

Abstract
The innate immune system plays an important role in cerebral health and disease. In recent years the role of innate immune regulation by toll-like receptors in the brain has been highlighted. In this paper the expression of toll-like receptors and endogenous toll-like receptor ligands in the brain and their role in cerebral ischemia will be discussed. Further, the ability of systemic toll-like receptor ligands to induce cerebral inflammation will be reviewed. Finally, the capacity of toll-like receptors to both increase (sensitization) and decrease (preconditioning/tolerance) the vulnerability of the brain to damage will be disclosed. Studies investigating the role of toll-like receptors in the developing brain will be emphasized.
1. Introduction
Toll-like receptors (TLRs) were first discovered as the product of the Drosophila gene, Toll. It was shown by Dr Nüsslein-Volhard and colleagues that the product of Toll in the mother controls the establishment of the dorsal-ventral pattern of the Drosophila embryo [1]. However, 10 years later it was discovered that Toll signaling was also involved in the antifungal defence in fruit flies and thereby a link between Toll and immunological functions in Drosophila was established [2].
The first identified and most extensively studied TLR in mammals is TLR4, which is the receptor that mediates effects by lipopolysaccharide (LPS), the endotoxin secreted by Gram-negative bacteria. It has long been known that the C3H/HeJ strain of mice is resistant to LPS [3, 4]. It was later found that another mouse strain (C57BL/10ScCr) also was resistant to LPS [5]. Mutations in both of these strains were associated with a defect in the Lps locus. In 1998 the Lps locus was identified to encode the TLR4 receptor [6] and mice with mutations in the Tlr4 gene were shown to have defective LPS signaling [7]. Since then 13 TLRs have been identified in mouse and human, each reacting to a specific set of signature molecules on different microbes [8]. However, not all TLRs are expressed in both human and mouse. TLR10 exists in humans but not in mouse and the ligand is currently unknown [9]. TLR11, TLR12, and TLR13 are present in mouse but not in human. TLRs are now among the most well-characterized pattern recognition receptors (PRRs) and have been shown to detect a variety of pathogen-associated molecular patterns (PAMPs) as well as endogenous danger signals, so-called damage-associated molecular patterns (DAMPs) [10].
2. Toll-Like Receptor Signaling
TLRs are transmembrane receptors containing an extracellular domain, a trans-membrane domain, and an intracellular domain. Upon stimulation the receptors form homodimers or heterodimers. For example, TLR2 can form heterodimer complexes with TLR1 or TLR6 and specific ligands have been identified that can differentiate functions between these TLR1/2 and TLR2/6 complexes. Some receptors are also dependent on non-TLR comolecules. For example, TLR4 activation by LPS requires the comolecules MD2 and cluster of differentiation (CD) 14 [11]. Also TLR2 use accessory molecules such as CD36 [12] and Dectin-1 [13].
TLR1, 2, 4, and 5 are localized to the outer cell surface membrane, while TLR3, TLR7, TLR8, and TLR9 are intracellular receptors positioned on endosomes or lysosomes. However, it has become clear over the last few years that extensive intracellular trafficking of the TLRs occurs inside the cell. The endosomal receptors travel from the endoplasmatic reticulum aided by the “helper” protein Unc-93 homolog B1 [14], to the Golgi apparatus and then become activated by proteolytic cleavage by intracellular proteases such as cathepsins in the endosome [15–18]. The compartmentalization of the receptors is believed to be an important mechanism for the organism to distinguish “self” from “nonself” [19].
Downstream of the TLRs different toll/interleukin-1 receptor (TIR) domain containing adaptor molecules links the recognition of microbes to cellular immune responses. The family of TLR-linked adapter proteins is growing and to date includes myeloid differentiation factor-88 (MyD88), MyD88 adaptor-like protein (MAL/TIRAP), TIR domain containing adaptor protein inducing interferon beta (TRIF), TRIF-related adaptor molecule (TRAM), and sterile α- and HEAT/armadillo-motif-containing protein (SARM). MyD88 is a common adapter for all TLRs except TLR3 [20]. The TRIF adaptor protein is the sole adaptor protein for TLR3 [21] but is also coupled to TLR4 signaling via the adaptor TRAM [22]. MAL/TIRAP is a bridging adaptor for TLR2 and TLR4 [23]. The fifth adaptor protein, SARM, has been suggested to negatively regulate TRIF [24]. However, with respect to the mouse brain, SARM has been mainly localized to mitochondria in neurons and has been suggested to contribute to cell death [25], which was recently further confirmed [26].
3. Toll-Like Receptor Expression in the Brain
Both mRNA and protein expression of TLRs have been identified in the brain. The gene expression for TLR2 and TLR4 was first characterized in the adult rodent brain [27]. Using in situ hybridization, it was shown that the mRNA expression for TLR2 and TLR4, under basal conditions, was mainly found in the leptomeninges, choroid plexus, and circumventricular organs (CVOs). These findings were later extended to show an even more widespread expression of TLR4 transcript in the brain, almost exclusively in nonneuronal cells [28]. However, immunoreactivity for TLR2 and TLR4 has later been identified in neural progenitor cells in the hippocampus in adult rodents [29]. Widespread expression of TLRs has also been detected in human cerebral tissue [30]. In this study, microglia, astrocytes, and oligodendrocytes were cultured from human brain and it was found that primary microglia express mRNA encoding for most TLR family members while astrocytes and oligodendrocytes primarily express TLR2 and TLR3. By examining brain and spinal cord tissue sections from control and multiple sclerosis brains, it was observed that TLR3 and TLR4 expression was enhanced in inflamed central nervous system (CNS) tissues.
TLRs have been studied to a lesser extent in the immature brain; however, recent data suggest that several of the TLRs play a role during brain development. We were the first to demonstrate TLR4 and CD14 gene transcripts in the newborn rat brain [31]. Using reverse transcription-polymerase chain reaction (RT-PCR) we found that while CD14 expression in the brain was strongly induced by systemic LPS, the gene transcript for TLR4 decreased. Later studies have emphasized a role for TLR3 and TLR8 in embryonic brain development. TLR8 immunopositive cells were identified as neurons and stimulation of TLR8 of cortical neurons in vitro resulted in inhibition of neurite outgrowth and induced apoptosis [32]. Treatment of cultured embryonic cortical neurospheres with the TLR3 ligand polyinosinic:polycytidylic acid (poly I:C) reduced cell proliferation in a TLR3-dependent manner [33]. In contrast to TLR3 and TLR8, expression of TLR2 is low before birth and increases during the first 2 weeks of life after birth [33]. We showed that mRNA for TLR1–9 is expressed in the neonatal mouse brain and particularly TLR1 and TLR2 transcripts are regulated by hypoxia-ischemia [34]. Further we found that TLR1 protein is expressed in neurons and that TLR2 protein expression is present in astrocytes in the cerebral white matter and in a specific neuronal population in the paraventricular nucleus (PVN) in neonatal mice. In support, the mRNA for TLR1–9 was recently described during mouse development. In this study TLR7 and TLR9 were suggested to be the most developmentally regulated TLRs and the protein expression of TLR7 and TLR9 was identified in neurons in cortex and hippocampus during mouse brain development [35].
Taken together, current evidence strongly suggests the presence of TLR gene regulation in cerebral tissue, both during development and after various provocations to the developing CNS. However, the protein expression of TLRs in the brain described in different studies is variable and there appears to be no consensus of which TLRs are expressed and in which cell types. Our own experience is that many of the commercially available antibodies towards TLRs are nonspecific and need further development.
4. Toll-Like Receptors in Ischemic Brain Damage
As indicated above most of the TLRs are expressed in the brain and are regulated during embryonic and neonatal life, suggesting that they may play a role in brain development. However, it is also becoming increasingly clear that brain TLRs may be important regulators of cerebral ischemia. Tang and colleagues demonstrated that both TLR2 and TLR4 expression is upregulated in cerebral cortical neurons in response to ischemia/reperfusion injury [36]. Similarly, TLR2 is increased in the brain after both permanent and transient focal ischemia [37, 38].
Further evidence to indicate that TLRs may contribute to brain ischemia comes from studies demonstrating neuroprotection in TLR-deficient mice. Both the degree of brain damage and neurological deficits observed following permanent and transient middle cerebral artery occlusion (MCAO) are reduced in mice deficient in TLR2 or TLR4 [36, 38–41]. This was later confirmed also in other models of brain ischemia [42, 43]. However, there is one conflicting study reporting that in a focal cerebral ischemia/reperfusion model TLR2 knockout (KO) mice had higher mortality, decreased neurological function, and increased brain infarct size, while TLR4 KO mice were protected from these detrimental processes following ischemia [44]. Interestingly, recent clinical studies have shown that TLR2, TLR4, TLR7, and TLR8 expression in blood was associated with poor outcome following ischemic stroke [45, 46].
In contrast to TLR2 and TLR4, neither TLR3 nor TLR9 deficiency, appears to provide protection from ischemic insults [42]. Neither is there an apparent neuroprotective effect by disruption of the TLR downstream adaptor proteins MyD88 or TRIF against cerebral ischemia [47]. It is intriguing that despite that there are several studies demonstrating neuroprotection in TLR2 and TLR4-deficient animals following cerebral ischemia, KO of the gene for the downstream adaptor molecule, MyD88, does not provide protection. These results open up for the possibility that these receptors might be able to use different downstream adaptors during TLR signaling in the brain. It has for example been suggested that activation of TLR4 results in phosphatidylinositol-3-kinase-(PI3 K-) MyD88 complex formation, and that PI3 K activity selectively leads to cytokine induction downstream of TLR4 [48].
We have studied the role of TLR1, TLR2, TLR4, TRIF, and MyD88 in hypoxic-ischemic brain injury in neonatal mice. In contrast to the adult, we find no neuroprotection in TLR4 deficient mice [49]. Neither have we observed any protection in TLR1 KO [34], MyD88 KO [49], or TRIF KO (Stridh et al., unpublished) animals. However, we did find that mice that were subjected to hypoxia-ischemia on postnatal day 9 demonstrate increased mRNA expression of TLR2 at 6 h and 24 h after ischemia. Immunohistochemical staining of the brains showed that TLR2 was expressed in astrocytes and in a specific population of neurons in the PVN in the hypothalamus. Furthermore, TLR2 KO mice developed smaller infarcts compared to wild-type mice after hypoxia-ischemia [34], indicating a role for TLR2 in the immature ischemic brain.
5. Endogenous Toll-Like Receptor Ligands
The presence of TLRs in the brain and their regulation following cerebral ischemia suggest that TLRs can be activated by endogenous TLR ligands. Tissue damage causing cell death and tissue remodeling has been shown to generate endogenous danger molecules, which can act as endogenous TLR ligands [50]. Such endogenous danger molecules, so called DAMPs, have been noted after brain ischemia and include heat shock proteins (HSPs), hyaluronan, nucleic acids, and high mobility group box 1 (HMGB1) [51, 52]. These will be described in more detail below.
5.1. Heat Shock Proteins
HSPs are a family of molecular chaperones, which support the correct folding of proteins. HSPs are normally localized in the cytoplasm but are released from necrotic cells. It has been reported that necrotic, but not apoptotic, cell death leads to the release of chaperones such as HSP70, HSP90, calreticulin, and Gp96 [53]. Once outside the cell, extracellular HSPs can have immune-stimulatory properties and have been shown to act as endogenous ligands for TLRs [54–56] and there appears to exist a TLR4-dependent link between the release of HSP60 from damaged cells in the brain and microglia activation [57].
5.2. Hyaluronan
Hyaluronan (or hyaluronic acid) is a major component of the extracellular matrix. It is widely distributed in many tissues in the body. Under physiological conditions, hyaluronan exists as a high molecular weight form; however, hyaluronan is actively broken down into lower molecular weight fragments following tissue damage. It has become clear that the function of hyaluronan depends on in which form it exists and low molecular fragments are often seen in pathological situations. The fragmentation of hyaluronan in disease may be a consequence of dysregulation of hyaluronan degradation enzymes [58] or release of reactive oxygen species during tissue damage [59, 60].
The low molecular weight hyaluronan is known to interact with innate immune receptors. Hyaluronan stimulation of dendritic cells is dependent on TLR4 [61] and chemokine and cytokine production in macrophages is abolished in a MyD88- or TLR2/TLR4-dependent manner [62, 63]. Interestingly, although both hyaluronan and LPS activate TLR4, they act via different coreceptors. It was discovered that a unique complex of TLR4, MD2, and CD44 recognizes hyaluronan in noninfectious inflammation, which is different from the TLR4, MD2, and CD14 complex that recognizes LPS during infection [64]. It was further noted that hyaluronan and LPS induce different sets of gene expression, with hyaluronan generating a pattern of gene induction that mimics the response seen after sterile injury with an increase in molecules such as transforming growth factor (TGF) beta 2 and matrix metalloproteinase (MMP) 13. Such genes were not regulated following LPS, suggesting distinct differences between infectious versus noninfectious stimulation of TLR4.
The effect of high and low molecular weight hyaluronan in brain damage is unclear. High molecular weight hyaluronan accumulates in astrocytes in demyelinated lesions from individuals with multiple sclerosis and in mice with experimental autoimmune encephalomyelitis [65]. It was also shown that the addition of high molecular weight hyaluronan to oligodendrocyte progenitor cultures reversibly inhibits progenitor cell maturation, whereas degrading hyaluronan in astrocyte-oligodendrocyte progenitor cocultures promotes oligodendrocyte maturation. In contrast, in the human brain following cerebral ischemia the low molecular weight hyaluronan form is found [66] and a recent report shows that TLR2 activation by low molecular weight hyaluronan inhibits neurosphere formation in vitro [67]. These reports suggest that high and low molecular weight hyaluronan may have differential effects depending on cell type and developmental stage.
5.3. High Mobility Group Box 1
HMGB1 is released extracellularly during acute inflammatory responses and when membrane integrity is lost in permeabilized or necrotic cells [68, 69]. Thus HMGB1 release is thought to be an important mechanism whereby necrotic cells can trigger inflammation [70]. Park and colleagues reported that stimulation of neutrophils, monocytes, or macrophages by HMGB1 required both TLR2 and TLR4 resulting in increased nuclear translocation of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and enhanced expression of proinflammatory cytokines [71]. However, in vitro studies using primary cells and cell lines demonstrated that the usage of TLR2 and TLR4 in HMGB1 signaling is complex and context dependent [72]. Neutralizing antibodies against TLR4, but not TLR2, dose-dependently attenuated HMGB1-induced interleukin (IL) 8 release in human blood and tumor necrosis factor alpha (TNF-α) release in primary macrophages. In contrast, in human embryonic kidney 293 cells transfected with TLR2 or TLR4, HMGB1 effectively induced IL-8 release only from TLR2 overexpressing cells.
There is evidence to suggest that HMGB1 also acts as a DAMP in the brain following ischemia. Treatment with neutralizing anti-HMGB1 monoclonal antibody ameliorated brain infarction and reduced deficits in locomotor function following 2-hour occlusion of the middle cerebral artery in rats [73]. Anti-HMGB1 treatment inhibited the increased permeability of the blood-brain barrier, activation of microglia, the expression of TNF-α, and inducible nitric oxide synthase (iNOS) and suppressed the activity of MMP9. Ischemia-induced release of HMGB1 in brain has later been confirmed by several laboratories [74–76] and appears to be dependent on TLR4 [77]. Recently, another DAMP, peroxiredoxin, was identified in the ischemic brain [78]. Neutralization of peroxiredoxin with antibodies suppressed inflammatory cytokine expression and infarct volume following MCAO. These effects were more pronounced than those seen with inhibition of HMGB1, but interestingly, there were synergistic effects of peroxiredoxin and HMGB1.
5.4. Nucleic Acids
The intracellular TLRs (TLR3, TLR7, TLR8, and TLR9) are located in endolysosomes where they recognize microbial nucleic acids and initiate innate and adaptive immune responses [79, 80]. The subcellular compartmentalization of TLRs is believed to be an important mechanism to prevent autoimmune reactions and block responses to self-nucleic acids [81]. “Self-” nucleic acids, but not viral nuclei acids, are rapidly degraded before reaching endolysosomes, therefore by locating the receptors in the endosomes recognition of “self” is averted [19, 82]. However, both TLR7 and TLR9 are able to respond to endogenous RNA and DNA if they are expressed on the cell surface [83]. TLR3 recognizes viral double-stranded RNA [84], but endogenous mRNA released from necrotic cells has also been shown to activate TLR3 [85, 86]. Therefore, there are situations where possibly TLR3, TLR7, and TLR9 can be activated by endogenous molecules such as “self” RNA and DNA.
Several DAMPs have been recognized to originate from mitochondria following cellular stress [87]. Mitochondria share many features with microbes due to their bacterial origin, suggesting that release of mitochondrial content may act as a TLR activator. As such, mitochondrial DNA (mtDNA) released from dying cells has been identified as a DAMP [88]. mtDNA contains hypomethylated CpG motifs that are similar to bacterial CpG DNA and hence are potent stimulators of TLR9 [89]. mtDNA was recently shown to cause inflammation and heart failure [90]. Interestingly, cerebral hypoxia-ischemia rapidly increases mitochondrial biogenesis in neonatal rats [91]. In this study brain mtDNA content increased 6–24 hours after hypoxia-ischemia, raising the possibility that mtDNA may also be a stimulator of innate immunity in the brain.
6. Effects of Systemic Toll-Like Receptor Ligands on Brain Inflammation
As discussed above, TLRs are expressed in the brain and endogenous ligands are released following ischemic brain damage, suggesting that innate immune receptors may have a direct effect on brain damage. However, innate immune responses in the circulation also clearly play important roles in generating brain inflammation [28, 92]. Systemically applied LPS initiates a wide range of inflammatory responses in the brain, including increase in cytokines, chemokines, prostaglandins, and nitric oxide. The exposure paradigm seems to play a role as it was shown that in comparison to measurements taken from a time course after a single injection of LPS, repeated injections produced significantly higher cytokine levels in the brain [93].
LPS can also affect the integrity of the blood-brain barrier, which could alter interactions between circulating and central mediators. For example, altered immunostaining for junctional proteins b-catenin, ZO-1, and claudin-5; enlargement of intercellular spaces and redistribution of junctional proteins were found in brain endothelial cells in vitro after LPS exposure [94]. In the immature brain, Dr. Saunder's group has described the presence of plasma proteins within the white matter tracts of the brain after repeated intraperitoneal injections of LPS (0.2 mg/kg) given at postnatal day 0, 2, 4, 6, and 8. Interestingly, the permeability of the blood-brain barrier to 14C-sucrose and 14C-inulin was still apparent in adult animals that had received serial LPS injections during development. Such changes were not observed after only a single neonatal LPS injection [95]. In later studies, using marsupials, they showed that anti-inflammatory treatment with minocycline restored blood-brain barrier integrity following prolonged LPS-induced inflammation but did not improve LPS-induced damage to white matter. The authors conclude that long-term changes in blood-brain barrier permeability occur only after a prolonged period of inflammation during development; however, damage to white matter can result from even a short-lasting breakdown of the barrier [96].
7. Transfer of Signal from Circulating Toll-Like Receptors to the Brain
There are several potential mechanisms that can explain how TLR ligands induce brain inflammation. In the sections below the following possible scenarios will be discussed: TLR ligand transport across the blood-brain barrier, interaction with brain endothelial cells, interaction with circumventricular organs or epithelial cells in the choroid plexus, and immune transfer via vagal afferents (Figures 1 and 2).
Figure 1.
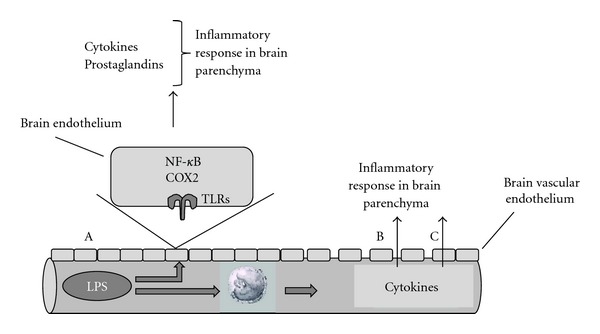
TLR ligand transport across the blood-brain barrier. (A) Circulating LPS binds to endothelial cells in the brain vasculature and transmits inflammatory signals to the brain via COX-2- and NF-κB-associated pathways. Alternatively, LPS in the circulation induces release of cytokines from circulating blood cells, which can either affect the integrity of the blood-brain barrier (B) or be transported across the intact blood-brain barrier (C) to induce further inflammation in the brain parenchyma.
Figure 2.
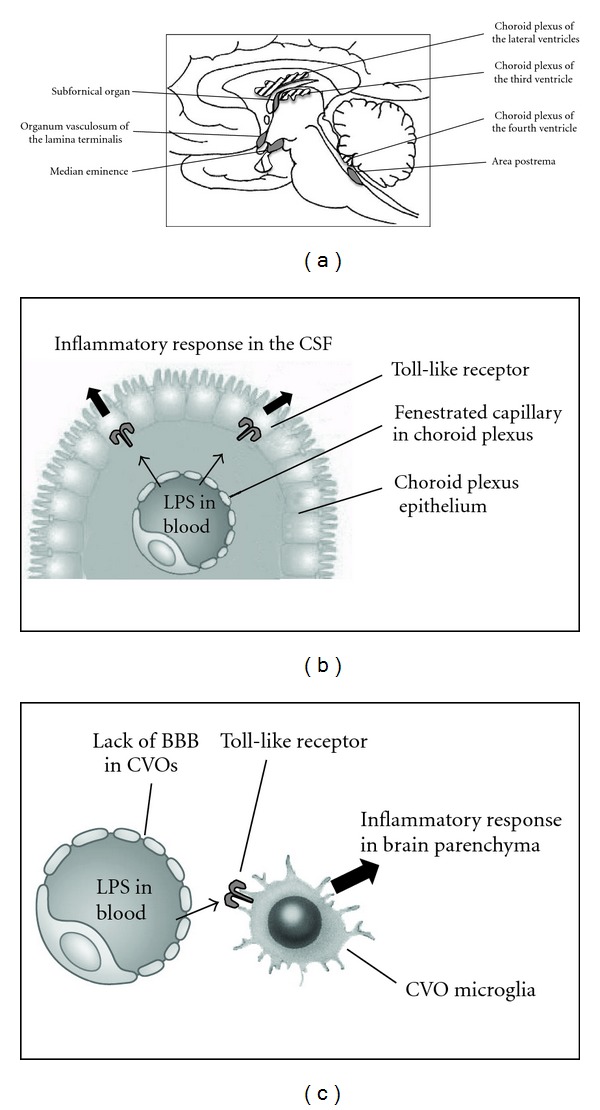
Interaction of systemic TLR ligands with circumventricular organs and choroid plexus. (a) Schematic illustrating the anatomical location of choroid plexus and circumventricular organs (CVO) in the rat brain. (b) Depicted image of suggested transfer of inflammatory stimuli from circulating LPS via the epithelial cells of the choroid plexus and induction of inflammatory responses in the cerebrospinal fluid (CSF) of the brain. (c) The circumventricular organs of the brain lack a fully developed blood-brain barrier (BBB). This allows LPS to instigate interactions with inflammatory cells in these brain regions, which initiates an inflammatory response in brain parenchyma.
7.1. TLR Ligand Transport across the Blood-Brain Barrier
It has long been debated whether circulating LPS, or other TLR ligands, cross the blood-brain barrier to induce brain inflammation. Several recent reports indicate that this is not the case. Iodine-labeled LPS is rapidly cleared (half-life < 30 minutes) from the blood, but there is substantial uptake of LPS by liver, spleen, and lung during this initial period following intravenous injection into rabbits [97]. Tissue-bound LPS was found to be concentrated in phagocytic vacuoles of hepatic Kupffer cells, splenic macrophages, and leukocytes. LPS remaining in plasma beyond 30 minutes was converted to a low-density form, which disappeared from the blood with a half-life of 12 hours. Intraperitoneally injected LPS reaches the circulation within 15 minutes of administration and could potentially also cross the blood-brain barrier [98]. However, after systemic LPS, ions associated with LPS lipids were tracked and found to be bound to brain endothelium but were not found inside the brain, suggesting that LPS does not cross the blood-brain barrier [99]. Similarly, in a carefully conducted study using iodine-labeled LPS, Banks and Robinson recently demonstrated that while intravenously administered LPS binds to endothelial cells of the blood-brain barrier, only minute (<0.025%) levels of LPS were detected inside the brain [100]. This is probably true also for other TLR ligands. A single peritoneal injection of Poly I:C (12 mg/kg) to 8-week-old mice resulted in strong mRNA regulation of a number of chemokines in the brain [101]. Interestingly, blood plasma collected 3 hours after Poly I:C and injected into naive animals also induced an inflammatory response in the brain, suggesting that the presence of Poly I:C itself was not necessary. Further, tracking Poly I:C after injection showed that only minute levels were detected in the blood after intraperitoneal injection. Thus, rather than crossing the blood-brain barrier and directly interact with the brain parenchyma, TLR ligands, such as LPS and Poly I:C, are more likely to induce brain inflammation via indirect mechanisms as outlined below.
7.2. Interaction of Systemic TLR Ligands with Brain Endothelial Cells
The blood-brain barrier, which is formed by the tight junctions of brain capillary endothelial cells, expresses various transporters to regulate exchange of compounds between the brain and the circulating blood [102]. Brain endothelial cells are polarized cells with a luminal (blood-facing) and abluminal (brain-facing) side of the cell membrane, allowing for substances applied to one side to affect release of molecules and regulate transport mechanisms on the other side. Although, TLR ligands, such as LPS and Poly I:C, do not seem to cross the blood-brain barrier, there is ample evidence to suggest that they interact with brain endothelial cells of the barrier and thereby initiate inflammatory responses inside the brain.
The mRNA for the central immune regulator, I-kappa-B-alpha (IκBα), is dramatically increased in the brain after intraperitoneal LPS injection (2.5 mg/kg) [103]. The transcript for IκBα was first detected in cells lining the blood side of the blood-brain barrier and then progressed to cells inside brain. The authors suggested that cells of the blood-brain barrier synthesize immune signal molecules to activate cells inside the CNS in response to peripheral LPS. In support, brain endothelial cells isolated from rhesus monkeys that are exposed in vitro to either an immune stimulus (IL-1β or LPS) or an oxidative challenge (hypoxia) release IL-6 [104]. There is also evidence to show that LPS applied to the abluminal side of brain endothelial cells evokes secretion of IL-6 on the luminal side [105], suggesting a bidirectional release of immune-stimulating mediators across the blood-brain barrier. Furthermore, both IL-1β and IL-6 are themselves able to be transported across the blood-brain barrier [106, 107]. Saturable transport systems have also been identified for several other proinflammatory cytokines, including TNF-α, leukemia inhibitory factor (LIF), several interleukins, and interferons [108, 109]. However, it is generally believed that the amount of cytokines that are transported across the blood-brain barrier is rather small and play a minor role in affecting brain inflammation in disease.
Prostaglandin production in cerebral vascular cells has been suggested to be another important interface between peripheral and central inflammation. There is no constitutive expression of cyclo-oxygenase 2 (COX2) mRNA in cerebral blood vessels. However, intravenous injection of LPS induces strong induction of COX2 in blood vessels and the leptomeninges over the entire brain, with the signal maximally enhanced by 50 to 80% over the basal level 1 hour after LPS injection [110]. The route of LPS administration, intraperitoneal or intravenous does not appear to affect the expression [111]. It was later found that it is the interaction between COX2 and microsomal prostaglandin E synthase in brain endothelial cells that is responsible for inducing prostaglandin in the brain [112].
Brain endothelial cells express TLR2, TLR4, and CD14 mRNAs [99] and later studies have also shown expression for TLR3 and TLR6 on rat and human cerebral endothelial cells [113]. Convincing data, using chimeric mice, have shown that it is the blood-brain barrier endothelial cells, rather than perivascular microglia, that are the main target of circulating inflammatory mediators to activate the brain response. While the systemic release of acute phase cytokines was dependent on TLR4 on hematopoietic cells, the presence of TLR4 on CNS-resident cells (i.e., nonhematopoietic cells like endothelial cells) was required for sustained inflammation in the brain after systemic LPS administration [28]. Later, these findings were confirmed and extended to show that systemic IL-1β caused a robust transcriptional activation of genes involved in prostaglandin E2 production by vascular cells of the brain. Upregulation of these genes was dependent on functional MyD88 signaling in the endothelium, as MyD88-deficient mice that received bone marrow stem cells from wild-type animals (including functional perivascular microglia) exhibited no response to systemic IL-1β administration [114].
7.3. Interaction of Systemic TLR Ligands with Circumventricular Organs and Choroid Plexus
The CVOs and choroid plexus of the brain have been indicated as important links between systemic inflammation and cerebral innate immune responses. Neurons within the CVOs are activated by intravenous LPS injection [115]. In an early phase following systemic administration of LPS, production of TNF-α mRNA was observed in perivascular cells and neurons in CVOs, including the vascular organ of the lamina terminalis, median eminence, and area postrema, as well as along the ventral surface of the medulla in the mouse brain [116]. Later TNF-α hybridization was observed over neurons in the hypothalamus and the nucleus of the solitary tract. In support, it has been shown that LPS causes a fast transient rise in intracellular calcium concentrations in the microglial cells in a primary culture of the rat area postrema, with limited responses of neurons, astrocytes, and oligodendrocytes [117]. Similar to TNF-α, IL-1β production was demonstrated in organum vasculosum laminae terminalis and some cells around the blood vessels in the parenchyma 1 hour after intravenous LPS (4 μg/kg) [118]. Also mRNA expression of IL-6 was detected in the CVOs and choroid plexus following intraperitoneal LPS injection [119]. It was subsequently shown that also TLR4 is present in CVOs and choroid plexus and mediates signals from the periphery by intracellular signaling and then rapid transcription of proinflammatory cytokines, first within these organs and thereafter throughout the brain parenchyma [27].
A recent microarray analysis revealed that the mouse choroid plexus displays an acute-phase response after an inflammatory stimulus induced in the periphery by LPS [120]. Genes implicated in immune-mediated cascades and in extracellular matrix remodeling were upregulated, whereas genes that code for protein that participate in maintenance of the barrier function were downregulated. We have evidence to indicate that systemic LPS induces a downregulation of endogenous antioxidant systems in the choroid plexus in neonatal mice (D'Angelo et al., publication under revision). Thus an important mechanism for transducing peripheral inflammation into the brain appears to be by LPS interacting with TLRs in CVOs and choroid plexus.
7.4. Vagal Stimulation by Systemic LPS and Cytokines
Neural afferents, via the vagus nerve, transmit immune messages from the periphery to the brain and contribute to the hyperalgesia, fever, anorexia, taste aversions, increased levels of plasma corticosteroid, and brain norepinephrine changes produced by intraperitoneal injections of IL-1β, TNF-α, and LPS [121, 122]. These effects seem to be specific to the intraperitoneal route of administration of cytokines because vagotomized animals are still able to respond to IL-1β injected intravenously, subcutaneously, and into the lateral ventricle of the brain, but not intraperitoneally [123, 124]. Presently, it is not known whether central cytokine induction via peripheral nerves has any impact on brain damage.
In summary, circulating mediators are unlikely to move in sufficient concentrations across barriers of the central nervous system to directly induce major inflammatory processes in the brain. Instead, evidence points towards that peripheral TLR ligands or peripheral cytokines interact with receptors on brain vascular endothelial cells, epithelial cells of the choroid plexus, or cells in the CVOs. The subsequent release of prostaglandin E2 or cytokines into the adjacent brain parenchymal environment appears to be a fundamental step in the relay of blood-borne immune signals to the CNS.
8. Role of TLRs in Perinatal Brain Damage
More than 35 years ago, Gilles and Leviton presented the first evidence that LPS endotoxin can cause injury to the developing brain [125, 126]. They showed that a single peritoneal injection of Escherichia coli LPS resulted in brain lesions in kittens, neonatal monkeys, and rabbits. The authors further went on and studied infants who died with perinatal telencephalic white matter injury and found that this type of neuropathology was more common in infants who had bacteria isolated from blood at autopsy [127]. These original observations initiated a new field of research and have led to numerous studies over the last few decades into the relationship between inflammation, preterm birth, and perinatal brain damage. Epidemiological and clinical studies have convincingly shown a link between intrauterine infection, neonatal sepsis, and brain damage and the development of cerebral palsy or neurodevelopmental disabilities in children [128–132]. Thus inflammation, either before birth or in the neonate, is now recognized as an important contributing factor in what has been termed “encephalopathy of prematurity” [133].
8.1. Maternal Exposure to TLR Ligands
In order to better understand how infection/inflammation can affect the immature brain a number of studies have been performed where the mother, fetus, or newborn animals have been exposed to microbes or bacterial products that act as TLR ligands. Pregnant rabbits that survived inoculation with Escherichia coli developed extensive white matter damage [134, 135]. Animal models of maternal infection have also been developed in rodents. Cai and colleagues demonstrated that cytokines were induced in the rat fetus following maternal LPS administration and neonatal offspring displayed brain damage [136]. Later it was shown that offspring to LPS-treated mothers showed decreased staining for myelin and an increase in astrogliosis [137, 138]. LPS administration to pregnant mice results in extensive gene regulation in the fetal brain, including altered expression of proinflammatory and developmentally regulated genes [139]. A recent study demonstrates that offspring born to LPS-treated dams exhibit reduced social preference and exploration behaviors as juveniles and young adults. In this study, maternal LPS induced dysregulation of genes in the fetal brain belonging to specific functional categories, including increased mRNA expression of cellular stress and cell death genes and reduced expression of developmentally regulated and brain-specific genes, specifically those that regulate neuronal migration of γ-aminobutyric acid (GABA)-ergic interneurons [140].
Several studies have been performed to mimic viral infections during pregnancy by giving TLR3 ligands as models for developmentally induced psychiatric disorders [141, 142]. It is now well established that stimulation of the TLR3 receptor during embryonic or fetal development can adversely affect brain development in the offspring. However, the timing of the exposure determines the outcome to some extent [143]. Poly I:C-induced prenatal immune challenge on gestation day 9 but not gestation day 17 significantly impaired sensorimotor gating and reduced prefrontal dopamine D1 receptors in adulthood, whereas prenatal immune activation specifically in late gestation impaired working memory, potentiated the locomotor reaction to the N-Methyl-D-aspartate- (NMDA-) receptor antagonist dizocilpine, and reduced hippocampal NMDA-receptor subunit 1 expression. Pregnant rats given Poly I:C (10 mg/kg), repeatedly during late gestation (E14, E16, and E18), resulted in an increase in monocyte chemotactic protein-1 (MCP-1) in maternal blood 5 hours after injection [144]. In the offspring, the expression of the GluN1 subunits of the NMDA receptors was decreased, but without changes in GluN2A or GluN2B subunits, the postsynaptic density protein 95, or the NMDA receptor modulator EphA4. Also an increase was noted in presynaptic markers such as vesicle-associated membrane protein 1 and synaptobrevin. In contrast, there were no changes in cell proliferation as detected by proliferating cell nuclear antigen or doublecortin. Interestingly, neuropathological consequences of prenatal Poly I:C exposure are exacerbated in offspring with genetic predisposition to dopaminergic abnormalities induced by mutations in the nuclear receptor-related 1 protein [145]. These findings emphasize the importance of gene-environment interactions in these situations.
8.2. Fetal Exposure to TLR Ligands
Direct injection of LPS to fetal sheep results in white matter damage, both in the forebrain [146, 147] and in the cerebellum [148], which is very similar to that observed in preterm infants. Recently, we observed reductions in both white matter volume (~21%) and cortical tissue (~18%) when brains were examined 10 days after LPS exposure in fetal sheep [149]. These neuropathological changes were also confirmed by ex vivo magnetic resonance imaging analysis [150]. Importantly, we found that there was loss of the normal maturational increase in cortical electroencephalography amplitude, which correlated with reduced cortical volumes. In the same animal model, we used a global metabolomics approach to examine plasma metabolites differentially regulated by intrauterine inflammation [151]. We detected both acute and delayed effects of LPS on fetal metabolism, with a long-term down-regulation of fetal energy metabolism. The characteristics of the metabolite response to LPS were strongly correlated with white and grey matter volumes at 10 days recovery, suggesting the potential to use metabolomics analysis as biomarker for injury and for identification of therapeutic targets.
8.3. Neonatal TLR Exposure
We have studied the effect of inflammation during a developmental stage in the mouse that corresponds to the sensitive period of myelination by injection of either LPS or Pam3CSK4 (TLR1/2 ligand) to mice from postnatal day 3 to postnatal day 11. LPS decreased the serum insulin-like growth factor 1 level on postnatal day 12 and quantification of immunohistochemical staining for axonal, myelin, and oligodendrocyte markers revealed impaired myelination in subcortical white matter. In addition, brain gray matter volume decreased and spleen and liver weight increased at postnatal day 12 [152]. Similarly, mice injected with Pam3CSK4 (5 mg/kg) displayed decreased volume of cerebral gray matter, white matter in the forebrain, and cerebellar molecular layer at PND12 [153]. Such effects were not observed in Pam3CSK4-treated TLR2-deficient mice, indicating a specific TLR2 effect. Systemic Pam3CSK4 injection significantly increased the levels of IL-1β, IL-6, chemokine (C-X-C motif) ligand 1, and MCP-1 protein in the brain. The neuropathological changes appear to be transient as there were no long-term effects on memory function, assessed by the trace fear conditioning test at postnatal day 50, nor on the volume of gray or white matter.
9. Cerebral Consequences of TLR Ligand Interactions with Other Stimuli
It has long been known that endotoxin can induce a state of tolerance to further infections[154, 155] and numerous studies have shown that different TLR agonists alter inflammatory responses to one another [156, 157]. On the other hand, synergestic induction of TNF production by simultaneous activation of TLR2 and TLR4 has been shown [158, 159] and stimulation of mouse macrophages with both Poly I:C (TLR3 ligand) and CpG DNA (TLR9 ligand) induced more-than-additive levels of TNF, IL-6, and IL-12 p40 [160]. Therefore, it is clear that TLR ligands can induce both priming, synergistic effects on cytokine production as well as tolerance phenomena. The priming or tolerance to cytokine production has been suggested to depend on the TLR adaptor proteins. Thus it was reported that simultaneous and sequential activation of the MyD88- and TRIF-dependent pathways causes synergy and priming, while tolerance is induced by agonists that act through the same pathway [161]. Further, regulation of signaling pathways such as NF-κB [162] and IFN-γ [163] has been implicated in the interaction between TLRs.
Compared to systemic effects of TLR cross-reactions, little is known about these processes in perinatal brain damage. Clinical studies indicate that a combination of different etiologies is often present in infants with brain injury [164, 165]. Birth complications are commonly preceded by antenatal infections [166] and the combination of such events dramatically increases the risk of spastic cerebral palsy [132]. However, in a recent study, that performed a systematic review of the literature, it was found that there are both benefits and risks with regard to the effects of chorioamnionitis on brain development of preterm infants [167]. The possibility that TLR stimulation alters the vulnerability of the immature brain [168, 169] and adult brain [170] to injury has been postulated.
9.1. TLR-Induced Increase in Vulnerability of the Brain
In order to investigate the interaction between LPS and hypoxic-ischemic brain injury we combined a low sub-sepsis dose of LPS with a subinjury hypoxic-ischemic insult (LPS/HI) in neonatal rats [31]. LPS (0.3 mg/kg) was administered to 7-day-old rats 4 hours prior to 20 minutes of unilateral hypoxia-ischemia. LPS dramatically increased the vulnerability of the immature brain to injury, which could not be explained by a reduction in cerebral blood flow or hyperthermia. In association with the sensitization of injury we found an altered mRNA expression for CD14 and TLR4 in the brain. Subsequently, also direct application of LPS into the brain was shown to increase the vulnerability to hypoxia-ischemia. The combination of intracisternal administration of LPS to 7-day-old rats and hypoxia-ischemia 2 hours later resulted in marked expression of TNF-α in the leptomeninges and neuronal injury in the cerebral cortex that was significantly higher than in animals that were subjected to hypoxia-ischemia after intracisternal application of saline [171].
The priming effect of LPS on neonatal brain damage was later shown to be reliant on TLR4 [172] and MyD88 adaptor protein, via microglia stimulation [173, 174]. Several subsequent studies have confirmed the concept of LPS priming on hypoxic-ischemic brain injury in neonatal rodents [175–178]. It remains to investigate how other TLR ligands affect perinatal hypoxic-ischemic brain damage. Our own preliminary observations suggest that stimulation of both TLR2 and TLR3 induces increased vulnerability to subsequent hypoxia-ischemia (Stridh et al., unpublished).
The precise underlying molecular mechanisms of LPS-induced sensitization of brain damage remain unclear. We showed that there is marked gene regulation in the brain following systemic LPS injection in neonatal rats [179]. Gene ontology analysis demonstrated that within the first few hours after LPS, genes associated with protein metabolism, immune and inflammatory responses, chemotaxis, and cell death were overrepresented. We further showed that caspase-3 activity increased and phosphorylation of the Akt kinase decreased in the brain after systemic LPS exposure. Others have shown an imbalance between agonist and antagonist in the IL-1 system, with a shift towards inflammation in LPS/HI brains [180]. The same group has reported that LPS/HI also enhance IL-2 in microglia, but T lymphocytes were not found in the brain [181]. The role of TNF-α in LPS/HI-induced brain damage is debated. While the Sebire group has suggested little involvement of this cytokine [182], Kendall and colleagues demonstrated complete prevention of the LPS-induced sensitization of hypoxia-ischemia by deletion of the TNF gene cluster [183]. Interestingly, it was recently shown that LPS preexposure significantly decreased the hypoxia-ischemia-induced tissue-type plasminogen activator (tPA) proteolytic activity but amplified the NF-κB signaling pathway. Anti-tPA therapy lessened microglia activation and brain injury [184].
Treatment with corticosteroid improves long-lasting learning impairment following LPS/HI [185]. We later demonstrated that multiple injections of the antioxidant and glutathione precursor N-acetylcysteine (NAC, 200 mg/kg) provided marked neuroprotection, with up to 78% reduction of brain injury, when given both before and after LPS/HI [186]. Protection by NAC was associated with improvement of the redox state and inhibition of apoptosis, suggesting that these events play critical roles in the development of LPS-sensitized hypoxic-ischemic brain injury. We have further investigated the effects of LPS on redox states in vitro and showed that conditioned medium from LPS-stimulated microglia induces death of astrocytes, which was associated with down-regulation of the endogenous antioxidant nuclear factor (erythroid-derived 2)-like 2 (Nrf2) system, while there was sustained activation of glycogen synthase kinase 3 beta β and p38 mitogen-activated protein kinase [187]. In parallel we noticed decreased acetylation of histone 3 and elevated trimethylation of H3-K9. These effects of microglia-conditioned medium on both the Nrf2 system and the histone acetylation levels were reversed by histone deacetylase inhibitors (HDACs) [188]. Strengthening the possibility that LPS-induced brain damage may cause epigenetic alterations, we recently showed that treatment with the HDAC inhibitor, trichostatin A (TSA), increased acetylation in females after neonatal LPS exposure and reduced grey matter and white matter injury at 5 days post-LPS/HI. Further, TSA treatment altered animal behaviour in the open field and improved learning in the fear-conditioning test in adult females following LPS/HI [189].
9.2. TLR-Induced Tolerance in the Brain
In contrast to the tolerance phenomena on cytokine production that develops in the circulation, similar events are not necessarily seen in the brain. In one study, TNF-α was repeatedly infused into the lateral ventricle of guinea pig brains. Fever developed after each of the 4 infusions indicating no diminished response to TNF-α in the brain [190]. The differential cytokine response in circulation and brain during tolerance has later been confirmed by systemic LPS administration. During endotoxin tolerance, elevation of cytokine expression still occurred in the brain, even when cytokines in the periphery were no longer induced [191]. Similarly, it was recently concluded that innate immune cells in the brain do not become tolerant to systemic infection, but are primed instead, which may lead to prolonged and damaging cytokine production that may have a profound effect on the onset and/or progression of preexisting disease [192].
However, despite the data indicating a lack of tolerance in the brain several studies have demonstrated the development of cross-tolerance between systemic LPS and cerebral ischemia, also called LPS-induced preconditioning. This was first described in adult spontaneously hypertensive rats where LPS was injected prior to permanent MCAO [193]. Infarct volume was significantly reduced by LPS administration 2, 3, or 4 days prior to MCAO. The protective effect of LPS was blocked by coadministration of TNF-binding protein, but not IL-1 receptor antagonist, suggesting that the LPS-induced tolerance to ischemia was mediated by TNF-α. It was further shown that the LPS-induced tolerance was not due to attenuation of the ischemic insult by augmenting collateral blood flow, local cerebral blood flow [194].
We demonstrated that LPS-induced tolerance to hypoxia-ischemia in neonatal rats was dependent on the exposure paradigm. Thus, rats pretreated with LPS either 6 hours or 72 hours before a short episode of hypoxia-ischemia suffered increased brain damage compared to animals pretreated with saline [195]. In contrast if LPS was administered 24 hours prior to hypoxia-ischemia, brain injury was reduced. Also when hypoxic-ischemic injury was induced 48 hours following LPS, infarct volume was smaller in LPS pretreated animals compared with saline-treated pups [196]. The preconditioning effect of LPS was age related as it was observed in postnatal day 7, 9, and 14 rat pups but not in postnatal day 3 and 5 rats. The effects of neonatal LPS preconditioning are long lasting as long-term followup showed significantly better learning and memory and less brain damage in adult [197].
Many potential mechanisms of LPS-induced tolerance to ischemic brain injury have been suggested. In one study, an increase in superoxide dismutase was observed in association with LPS-induced tolerance in the brain. The beneficial effect of LPS was suppressed by dexamethasone and indomethacin administered 1 hour before LPS, and it was concluded that activation of inflammatory pathways is involved in the development of LPS-induced tolerance [198]. In support of a role for inflammatory mediators in LPS-induced tolerance against ischemia, ceramide, a downstream messenger in TNF-α signaling, was shown to be upregulated in the tolerant brain [199]. Also in cultured cerebellar granule neurons, endogenous TNF-α seems to be a critical mediator of the neuroprotective actions of LPS independently of the presence of endogenous IL-1β [200]. In particular TNF-receptor (TNFR) 1 appears to be important for LPS-induced tolerance as the protective effect of LPS in a model of cell death, induced by oxygen-glucose deprivation in hippocampal slices, was present in tissue from wild-type and TNFR 2-deficient mice, but not in TNFR 1-deficient mice [201]. LPS preconditioning has also been shown to preserve neurovascular function following ischemia [202] and prevent neutrophil infiltration into the brain and microglia/macrophage activation in the ischemic hemisphere [203]. Further, the involvement of adenosine, an endogenous neuroprotectant in the brain after ischemia, has been proposed. In mice that overexpress adenosine kinase, which is the major negative metabolic regulator of adenosine, LPS-induced ischemic preconditioning was abolished [204].
In an interesting series of experiments, Dr. Stenzel-Poore and colleagues have shown the importance of type-1-IFN-related mechanisms in preconditioning of ischemic brain damage in the adult. Microarray analysis of brains collected 24 hours after stroke identified an overrepresentation of type-I-IFN-associated transcriptional regulatory elements in LPS-pretreated animals. These findings were linked to the TRIF pathway as mice that lack TRIF or a TRIF-dependent transcription factor, interferon regulatory factor 3 (IRF3), were not protected by LPS preconditioning [205, 206]. They also investigated the importance of other TLRs in ischemic tolerance and found that systemic administration of the TLR9 ligand CpG oligodeoxynucleotide [207] or TLR3 ligand Poly I:C [208] prior to brain ischemia reduced brain damage. The same group showed that the common denominator in preconditioning by TLR4 and TLR9 ligands as well as brief ischemia, induced genomic changes in the brain characteristic of sequences required for IRF-mediated transcription [209].
In neonatal rats, we have found that LPS-induced tolerance was mediated by upregulation of corticosterone in the circulation as RU486, a glucocorticoid receptor blocker, counteracted the LPS-induced tolerance effect and aggravated the hypoxia-ischemia-induced brain injury compared with the vehicle-LPS-treated group [210]. We also found, by gene ontology analysis, that the expression profile in association with tolerance was characterized by over-represented genes belonging to immune and inflammatory processes and cell death/survival genes, including complement component 1, complement component 3, aquaporin 4, epidermal growth factor receptor pathway substrate 15, and PYD and CARD domain containing adaptor protein. Interestingly, there was no indication of a marked type I IFN response in the LPS-preconditioned brain [169]. Hence, this indicates that the molecular cues that mediate preconditioning mechanisms in the immature brain likely differ from those observed in the adult.
10. Effects of Perinatal Inflammation on the Adult Brain
Neonatal LPS alters the neuroendocrine, neurochemical and febrile responses to a subsequent, homotypic (LPS) immune challenge in adults [211–214]. Similarly, animals treated neonatally with Poly I:C have significantly attenuated febrile responses to an adult Poly I:C challenge, which coincided with a heightened corticosteroid response [215]. However, neither neonatal Poly I:C nor neonatal LPS challenges lead to an alteration in the adult febrile or corticosteroid responses to a heterotypic adult immune challenge, indicating that the programming effects of the neonatal immune environment are stimulus specific and do not alter the adult responses to other immune stimuli.
The effect of perinatal immune challenge on adult brain ischemia differs between experimental models. In one study, male Sprague-Dawley rats were subjected to a single injection of LPS at postnatal day 14 and were examined as adults for neuronal cell loss associated with global cerebral ischemia after a two-vessel occlusion. Neonatally LPS-treated rats showed increased cell loss in the central nucleus of the amygdala, a region that is important in the processing of emotional responses. No differences were seen in the CA1, CA3, or dentate gyrus regions of the hippocampus [216]. We subjected mice to intrauterine injection of LPS on gestational day 15. On postnatal day 5, 9, and 70, the offspring were subjected to hypoxia-ischemia. LPS preexposure markedly enhanced brain injury after hypoxia-ischemia in neonatal mice. In contrast, in adult mice, LPS preexposure prevented overall tissue loss after hypoxia-ischemia, but there was still injury to white matter [217]. Neonatal exposure to LPS also impact on experimental autoimmune encephalomyelitis (EAE). Mice exposed to LPS at 2 weeks of age showed a delayed onset and diminished severity of myelin-oligodendrocyte- glycoprotein- (MOG-) induced EAE, induced at 12 weeks. Neuroprotection was associated with an increased number of CD3/forkhead box P3 immunoreactive cells, suggesting early-life microbial exposure influencing the generation of neuroprotective regulatory T cells [218].
Early-life exposure to TLR ligands have also been shown to affect the response to a second immune challenge later in life, which can impact the neural processes underlying memory. In fact, caspase-1 inhibition [219] or inhibiting brain IL-1β or microglia activation before the LPS challenge [220] prevents memory impairment in neonatally infected rats. Neonatal LPS exposure also enhances the vulnerability of nigrostriatal dopaminergic neurons to rotenone neurotoxicity in later life suggesting that perinatal brain inflammation may increase adult susceptibility to the development of neurodegenerative disorders [221]. Taken together, there is considerable evidence to indicate that exposure to immune events early in life can impact on a wide range of neurological processes when challenged by similar or different stimuli again in adulthood.
Acknowledgments
Dr. Carina Mallard receives financial support from the Swedish Medical Research Council (VR 2009-2630), Governmental Grants to University Hospitals in Sweden (ALFGBG-142881), European Union grant FP6 (NEOBRAIN, 2006-036534), European Union Grant FP7 (NEUROBID, HEALTH-F2-2009-241778), the Foundation Leducq (DSRR_P34404), Åhlén Foundation, and Wilhelm and Martina Lundgren Foundation. The author thanks Ms Anna-Jean Mallard for excellent help with illustrations.
Abbreviations
- BBB:
Blood-brain barrier
- CD:
Cluster of differentiation
- CNS:
Central nervous system
- COX2:
Cyclo-oxygenase 2
- CVO:
Circumventricular organ
- DAMP:
Danger-associated molecular pattern
- EAE:
Experimental autoimmune encephalomyelitis
- GABA:
γ-aminobutyric acid
- HDAC:
Histone deacetylase inhibitor
- HSP:
Heat shock protein
- HMGB1:
High mobility group box 1
- IFN:
Interferon
- IL:
Interleukin
- IκBα:
I-kappa-B-alpha
- iNOS:
Inducible nitric oxide synthase
- IRF3:
Interferon regulatory factor 3
- KO:
Knockout
- LIF:
Leukemia inhibitory factor
- LPS:
Lipopolysaccharide
- LPS/HI:
LPS and hypoxia-ischemia
- MAL/TIRAP:
MyD88 adaptor-like protein
- MCAO:
Middle cerebral artery occlusion
- MCP1:
Monocyte chemotactic protein-1
- MMP:
Matrix metalloproteinase
- MOG:
Myelin oligodendrocyte glycoprotein
- mtDNA:
Mitochondrial DNA
- MyD88:
Myeloid differentiation factor-88
- NAC:
N-acetylcysteine
- NF-κB:
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NMDA:
N-Methyl-D-aspartate
- Nrf2:
Nuclear factor (erythroid-derived 2)-like 2
- PAMP:
Pathogen-associated molecular pattern
- PI3 K:
Phosphatidylinositol 3-kinase
- Poly I: C:
polyinosinic:polycytidylic acid
- PRR:
Pattern recognition receptor
- PVN:
Paraventricular nucleus
- RT-PCR:
Reverse transcription-polymerase chain reaction
- SARM:
Sterile α- and HEAT/armadillo-motif-containing protein
- TGF:
Transforming growth factor
- TIR:
Toll/interleukin-1 receptor
- TLR:
Toll-like receptor
- TNF-α:
Tumor necrosis factor alpha
- TNFR:
TNF receptor
- tPA:
Tissue-type plasminogen activator
- TRAM:
TRIF-related adaptor molecule
- TRIF:
TIR domain containing adaptor protein inducing IFN beta
- TSA:
Trichostatin A.
References
- 1.Anderson KV, Bokla L, Nusslein-Volhard C. Establishment of dorsal-ventral polarity in the Drosophila embryo: the induction of polarity by the Toll gene product. Cell . 1985;42(3):791–798. doi: 10.1016/0092-8674(85)90275-2. [DOI] [PubMed] [Google Scholar]
- 2.Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spätzle/Toll/Cactus controls the potent antifungal response in Drosophila adults. Cell . 1996;86(6):973–983. doi: 10.1016/s0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- 3.Heppner DW, Weiss G. High susceptibility of atrain a mice to endotoxin and endotoxin-red blood cell mixtures. Journal of Bacteriology . 1965;90:696–703. doi: 10.1128/jb.90.3.696-703.1965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sultzer BM. Genetic control of leucocyte responses to endotoxin. Nature . 1968;219(5160):1253–1254. doi: 10.1038/2191253a0. [DOI] [PubMed] [Google Scholar]
- 5.Coutinho A, Meo T. Genetic basis for unresponsiveness to lipopolysaccharide in C57BL/10Cr mice. Immunogenetics . 1978;7(1):17–24. doi: 10.1007/BF01843983. [DOI] [PubMed] [Google Scholar]
- 6.Poltorak A, Smirnova I, He X, et al. Genetic and physical mapping of the Lps locus: identification of the toll-4 receptor as a candidate gene in the critical region. Blood Cells, Molecules, and Diseases . 1998;24(3):340–355. doi: 10.1006/bcmd.1998.0201. [DOI] [PubMed] [Google Scholar]
- 7.Poltorak A, He X, Smirnova I, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science . 1998;282(5396):2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 8.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on toll-like receptors. Nature Immunology . 2010;11(5):373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 9.Govindaraj RG, Manavalan B, Lee G, Choi S. Molecular modeling-based evaluation of hTLR10 and identification of potential ligands in Toll-like receptor signaling. PloS ONE . 2010;5(9):p. e12713. doi: 10.1371/journal.pone.0012713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Miyake Y, Yamasaki S. Sensing necrotic cells. Advances in Experimental Medicine and Biology . 2012;738:144–152. doi: 10.1007/978-1-4614-1680-7_9. [DOI] [PubMed] [Google Scholar]
- 11.Da Silva Correia J, Soldau K, Christen U, Tobias PS, Ulevitch RJ. Lipopolysaccharide is in close proximity to each of the proteins in its membrane receptor complex. Transfer from CD14 to TLR4 and MD-2. The Journal of Biological Chemistry . 2001;276(24):21129–21135. doi: 10.1074/jbc.M009164200. [DOI] [PubMed] [Google Scholar]
- 12.Hoebe K, Georgel P, Rutschmann S, et al. CD36 is a sensor of diacylglycerides. Nature . 2005;433(7025):523–527. doi: 10.1038/nature03253. [DOI] [PubMed] [Google Scholar]
- 13.Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. Collaborative induction of inflammatory responses by dectin-1 and toll-like receptor 2. The Journal of Experimental Medicine . 2003;197(9):1107–1117. doi: 10.1084/jem.20021787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brinkmann MM, Spooner E, Hoebe K, Beutler B, Ploegh HL, Kim YM. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. Journal of Cell Biology . 2007;177(2):265–275. doi: 10.1083/jcb.200612056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Asagiri M, Hirai T, Kunigami T, et al. Cathepsin K-dependent toll-like receptor 9 signaling revealed in experimental arthritis. Science . 2008;319(5863):624–627. doi: 10.1126/science.1150110. [DOI] [PubMed] [Google Scholar]
- 16.Ewald SE, Lee BL, Lau L, et al. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature . 2008;456(7222):658–662. doi: 10.1038/nature07405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Matsumoto F, Saitoh SI, Fukui R, et al. Cathepsins are required for Toll-like receptor 9 responses. Biochemical and Biophysical Research Communications . 2008;367(3):693–699. doi: 10.1016/j.bbrc.2007.12.130. [DOI] [PubMed] [Google Scholar]
- 18.Park B, Brinkmann MM, Spooner E, Lee CC, Kim YM, Ploegh HL. Proteolytic cleavage in an endolysosomal compartment is required for activation of Toll-like receptor 9. Nature Immunology . 2008;9(12):1407–1414. doi: 10.1038/ni.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barton GM, Kagan JC. A cell biological view of toll-like receptor function: regulation through compartmentalization. Nature Reviews Immunology . 2009;9(8):535–541. doi: 10.1038/nri2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Medzhitov R, Preston-Hurlburt P, Kopp E, et al. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Molecular Cell . 1998;2(2):253–258. doi: 10.1016/s1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- 21.Hoebe K, Du X, Georgel P, et al. Identification of Lps2 as a key transducer of MyD88-independent TIR signalling. Nature . 2003;424(6950):743–748. doi: 10.1038/nature01889. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto M, Sato S, Hemmi H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science . 2003;301(5633):640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 23.Yamamoto M, Sato S, Hemmi H, et al. Essential role for TIRAP in activation of the signalling cascade shared by TLR2 and TLR4. Nature . 2002;420(6913):324–329. doi: 10.1038/nature01182. [DOI] [PubMed] [Google Scholar]
- 24.Carty M, Goodbody R, Schröder M, Stack J, Moynagh PN, Bowie AG. The human adaptor SARM negatively regulates adaptor protein TRIF-dependent Toll-like receptor signaling. Nature Immunology . 2006;7(10):1074–1081. doi: 10.1038/ni1382. [DOI] [PubMed] [Google Scholar]
- 25.Kim Y, Zhou P, Qian L, et al. MyD88-5 links mitochondria, microtubules, and JNK3 in neurons and regulates neuronal survival. The Journal of Experimental Medicine . 2007;204(9):2063–2074. doi: 10.1084/jem.20070868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Panneerselvam P, Singh LP, Ho B, Chen J, Ding JL. Targeting of pro-apoptotic TLR adaptor SARM to mitochondria: definition of the critical region and residues in the signal sequence. Biochemical Journal . 2012;442(2):263–271. doi: 10.1042/BJ20111653. [DOI] [PubMed] [Google Scholar]
- 27.Laflamme N, Rivest S. Toll-like receptor 4: the missing link of the cerebral innate immune response triggered by circulating gram-negative bacterial cell wall components. The FASEB Journal . 2001;15(1):155–163. doi: 10.1096/fj.00-0339com. [DOI] [PubMed] [Google Scholar]
- 28.Chakravarty S, Herkenham M. Toll-like receptor 4 on nonhematopoietic cells sustains CNS inflammation during endotoxemia, independent of systemic cytokines. Journal of Neuroscience . 2005;25(7):1788–1796. doi: 10.1523/JNEUROSCI.4268-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rolls A, Shechter R, London A, et al. Toll-like receptors modulate adult hippocampal neurogenesis. Nature Cell Biology . 2007;9(9):1081–1088. doi: 10.1038/ncb1629. [DOI] [PubMed] [Google Scholar]
- 30.Bsibsi M, Ravid R, Gveric D, Van Noort JM. Broad expression of Toll-like receptors in the human central nervous system. Journal of Neuropathology and Experimental Neurology . 2002;61(11):1013–1021. doi: 10.1093/jnen/61.11.1013. [DOI] [PubMed] [Google Scholar]
- 31.Eklind S, Mallard C, Leverin AL, et al. Bacterial endotoxin sensitizes the immature brain to hypoxic-ischaemic injury. European Journal of Neuroscience . 2001;13(6):1101–1106. doi: 10.1046/j.0953-816x.2001.01474.x. [DOI] [PubMed] [Google Scholar]
- 32.Ma Y, Li J, Chiu I, et al. Toll-like receptor 8 functions as a negative regulator of neurite outgrowth and inducer of neuronal apoptosis. Journal of Cell Biology . 2006;175(2):209–215. doi: 10.1083/jcb.200606016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lathia JD, Okun E, Tang SC, et al. Toll-like receptor 3 is a negative regulator of embryonic neural progenitor cell proliferation. Journal of Neuroscience . 2008;28(51):13978–13984. doi: 10.1523/JNEUROSCI.2140-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stridh L, Smith PLP, Naylor AS, Wang X, Mallard C. Regulation of Toll-like receptor 1 and -2 in neonatal mice brains after hypoxia-ischemia. Journal of Neuroinflammation . 2011;8, article 45 doi: 10.1186/1742-2094-8-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaul D, Habbel P, Derkow K, et al. Expression of toll-like receptors in the developing brain. PLoS ONE . 2012;7(5) doi: 10.1371/journal.pone.0037767.37767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang SC, Arumugam TV, Xu X, et al. Pivotal role for neuronal Toll-like receptors in ischemic brain injury and functional deficits. Proceedings of the National Academy of Sciences of the United States of America . 2007;104(34):13798–13803. doi: 10.1073/pnas.0702553104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tu XK, Yang WZ, Shi SS, et al. Spatio-temporal distribution of inflammatory reaction and expression of TLR2/4 signaling pathway in rat brain following permanent focal cerebral ischemia. Neurochemical Research . 2010;35(8):1147–1155. doi: 10.1007/s11064-010-0167-6. [DOI] [PubMed] [Google Scholar]
- 38.Ziegler G, Harhausen D, Schepers C, et al. TLR2 has a detrimental role in mouse transient focal cerebral ischemia. Biochemical and Biophysical Research Communications . 2007;359(3):574–579. doi: 10.1016/j.bbrc.2007.05.157. [DOI] [PubMed] [Google Scholar]
- 39.Cao CX, Yang QW, Lv FL, Cui J, Fu HB, Wang JZ. Reduced cerebral ischemia-reperfusion injury in Toll-like receptor 4 deficient mice. Biochemical and Biophysical Research Communications . 2007;353(2):509–514. doi: 10.1016/j.bbrc.2006.12.057. [DOI] [PubMed] [Google Scholar]
- 40.Caso JR, Pradillo JM, Hurtado O, Lorenzo P, Moro MA, Lizasoain I. Toll-like receptor 4 is involved in brain damage and inflammation after experimental stroke. Circulation . 2007;115(12):1599–1608. doi: 10.1161/CIRCULATIONAHA.106.603431. [DOI] [PubMed] [Google Scholar]
- 41.Lehnardt S, Lehmann S, Kaul D, et al. Toll-like receptor 2 mediates CNS injury in focal cerebral ischemia. Journal of Neuroimmunology . 2007;190(1-2):28–33. doi: 10.1016/j.jneuroim.2007.07.023. [DOI] [PubMed] [Google Scholar]
- 42.Hyakkoku K, Hamanaka J, Tsuruma K, et al. Toll-like receptor 4 (TLR4), but not TLR3 or TLR9, knock-out mice have neuroprotective effects against focal cerebral ischemia. Neuroscience . 2010;171(1):258–267. doi: 10.1016/j.neuroscience.2010.08.054. [DOI] [PubMed] [Google Scholar]
- 43.Kilic U, Kilic E, Matter CM, Bassetti CL, Hermann DM. TLR-4 deficiency protects against focal cerebral ischemia and axotomy-induced neurodegeneration. Neurobiology of Disease . 2008;31(1):33–40. doi: 10.1016/j.nbd.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 44.Hua F, Ma J, Ha T, et al. Differential roles of TLR2 and TLR4 in acute focal cerebral ischemia/reperfusion injury in mice. Brain Research . 2009;1262:100–108. doi: 10.1016/j.brainres.2009.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brea D, Blanco M, Ramos-Cabrer P, et al. Toll-like receptors 2 and 4 in ischemic stroke: outcome and therapeutic values. Journal of Cerebral Blood Flow and Metabolism . 2011;31(6):1424–1431. doi: 10.1038/jcbfm.2010.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brea D, Sobrino T, Rodríguez-Yáñez M, et al. Toll-like receptors 7 and 8 expression is associated with poor outcome and greater inflammatory response in acute ischemic stroke. Clinical Immunology . 2011;139(2):193–198. doi: 10.1016/j.clim.2011.02.001. [DOI] [PubMed] [Google Scholar]
- 47.Famakin BM, Mou Y, Ruetzler CA, Bembry J, Maric D, Hallenbeck JM. Disruption of downstream MyD88 or TRIF Toll-like receptor signaling does not protect against cerebral ischemia. Brain Research . 2011;1388:148–156. doi: 10.1016/j.brainres.2011.02.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ojaniemi M, Glumoff V, Harju K, Liljeroos M, Vuori K, Hallman M. Phosphatidylinositol 3-kinase is involved in Toll-like receptor 4-mediated cytokine expression in mouse macrophages. European Journal of Immunology . 2003;33(3):597–605. doi: 10.1002/eji.200323376. [DOI] [PubMed] [Google Scholar]
- 49.Mallard C, Wang X, Hagberg H. The role of Toll-like receptors in perinatal brain injury. Clinics in Perinatology . 2009;36(4):763–772. doi: 10.1016/j.clp.2009.07.009. [DOI] [PubMed] [Google Scholar]
- 50.Chen GY, Nuñez G. Sterile inflammation: sensing and reacting to damage. Nature Reviews Immunology . 2010;10(12):826–837. doi: 10.1038/nri2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beg AA. Endogenous ligands of Toll-like receptors: implications for regulating inflammatory and immune responses. Trends in Immunology . 2002;23(11):509–512. doi: 10.1016/s1471-4906(02)02317-7. [DOI] [PubMed] [Google Scholar]
- 52.Hanisch UK, Johnson TV, Kipnis J. Toll-like receptors: roles in neuroprotection? Trends in Neurosciences . 2008;31(4):176–182. doi: 10.1016/j.tins.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 53.Basu S, Binder RJ, Suto R, Anderson KM, Srivastava PK. Necrotic but not apoptotic cell death releases heat shock proteins, which deliver a partial maturation signal to dendritic cells and activate the NF-κB pathway. International Immunology . 2000;12(11):1539–1546. doi: 10.1093/intimm/12.11.1539. [DOI] [PubMed] [Google Scholar]
- 54.Asea A, Kraeft SK, Kurt-Jones EA, et al. HSP70 stimulates cytokine production through a CD 14-dependant pathway, demonstrating its dual role as a chaperone and cytokine. Nature Medicine . 2000;6(4):435–442. doi: 10.1038/74697. [DOI] [PubMed] [Google Scholar]
- 55.Ohashi K, Burkart V, Flohé S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. The Journal of Immunology . 2000;164(2):558–561. doi: 10.4049/jimmunol.164.2.558. [DOI] [PubMed] [Google Scholar]
- 56.Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H. HSP70 as endogenous stimulus of the toll/interleukin-1 receptor signal pathway. The Journal of Biological Chemistry . 2002;277(17):15107–15112. doi: 10.1074/jbc.M111204200. [DOI] [PubMed] [Google Scholar]
- 57.Lehnardt S, Schott E, Trimbuch T, et al. A vicious cycle involving release of heat shock protein 60 from injured cells and activation of toll-like receptor 4 mediates neurodegeneration in the CNS. Journal of Neuroscience . 2008;28(10):2320–2331. doi: 10.1523/JNEUROSCI.4760-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chajara A, Raoudi M, Delpech B, Leroy M, Basuyau JP, Levesque H. Increased hyaluronan and hyaluronidase production and hyaluronan degradation in injured aorta of insulin-resistant rats. Arteriosclerosis, Thrombosis, and Vascular Biology . 2000;20(6):1480–1487. doi: 10.1161/01.atv.20.6.1480. [DOI] [PubMed] [Google Scholar]
- 59.Panasyuk A, Frati E, Ribault D, Mitrovic D. Effect of reactive oxygen species on the biosynthesis and structure of newly synthesized proteoglycans. Free Radical Biology and Medicine . 1994;16(2):157–167. doi: 10.1016/0891-5849(94)90139-2. [DOI] [PubMed] [Google Scholar]
- 60.Šoltés L, Mendichi R, Kogan G, Schiller J, Stankovská M, Arnhold J. Degradative action of reactive oxygen species on hyaluronan. Biomacromolecules . 2006;7(3):659–668. doi: 10.1021/bm050867v. [DOI] [PubMed] [Google Scholar]
- 61.Termeer C, Benedix F, Sleeman J, et al. Oligosaccharides of hyaluronan activate dendritic cells via Toll-like receptor 4. The Journal of Experimental Medicine . 2002;195(1):99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jiang D, Liang J, Fan J, et al. Regulation of lung injury and repair by Toll-like receptors and hyaluronan. Nature Medicine . 2005;11(11):1173–1179. doi: 10.1038/nm1315. [DOI] [PubMed] [Google Scholar]
- 63.Scheibner KA, Lutz MA, Boodoo S, Fenton MJ, Powell JD, Horton MR. Hyaluronan fragments act as an endogenous danger signal by engaging TLR2. The Journal of Immunology . 2006;177(2):1272–1281. doi: 10.4049/jimmunol.177.2.1272. [DOI] [PubMed] [Google Scholar]
- 64.Taylor KR, Yamasaki K, Radek KA, et al. Recognition of hyaluronan released in sterile injury involves a unique receptor complex dependent on toll-like receptor 4, CD44, and MD-2. The Journal of Biological Chemistry . 2007;282(25):18265–18275. doi: 10.1074/jbc.M606352200. [DOI] [PubMed] [Google Scholar]
- 65.Back SA, Tuohy TMF, Chen H, et al. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nature Medicine . 2005;11(9):966–972. doi: 10.1038/nm1279. [DOI] [PubMed] [Google Scholar]
- 66.Al’Qteishat A, Gaffney J, Krupinski J, et al. Changes in hyaluronan production and metabolism following ischaemic stroke in man. Brain . 2006;129(8):2158–2176. doi: 10.1093/brain/awl139. [DOI] [PubMed] [Google Scholar]
- 67.Okun E, Griffioen KJ, Gen Son T, et al. TLR2 activation inhibits embryonic neural progenitor cell proliferation. Journal of Neurochemistry . 2010;114(2):462–474. doi: 10.1111/j.1471-4159.2010.06778.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Degryse B, Bonaldi T, Scaffidi P, et al. The high mobility group (HMG) boxes of the nuclear protein HMG1 induce chemotaxis and cytoskeleton reorganization in rat smooth muscle cells. Journal of Cell Biology . 2001;152(6):1197–1206. doi: 10.1083/jcb.152.6.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Müller S, Scaffidi P, Degryse B, et al. The double life of HMGB1 chromatin protein: architectural factor and extracellular signal. The EMBO Journal . 2001;20(16):4337–4340. doi: 10.1093/emboj/20.16.4337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature . 2002;418(6894):191–195. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 71.Park JS, Svetkauskaite D, He Q, et al. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. The Journal of Biological Chemistry . 2004;279(9):7370–7377. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 72.Yu M, Wang H, Ding A, et al. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock . 2006;26(2):174–179. doi: 10.1097/01.shk.0000225404.51320.82. [DOI] [PubMed] [Google Scholar]
- 73.Liu K, Mori S, Takahashi HK, et al. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. The FASEB Journal . 2007;21(14):3904–3916. doi: 10.1096/fj.07-8770com. [DOI] [PubMed] [Google Scholar]
- 74.Faraco G, Fossati S, Bianchi ME, et al. High mobility group box 1 protein is released by neural cells upon different stresses and worsens ischemic neurodegeneration in vitro and in vivo. Journal of Neurochemistry . 2007;103(2):590–603. doi: 10.1111/j.1471-4159.2007.04788.x. [DOI] [PubMed] [Google Scholar]
- 75.Kim JB, Lim CM, Yu YM, Lee JK. Induction and subcellular localization of high-mobility group box-1 (HMGB1) in the postischemic rat brain. Journal of Neuroscience Research . 2008;86(5):1125–1131. doi: 10.1002/jnr.21555. [DOI] [PubMed] [Google Scholar]
- 76.Qiu J, Nishimura M, Wang Y, et al. Early release of HMGB-1 from neurons after the onset of brain ischemia. Journal of Cerebral Blood Flow and Metabolism . 2008;28(5):927–938. doi: 10.1038/sj.jcbfm.9600582. [DOI] [PubMed] [Google Scholar]
- 77.Yang QW, Lu FL, Zhou Y, et al. HMBG1 mediates ischemia-reperfusion injury by TRIF-adaptor independent Toll-like receptor 4 signaling. Journal of Cerebral Blood Flow and Metabolism . 2011;31(2):593–605. doi: 10.1038/jcbfm.2010.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shichita T, Hasegawa E, Kimura A, et al. Peroxiredoxin family proteins are key initiators of post-ischemic inflammation in the brain. Nature Medicine . 2012;18(6):911–917. doi: 10.1038/nm.2749. [DOI] [PubMed] [Google Scholar]
- 79.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell . 2006;124(4):783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 80.Heil F, Hemmi H, Hochrein H, et al. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science . 2004;303(5663):1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 81.Barton GM, Kagan JC, Medzhitov R. Intracellular localization of Toll-like receptor 9 prevents recognition of self DNA but facilitates access to viral DNA. Nature Immunology . 2006;7(1):49–56. doi: 10.1038/ni1280. [DOI] [PubMed] [Google Scholar]
- 82.Fukui R, Saitoh SI, Matsumoto F, et al. Unc93B1 biases Toll-like receptor responses to nucleic acid in dendritic cells toward DNA- but against RNA-sensing. The Journal of Experimental Medicine . 2009;206(6):1339–1350. doi: 10.1084/jem.20082316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Diebold SS, Massacrier C, Akira S, Paturel C, Morel Y, Reis e Sousa C. Nucleic acid agonists for Toll-like receptor 7 are defined by the presence of uridine ribonucleotides. European Journal of Immunology . 2006;36(12):3256–3267. doi: 10.1002/eji.200636617. [DOI] [PubMed] [Google Scholar]
- 84.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature . 2001;413(6857):732–738. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 85.Cavassani KA, Ishii M, Wen H, et al. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. The Journal of Experimental Medicine . 2008;205(11):2609–2621. doi: 10.1084/jem.20081370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Karikó K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for Toll-like receptor 3. The Journal of Biological Chemistry . 2004;279(13):12542–12550. doi: 10.1074/jbc.M310175200. [DOI] [PubMed] [Google Scholar]
- 87.West AP, Shadel GS, Ghosh S. Mitochondria in innate immune responses. Nature Reviews Immunology . 2011;11(6):389–402. doi: 10.1038/nri2975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Arnoult D, Soares F, Tattoli I, Girardin SE. Mitochondria in innate immunity. EMBO Reports . 2011;12:901–910. doi: 10.1038/embor.2011.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hemmi H, Takeuchi O, Kawai T, et al. A Toll-like receptor recognizes bacterial DNA. Nature . 2000;408(6813):740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 90.Oka T, Hikoso S, Yamaguchi O, et al. Mitochondrial DNA that escapes from autophagy causes inflammation and heart failure. Nature . 2012;484(7397):251–255. doi: 10.1038/nature10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Yin W, Signore AP, Iwai M, Cao G, Gao Y, Chen J. Rapidly increased neuronal mitochondrial biogenesis after hypoxic-ischemic brain injury. Stroke . 2008;39(11):3057–3063. doi: 10.1161/STROKEAHA.108.520114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Cunningham C, Campion S, Teeling J, Felton L, Perry VH. The sickness behaviour and CNS inflammatory mediator profile induced by systemic challenge of mice with synthetic double-stranded RNA (poly I:C) Brain, Behavior, and Immunity . 2007;21(4):490–502. doi: 10.1016/j.bbi.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 93.Erickson MA, Banks WA. Cytokine and chemokine responses in serum and brain after single and repeated injections of lipopolysaccharide: multiplex quantification with path analysis. Brain, Behavior, and Immunity . 2011 doi: 10.1016/j.bbi.2011.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cardoso FL, Kittel A, Veszelka S, et al. Exposure to lipopolysaccharide and/or unconjugated bilirubin impair the integrity and function of brain microvascular endothelial cells. PLoS ONE . 2012;7(5) doi: 10.1371/journal.pone.0035919.35919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Stolp HB, Dziegielewska KM, Ek CJ, Potter AM, Saunders NR. Long-term changes in blood-brain barrier permeability and white matter following prolonged systemic inflammation in early development in the rat. European Journal of Neuroscience . 2005;22(11):2805–2816. doi: 10.1111/j.1460-9568.2005.04483.x. [DOI] [PubMed] [Google Scholar]
- 96.Stolp HB, Ek CJ, Johansson PA, et al. Effect of minocycline on inflammation-induced damage to the blood-brain barrier and white matter during development. European Journal of Neuroscience . 2007;26(12):3465–3474. doi: 10.1111/j.1460-9568.2007.05973.x. [DOI] [PubMed] [Google Scholar]
- 97.Mathison JC, Ulevitch RJ. The clearance, tissue distribution, and cellular localization of intravenously injected lipopolysaccharide in rabbits. The Journal of Immunology . 1979;123(5):2133–2143. [PubMed] [Google Scholar]
- 98.Lenczowski MJP, Van Dam AM, Poole S, Larrick JW, Tilders FJH. Role of circulating endotoxin and interleukin-6 in the ACTH and corticosterone response to intraperitoneal LPS. American Journal of Physiology . 1997;273(6):R1870–R1877. doi: 10.1152/ajpregu.1997.273.6.R1870. [DOI] [PubMed] [Google Scholar]
- 99.Singh AK, Jiang Y. How does peripheral lipopolysaccharide induce gene expression in the brain of rats? Toxicology . 2004;201(1–3):197–207. doi: 10.1016/j.tox.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 100.Banks WA, Robinson SM. Minimal penetration of lipopolysaccharide across the murine blood-brain barrier. Brain, Behavior, and Immunity . 2010;24(1):102–109. doi: 10.1016/j.bbi.2009.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fil D, Borysiewicz E, Konat GW. A broad upregulation of cerebral chemokine genes by peripherally-generated inflammatory mediators. Metabolic Brain Disease . 2011;26(1):49–59. doi: 10.1007/s11011-010-9231-9. [DOI] [PubMed] [Google Scholar]
- 102.Abbott NJ, Patabendige AAK, Dolman DEM, Yusof SR, Begley DJ. Structure and function of the blood-brain barrier. Neurobiology of Disease . 2010;37(1):13–25. doi: 10.1016/j.nbd.2009.07.030. [DOI] [PubMed] [Google Scholar]
- 103.Quan N, Whiteside M, Kim L, Herkenham M. Induction of inhibitory factor κBα mRNA in the central nervous system after peripheral lipopolysaccharide administration: an in situ hybridization histochemistry study in the rat. Proceedings of the National Academy of Sciences of the United States of America . 1997;94(20):10985–10990. doi: 10.1073/pnas.94.20.10985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Reyes TM, Fabry Z, Coe CL. Brain endothelial cell production of a neuroprotective cytokine, interleukin-6, in response to noxious stimuli. Brain Research . 1999;851(1-2):215–220. doi: 10.1016/s0006-8993(99)02189-7. [DOI] [PubMed] [Google Scholar]
- 105.Verma S, Nakaoke R, Dohgu S, Banks WA. Release of cytokines by brain endothelial cells: a polarized response to lipopolysaccharide. Brain, Behavior, and Immunity . 2006;20(5):449–455. doi: 10.1016/j.bbi.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 106.Banks WA, Kastin AJ, Durham DA. Bidirectional transport of interleukin-1 alpha across the blood-brain barrier. Brain Research Bulletin . 1989;23(6):433–437. doi: 10.1016/0361-9230(89)90185-8. [DOI] [PubMed] [Google Scholar]
- 107.Banks WA, Kastin AJ, Gutierrez EG. Penetration of interleukin-6 across the murine blood-brain barrier. Neuroscience Letters . 1994;179(1-2):53–56. doi: 10.1016/0304-3940(94)90933-4. [DOI] [PubMed] [Google Scholar]
- 108.Banks WA. Blood-brain barrier transport of cytokines: a mechanism for neuropathology. Current Pharmaceutical Design . 2005;11(8):973–984. doi: 10.2174/1381612053381684. [DOI] [PubMed] [Google Scholar]
- 109.Pan W, Hsuchou H, Yu C, Kastin AJ. Permeation of blood-borne IL15 across the blood-brain barrier and the effect of LPS. Journal of Neurochemistry . 2008;106(1):313–319. doi: 10.1111/j.1471-4159.2008.05390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Cao C, Matsumura K, Yamagata K, Watanabe Y. Endothelial cells of the rat brain vasculature express cyclooxygenase-2 mRNA in response to systemic interleukin-1β: a possible site of prostaglandin synthesis responsible for fever. Brain Research . 1996;733(2):263–272. doi: 10.1016/0006-8993(96)00575-6. [DOI] [PubMed] [Google Scholar]
- 111.Quan N, Whiteside M, Herkenham M. Cyclooxygenase 2 mRNA expression in rat brain after peripheral injection of lipopolysaccharide. Brain Research . 1998;802(1-2):189–197. doi: 10.1016/s0006-8993(98)00402-8. [DOI] [PubMed] [Google Scholar]
- 112.Ek M, Engblom D, Saha S, Blomqvist A, Jakobsson PJ, Ericsson-Dahlstrand A. Pathway across the blood-brain barrier. Nature . 2001;410(6827):430–431. doi: 10.1038/35068632. [DOI] [PubMed] [Google Scholar]
- 113.Nagyoszi P, Wilhelm I, Farkas AE, et al. Expression and regulation of toll-like receptors in cerebral endothelial cells. Neurochemistry International . 2010;57(5):556–564. doi: 10.1016/j.neuint.2010.07.002. [DOI] [PubMed] [Google Scholar]
- 114.Gosselin D, Rivest S. MyD88 signaling in brain endothelial cells is essential for the neuronal activity and glucocorticoid release during systemic inflammation. Molecular Psychiatry . 2008;13(5):480–497. doi: 10.1038/sj.mp.4002122. [DOI] [PubMed] [Google Scholar]
- 115.Elmquist JK, Scammell TE, Jacobson CD, Saper CB. Distribution of Fos-like immunoreactivity in the rat brain following intravenous lipopolysaccharide administration. Journal of Comparative Neurology . 1996;371(1):85–103. doi: 10.1002/(SICI)1096-9861(19960715)371:1<85::AID-CNE5>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- 116.Breder CD, Hazuka C, Ghayur T, et al. Regional induction of tumor necrosis factor α expression in the mouse brain after systemic lipopolysaccharide administration. Proceedings of the National Academy of Sciences of the United States of America . 1994;91(24):11393–11397. doi: 10.1073/pnas.91.24.11393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Wuchert F, Ott D, Murgott J, et al. Rat area postrema microGlial cells act as sensors for the toll-like receptor-4 agonist lipopolysaccharide. Journal of Neuroimmunology . 2008;204(1-2):66–74. doi: 10.1016/j.jneuroim.2008.07.017. [DOI] [PubMed] [Google Scholar]
- 118.Nakamori T, Morimoto A, Yamaguchi K, Watanabe T, Murakami N. Interleukin-1β production in the rabbit brain during endotoxin-induced fever. Journal of Physiology . 1994;476(1):177–186. [PMC free article] [PubMed] [Google Scholar]
- 119.Vallières L, Rivest S. Regulation of the genes encoding interleukin-6, its receptor, and gp130 in the rat brain in response to the immune activator lipopolysaccharide and the proinflammatory cytokine interleukin-1β . Journal of Neurochemistry . 1997;69(4):1668–1683. doi: 10.1046/j.1471-4159.1997.69041668.x. [DOI] [PubMed] [Google Scholar]
- 120.Marques F, Sousa JC, Coppola G, et al. Kinetic profile of the transcriptome changes induced in the choroid plexus by peripheral inflammation. Journal of Cerebral Blood Flow and Metabolism . 2009;29(5):921–932. doi: 10.1038/jcbfm.2009.15. [DOI] [PubMed] [Google Scholar]
- 121.Goehler LE, Gaykema RPA, Nguyen KT, et al. Interleukin-1β in immune cells of the abdominal vagus nerve: a link between the immune and nervous systems? Journal of Neuroscience . 1999;19(7):2799–2806. doi: 10.1523/JNEUROSCI.19-07-02799.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Maier SF, Goehler LE, Fleshner M, Watkins LR. The role of the vagus nerve in cytokine-to-brain communication. Annals of the New York Academy of Sciences . 1998;840:289–300. doi: 10.1111/j.1749-6632.1998.tb09569.x. [DOI] [PubMed] [Google Scholar]
- 123.Dantzer R, Bluthé RM, Layé S, Bret-Dibat JL, Parnet P, Kelley KW. Cytokines and sickness behavior. Annals of the New York Academy of Sciences . 1998;840:586–590. doi: 10.1111/j.1749-6632.1998.tb09597.x. [DOI] [PubMed] [Google Scholar]
- 124.Laye S, Bluthe RM, Kent S, et al. Subdiaphragmatic vagotomy blocks induction of IL-1β mRNA in mice brain in response to peripheral LPS. American Journal of Physiology . 1995;268(5):R1327–R1331. doi: 10.1152/ajpregu.1995.268.5.R1327. [DOI] [PubMed] [Google Scholar]
- 125.Gilles FH, Averill DR, Kerr CS. Neonatal endotoxin encephalopathy. Annals of Neurology . 1977;2(1):49–56. doi: 10.1002/ana.410020108. [DOI] [PubMed] [Google Scholar]
- 126.Gilles FH, Leviton A, Kerr CS. Endotoxin leucoencephalopathy in the telencephalon of the newborn kitten. Journal of the Neurological Sciences . 1976;27(2):183–191. doi: 10.1016/0022-510x(76)90060-5. [DOI] [PubMed] [Google Scholar]
- 127.Leviton A, Gilles F, Neff R, Yaney P. Multivariate analysis of risk of perinatal telencephalic leucoencephalopathy. American Journal of Epidemiology . 1976;104(6):621–626. doi: 10.1093/oxfordjournals.aje.a112340. [DOI] [PubMed] [Google Scholar]
- 128.Chau V, Brant R, Poskitt KJ, Tam EWY, Synnes A, Miller SP. Postnatal infection is associated with widespread abnormalities of brain development in premature newborns. Pediatric Research . 2012;71(3):274–279. doi: 10.1038/pr.2011.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dammann O, Leviton A. Maternal intrauterine infection, cytokines, and brain damage in the preterm newborn. Pediatric Research . 1997;42(1):1–8. doi: 10.1203/00006450-199707000-00001. [DOI] [PubMed] [Google Scholar]
- 130.Marlow N, Wolke D, Bracewell MA, Samara M. Neurologic and developmental disability at six years of age after extremely preterm birth. The New England Journal of Medicine . 2005;352(1):9–19. doi: 10.1056/NEJMoa041367. [DOI] [PubMed] [Google Scholar]
- 131.Mwaniki MK, Atieno M, Lawn JE, Newton CRJC. Long-term neurodevelopmental outcomes after intrauterine and neonatal insults: a systematic review. The Lancet . 2012;379(9814):445–452. doi: 10.1016/S0140-6736(11)61577-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nelson KB, Dambrosia JM, Grether JK, Phillips TM. Neonatal cytokines and coagulation factors in children with cerebral palsy. Annals of Neurology . 1998;44(4):665–675. doi: 10.1002/ana.410440413. [DOI] [PubMed] [Google Scholar]
- 133.Volpe JJ. Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. The Lancet Neurology . 2009;8(1):110–124. doi: 10.1016/S1474-4422(08)70294-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Debillon T, Gras-Leguen C, Vérielle V, et al. Intrauterine infection induces programmed cell death in rabbit periventricular white matter. Pediatric Research . 2000;47(6):736–742. doi: 10.1203/00006450-200006000-00009. [DOI] [PubMed] [Google Scholar]
- 135.Bo Hyun Yoon, Chong Jai Kim, Romero R, et al. Experimentally induced intrauterine infection causes fetal brain white matter lesions in rabbits. American Journal of Obstetrics and Gynecology . 1997;177(4):797–802. doi: 10.1016/s0002-9378(97)70271-0. [DOI] [PubMed] [Google Scholar]
- 136.Cai Z, Pan ZL, Pang Y, Evans OB, Rhodes PG. Cytokine induction in fetal rat brains and brain injury in neonatal rats after maternal lipopolysaccharide administration. Pediatric Research . 2000;47(1):64–72. doi: 10.1203/00006450-200001000-00013. [DOI] [PubMed] [Google Scholar]
- 137.Bell MJ, Hallenbeck JM. Effects of intrauterine inflammation on developing rat brain. Journal of Neuroscience Research . 2002;70(4):570–579. doi: 10.1002/jnr.10423. [DOI] [PubMed] [Google Scholar]
- 138.Rousset CI, Chalon S, Cantagrel S, et al. Maternal exposure to LPS induces hypomyelination in the internal capsule and programmed cell death in the deep gray matter in newborn rats. Pediatric Research . 2006;59(3):428–433. doi: 10.1203/01.pdr.0000199905.08848.55. [DOI] [PubMed] [Google Scholar]
- 139.Liverman CS, Kaftan HA, Cui L, et al. Altered expression of pro-inflammatory and developmental genes in the fetal brain in a mouse model of maternal infection. Neuroscience Letters . 2006;399(3):220–225. doi: 10.1016/j.neulet.2006.01.064. [DOI] [PubMed] [Google Scholar]
- 140.Oskvig DB, Elkahloun AG, Johnson KR, Phillips TM, Herkenham M. Maternal immune activation by LPS selectively alters specific gene expression profiles of interneuron migration and oxidative stress in the fetus without triggering a fetal immune response. Brain, Behavior, and Immunity . 2012;26(4):623–634. doi: 10.1016/j.bbi.2012.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Hagberg H, Gressens P, Mallard C. Inflammation during fetal and neonatal life: implications for neurologic and neuropsychiatric disease in children and adults. Annals of Neurology . 2012;71(4):444–457. doi: 10.1002/ana.22620. [DOI] [PubMed] [Google Scholar]
- 142.Meyer U, Feldon J, Dammann O. Schizophrenia and autism: both shared and disorder-specific pathogenesis via perinatal inflammation? Pediatric Research . 2011;69(5):26R–33R. doi: 10.1203/PDR.0b013e318212c196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Meyer U, Nyffeler M, Yee BK, Knuesel I, Feldon J. Adult brain and behavioral pathological markers of prenatal immune challenge during early/middle and late fetal development in mice. Brain, Behavior, and Immunity . 2008;22(4):469–486. doi: 10.1016/j.bbi.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 144.Forrest CM, Khalil OS, Pisar M, Smith RA, Darlington LG, Stone TW. Prenatal activation of Toll-like receptors-3 by administration of the viral mimetic poly(I:C) changes synaptic proteins, N-methyl-D-aspartate receptors and neurogenesis markers in offspring. Molecular Brain . 2012;5, article 22 doi: 10.1186/1756-6606-5-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Vuillermot S, Joodmardi E, Perlmann T, Ögren SO, Feldon J, Meyer U. Prenatal immune activation interacts with Genetic Nurr1 deficiency in the development of attentional impairments. Journal of Neuroscience . 2012;32(2):436–451. doi: 10.1523/JNEUROSCI.4831-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Duncan JR, Cock ML, Scheerlinck JPY, et al. White matter injury after repeated endotoxin exposure in the preterm ovine fetus. Pediatric Research . 2002;52(6):941–949. doi: 10.1203/00006450-200212000-00021. [DOI] [PubMed] [Google Scholar]
- 147.Mallard C, Welin AK, Peebles D, Hagberg H, Kjellmer I. White matter injury following systemic endotoxemia or asphyxia in the fetal sheep. Neurochemical Research . 2003;28(2):215–223. doi: 10.1023/a:1022368915400. [DOI] [PubMed] [Google Scholar]
- 148.Dean JM, Farrag D, Zahkouk SAM, et al. Cerebellar white matter injury following systemic endotoxemia in preterm fetal sheep. Neuroscience . 2009;160(3):606–615. doi: 10.1016/j.neuroscience.2009.02.071. [DOI] [PubMed] [Google Scholar]
- 149.Dean JM, Van De Looij Y, Sizonenko SV, et al. Delayed cortical impairment following lipopolysaccharide exposure in preterm fetal sheep. Annals of Neurology . 2011;70(5):846–856. doi: 10.1002/ana.22480. [DOI] [PubMed] [Google Scholar]
- 150.Van De Looij Y, Lodygensky GA, Dean J, et al. High-field diffusion tensor imaging characterization of cerebral white matter injury in lipopolysaccharide-exposed fetal sheep. Pediatric Research . 2012;72(3):285–292. doi: 10.1038/pr.2012.72. [DOI] [PubMed] [Google Scholar]
- 151.Keller M, Enot DP, Hodson MP, et al. Inflammatory-induced hibernation in the fetus: priming of fetal sheep metabolism correlates with developmental brain injury. PLoS ONE . 2011;6(12) doi: 10.1371/journal.pone.0029503.29503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Wang X, Hellgren G, Löfqvist C, et al. White matter damage after chronic subclinical inflammation in newborn mice. Journal of Child Neurology . 2009;24(9):1171–1178. doi: 10.1177/0883073809338068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Du X, Fleiss B, Li H, et al. Systemic stimulation of TLR2 impairs neonatal mouse brain development. PLoS ONE . 2011;6(5) doi: 10.1371/journal.pone.0019583.e19583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Berger FM. The effect of endotoxin on resistance to infection and disease. Advances in Pharmacology . 1967;5:19–46. doi: 10.1016/s1054-3589(08)60653-5. [DOI] [PubMed] [Google Scholar]
- 155.Neva FA, Morgan HR. Tolerance to the action of endotoxins of enteric bacilli in patients convalescent from typhoid and paratyphoid fevers. Journal of Laboratory and Clinical Medicine . 1950;35:911–922. [PubMed] [Google Scholar]
- 156.Kreutz M, Ackermann U, Hauschildt S, et al. A comparative analysis of cytokine production and tolerance induction by bacterial lipopeptides, lipopolysaccharides and staphyloccocus aureus in human monocytes. Immunology . 1997;92(3):396–401. doi: 10.1046/j.1365-2567.1997.00365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.West MA, Bennet T, Clair L. Reprogrammed macrophage tumor necrosis factor and interleukin-1 release with inflammatory pretreatment: differential regulation by endotoxin and zymosan. Journal of Trauma . 1995;39(3):404–410. doi: 10.1097/00005373-199509000-00002. [DOI] [PubMed] [Google Scholar]
- 158.Beutler E, Gelbart T, West C. Synergy between TLR2 and TLR4: a safety mechanism. Blood Cells, Molecules, and Diseases . 2001;27(4):728–730. doi: 10.1006/bcmd.2001.0441. [DOI] [PubMed] [Google Scholar]
- 159.Sato S, Nomura F, Kawai T, et al. Synergy and cross-tolerance between Toll-like receptor (TLR) 2- and TLR4-mediated signaling pathways. The Journal of Immunology . 2000;165(12):7096–7101. doi: 10.4049/jimmunol.165.12.7096. [DOI] [PubMed] [Google Scholar]
- 160.Whitmore MM, DeVeer MJ, Edling A, et al. Synergistic activation of innate immunity by double-stranded RNA and CpG DNA promotes eahanced antitumor activity. Cancer Research . 2004;64(16):5850–5860. doi: 10.1158/0008-5472.CAN-04-0063. [DOI] [PubMed] [Google Scholar]
- 161.Bagchi A, Herrup EA, Warren HS, et al. MyD88-dependent and MyD88-independent pathways in synergy, priming, and tolerance between TLR agonists. The Journal of Immunology . 2007;178(2):1164–1171. doi: 10.4049/jimmunol.178.2.1164. [DOI] [PubMed] [Google Scholar]
- 162.Dobrovolskaia MA, Medvedev AE, Thomas KE, et al. Induction of in vitro reprogramming by toll-like receptor (TLR)2 and TLR4 agonists in murine macrophages: effects of TLR “homotolerance” versus “heterotolerance” on NF-κB signaling pathway components. The Journal of Immunology . 2003;170(1):508–519. doi: 10.4049/jimmunol.170.1.508. [DOI] [PubMed] [Google Scholar]
- 163.Peck OM, Fan H, Tempel GE, Teti G, Halushka PV, Cook JA. Staphylococcus aureus and lipopolysaccharide induce homologous tolerance but heterologous priming: role of interferon-gamma. Shock . 2004;21(3):254–260. doi: 10.1097/01.shk.0000111662.09279.59. [DOI] [PubMed] [Google Scholar]
- 164.Badawi N, Kurinczuk JJ, Keogh JM, et al. Antepartum risk factors for newborn encephalopathy: the Western Australian case-control study. BMJ . 1998;317(7172):1549–1553. doi: 10.1136/bmj.317.7172.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Badawi N, Kurinczuk JJ, Keogh JM, et al. Intrapartum risk factors for newborn encephalopathy: the Western Australian case-control study. BMJ . 1998;317(7172):1554–1558. doi: 10.1136/bmj.317.7172.1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Grether JK, Nelson KB. Maternal infection and cerebral palsy in infants of normal birth weight. JAMA . 1997;278(3):207–211. [PubMed] [Google Scholar]
- 167.Ylijoki M, Ekholm E, Haataja L, Lehtonen L. Is chorioamnionitis harmful for the brain of preterm infants? A clinical overview. Acta Obstetricia et Gynecologica Scandinavica . 2012;91(4):403–419. doi: 10.1111/j.1600-0412.2012.01349.x. [DOI] [PubMed] [Google Scholar]
- 168.Hagberg H, Dammann O, Mallard C, Leviton A. Preconditioning and the developing brain. Seminars in Perinatology . 2004;28(6):389–395. doi: 10.1053/j.semperi.2004.10.006. [DOI] [PubMed] [Google Scholar]
- 169.Mallard C, Hagberg H. Inflammation-induced preconditioning in the immature brain. Seminars in Fetal and Neonatal Medicine . 2007;12(4):280–286. doi: 10.1016/j.siny.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 170.Karikó K, Weissman D, Welsh FA. Inhibition of Toll-like receptor and cytokine signaling—a unifying theme in ischemic tolerance. Journal of Cerebral Blood Flow and Metabolism . 2004;24(11):1288–1304. doi: 10.1097/01.WCB.0000145666.68576.71. [DOI] [PubMed] [Google Scholar]
- 171.Coumans ABC, Middelanis J, Garnier Y, et al. Intracisternal application of endotoxin enhances the susceptibility to subsequent hypoxic-ischemic brain damage in neonatal rats. Pediatric Research . 2003;53(5):770–775. doi: 10.1203/01.PDR.0000059221.40073.82. [DOI] [PubMed] [Google Scholar]
- 172.Lehnardt S, Massillon L, Follett P, et al. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proceedings of the National Academy of Sciences of the United States of America . 2003;100(14):8514–8519. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Dean JM, Wang X, Kaindl AM, et al. MicroGlial MyD88 signaling regulates acute neuronal toxicity of LPS-stimulated microGlia in vitro. Brain, Behavior, and Immunity . 2010;24(5):776–783. doi: 10.1016/j.bbi.2009.10.018. [DOI] [PubMed] [Google Scholar]
- 174.Wang X, Stridh L, Li W, et al. Lipopolysaccharide sensitizes neonatal hypoxic-ischemic brain injury in a MyD88-dependent manner. The Journal of Immunology . 2009;183(11):7471–7477. doi: 10.4049/jimmunol.0900762. [DOI] [PubMed] [Google Scholar]
- 175.Girard S, Kadhim H, Beaudet N, Sarret P, Sébire G. Developmental motor deficits induced by combined fetal exposure to lipopolysaccharide and early neonatal hypoxia/ischemia: a novel animal model for cerebral palsy in very premature infants. Neuroscience . 2009;158(2):673–682. doi: 10.1016/j.neuroscience.2008.10.032. [DOI] [PubMed] [Google Scholar]
- 176.Ikeda T, Mishima K, Aoo N, et al. Combination treatment of neonatal rats with hypoxia-ischemia and endotoxin induces long-lasting memory and learning impairment that is associated with extended cerebral damage. American Journal of Obstetrics and Gynecology . 2004;191(6):2132–2141. doi: 10.1016/j.ajog.2004.04.039. [DOI] [PubMed] [Google Scholar]
- 177.Wang LW, Chang YC, Lin CY, Hong JS, Huang CC. Low-dose lipopolysaccharide selectively sensitizes hypoxic ischemia-induced white matter injury in the immature brain. Pediatric Research . 2010;68(1):41–47. doi: 10.1203/PDR.0b013e3181df5f6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Yang L, Sameshima H, Ikeda T, Ikenoue T. Lipopolysaccharide administration enhances hypoxic-ischemic brain damage in newborn rats. Journal of Obstetrics and Gynaecology Research . 2004;30(2):142–147. doi: 10.1111/j.1447-0756.2003.00174.x. [DOI] [PubMed] [Google Scholar]
- 179.Eklind S, Hagberg H, Wang X, et al. Effect of lipopolysaccharide on global gene expression in the immature rat brain. Pediatric Research . 2006;60(2):161–168. doi: 10.1203/01.pdr.0000228323.32445.7d. [DOI] [PubMed] [Google Scholar]
- 180.Girard S, Kadhim H, Larouche A, Roy M, Gobeil F, Sébire G. Pro-inflammatory disequilibrium of the IL-1β/IL-1ra ratio in an experimental model of perinatal brain damages induced by lipopolysaccharide and hypoxia-ischemia. Cytokine . 2008;43(1):54–62. doi: 10.1016/j.cyto.2008.04.007. [DOI] [PubMed] [Google Scholar]
- 181.Girard S, Larouche A, Kadhim H, Rola-Pleszczynski M, Gobeil F, Sébire G. Lipopolysaccharide and hypoxia/ischemia induced IL-2 expression by microGlia in neonatal brain. NeuroReport . 2008;19(10):997–1002. doi: 10.1097/WNR.0b013e3283036e88. [DOI] [PubMed] [Google Scholar]
- 182.Roy M, Girard S, Larouche A, Kadhim H, Sébire G. TNF-α system response in a rat model of very preterm brain injuries induced by lipopolysaccharide and/or hypoxia-ischemia. American Journal of Obstetrics and Gynecology . 2009;201(5):493.e1–493.e10. doi: 10.1016/j.ajog.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 183.Kendall GS, Hirstova M, Horn S, et al. TNF gene cluster deletion abolishes lipopolysaccharide-mediated sensitization of the neonatal brain to hypoxic ischemic insult. Laboratory Investigation . 2011;91(3):328–341. doi: 10.1038/labinvest.2010.192. [DOI] [PubMed] [Google Scholar]
- 184.Yang D, Sun YY, Nemkul N, et al. Plasminogen activator inhibitor-1 mitigates brain injury in a rat model of infection-sensitized neonatal hypoxia-ischemia. doi: 10.1093/cercor/bhs115. Cerebral Cortex. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Ikeda T, Mishima K, Aoo N, et al. Dexamethasone prevents long-lasting learning impairment following a combination of lipopolysaccharide and hypoxia-ischemia in neonatal rats. American Journal of Obstetrics and Gynecology . 2005;192(3):719–726. doi: 10.1016/j.ajog.2004.12.048. [DOI] [PubMed] [Google Scholar]
- 186.Wang X, Svedin P, Nie C, et al. N-Acetylcysteine reduces lipopolysaccharide-sensitized hypoxic-ischemic brain injury. Annals of Neurology . 2007;61(3):263–271. doi: 10.1002/ana.21066. [DOI] [PubMed] [Google Scholar]
- 187.Correa F, Ljunggren E, Mallard C, Nilsson M, Weber SG, Sandberg M. The Nrf2-inducible antioxidant defense in astrocytes can be both up- and down-regulated by activated microGlia:Involvement of p38 MAPK. Glia . 2011;59(5):785–799. doi: 10.1002/glia.21151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Correa F, Mallard C, Nilsson M, Sandberg M. Activated microGlia decrease histone acetylation and Nrf2-inducible anti-oxidant defence in astrocytes: restoring effects of inhibitors of HDACs, p38 MAPK and GSK3β . Neurobiology of Disease . 2011;44(1):142–151. doi: 10.1016/j.nbd.2011.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Fleiss B, Nilsson MKL, Blomgren K, Mallard C. Neuroprotection by the histone deacetylase inhibitor trichostatin A in a model of lipopolysaccharide-sensitised neonatal hypoxic-ischaemic brain injury. Journal of Neuroinflammation . 2012;9, article 70 doi: 10.1186/1742-2094-9-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Goldbach JM, Roth J, Störr B, Zeisberger E. Lack of tolerance development to tumor necrosis factor α inside the central nervous system. Experientia . 1996;52(8):774–777. doi: 10.1007/BF01923988. [DOI] [PubMed] [Google Scholar]
- 191.Chen R, Zhou H, Beltran J, Malellari L, Chang SL. Differential expression of cytokines in the brain and serum during endotoxin tolerance. Journal of Neuroimmunology . 2005;163(1-2):53–72. doi: 10.1016/j.jneuroim.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 192.Puentener U, Booth SG, Perry VH, Teeling JL. Long-term impact of systemic bacterial infection on the cerebral vasculature and microGlia. Journal of Neuroinflammation . 2012;9, article 146 doi: 10.1186/1742-2094-9-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Tasaki K, Ruetzler CA, Ohtsuki T, Martin D, Nawashiro H, Hallenbeck JM. Lipopolysaccharide pre-treatment induces resistance against subsequent focal cerebral ischemic damage in spontaneously hypertensive rats. Brain Research . 1997;748(1-2):267–270. doi: 10.1016/s0006-8993(96)01383-2. [DOI] [PubMed] [Google Scholar]
- 194.Dawson DA, Furuya K, Gotoh J, Nakao Y, Hallenbeck JM. Cerebrovascular hemodynamics and ischemic tolerance: lipopolysaccharide- induced resistance to focal cerebral ischemia is not due to changes in severity of the initial ischemic insult, but is associated with preservation of microvascular perfusion. Journal of Cerebral Blood Flow and Metabolism . 1999;19(6):616–623. doi: 10.1097/00004647-199906000-00004. [DOI] [PubMed] [Google Scholar]
- 195.Eklind S, Mallard C, Arvidsson P, Hagberg H. Lipopolysaccharide induces both a primary and a secondary phase of sensitization in the developing rat brain. Pediatric Research . 2005;58(1):112–116. doi: 10.1203/01.PDR.0000163513.03619.8D. [DOI] [PubMed] [Google Scholar]
- 196.Hickey E, Shi H, Van Arsdell G, Askalan R. Lipopolysaccharide-induced preconditioning against ischemic injury is associated with changes in Toll-like receptor 4 expression in the rat developing brain. Pediatric Research . 2011;70(1):10–14. doi: 10.1203/PDR.0b013e31821d02aa. [DOI] [PubMed] [Google Scholar]
- 197.Lin HY, Wu CL, Huang CC. The akt-endothelial nitric oxide synthase pathway in lipopolysaccharide preconditioning-induced hypoxic-ischemic tolerance in the neonatal rat brain. Stroke . 2010;41(7):1543–1551. doi: 10.1161/STROKEAHA.109.574004. [DOI] [PubMed] [Google Scholar]
- 198.Bordet R, Deplanque D, Maboudou P, et al. Increase in endogenous brain superoxide dismutase as a potential mechanism of lipopolysaccharide-induced brain ischemic tolerance. Journal of Cerebral Blood Flow and Metabolism . 2000;20(8):1190–1196. doi: 10.1097/00004647-200008000-00004. [DOI] [PubMed] [Google Scholar]
- 199.Zimmermann C, Ginis I, Furuya K, et al. Lipopolysaccharide-induced ischemic tolerance is associated with increased levels of ceramide in brain and in plasma. Brain Research . 2001;895(1-2):59–65. doi: 10.1016/s0006-8993(01)02028-5. [DOI] [PubMed] [Google Scholar]
- 200.Lastres-Becker I, Cartmell T, Molina-Holgado F. Endotoxin preconditioning protects neurones from in vitro ischemia: role of endogenous IL-1β and TNF-α . Journal of Neuroimmunology . 2006;173(1-2):108–116. doi: 10.1016/j.jneuroim.2005.12.006. [DOI] [PubMed] [Google Scholar]
- 201.Markus T, Cronberg T, Cilio C, Pronk C, Wieloch T, Ley D. Tumor necrosis factor receptor-1 is essential for LPS-induced sensitization and tolerance to oxygen-glucose deprivation in murine neonatal organotypic hippocampal slices. Journal of Cerebral Blood Flow and Metabolism . 2009;29(1):73–86. doi: 10.1038/jcbfm.2008.90. [DOI] [PubMed] [Google Scholar]
- 202.Kunz A, Park L, Abe T, et al. Neurovascular protection by ischemic tolerance: role of nitric oxide and reactive oxygen species. Journal of Neuroscience . 2007;27(27):7083–7093. doi: 10.1523/JNEUROSCI.1645-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Rosenzweig HL, Lessov NS, Henshall DC, Minami M, Simon RP, Stenzel-Poore MP. Endotoxin preconditioning prevents cellular inflammatory response during ischemic neuroprotection in mice. Stroke . 2004;35(11):2576–2581. doi: 10.1161/01.STR.0000143450.04438.ae. [DOI] [PubMed] [Google Scholar]
- 204.Shen HY, Lusardi TA, Williams-Karnesky RL, Lan JQ, Poulsen DJ, Boison D. Adenosine kinase determines the degree of brain injury after ischemic stroke in mice. Journal of Cerebral Blood Flow and Metabolism . 2011;31(7):1648–1659. doi: 10.1038/jcbfm.2011.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Marsh B, Stevens SL, Packard AEB, et al. Systemic lipopolysaccharide protects the brain from ischemic injury by reprogramming the response of the brain to stroke: a critical role for IRF3. Journal of Neuroscience . 2009;29(31):9839–9849. doi: 10.1523/JNEUROSCI.2496-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Vartanian KB, Stevens SL, Marsh BJ, Williams-Karnesky R, Lessov NS, Stenzel-Poore MP. LPS preconditioning redirects TLR signaling following stroke: TRIF-IRF3 plays a seminal role in mediating tolerance to ischemic injury. Journal of Neuroinflammation . 2011;8, article 140 doi: 10.1186/1742-2094-8-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Stevens SL, Ciesielski TMP, Marsh BJ, et al. Toll-like receptor 9: a new target of ischemic preconditioning in the brain. Journal of Cerebral Blood Flow and Metabolism . 2008;28(5):1040–1047. doi: 10.1038/sj.jcbfm.9600606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Packard AEB, Hedges JC, Bahjat FR, et al. Poly-IC preconditioning protects against cerebral and renal ischemia-reperfusion injury. Journal of Cerebral Blood Flow and Metabolism . 2012;32(2):242–247. doi: 10.1038/jcbfm.2011.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Stevens SL, Leung PY, Vartanian KB, et al. Multiple preconditioning paradigms converge on interferon regulatory factor-dependent signaling to promote tolerance to ischemic brain injury. Journal of Neuroscience . 2011;31(23):8456–8463. doi: 10.1523/JNEUROSCI.0821-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Ikeda T, Yang L, Ikenoue T, Mallard C, Hagberg H. Endotoxin-induced hypoxic-ischemic tolerance is mediated by up-regulation of corticosterone in neonatal rat. Pediatric Research . 2006;59(1):56–60. doi: 10.1203/01.pdr.0000191140.87314.ce. [DOI] [PubMed] [Google Scholar]
- 211.Boissé L, Mouihate A, Ellis S, Pittman QJ. Long-term alterations in neuroimmune responses after neonatal exposure to lipopolysaccharide. Journal of Neuroscience . 2004;24(21):4928–4934. doi: 10.1523/JNEUROSCI.1077-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Ellis S, Mouihate A, Pittman QJ. Early life immune challenge alters innate immune responses to lipopolysaccharide: implications for host defense as adults. The FASEB Journal . 2005;19(11):1519–1521. doi: 10.1096/fj.04-3569fje. [DOI] [PubMed] [Google Scholar]
- 213.Shanks N, Larocque S, Meaney MJ. Neonatal endotoxin exposure alters the development of the hypothalamic- pituitary-adrenal axis: early illness and later responsivity to stress. Journal of Neuroscience . 1995;15(1):376–384. doi: 10.1523/JNEUROSCI.15-01-00376.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Spencer SJ, Heida JG, Pittman QJ. Early life immune challenge—effects on behavioural indices of adult rat fear and anxiety. Behavioural Brain Research . 2005;164(2):231–238. doi: 10.1016/j.bbr.2005.06.032. [DOI] [PubMed] [Google Scholar]
- 215.Ellis S, Mouihate A, Pittman QJ. Neonatal programming of the rat neuroimmune response: stimulus specific changes elicited by bacterial and viral mimetics. Journal of Physiology . 2006;571(3):695–701. doi: 10.1113/jphysiol.2005.102939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Spencer SJ, Auer RN, Pittman QJ. Rat neonatal immune challenge alters adult responses to cerebral ischaemia. Journal of Cerebral Blood Flow and Metabolism . 2006;26(4):456–467. doi: 10.1038/sj.jcbfm.9600206. [DOI] [PubMed] [Google Scholar]
- 217.Wang X, Hagberg H, Nie C, Zhu C, Ikeda T, Mallard C. Dual role of intrauterine immune challenge on neonatal and adult brain vulnerability to hypoxia-ischemia. Journal of Neuropathology and Experimental Neurology . 2007;66(6):552–561. doi: 10.1097/01.jnen.0000263870.91811.6f. [DOI] [PubMed] [Google Scholar]
- 218.Ellestad KK, Tsutsui S, Noorbakhsh F, et al. Early life exposure to lipopolysaccharide suppresses experimental autoimmune encephalomyelitis by promoting tolerogenic dendritic cells and regulatory T cells. The Journal of Immunology . 2009;183(1):298–309. doi: 10.4049/jimmunol.0803576. [DOI] [PubMed] [Google Scholar]
- 219.Bilbo SD, Biedenkapp JC, Der-Avakian A, Watkins LR, Rudy JW, Maier SF. Neonatal infection-induced memory impairment after lipopolysaccharide in adulthood is prevented via caspase-1 inhibition. Journal of Neuroscience . 2005;25(35):8000–8009. doi: 10.1523/JNEUROSCI.1748-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Williamson LL, Sholar PW, Mistry RS, Smith SH, Bilbo SD. MicroGlia and memory: modulation by early-life infection. Journal of Neuroscience . 2011;31(43):15511–15521. doi: 10.1523/JNEUROSCI.3688-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Fan LW, Tien LT, Lin RCS, Simpson KL, Rhodes PG, Cai Z. Neonatal exposure to lipopolysaccharide enhances vulnerability of nigrostriatal dopaminergic neurons to rotenone neurotoxicity in later life. Neurobiology of Disease . 2011;44:304–316. doi: 10.1016/j.nbd.2011.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]