1111/j.1365-2265.1993.tb01767.x. [DOI] [PubMed] [Google Scholar]
- [23].Berga SL, Guzick DS, Winters SJ. Increased luteinizing hormone and a-subunit secretion in women with hyperandrogenic anovulation. J Clin Endocrinol Metab. 1993;77:895. doi: 10.1210/jcem.77.4.7691863. [DOI] [PubMed] [Google Scholar]
- [24].Eagleson CA, Gingrich MB, Pastor CL, et al. Polycystic ovarian syndrome: evidence that flutamide restores sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J Clin Endocrinol Metab. 2000;85:4047. doi: 10.1210/jcem.85.11.6992. [DOI] [PubMed] [Google Scholar]
- [25].Pastor CL, Griffin-Korf ML, Aloi JA, et al. Polycystic ovary syndrome: evidence for reduced sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J Clin Endocrinol Metab. 1998;83:582. doi: 10.1210/jcem.83.2.4604. [DOI] [PubMed] [Google Scholar]
- [26].Patel K, Coffler MS, Dahan MH, et al. Increased luteinizing hormone secretion in women with polycystic ovary syndrome is unaltered by prolonged insulin infusion. J Clin Endocrinol Metab. 2003;88:5456. doi: 10.1210/jc.2003-030816. [DOI] [PubMed] [Google Scholar]
- [27].Buggs C, Weinberg F, Wolfe A, et al. Insulin together with GnRH modulates expression of LH beta gene expression. Presented at the Endocrine Society’s 85th Annual Meeting; Philadelphia. Jun 19–22, 2003. [Google Scholar]
- [28].Dorn C, Mouillet JF, Yan X, et al. Insulin enhances the transcription of luteinizing hormone-beta gene. Am J Obstet Gynecol. 2004;191:132. doi: 10.1016/j.ajog.2004.01.054. [DOI] [PubMed] [Google Scholar]
- [29].Maciel GA, Baracat EC, Benda JA, et al. Stockpiling of transitional and classic primary follicles in ovaries of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2004;89:5321. doi: 10.1210/jc.2004-0643. [DOI] [PubMed] [Google Scholar]
- [30].Jakimiuk AJ, Weitsman SR, Navab A, et al. Luteinizing hormone receptor, steroidogenesis acute regulatory protein, and steroidogenic enzyme messenger ribonucleic acids are overexpressed in thecal and granulosa cells from polycystic ovaries. J Clin Endocrinol Metab. 2001;86:1318. doi: 10.1210/jcem.86.3.7318. [DOI] [PubMed] [Google Scholar]
- [31].Nelson VL, Qin Kn K, Rosenfield RL, et al. The biochemical basis for increased testosterone production in theca cells propagated from patients with polycystic ovary syndrome. J Clin Endocrinol Metab. 2001;86:5925. doi: 10.1210/jcem.86.12.8088. [DOI] [PubMed] [Google Scholar]
- [32].Wickenheisser JK, Quinn PG, Nelson VL, et al. Differential activity of the cytochrome P450 17α-hydroxylase and steroidogenic acute regulatory protein gene promoters in normal and polycystic ovary syndrome theca cells. J Clin Endocrinol Metab. 2000;85:2304. doi: 10.1210/jcem.85.6.6631. [DOI] [PubMed] [Google Scholar]
- [33].Coffler MS, Patel K, Dahan MH, et al. Evidence for abnormal granulosa cell responsiveness to follicle-stimulating hormone in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2003;88:1742. doi: 10.1210/jc.2002-021280. [DOI] [PubMed] [Google Scholar]
- [34].Fassnacht M, Schlenz N, Schneider SB, et al. Beyond adrenal and ovarian androgen generation: Increased peripheral 5 alpha-reductase activity in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2003;88:2760. doi: 10.1210/jc.2002-021875. [DOI] [PubMed] [Google Scholar]
- [35].Quinkler M, Sinha B, Tomlinson JW, et al. Androgen generation in adipose tissue in women with simple obesity - a site-specific role for 17β-hydroxysteroid dehydrogenase type 5. J Endocrinol. 2004;183:331. doi: 10.1677/joe.1.05762. [DOI] [PubMed] [Google Scholar]
- [36].Book CB, Dunaif A. Selective insulin resistance in the polycystic ovary syndrome. J Clin Endocrinol Metab. 1999;84:3110. doi: 10.1210/jcem.84.9.6010. [DOI] [PubMed] [Google Scholar]
- [37].Bruning JC, Gautam D, Burks DJ, et al. Role of brain insulin receptor in control of body weight and reproduction. Science. 2000;289:2122. doi: 10.1126/science.289.5487.2122. [DOI] [PubMed] [Google Scholar]
- [38].Roldan B, Escobar-Morreale HF, Barrio R, et al. Identification of the source of androgen excess in hyperandrogenic type 1 diabetic patients. Diabetes Care. 2001;24:1297. doi: 10.2337/diacare.24.7.1297. [DOI] [PubMed] [Google Scholar]
- [39].Legro RS. Polycystic ovary syndrome and cardiovascular disease: a premature association? Endocr Rev. 2003;24:302. doi: 10.1210/er.2003-0004. [DOI] [PubMed] [Google Scholar]
- [40].Guzick DS. Cardiovascular risk in PCOS. J Clin Endocrinol Metab. 2004;89:3694. doi: 10.1210/jc.2004-1136. [DOI] [PubMed] [Google Scholar]
- [41].Legro RS, Strauss JF. Molecular progress in infertility: polycystic ovary syndrome. Fertil Steril. 2002;78:569. doi: 10.1016/s0015-0282(02)03275-2. [DOI] [PubMed] [Google Scholar]
- [42].Kahsar-Miller MD, Nixon C, Boots LR, et al. Prevalence of polycystic ovary syndrome (PCOS) in first-degree relatives of patients with PCOS. Fertil Steril. 2001;75:53. doi: 10.1016/s0015-0282(00)01662-9. [DOI] [PubMed] [Google Scholar]
- [43].Legro RS, Driscoll D, Strauss JF, III, et al. Evidence for a genetic basis for hyperandrogenemia in polycystic ovary syndrome. Proc Natl Acad Sci U S A. 1998;95:14956. doi: 10.1073/pnas.95.25.14956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Carey A, Waterworth D, Patel K, et al. Polycystic ovaries and premature male-pattern baldness are associated with one allele of the steroid metabolism gene CYP 17. Hum Mol Genet. 1994;3:1873. doi: 10.1093/hmg/3.10.1873. [DOI] [PubMed] [Google Scholar]
- [45].Govind A, Obhrai MS, Clayton RN. Polycystic ovaries are inherited as an autosomal dominant trait: analysis of 29 polycystic ovary syndrome and 10 control families. J Clin Endocrinol Metab. 1999;84:38. doi: 10.1210/jcem.84.1.5382. [DOI] [PubMed] [Google Scholar]
- [46].Gilling-Smith C, Willis DS, Beard RW, et al. Hypersecretion of androstenedione by isolated theca cells from polycystic ovaries. J Clin Endocrinol Metab. 1994;79:1158. doi: 10.1210/jcem.79.4.7962289. [DOI] [PubMed] [Google Scholar]
- [47].Adams JM, Taylor AE, Crowley WF, Jr, et al. Polycystic ovarian morphology with regular ovulatory cycles: insights into the pathophysiology of polycystic ovarian syndrome. J Clin Endocrinol Metab. 2004;89:4343. doi: 10.1210/jc.2003-031600. [DOI] [PubMed] [Google Scholar]
- [48].Gilling-Smith C, Story H, Rogers V, et al. Evidence for a primary abnormality of thecal cell steroidogenesis in the polycystic ovary syndrome. Clin Endocrinol. 1997;47:93. doi: 10.1046/j.1365-2265.1997.2321049.x. [DOI] [PubMed] [Google Scholar]
- [49].Polson D, Adams J, Wadsworth J, et al. Polycystic ovaries—a common finding in normal women. Lancet. 1988;1:870. doi: 10.1016/s0140-6736(88)91612-1. [DOI] [PubMed] [Google Scholar]
- [50].Nayak S, Lee PA, Witchel SF. Variants of the type II 3β-hydroxysteroid dehydrogenase gene in children with premature pubic hair and hyperandrogenic adolescents. Mol Genet Metab. 1998;64:184. doi: 10.1006/mgme.1998.2715. [DOI] [PubMed] [Google Scholar]
- [51].Witchel SF, Lee PA, Suda-Hartman M, et al. Hyperandrogenism and manifesting heterozygotes for 21-hydroxylase deficiency. Biochem Mol Med. 1997;62:151. doi: 10.1006/bmme.1997.2632. [DOI] [PubMed] [Google Scholar]
- [52].Nelson VL, Legro RS, Strauss JF, III, et al. Augmented androgen production is a stable steroidogenic phenotype of propagated theca cells from polycystic ovaries. Mol Endocrinol. 1999;13:946. doi: 10.1210/mend.13.6.0311. [DOI] [PubMed] [Google Scholar]
- [53].Zhang LH, Rodriguez H, Ohno S, et al. Serine phosphorylation of human P450c17 increases 17,20-lyase activity: implications for adrenarche and the polycystic ovary syndrome. Proc Natl Acad Sci U S A. 1995;92:10619. doi: 10.1073/pnas.92.23.10619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Abbott DH, Dumesic DA, Franks S, et al. Developmental origin of polycystic ovary syndrome—a hypothesis. J Endocrinol. 2002;174:1. doi: 10.1677/joe.0.1740001. [DOI] [PubMed] [Google Scholar]
- [55].Barnes RB, Rosenfield RL, Ehrmann DA, et al. Ovarian hyperandrogenism as a result of congenital adrenal virilizing disorders: Evidence for perinatal masculinization of neuroendocrine function in women. J Clin Endocrinol Metab. 1994;79:1328. doi: 10.1210/jcem.79.5.7962325. [DOI] [PubMed] [Google Scholar]
- [56].Eisner JR, Dumesic DA, Kemnitz JW, et al. Increased adiposity in female rhesus monkeys exposed to androgen excess during early gestation. Obes Res. 2003;11:279. doi: 10.1038/oby.2003.42. [DOI] [PubMed] [Google Scholar]
- [57].Dunaif A. Insulin resistance and the polycystic ovary syndrome: mechanism and implications for pathogenesis. Endocr Rev. 1997;18:774. doi: 10.1210/edrv.18.6.0318. [DOI] [PubMed] [Google Scholar]
- [58].Glueck CJ, Papanna R, Wang P, et al. Incidence and treatment of metabolic syndrome in newly referred women with confirmed polycystic ovarian syndrome. Metabolism. 2003;52:908. doi: 10.1016/s0026-0495(03)00104-5. [DOI] [PubMed] [Google Scholar]
- [59].Orio F, Jr, Palomba S, Spinelli L, et al. The cardiovascular risk of young women with polycystic ovary syndrome: an observational, analytical, prospective case-control study. J Clin Endocrinol Metab. 2004;89:3696. doi: 10.1210/jc.2003-032049. [DOI] [PubMed] [Google Scholar]
- [60].Ehrmann DA, Barnes RB, Rosenfield RL, et al. Prevalence of impaired glucose tolerance and diabetes in women with polycystic ovary syndrome. Diabetes Care. 1999;22:141. doi: 10.2337/diacare.22.1.141. [DOI] [PubMed] [Google Scholar]
- [61].Legro RS, Kunselman AR, Dodson WC, et al. Prevalence and predictors of risk for type 2 diabetes mellitus and impaired glucose tolerance in polycystic ovary syndrome: a prospective, controlled study in 254 affected women. J Clin Endocrinol Metab. 1999;84:165. doi: 10.1210/jcem.84.1.5393. [DOI] [PubMed] [Google Scholar]
- [62].Leibel N, Baumann E, Rosenfield RL. Metabolic syndrome features are the typical parental phenotype of adolescent polycystic ovary syndrome (PCOS) Pediatr Res. 2004;55:A818. [Google Scholar]
- [63].Freeman MS, Mansfield MW, Barrett JH, et al. Heritability of features of the insulin resistance syndrome in a community-based study of healthy families. Diabet Med. 2002;19:994. doi: 10.1046/j.1464-5491.2002.00843.x. [DOI] [PubMed] [Google Scholar]
- [64].Mills GW, Avery PJ, McCarthy MI, et al. Heritability estimates for beta cell function and features of the insulin resistance syndrome in UK families with an increased susceptibility to type 2 diabetes. Diabetologia. 2004;47:732. doi: 10.1007/s00125-004-1338-2. [DOI] [PubMed] [Google Scholar]
- [65].Ehrmann DA. Medical progress: polycystic ovary syndrome. N Engl J Med. 2004;352:1223–36. doi: 10.1056/NEJMra041536. [DOI] [PubMed] [Google Scholar]
- [66].Deplewski D, Rosenfield RL. Role of hormones in pilosebaceous unit development. Endocr Rev. 2000;21:363. doi: 10.1210/edrv.21.4.0404. [DOI] [PubMed] [Google Scholar]
- [67].Hatch R, Rosenfield RL, Kim MH, et al. Hirsutism: implications, etiology, and management. Am J Obstet Gynecol. 1981;140:815. doi: 10.1016/0002-9378(81)90746-8. [DOI] [PubMed] [Google Scholar]
- [68].Kim SS, Rosenfield RL. Hyperhidrosis as the only manifestation of hyperandrogenism in an adolescent girl [letter] Arch Dermatol. 2000;136:430. doi: 10.1001/archderm.136.3.430. [DOI] [PubMed] [Google Scholar]
- [69].Orfanos C, Adler Y, Zouboulis CC. The SAHA syndrome. Horm Res. 2000;54:251. doi: 10.1159/000053267. [DOI] [PubMed] [Google Scholar]
- [70].Treloar A, Boynton R, Benn B, et al. Variation of human menstrual cycle through reproductive life. Int J Fertil. 1967;12:77. [PubMed] [Google Scholar]
- [71].Flier JS, Moller DE, Moses AC, et al. Insulin-mediated pseudoacromegaly: clinical and biochemical characterization of a syndrome of selective insulin resistance. J Clin Endocrinol Metab. 1993;76:1533. doi: 10.1210/jcem.76.6.8388881. [DOI] [PubMed] [Google Scholar]
- [72].Invitti C, De Martin M, Delitala G, et al. Altered morning and nighttime pulsatile corticotropin and cortisol release in polycystic ovary syndrome. Metabolism. 1998;47:143. doi: 10.1016/s0026-0495(98)90210-4. [DOI] [PubMed] [Google Scholar]
- [73].Salami DJ, Zisser HC, Jovanovic L. Screening for and treatment of polycystic ovary syndrome in teenagers. Exp Biol Med. 2004;229:369. doi: 10.1177/153537020422900504. [DOI] [PubMed] [Google Scholar]
- [74].Lucky AW, Biro FM, Huster GA, et al. Acne vulgaris in premenarchal girls. Arch Dermatol. 1994;130:308. doi: 10.1001/archderm.130.3.308. [DOI] [PubMed] [Google Scholar]
- [75].Apter D, Vihko R. Serum pregnenolone, progesterone, 17-hydroxyprogesterone, testosterone, and 5α-dihydrotestosterone during female puberty. J Clin Endocrinol Metab. 1977;45:1039. doi: 10.1210/jcem-45-5-1039. [DOI] [PubMed] [Google Scholar]
- [76].Cook S, Weitzman M, Auinger P, et al. Prevalence of a metabolic syndrome phenotype in adolescents: findings from the third National Health and Nutrition Examination Survey, 1988–1994. Arch Pediatr Adolesc Med. 2003;157:821. doi: 10.1001/archpedi.157.8.821. [DOI] [PubMed] [Google Scholar]
- [77].Azziz R, Carmina E, Sawaya ME. Idiopathic hirsutism. Endocr Rev. 2000;21:347. doi: 10.1210/edrv.21.4.0401. [DOI] [PubMed] [Google Scholar]
- [78].Baumann EE, Rosenfield RL. Polycystic ovary syndrome in adolescence. Endocrinologist. 2002;12:333. [Google Scholar]
- [79].Joffe H, Taylor AE, Hall JE. Polycystic ovarian syndrome–relationship to epilepsy and antiepileptic drug therapy. J Clin Endocrinol Metab. 2001;86:2946. doi: 10.1210/jcem.86.7.7788. [DOI] [PubMed] [Google Scholar]
- [80].Azziz R, Dewailly D, Owerbach D. Nonclassic adrenal hyperplasia: current concepts. J Clin Endocrinol Metab. 1994;78:810. doi: 10.1210/jcem.78.4.8157702. [DOI] [PubMed] [Google Scholar]
- [81].Azziz R, Sanchez LA, Knochenhauer ES, et al. Androgen excess in women: experience with over 1000 consecutive patients. J Clin Endocrinol Metab. 2004;89:453. doi: 10.1210/jc.2003-031122. [DOI] [PubMed] [Google Scholar]
- [82].Kaltsas GA, Isidori AM, Kola BP, et al. The value of the low-dose dexamethasone suppression test in the differential diagnosis of hyperandrogenism in women. J Clin Endocrinol Metab. 2003;88:2634. doi: 10.1210/jc.2002-020922. [DOI] [PubMed] [Google Scholar]
- [83].Futterweit W, Krieger DT. Pituitary tumors associated with hyperprolactinemia and polycystic ovary disease. Fertil Steril. 1979;31:608. doi: 10.1016/s0015-0282(16)44049-5. [DOI] [PubMed] [Google Scholar]
- [84].Glickman SP, Rosenfield RL, Bergenstal RM, et al. Multiple androgenic abnormalities, including elevated free testosterone, in hyperprolactinemic women. J Clin Endocrinol Metab. 1982;55:251. doi: 10.1210/jcem-55-2-251. [DOI] [PubMed] [Google Scholar]
- [85].Kaltsas GA, Korbonits M, Isidori AM, et al. How common are polycystic ovaries and the polycystic ovarian syndrome in women with Cushing’s syndrome? Clin Endocrinol (Oxf) 2000;53:493. doi: 10.1046/j.1365-2265.2000.01117.x. [DOI] [PubMed] [Google Scholar]
- [86].Kaltsas GA, Mukherjee JJ, Jenkins PJ, et al. Menstrual irregularity in women with acromegaly. J Clin Endocrinol Metab. 1999;84:2731. doi: 10.1210/jcem.84.8.5858. [DOI] [PubMed] [Google Scholar]
- [87].Rosenfield RL. Current concepts of polycystic ovary syndrome. Baillieres Clin Obstet Gynaecol. 1997;11:307. doi: 10.1016/s0950-3552(97)80039-9. [DOI] [PubMed] [Google Scholar]
- [88].Charmandari E, Kino T, Souvatzoglou E, et al. Natural glucocorticoid receptor mutants causing generalized glucocorticoid resistance: molecular genotype, genetic transmission, and clinical phenotype. J Clin Endocrinol Metab. 2004;89:1939. doi: 10.1210/jc.2003-030450. [DOI] [PubMed] [Google Scholar]
- [89].Draper N, Walker EA, Bujalska IJ, et al. Mutations in the genes encoding 11β-hydroxysteroid dehydrogenase type 1 and hexose-6-phosphate dehydrogenase interact to cause cortisone reductase deficiency. Nat Genet. 2003;34:434. doi: 10.1038/ng1214. [DOI] [PubMed] [Google Scholar]
- [90].Southam A, Richart E. The prognosis for adolescents with menstrual abnormalities. Am J Obstet Gynecol. 1966;94:637. doi: 10.1016/0002-9378(66)90398-x. [DOI] [PubMed] [Google Scholar]
- [91].Taieb J, Mathian B, Millot F, et al. Testosterone measured by 10 immunoassays and by isotope-dilution gas chromatography-mass spectrometry in sera from 116 men, women, and children. Clin Chem. 2003;49:1381. doi: 10.1373/49.8.1381. [DOI] [PubMed] [Google Scholar]
- [92].Miller KK, Rosner W, Lee H, et al. Measurement of free testosterone in normal women and women with androgen deficiency: comparison of methods. J Clin Endocrinol Metab. 2004;89:525. doi: 10.1210/jc.2003-030680. [DOI] [PubMed] [Google Scholar]
- [93].Goodman NF, Bledsoe MB, Futterweit W, et al. American Association of Clinical Endocrinologists medical guidelines for clinical practice for the diagnosis and treatment of hyperandrogenic disorders. Endocr Pract. 2001;7:120. [PubMed] [Google Scholar]
- [94].Rosenfield RL. Plasma free androgen patterns in hirsute women and their diagnostic implications. Am J Med. 1979;66:417. doi: 10.1016/0002-9343(79)91061-1. [DOI] [PubMed] [Google Scholar]
- [95].Moll G, Jr, Rosenfield R. Testosterone binding and free plasma androgen concentrations under physiologic conditions: characterization by flow dialysis technique. J Clin Endocrinol Metab. 1979;49:730. doi: 10.1210/jcem-49-5-730. [DOI] [PubMed] [Google Scholar]
- [96].Wild RA, Umstot ES, Andersen RN, et al. Androgen parameters and their correlation with body weight in one hundred thirty-eight women thought to have hyperandrogenism. Am J Obstet Gynecol. 1983;146:602. doi: 10.1016/0002-9378(83)90998-5. [DOI] [PubMed] [Google Scholar]
- [97].Nestler J. Sex hormone binding globulin: a marker for hyperinsulinemia and/or insulin resistance? J Clin Endocrinol Metab. 1993;76:273. doi: 10.1210/jcem.76.2.8432767. [DOI] [PubMed] [Google Scholar]
- [98].Rosenfield RL, Moll GW. The role of proteins in the distribution of plasma androgens and estradiol. In: Molinatti G, Martini L, James V, editors. Androgenization in women. Raven Press; New York: 1983. p. 25. [Google Scholar]
- [99].Rosner W. The functions of corticosteroid-binding globulin and sex hormone binding-globulin: recent advances. Endocr Rev. 1990;11:80. doi: 10.1210/edrv-11-1-80. [DOI] [PubMed] [Google Scholar]
- [100].Moll G, Jr, Rosenfield RL. Plasma-free testosterone in the diagnosis of adolescent polycystic ovary syndrome. J Pediatr. 1983;102:461. doi: 10.1016/s0022-3476(83)80678-7. [DOI] [PubMed] [Google Scholar]
- [101].American College of Obstetrics and Gynecology ACOG practice bulletin. Clinical management guidelines for obstetrician-gynecologists.Polycystic ovary syndrome. Obstet Gynecol. 2002;100:1389. doi: 10.1016/s0029-7844(02)02628-5. [DOI] [PubMed] [Google Scholar]
- [102].American College of Obstetrics and Gynecology ACOG technical bulletin. Evaluation and treatment of hirsute women. Int J Gynaecol Obstet. 1995;49:341. [PubMed] [Google Scholar]
- [103].Balen AH, Laven JS, Tan SL, et al. Ultrasound assessment of the polycystic ovary: international consensus definitions. Hum Reprod Update. 2003;9:505. doi: 10.1093/humupd/dmg044. [DOI] [PubMed] [Google Scholar]
- [104].Venturoli S, Porcu E, Fabbri R, et al. Longitudinal change of sonographic ovarian aspects and endocrine parameters in irregular cycles of adolescence. Pediatr Res. 1995;38:974. doi: 10.1203/00006450-199512000-00024. [DOI] [PubMed] [Google Scholar]
- [105].Adams J, Franks S, Polson DW, et al. Multi-follicular ovaries: clinical and endocrine features and response to pulsatile gonadotrophin-releasing hormone. Lancet. 1985;2:1375. doi: 10.1016/s0140-6736(85)92552-8. [DOI] [PubMed] [Google Scholar]
- [106].Rosenfield RL, Barnes RB, Ehrmann DA, et al. The value of the low-dose dexamethasone suppression test in the differential diagnosis of hyperandrogenism in women. J Clin Endocrinol Metab. 2003;88:6115. doi: 10.1210/jc.2003-031357. [DOI] [PubMed] [Google Scholar]
- [107].Joehrer K, Geley S, Strasser-Wozak E, et al. CYP11B1 mutations causing nonclassic adrenal hyperplasia due to 11β-hydroxylase deficiency. Hum Mol Genet. 1997;6:1829. doi: 10.1093/hmg/6.11.1829. [DOI] [PubMed] [Google Scholar]
- [108].Lutfallah C, Wang W, Mason JI, et al. Newly proposed hormonal criteria via genotypic proof for type II 3β-hydroxysteroid dehydrogenase deficiency. J Clin Endocrinol Metab. 2002;87:2611. doi: 10.1210/jcem.87.6.8615. [DOI] [PubMed] [Google Scholar]
- [109].Dierickx CC. Hair removal by lasers and intense pulsed light sources. Dermatol Clin. 2002;20:135. doi: 10.1016/s0733-8635(03)00052-4. [DOI] [PubMed] [Google Scholar]
- [110].Petitti DB. Clinical practice. Combination estrogen-progestin oral contraceptives. N Engl J Med. 2003;349:1443. doi: 10.1056/NEJMcp030751. [DOI] [PubMed] [Google Scholar]
- [111].Kuhl H. Comparative pharmacology of newer progestogens. Drugs. 1996;51:188. doi: 10.2165/00003495-199651020-00002. [DOI] [PubMed] [Google Scholar]
- [112].Akert J. A new generation of contraceptives. RN. 2003;66:54. [PubMed] [Google Scholar]
- [113].Kannisto S, Korppi M, Remes K, et al. Adrenal suppression, evaluated by a low dose adrenocorticotropin test, and growth in asthmatic children treated with inhaled steroids. J Clin Endocrinol Metab. 2000;85:652. doi: 10.1210/jcem.85.2.6336. [DOI] [PubMed] [Google Scholar]
- [114].Ibañez L, Potau N, Marcos MV, et al. Treatment of hirsutism, hyperandrogenism, oligomenorrhea, dyslipidemia, and hyperinsulinism in nonobese, adolescent girls: effect of flutamide. J Clin Endocrinol Metab. 2000;85:3251. doi: 10.1210/jcem.85.9.6814. [DOI] [PubMed] [Google Scholar]
- [115].De Leo V, Lanzetta D, D’Antona D, et al. Hormonal effects of flutamide in young women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1998;83:99. doi: 10.1210/jcem.83.1.4500. [DOI] [PubMed] [Google Scholar]
- [116].Farquhar C, Lee O, Toomath R, et al. Spironolactone versus placebo or in combination with steroids for hirsutism and/or acne. Cochrane Database Syst Rev. 2003;(4):CD000194. doi: 10.1002/14651858.CD000194. [DOI] [PubMed] [Google Scholar]
- [117].Olsen EA. Female-pattern hair loss. J Am Acad Dermatol. 2001;45:S70. doi: 10.1067/mjd.2001.117426. [DOI] [PubMed] [Google Scholar]
- [118].Vexiau P, Chaspoux C, Boudou P, et al. Effects of minoxidil 2% vs. cyproterone acetate treatment on female androgenetic alopecia: a controlled, 12-month randomized trial. Br J Dermatol. 2002;146:992. doi: 10.1046/j.1365-2133.2002.04798.x. [DOI] [PubMed] [Google Scholar]
- [119].Futterweit W, Deligdisch L. Histopathological effects of exogenously administered testosterone in 19 female to male transsexuals. J Clin Endocrinol Metab. 1986;62:16. doi: 10.1210/jcem-62-1-16. [DOI] [PubMed] [Google Scholar]
- [120].Vandenbroucke JP, Rosing J, Bloemenkamp KW, et al. Oral contraceptives and the risk of venous thrombosis. N Engl J Med. 2001;344:1527. doi: 10.1056/NEJM200105173442007. [DOI] [PubMed] [Google Scholar]
- [121].Lidegaard O. Oral contraceptives, pregnancy, and the risk of cerebral thromboembolism: The influence of diabetes, hypertension, migraine, and previous thrombotic disease. Br J Obstet Gynaecol. 1995;102:153. doi: 10.1111/j.1471-0528.1995.tb09070.x. [DOI] [PubMed] [Google Scholar]
- [122].Venn A, Bruinsma F, Werther G, et al. Oestrogen treatment to reduce the adult height of tall girls: long-term effects on fertility. Lancet. 2004;364:1513. doi: 10.1016/S0140-6736(04)17274-7. [DOI] [PubMed] [Google Scholar]
- [123].Huber-Buchholz M, Carey DG, Norman R. Restoration of reproductive potential by lifestyle modification in obese polycystic ovary syndrome: role of insulin sensitivity and luteinizing hormone. J Clin Endocrinol Metab. 1999;84:1470. doi: 10.1210/jcem.84.4.5596. [DOI] [PubMed] [Google Scholar]
- [124].Kiddy DS, Hamilton-Fairley D, Bush A, et al. Improvement in endocrine and ovarian function during dietary treatment of obese women with polycystic ovary syndrome. Clin Endocrinol. 1992;36:105. doi: 10.1111/j.1365-2265.1992.tb02909.x. [DOI] [PubMed] [Google Scholar]
- [125].Pasquali R, Antenucci D, Casimirri F, et al. Clinical and hormonal characteristics of obese amenorrheic hyperandrogenic women before and after weight loss. J Clin Endocrinol Metab. 1989;68:173. doi: 10.1210/jcem-68-1-173. [DOI] [PubMed] [Google Scholar]
- [126].Kirpichnikov D, McFarlane SI, Sowers JR. Metformin: an update. Ann Intern Med. 2002;137:25. doi: 10.7326/0003-4819-137-1-200207020-00009. [DOI] [PubMed] [Google Scholar]
- [127].Yki-Jarvinen H. Thiazolidinediones. N Engl J Med. 2004;351:1106. doi: 10.1056/NEJMra041001. [DOI] [PubMed] [Google Scholar]
- [128].Harborne L, Fleming R, Lyall H, et al. Descriptive review of the evidence for the use of metformin in polycystic ovary syndrome. Lancet. 2003;361:1894. doi: 10.1016/S0140-6736(03)13493-9. [DOI] [PubMed] [Google Scholar]
- [129].Lord JM, Flight IH, Norman RJ. Insulin-sensitising drugs (metformin, troglitazone, rosiglitazone, pioglitazone, D-chiro-inositol) for polycystic ovary syndrome. Cochrane Database Syst Rev. 2003;3:CD003053. doi: 10.1002/14651858.CD003053. [DOI] [PubMed] [Google Scholar]
- [130].Arlt W, Auchus RJ, Miller WL. Thiazolidinediones but not metformin directly inhibit the steroidogenic enzymes P450c17 and 3β-hydroxysteroid dehydrogenase. J Biol Chem. 2001;276:16767. doi: 10.1074/jbc.M100040200. [DOI] [PubMed] [Google Scholar]
- [131].Mansfield R, Galea R, Brincat M, et al. Metformin has direct effects on human ovarian steroidogenesis. Fertil Steril. 2003;79:956. doi: 10.1016/s0015-0282(02)04925-7. [DOI] [PubMed] [Google Scholar]
- [132].Baillargeon JP, Jakubowicz DJ, Iuorno MJ, et al. Effects of metformin and rosiglitazone, alone and in combination, in nonobese women with polycystic ovary syndrome and normal indices of insulin sensitivity. Fertil Steril. 2004;82:893. doi: 10.1016/j.fertnstert.2004.02.127. [DOI] [PubMed] [Google Scholar]
- [133].Ibanez L, Valls C, Marcos MV, et al. Insulin sensitization for girls with precocious pubarche and with risk for polycystic ovary syndrome: effects of prepubertal initiation and postpubertal discontinuation of metformin treatment. J Clin Endocrinol Metab. 2004;89:4331. doi: 10.1210/jc.2004-0463. [DOI] [PubMed] [Google Scholar]
- [134].Ehrmann DA. Relation of functional ovarian hyperandrogenism to noninsulin-dependent diabetes mellitus. Baillières Clin Obstet Gynaecol. 1997;11:335. doi: 10.1016/s0950-3552(97)80040-5. [DOI] [PubMed] [Google Scholar]
- [135].Brettenthaler N, De Geyter C, Huber PR, et al. Effect of the insulin sensitizer pioglitazone on insulin resistance, hyperandrogenism, and ovulatory dysfunction in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2004;89:3835. doi: 10.1210/jc.2003-031737. [DOI] [PubMed] [Google Scholar]