

Abstract
Functional analysis has shown that the missense gain-in-function KCNQ1 S140G mutation associated with familial atrial fibrillation produces an increase of the slow delayed rectifier potassium current (IKs). Through computer modelling, this study investigated mechanisms by which the KCNQ1 S140G mutation promotes and perpetuates atrial fibrillation. In simulations, Courtemanche et al.'s model of human atrial cell action potentials (APs) was modified to incorporate experimental data on changes of IKs induced by the KCNQ1 S140G mutation. The cell models for wild type (WT) and mutant type (MT) IKs were incorporated into homogeneous multicellular 2D and 3D tissue models. Effects of the mutation were quantified on AP profile, AP duration (APD) restitution, effective refractory period (ERP) restitution, and conduction velocity (CV) restitution. Temporal and spatial vulnerabilities of atrial tissue to genesis of re-entry were computed. Dynamic behaviours of re-entrant excitation waves (lifespan (LS), tip meandering patterns and dominant frequency) in 2D and 3D models were characterised. It was shown that the KCNQ1 S140G mutation abbreviated atrial APD and ERP and flattened APD and ERP restitution curves. It reduced atrial CV at low excitation rates, but increased it at high excitation rates that facilitated the conduction of high rate atrial excitation waves. Although it increased slightly tissue temporal vulnerability for initiating re-entry, it reduced markedly the minimal substrate size necessary for sustaining re-entry (increasing the tissue spatial vulnerability). In the 2D and 3D models, the mutation also stabilized and accelerated re-entrant excitation waves, leading to rapid and sustained re-entry. In the 3D model, scroll waves under the mutation condition MT conditions also degenerated into persistent and erratic wavelets, leading to fibrillation. In conclusion, increased IKs due to the KCNQ1 S140G mutation increases atrial susceptibility to arrhythmia due to increased tissue vulnerability, shortened ERP and altered atrial conduction velocity, which, in combination, facilitate initiation and maintenance of re-entrant excitation waves.
Key points
A previous study has identified a gene mutation (KCNQ1 S140G) in some patients with a familial form of atrial fibrillation, one of the most common cardiac rhythm disturbances causing morbidity and mortality. A causal link between the mutation and genesis of atrial fibrillation has not yet been directly demonstrated.
Increased IKs arising from the KCNQ1 S140G mutation abbreviated atrial action potential duration (APD) and effective refractory period (ERP) and flattened APD and ERP restitution curves. It reduced atrial conduction velocity at low excitation rates, but increased it at high excitation rates that facilitated the conduction of high rate atrial excitation waves.
The mutation increased tissue susceptibility for initiation and maintenance of atrial arrhythmias.
The mutation stabilizes and accelerates re-entrant excitation waves, leading to rapid and sustained re-entry.
This study provides novel insights towards understanding the mechanisms underlying the pro-arrhythmic effects of the KCNQ1 S140G mutation.
Introduction
Atrial fibrillation (AF) is a common age-related arrhythmia causing morbidity, mortality and a significant burden on health care systems (Khoo & Lip, 2009; Sanoski, 2009). AF is associated with different conditions including cardiomegaly, thromboembolism and heart failure (Novo et al. 2008; Bourke & Boyle, 2009). Characterized by high rate atrial excitation, AF is associated with sustained erratic re-entrant circuits or ectopic focal activity (Nattel et al. 2000; Shiroshita-Takeshita et al. 2005). Whilst the underlying mechanisms of AF are as yet incompletely understood, organic cardiac disease (Danicek et al. 2008; Tanabe et al. 2009) and AF-induced electrical remodelling of ion channels (Bosch et al. 1999; Workman et al. 2001, 2008; Ashcroft, 2006) are major contributory factors to initiating and sustaining AF. However, lone AF patients in some instances have characteristic solitary gene defects with no other underlying organic disease (Fox et al. 2004; Fatkin et al. 2007; Campuzano & Brugada, 2009). Recent studies have identified several gene mutations in familial AF patients (Yang et al. 2004; Xia et al. 2005; Olson et al. 2006; Otway et al. 2007), including mutations in the KCNQ1 gene that encodes the pore forming (α) subunit of channels mediating the cardiac slow delayed rectifier potassium current (IKs) (Lundby et al. 2007; Ravn et al. 2008; Restier et al. 2008). In their study, Chen et al. (2003) identified a familial missense mutation of KCNQ1 at nucleotide 418 from adenine to guanine, which results in a change of amino acid from serine to glycine at position 140 (S140G) of the KCNE1 protein. Functional analysis of co-expressed wild-type (WT) and S140G mutant (MT) KCNQ1 with KCNE1 (the IKs channel β-subunit) revealed an increased IKs (Chen et al. 2003).
IKs is an important outward potassium current regulating late repolarisation of action potentials (APs) in cardiac cells. Abnormalities in IKs have been found in various cardiac diseases including long QT (Lundby et al. 2007; Zhang et al. 2008b) and short QT (Bellocq et al. 2004) syndromes. In a previous simulation study it has been shown that increased IKs due to a gain-in-function (V307L) KCNQ1 mutation in Short QT-2 syndrome dramatically abbreviates human ventricular AP duration (APD) and augments intra-ventricular heterogeneity, leading to increased ventricular susceptibility to arrhythmia (Zhang et al. 2008a). By contrast, whilst increased IKs due to the S140G KCNQ1 mutation has been suggested to increase the likelihood of atrial arrhythmia (Chen et al. 2003), this link remains to be demonstrated directly. Transgenic mice expressing S140G KCNQ1 have been reported to exhibit atrio-ventricular block, but failed to show AF (Yang et al. 2007), and at present there is no phenotypically accurate experimental model of S140G KCNQ1. Consequently, the mechanisms by which altered IKs caused by the S140G KCNQ1 mutation promote and perpetuate atrial arrhythmia have not yet been elucidated. Therefore, utilizing a computer modelling approach, the aim of this study was to quantify the pro-arrythmogenic effects of the KCNQ1 S140G mutation in the human atrium at cell, tissue and whole organ levels.
Methods
Model of IKs and atrial cell APs
The Courtemanche–Ramirez–Nattel (CRN) model for the human atrial cell AP (Courtemanche et al. 1998) was modified to incorporate the experimental data of Chen et al. (2003) on the changes to IKs due to the KCNQ1 S140G mutation. The CRN model was chosen as it incorporates available human atrial cell data and reproduces human atrial APs and APD rate dependence (Courtemanche et al. 1998). The model has been found to be well suited to the study of re-entrant arrhythmias in human atrium (e.g. Pandit et al. 2005; Seemann et al. 2006; Kharche et al. 2008a).
In the original CRN model, the IKs potassium (K+) current is modelled as:
| (1) |
where gKs is the current conductance (gKs= 0.129 nS pF−1), n the activation gate variable, V the membrane voltage, and EK the K+ reversal potential. The equations and parameters for the steady state properties of n are described in the original CRN model (Courtemanche et al. 1998).
The model was modified to incorporate experimental data of changes in IKs due to the KCNQ1 S140G mutation (Chen et al. 2003). Upon analysis of the experimental data of Chen et al. (2003) in terms of IKs records during voltage-clamp and the current–voltage (I–V) relationship, three major changes to IKs due to the mutation were identified. These changes include: (i) a large increase in the amplitude of IKs current at the end of voltage-clamp pulses; (ii) a substantial instantaneous component of IKs through the whole time course of voltage commands; and (iii) a substantial inward IKs at more hyperpolarized potentials that is not seen in the WT condition.
To model these changes to IKs due to the S140G KCNQ1 mutation as observed experimentally (Chen et al. 2003), equation (1) was modified by adding a non-gated but voltage-dependent instantaneous component. Therefore, the model for mutant 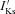 is described as:
is described as:
| (2) |
where gKs has the same value as the original CRN model, 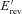 is the reversal potential of the instantaneous component, which was estimated to be −75.3 mV based on the experimental data of Chen et al. (2003), and ϕ is a scaling factor between 0 (WT; Control) and 1 (MT; homozygous) enabling simulation of possible intermediate mutant conditions (ϕ changes from 0 to 1).
is the reversal potential of the instantaneous component, which was estimated to be −75.3 mV based on the experimental data of Chen et al. (2003), and ϕ is a scaling factor between 0 (WT; Control) and 1 (MT; homozygous) enabling simulation of possible intermediate mutant conditions (ϕ changes from 0 to 1).
Modifications to IKs for the mutant condition were validated by reproducing the experimental data of Chen et al. on IKs I–V relationships (Chen et al. 2003) and IKs traces during voltage clamp commands under the WT (ϕ= 0) and the MT (ϕ= 1) conditions. Results are shown in Fig. 1. Figure 1A shows the voltage clamp protocol (Fig. 1Aa) as implemented by Chen et al. (Chen et al. 2003) and simulated IKs time courses during the voltage-clamp for the WT (ϕ= 0; Fig. 1Ab) and the MT (ϕ= 1; Fig. 1Ac) conditions. During the voltage clamp, the cell membrane potential was first held at −80 mV for 1 s, and then clamped at test potentials ranging from −130 mV to 50 mV by a step of 10 mV, each for a duration of 3 s before being clamped back to −40 mV for 1.5 s. Amplitudes of IKs at the end of the voltage clamp pulses were plotted as a function of the step voltages to obtain I–V relationships (Fig. 1B). In the MT condition, the simulated IKs shows an instantaneous component at the beginning of the test pulses, greater amplitude at the end of the test pulses and substantial component at more hyperpolarized potentials. These simulated changes of IKs under the MT condition match experimental data of Chen et al. (2003), as well as the simulated I–V relationships. In the following, simulation results are presented for ϕ= 0 (WT), ϕ= 0.1, ϕ= 0.25 for intermediate WT-MT states and ϕ= 1 for MT conditions.
Figure 1. Experimental and simulated IKs under WT and MT conditions, where experimental data are from Chen et al. (2003).
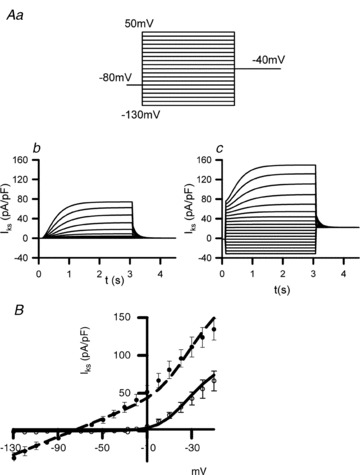
Aa, voltage-clamp protocol. Ab, simulated IKs traces under WT conditions. Ac, simulated IKs traces under MT conditions. B, experimental I–V relationships for IKs for WT (open circles) and S140G mutant IKs (filled circles) superimposed upon simulated I–V relationships for WT and MT cases (continuous lines).
Single cell simulations: APD and ERP restitution curves
Single cell APs were computed by conditioning the cell models 10 times at a pacing cycle length of 1 s (PCL) with supra-threshold stimuli of 2 nA pF−1 (duration of 2 ms). Such conditioning was sufficient to allow further elicitation of stable AP profiles and the 11th AP was used in analyses. APD90 was defined as the time duration between the AP upstroke and 90% repolarisation.
APD restitution (APDr) was computed using a standard S1–S2 protocol where a premature stimulus (S2) was applied after 10 conditioning stimuli (S1) at a PCL of 1 s. Diastolic interval (DI) was defined as the time interval between 90% repolarisation of the previous AP and the upstroke of final AP. A plot of APD90 against DI gave APDr. Maximum slopes of APDr curves were computed as indicators of spiral wave stability in tissue models (Qu et al. 1999; Kim et al. 2002).
Effective refractory period (ERP) was computed using the cell models with a modified S1–S2 stimulus protocol (Kharche et al. 2008a). For a variable PCL (S1), a premature S2 stimulus was applied and ERP was defined as the minimum S1–S2 interval that produced an AP peak potential over 80% of the last S1 AP peak potential (Workman et al. 2001). ERP restitution was determined for a large range of S1 values and ERP restitution curves were constructed by plotting ERP against the S1–S2 interval.
Multicellular 1D, 2D and 3D tissue models
The cell models were incorporated into a parabolic partial differential equation (PDE) to construct mono-domain spatial models of virtual cardiac electrical propagation. The PDE has the form
| (3) |
where Cm is cell membrane capacitance, D is the electrotonic diffusion coupling between cells simulating the gap junctional coupling, and Iion is the total membrane current. The value of D was taken to be 0.031 mm2 ms−1 (Kharche et al. 2007) that gave a conduction velocity (CV) of 0.27 mm ms−1 for a solitary planar wave under WT conditions. In the 1D and 2D models, spatial resolution was taken to be 0.1 mm. Such a space step is close to the length of atrial cells and gives stable numerical solutions. The length of the 1D atrial strand models was 20 mm, and the size of the 2D atrial sheet models was 37.5 × 37.5 mm2. In the 3D model, the anatomical model was previously reconstructed from the Visible Human Female project (Ackerman, 1991; Ackerman & Banvard, 2000) with a spatial resolution of 0.33 mm (Seemann et al. 2006). For the purposes of the present study, the 3D anatomical model was assumed to be electrically homogeneous.
1D strand simulations: CV restitution and temporal vulnerability
1D atrial strand models were used to compute the conduction velocity (CV) restitution curve (CVr) and tissue vulnerability to genesis of uni-directional conduction block.
To compute CVr, a conditioning pulse stimulus S1 was first applied at one end of the 1D strand, with a spatial size of 0.3 mm, and after a time delay a premature S2 stimulus was applied at the same location with the same size and strength. The CV of the S2 evoked excitation wave was computed as a function of the S1–S2 interval.
The temporal vulnerability of cardiac tissue was quantified by the width of a time window, during which a premature stimulus applied to the refractory tail of a preceding excitation wave produces uni-directional propagation (Quan & Rudy, 1990; Zhang et al. 2008a). To compute the vulnerable window (VW), the 1D tissue mode was first conditioned with a S1 evoked excitation wave applied at one end of the strand. After a time delay, a S2 stimulus was applied in the middle of the strand. Depending on the time delay, the S2 evoked excitation wave can either propagates bi-directionally, or uni-directionally in the retrograde direction of the conditioning wave, or fail to propagate. The time interval during which the S2 evoked excitation wave propagates uni-directionally was defined as the VW.
Critical substrates to initiate and sustain re-entry: the spatial vulnerability
2D models of homogeneous atrial tissues were used to compute the critical size of the S2 stimulus required to initiate and sustain re-entry (Kharche et al. 2008a). A conditioning planar wave was initiated at one edge of the 2D tissue and after a time delay, a S2 stimulus with variable length was applied in a localized part of the tissue during its temporal vulnerable window. Such a S2 stimulus evoked a conduction wave leading to a figure-of-eight re-entrant circuit with two tips rotating in opposite directions (one clockwise, the other anti-clockwise). If the length of S2 was sufficiently long, each of the two tips has sufficient space to follow its circuit path and the two spiral waves survive. However, if the length of S2 was short, there was insufficient space for the two tips to follow their paths. Then the two tips collide and the spiral wave terminates. The critical size of tissue to support re-entry was quantified as half of the minimal length of S2 that supports the formation and sustainability of spiral waves, which provides a reciprocal representation of spatial vulnerability of atrial tissue to re-entry: the smaller the critical size, the easier the initiation of re-entry in the tissue (Zou et al. 2005).
Initiation of re-entry in 2D and 3D models of the human atria
Re-entrant waves in the 2D model were initiated using a standard S1–S2 protocol, as used in a previous study (Leon et al. 1994). After initiation, 10 s of re-entrant electrical activity in the 2D sheets was computed. The stability of the re-entry was computed as the tip meander path and area of tip meander. Life span (LS) of the re-entry was computed as the time duration from initiation till dissipation of spiral wave tips. Dominant frequencies of virtual monophasic AP profiles from localized regions of the tips were computed using standard fast Fourier transform techniques.
In the 3D model, scroll waves were initiated using a protocol similar to that used in the 2D case (Kharche et al. 2007, 2008b), where two consecutive cross-field stimuli gave birth to a phase singularity, thus initiating a scroll wave. The scroll waves were initiated in the right atrial region to allow the initial evolution of the electrical excitations to develop unhindered. Six seconds of electrical activity was simulated for each case. The dominant frequency of localized atrial excitation from the 3D simulations was computed.
Numerical algorithms
The PDE models were solved by an explicit forward Euler approximation of the Laplacian operator. In the 1D and 2D simulations, the time step was 0.005 ms and space step 0.1 mm giving accurate numerical solutions. In the 3D simulations with a space step of 0.33 mm, a time step of 0.05 ms was seen to give solutions similar to the smaller time step of 0.005 ms (Kharche et al. 2007). Simulations were carried out on a distributed memory Bull Itanium2 208 64-bit CPU system with single rail Quadrics QsNet II interconnect, and on the UK National Grid Service 256 Opteron 64-bit CPU distributed memory system. Efficient parallelization was implemented using MPI parallel solvers (Kharche et al. 2008b).
Results
Effects of S140G-KCNQ1 increased IKs on atrial APs
Increased IKs due to the S140G KCNQ1 mutation abbreviated human atrial APs as shown in Fig. 2A. The measured APD90 was 314.2 ms under WT condition and 22.4 ms under the MT condition. The APD90 reduction was due to the mutation induced increase in IKs (Fig. 2B), and was associated with a decrease in the delayed rectifier K+ current, IKr (Fig. 2C) and in Ito (Fig. 2D). A marginal reduction of the L-type calcium current, ICa,L, (Fig. 2F) was also observed. Since the mutation was seen to cause such a dramatic abbreviation of the APD90, effects of altered IKs (IKs in eqn (2)) with variant intermediate proportions of the instantaneous component were investigated by a progressive increase of ϕ. Increase in ϕ resulted in a monotonic decrease in the APD (Fig. 2G). At ϕ= 0.1 (10%) and ϕ= 0.25 (25%), the measured APD90 was 147.5 ms and 79.32 ms respectively.
Figure 2. Simulated AP profiles and current traces under WT (continuous line), and MT conditions where simulated data for ϕ= 0.1 (dashed line), ϕ= 0.25 (dotted line) and ϕ= 1 (grey line) are shown in panels A–F.
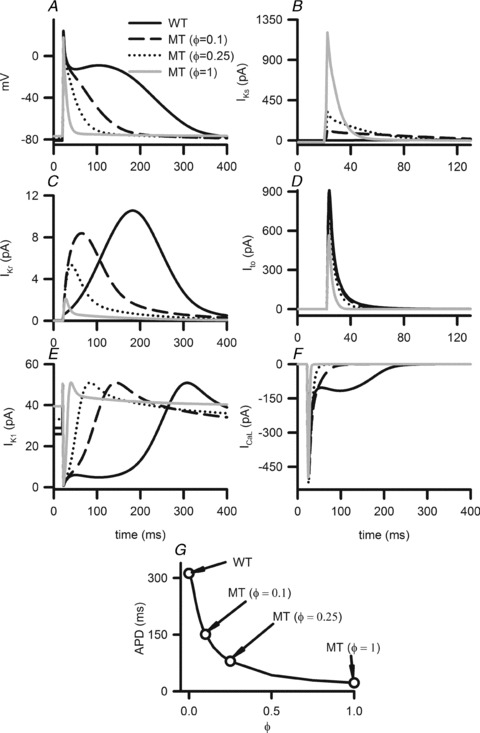
A, AP profiles. B, IKs profiles during APs. C, IKr profiles during APs. D, Ito profiles during APs. E, IK1 profiles during APs. F, ICaL profiles during APs. G, changes in APD as the modelling parameter ϕ is increased from 0 (WT) to 1 (MT) condition.
Effects of S140G-KCNQ1 increased IKs on APD and ERP restitution curves
Effects of the increased IKs due to the S140G KCNQ1 mutation on atrial APD restitution are shown in Fig. 3A. Across the range of DIs studied, the measured APD90 under the MT condition was considerably smaller than in the WT condition. MT conditions flattened APDr curves. The measured maximal APDr slopes were 1.3 (WT), 0.6 (ϕ= 0.1), 0.38 (ϕ= 0.25) and 0.05 (ϕ= 1) (Fig. 3B). Such a mutation induced loss of rate-dependent adaptation of APD manifested by flattened APDr curves was similar to that observed in chronic AF patients (Osaka et al. 2000; Workman et al. 2001; Watanabe et al. 2002) suggesting the increased ability of atrial cells to support high pacing rate electrical excitations that can be pro-arrhythmic.
Figure 3. APD and ERP restitution.
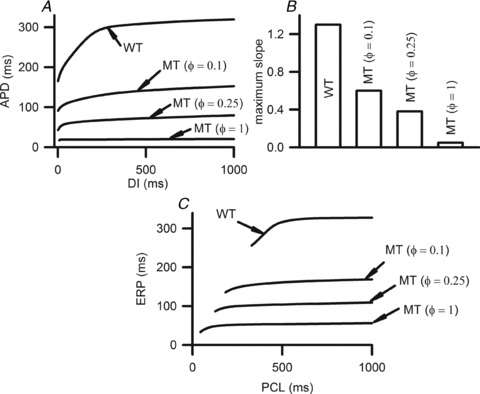
A, APDr curves under WT and MT conditions. B, maximum slopes of the APDr curves. C, ERP restitution curves under WT and MT conditions.
The mutation-induced increase in IKs reduced atrial ERP (Fig. 3C), which decreased from 319.6 ms in the WT condition to 169.1 ms, 109.5 ms and 56 ms in ϕ= 0.1, ϕ= 0.25 and ϕ= 1 conditions, respectively, as computed at a PCL of 1 s. The mutation shifted the ERP restitution curves leftward, demonstrating an increased ability of atrial cells to support high rate electrical activity.
Effects of S140G-KCNQ1 increased IKs on intra-atrial conduction velocity and tissue vulnerability to re-entry
Solitary wave conduction velocity (CV) in 1D atrial strands was decreased by the S140G mutation, markedly for the MT (ϕ= 1) case, but unnoticeably for the intermediate conditions studied. However, for all of the considered mutation conditions, the KCNQ1 S140G mutation facilitated conduction of excitation waves at higher rates that could not be conducted in WT tissue. This is shown in Fig. 4A by the leftward shift of the measured CVr under MT conditions. The measured maximum excitation rate was 185 beats min−1 under WT conditions, which increased to 393 beats min−1, 630 beats min−1, and 1298 beats min−1 in the ϕ= 0.1, ϕ= 0. 25 and ϕ= 1 conditions, respectively.
Figure 4. Simulated CVr and VW under WT and MT conditions.
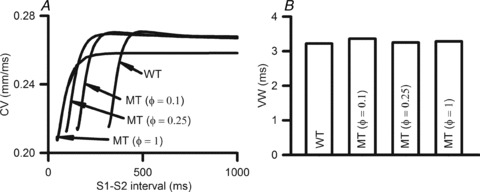
A, CVr curves under WT, ϕ= 0.1, ϕ= 0.25 and ϕ= 1 conditions. B, temporal VW under WT, ϕ= 0.1, ϕ= 0.25 and ϕ= 1 conditions.
Effects of S140G-KCNQ1 increased IKs on atrial tissue temporal and spatial vulnerability
The measured VW was increased (by 2.2%) by the mutation-induced increase of IKs. This is shown in Fig. 4B.
Measured spatial vulnerability of atrial tissue in the 2D model is shown in Fig. 5. Under the WT condition, the measured minimal substrate size was 49.6 mm. Under the MT conditions, the measured minimal size decreased to 7.45 mm (by 85%) when ϕ= 0.1, to 6.15 mm (by 87.6%) when ϕ= 0.25, and was lower than 0.6 mm in the ϕ= 1 case. Thus, mutant IKs caused a marked reduction in the substrate required to induce and sustaining re-entry, demonstrating a marked increase in tissue susceptibility to arrythmogenesis.
Figure 5. Critical length of the minimal substrate size for re-entry (MS) in 2D sheets.

Aa and b, Ba and b, illustration of MS required to induce a pair of re-entrant circuits in homogeneous 2D sheets. C, critical length under WT, ϕ= 0.1, ϕ= 0.25 and ϕ= 1 conditions. The critical length under mutant conditions was dramatically shorter than under Control conditions.
Effects of S140G-KCNQ1 increased IKs on dynamic behaviour of re-entrant excitation waves in 2D model
Figure 6 shows that increased IKs stabilized re-entrant spiral waves, which led to sustained re-entrant excitation in a limited atrial mass. In the WT condition, the initiated spiral wave was unstable. The tip meandered in a large area, which led to self-termination when it meandered out of the tissue boundaries or collided with its own repolarisation tail. The measured LS of spiral waves was 1.8 s in the WT condition. Power spectrum analysis of the recorded local electrical activity revealed a peak frequency of 3.5 Hz for WT. Under intermediate MT conditions (ϕ= 0.1 and ϕ= 0.25), re-entrant waves were stable, persistent and had limited meander throughout the 10 s simulation. Power spectrum analysis of the recorded electrical activity from the tissue revealed a peak frequency of 7.1 Hz and 9.8 Hz for ϕ= 0.1 and ϕ= 0.25, respectively. Similar behaviours of spiral waves were observed under the homozygous MT condition (ϕ= 1).
Figure 6. Simulation of spiral waves in 2D model of human atrium.
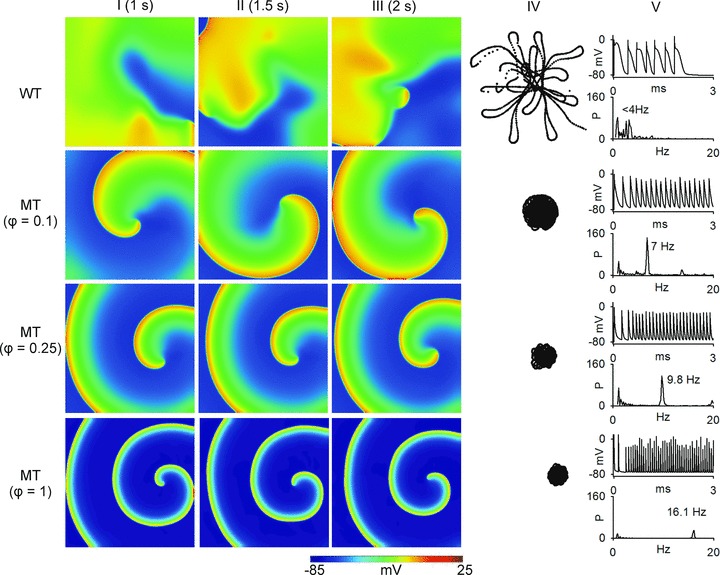
Top panels show results from 2D re-entrant wave simulation under WT condition; the second row of panels show results from ϕ= 0.1 conditions; the third row of panels show results from ϕ= 0.25 conditions; and the bottom row show results from ϕ= 1. Frames from the 2D simulation at time t= 1 s (column I), time t= 1.5 s (column II) and time t= 2 s (column III) are shown. Column IV shows the re-entrant wave tip trajectories. Column V shows time traces of localized electrical excitations (top) and dominant frequency (bottom).
Erratic re-entrant electrical excitation waves in 3D atria
Due to the complex geometry of atrial tissue, it cannot be assumed that accelerated and sustained re-entry under the mutation conditions necessarily translates into similar activity in a 3D tissue model. Therefore, further simulations were performed using 3D anatomical human atrial geometry. Results are shown in Fig. 7 for re-entrant excitation waves (scroll waves) under WT and intermediate MT (ϕ= 0.1) conditions. Under the WT condition, the scroll wave broke up into multiple re-entrant wavelets, with each meandering in a large area of the tissue leading to self-termination at 4.2 s. The observed self-termination of scroll waves was independent of their initiation location. Under the MT condition, the scroll wave also broke into multiple wavelets, each propagating erratically due to interaction with the complex atrial anatomy. The re-entrant excitations persisted throughout the duration of the simulation period of 6 s. The dominant frequency in the MT case was much higher (∼8.9 Hz at ϕ= 0.1) than under the WT condition (<3.5 Hz).
Figure 7. Simulation of scroll waves in 3D virtual human atrium.
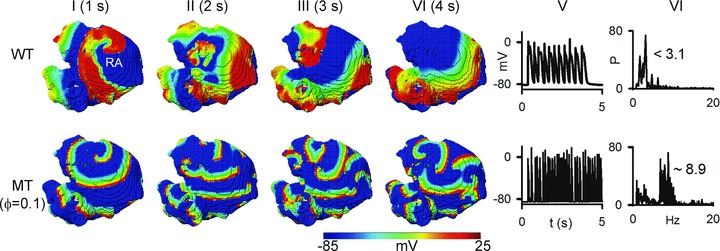
Top panels show representative frames from the simulation under WT condition and bottom panels show frames from the simulation under MT (ϕ= 0.1). Column I shows frames at time t= 1 s, Column II at time t= 2 s, Column III at time t= 3 s and Column IV at time t= 4 s. Column V shows time traces of localized electrical excitations. Column VI shows dominant frequencies.
A summary of the functional effects of KCNQ1 S140G mutated IKs on simulated human atrial electrical activity is listed in Table 1.
Table 1.
Quantitative summary of the effects of KCNQ1 (S140G) mutation on human atrial electrical activity
| Model | Quantity | Con (ϕ= 0) | MT (ϕ= 0.1) | MT (ϕ= 0.25) | MT (ϕ= 1) |
|---|---|---|---|---|---|
| Cell | Resting potential (mV) | −80.80 | −80.01 | −78.74 | −76.88 |
| APD (ms) | 314.2 | 147.5 | 79.3 | 22.4 | |
| Overshoot (mV) | 24.61 | 24.07 | 23.05 | 18.11 | |
| dV/dtmax (mV ms−1) | 217.10 | 214.17 | 208.58 | 193.44 | |
| APDr maximal slope | 1.3 | 0.6 | 0.38 | 0.05 | |
| ERP (ms) (stimulus interval ∼1 s) | 319.6 | 169.1 | 109.5 | 56.0 | |
| 1D | CV (mm ms−1) | 0.27 | 0.27 | 0.27 | 0.258 |
| VW (ms) | 3.22 | 3.36 | 3.25 | 3.22 | |
| Wavelength (mm) | 84.84 | 36.99 | 21.42 | 5.29 | |
| 2D | LS (s) | 1.42 | >10 | >10 | >10 |
| DF (Hz) | 3.5 | 7.1 | 9.8 | 16.1 | |
| Tip meander area (cm2) | 10.63 | 1.10 | 0.58 | 0.32 | |
| MS (mm) | 49.6 | 7.45 | 6.15 | 0.6 | |
| 3D | LS (s) | 4.2 | >6 | — | — |
| DF (Hz) | 3.1 | 8.9 | — | — |
Discussion
To our knowledge, this is the first study to investigate mechanisms underlying the genesis and maintenance of AF in patients with the KCNQ1 S140G mutation. Our major findings are the following. (i) The KCNQ1 S140G mutation increased IKs magnitude by inducing a voltage-dependent instantaneous component. This is similar to the constitutive component of IKs arising from the KCNQ1 V141M mutation responsible for atrial fibrillation and short QT syndrome (Hong et al. 2005). (ii) The mutation induced increase in IKs abbreviated and flattened APD and ERP restitution curves, leading to loss of rate-dependent adaptation of APD and ERP. (iii) The changes in cellular electrophysiology modulated atrial conduction at tissue level facilitating high rate conduction, and increased tissue temporal and spatial vulnerability. As a consequence, tissue susceptibility to AF genesis and maintenance was increased. (iv) The increase in IKs stabilized re-entry, allowing more wavelets to persist and co-exist in a limited size of tissue, leading to more irregular and frequent atrial excitation.
Mechanisms of pro-fibrillation of the KCNQ1 S140G mutation
Shortening of atrial ERP has been demonstrated to be pro-fibrillatory, as seen in the effects of vagal stimulation (Liu et al. 2009), acetylcholine (Voigt et al. 2008), or adenosine (Turley et al. 2008) on the initiation of AF. In the case of the KCNQ1 S140G mutation, the increased IKs abbreviated atrial APD and ERP. In turn, the shortened ERP led to reduced excitation wave wavelength facilitating high rate intra-atrial conduction, allowing more re-entrant wavelets to persist in a limited atrial mass. In the study of Chen et al. (2003), AF persisted in individual patients with the mutation once it appeared. This is consistent with our simulation that re-entry was sustained in the mutant condition when ϕ > 0.07.
The genesis of AF in patients with the KCNQ1 S140G mutation may be attributed to the increased susceptibility of atrial tissue to re-entry. Tissue susceptibility to re-entry can be indexed by both its temporal and spatial vulnerability. In the present study, the KCNQ1 S140G mutation has been shown to abbreviate atrial APD (and as a consequence ERP) and to be pro-arrhythmic at the tissue level. However, the pro-arrhythmic effects of the mutation were not reflected by significant changes to simulated atrial temporal vulnerability. The temporal vulnerability of cardiac tissue is measured as the width of a time window (vulnerable window; VW) during which a test stimulus applied to the refractory tail of a conditioning excitation wave evokes uni-directional conduction block that leads to re-entry. In this study, the VW was only slightly increased for atrial tissue incorporating the KCNQ1 S140G mutation. The VW is mainly determined by the repolarisation phase of APs (Shaw & Rudy, 1995). Therefore, whether or not an increase in tissue susceptibility to arrhythmogenesis is associated with an increase in tissue VW will be contingent upon whether the underlying condition is associated with APD (and ERP) prolongation, or with APD (and ERP) abbreviation. In the case of familial AF linked to the KCNQ1 S140G mutation, our simulations indicate that the S140G mutation abbreviates atrial APD (ERP), which results in a slightly increased temporal vulnerability (VW), despite the fact that the atrial susceptibility to arrhythmogenesis is increased. This observation is consistent with findings from a recent study by Moreno et al. (2011), that tissue temporal vulnerability is not, per se, a sufficient index for characterizing tissue susceptibility to arrhythmogenesis. On the other hand, tissue spatial vulnerability to arrhythmia is determined principally by the (reciprocal relationship to vulnerability of) critical tissue size required to accommodate the pathway of re-entry and thereby enable re-entrant excitation waves to become sustained. This index of arrhythmia susceptibility is related to the wavelength of the excitation wave, defined as the product of CV and APD. Therefore, an abbreviated APD (ERP) results in a shorter wavelength of excitation, requiring a smaller tissue substrate size to enable re-entry to become sustained. In our simulations, the KCNQ1 S140G mutation increased tissue spatial vulnerability by markedly decreasing the critical size of re-entrant substrate due to abbreviated APD and ERP. Consequently, atrial tissue susceptibility to maintain re-entry was increased. Clearly, therefore, to characterise the overall tissue susceptibility to arrhythmogenesis, both temporal and spatial vulnerability of cardiac tissue should be considered. In the case of the S140G KCNQ1 mutation, the increased spatial vulnerability appears to predominate over the slight increase in temporal VW in determining the overall pro-arrhythmic effect of the S140G mutation.
A pro-arrhythmic effect of abbreviated atrial APD and ERP with a single K+ channel subunit mutation has also been observed in simulations of the Kir2.1 V93I mutation (Kharche et al. 2008a) that has been identified in some familial AF patients (Xia et al. 2005). Computer modelling has shown that, in addition to abbreviating atrial APD and ERP, the Kir2.1 V93I mutation flattens atrial APD and ERP restitution curves (Kharche et al. 2008a). It also reduces atrial excitation wavelength, facilitating rapid and persistent re-entrant excitation waves. These observations for the Kir2.1 V93I mutant, which affects a molecularly and biophysically distinct K+ channel, are qualitatively similar to those observed here for KCNQ1 S140G mutant atrium. Therefore, the findings of the present study add to the growing weight of evidence implicating abbreviated APD and ERP in facilitating AF initiation and maintenance.
Limitations
The CRN model was used here to simulate cellular electrical activity (APs) of human atrial myocytes and its limitations have been discussed elsewhere (Courtemanche et al. 1998). In the multicellular tissue model, similar to other studies (Pandit et al. 2005; Kharche et al. 2008b), we assumed homogeneous cellular electrical properties and isotropic cell-to-cell electrical coupling. However, possible intrinsic electrical heterogeneity in cellular electrical properties (Seemann et al. 2006) and anisotropic intercellular electrical coupling may play important roles in initiation and perpetuation of re-entry. On the other hand, omitting such electrical heterogeneity and anisotropy can be useful (Pandit et al. 2005), in that changes to tissue behaviour observed in the present study (in terms of perpetuation and maintenance of re-entrant excitation) can be attributed with confidence to the implemented modifications to IKs. Another potential limitation is that we did not consider in our simulations AF-induced electrical remodelling (Bosch et al. 1999; Workman et al. 2001; Zhang et al. 2005; Kharche & Zhang, 2008) or gap junctional remodelling (van der Velden et al. 2000; Jongsma & Wilders, 2000), which influences the initiation and maintenance of AF. In addition, the tissue models in this study do not consider the effect of cardiac mechanics on tissue geometry, which feasibly might influence re-entry (Nash & Panfilov, 2004). Nevertheless, whilst it is important to make explicit the potential limitations of the approaches adopted in the present study, these potential limitations are not expected to influence fundamentally conclusions that can be drawn from our data on the mechanisms by which increased IKs due to the KCNQ1 S140G mutation can increase atrial arrhythmia risk.
Mutant IKs and cardiac arrhythmia – relevance to previous studies
Abnormality in IKs arising from KCNQ1 mutations has been identified in various cardiac diseases including the LQT syndrome (Lundby et al. 2007; Zhang et al. 2008a), the SQT syndrome (Bellocq et al. 2004) and atrial fibrillation (Brugada et al. 2004; Hong et al. 2005; Ellinor et al. 2006; Restier et al. 2008; Das et al. 2009; Abraham et al. 2010). Significantly, the gain-in-function of IKs associated KCNQ1 mutations can produce overlapped phenotypes of SQT and AF (Bellocq et al. 2004; Restier et al. 2008). Patients with these mutations that augment IKs exhibit shortened atrial and ventricular refractory periods, and increased susceptibility to atrial and ventricular fibrillation. It was hypothesized that a gain-in-function of IKs results in a shortened APD and ERP (Chen et al. 2003), which facilitate multiple re-entrant circuits in atrial and ventricular fibrillation. In a previous study (Zhang et al. 2009) we have shown that an augmented IKs arising from the (SQT-2) KCNQ1 V307L mutation shortened ventricular APD and ERP, increased intra-ventricular heterogeneity, led to abbreviated QT interval, and increased ventricular susceptibility to arrhythmia. Although, in principle the S140G KCNQ1 mutation would be predicted to increase IKs during both atrial and ventricular APs (El Harchi et al. 2010), patients with the S140G mutation were not reported to exhibit clear rate-corrected (QTC) interval abbreviation (Chen et al. 2003), possibly because irregularity in ventricular beats could influence QT representation and viability of simple QTC correction (Chen et al. 2003).
Accumulating evidence places particular emphasis on AF-induced electrical remodelling or electrophysiological modification due to genetic defects, in producing a pro-fibrillatory reduction in ERP (Yang et al. 1997; Hong et al. 2005; Olson et al. 2005, 2006; Xia et al. 2005; Otway et al. 2007; Restier et al. 2008; Ravens & Cerbai, 2008). The present study is both consistent with and extends this notion, indicating clearly a marked functional impact of the KCNQ1 S140G mutation on atrial cell and tissue electrophysiology that would promote susceptibility to AF. The general incidence of AF increases with age, with up to nearly 10% of octogenarians suffering from the condition (Nattel, 2002). However, ageing does not appear to have been a strong factor in the appearance of clinical symptoms in the family in whom the S140G KCNQ1 mutation was identified. The S140G KCNQ1 mutation was identified in a four-generation family, in whom structural heart disease and systemic disease were eliminated as the cause of AF (Chen et al. 2003). The proband was first identified at the age of 22, with an average age of diagnosis of AF for affected individuals of 24 (Chen et al. 2003). The average age of onset is likely to have been even younger than this (Chen et al. 2003). Thus, the substrate for arrhythmogensis in this heritable form of AF does not appear to be dependent upon ageing-related atrial remodelling. Our data are consistent with this. They indicate that in the case of the KCNQ1 S140G mutation, the ERP reduction and increase in spatial vulnerability to arrhythmia not only contribute to the substrate for maintaining AF through stabilizing re-entry, but also increase the susceptibility of human atrial tissue to the genesis of re-entry. Therefore, our study substantiates a causative link between the S140G KCNQ1 mutation and AF.
Acknowledgments
This work was supported by a Wellcome Trust project grant (081809/Z/06/Z). I.A. was supported by a basic sciences PhD studentship from the British Heart Foundation (FS/08/021 to H.Z. and J.C.H.).
Glossary
- AF
atrial fibrillation
- AP
action potential
- APD
action potential duration
- APD90
APD values at 90% repolarisation
- APDr
APD restitution curve
- CRN
Courtemanche–Ramirez–Nattel
- CV
conduction velocity
- DF
dominant frequency
- DI
diastolic interval
- ERP
effective refractory period
- ERPr
ERP restitution curve
- LS
lifespan
- MS
minimal substrate
- MT
mutant type
- PCL
pacing cycle length
- PV
pulmonary vein
- SI
stimulus time interval
- VW
vulnerable window (time)
Author contributions
H.Z., J.C.H. and M.B. conceived and designed the study. S.K., I.A., J.S. and P.L. performed numerical experiments and analysed data. All authors contributed to writing the paper and approved the manuscript.
References
- Abraham RL, Yang T, Blair M, Roden DM, Darbar D. Augmented potassium current is a shared phenotype for two genetic defects associated with familial atrial fibrillation. J Mol Cell Cardiol. 2010;48:181–190. doi: 10.1016/j.yjmcc.2009.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerman MJ. The Visible Human Project. J Biocommun. 1991;18:14. [PubMed] [Google Scholar]
- Ackerman MJ, Banvard RA. Imaging outcomes from the National Library of Medicine's Visible Human Project. Comput Med Imaging Graph. 2000;24:125–126. doi: 10.1016/s0895-6111(00)00012-4. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM. From molecule to malady. Nature. 2006;440:440–447. doi: 10.1038/nature04707. [DOI] [PubMed] [Google Scholar]
- Bellocq C, van Ginneken ACG, Bezzina CR, Alders M, Escande D, Mannens MMAM, Baró I, Wilde AAM. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation. 2004;109:2394–2397. doi: 10.1161/01.CIR.0000130409.72142.FE. [DOI] [PubMed] [Google Scholar]
- Bosch RF, Zeng X, Grammer JB, Popovic K, Mewis C, Kühlkamp V. Ionic mechanisms of electrical remodeling in human atrial fibrillation. Cardiovasc Res. 1999;44:121–131. doi: 10.1016/s0008-6363(99)00178-9. [DOI] [PubMed] [Google Scholar]
- Bourke T, Boyle NG. Atrial fibrillation and congestive heart failure. Minerva Med. 2009;100:137–143. [PubMed] [Google Scholar]
- Brugada R, Hong K, Dumaine R, Cordeiro J, Gaita F, Borggrefe M, Menendez TM, Brugada J, Pollevick GD, Wolpert C, Burashnikov E, Matsuo K, Wu YS, Guerchicoff A, Bianchi F, Giustetto C, Schimpf R, Brugada P, Antzelevitch C. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation. 2004;109:30–35. doi: 10.1161/01.CIR.0000109482.92774.3A. [DOI] [PubMed] [Google Scholar]
- Campuzano O, Brugada R. Genetics of familial atrial fibrillation. Europace. 2009;11:1267–1271. doi: 10.1093/europace/eup199. [DOI] [PubMed] [Google Scholar]
- Chen Y-H, et al. KCNQ1 gain-of-function mutation in familial atrial fibrillation. Science. 2003;299:251–254. doi: 10.1126/science.1077771. [DOI] [PubMed] [Google Scholar]
- Courtemanche M, Ramirez RJ, Nattel S. Ionic mechanisms underlying human atrial action potential properties: insights from a mathematical model. Am J Physiol Heart Circ Physiol. 1998;275:H301–H321. doi: 10.1152/ajpheart.1998.275.1.H301. [DOI] [PubMed] [Google Scholar]
- Danicek V, Theodorovich N, Bar-Chaim S, Miller A, Vered Z, Koren-Morag N, Uriel N, Czuriga I, Shopen A, Brantriss N, Kaluski E. Sinus rhythm restoration after atrial fibrillation: the clinical value of N-terminal pro-BNP measurements. Pacing Clin Electrophysiol. 2008;31:955–960. doi: 10.1111/j.1540-8159.2008.01121.x. [DOI] [PubMed] [Google Scholar]
- Das S, Makino S, Melman YF, Shea MA, Goyal SB, Rosenzweig A, Macrae CA, Ellinor PT. Mutation in the S3 segment of KCNQ1 results in familial lone atrial fibrillation. Heart Rhythm. 2009;6:1146–1153. doi: 10.1016/j.hrthm.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Harchi A, Zhang H, Hancox JC. The S140G KCNQ1 atrial fibrillation mutation affects “I(KS)” profile during both atrial and ventricular action potentials. J Physiol Pharmacol. 2010;61:759–764. [PubMed] [Google Scholar]
- Ellinor PT, Petrov-Kondratov VI, Zakharova E, Nam EG, MacRae CA. Potassium channel gene mutations rarely cause atrial fibrillation. BMC Med Genet. 2006;7:70. doi: 10.1186/1471-2350-7-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fatkin D, Otway R, Vandenberg JI. Genes and atrial fibrillation: a new look at an old problem. Circulation. 2007;116:782–792. doi: 10.1161/CIRCULATIONAHA.106.688889. [DOI] [PubMed] [Google Scholar]
- Fox CS, Parise H, D’Agostino RB, Sr, Lloyd-Jones DM, Vasan RS, Wang TJ, Levy D, Wolf PA, Benjamin EJ. Parental atrial fibrillation as a risk factor for atrial fibrillation in offspring. JAMA. 2004;291:2851–2855. doi: 10.1001/jama.291.23.2851. [DOI] [PubMed] [Google Scholar]
- Hong K, Piper DR, Diaz-Valdecantos A, Brugada J, Oliva A, Burashnikov E, Santos-de-Soto J, Grueso-Montero J, Diaz-Enfante E, Brugada P, Sachse F, Sanguinetti MC, Brugada R. De novo KCNQ1 mutation responsible for atrial fibrillation and short QT syndrome in utero. Cardiovasc Res. 2005;68:433–440. doi: 10.1016/j.cardiores.2005.06.023. [DOI] [PubMed] [Google Scholar]
- Jongsma HJ, Wilders R. Gap junctions in cardiovascular disease. Circ Res. 2000;86:1193–1197. doi: 10.1161/01.res.86.12.1193. [DOI] [PubMed] [Google Scholar]
- Kharche S, Zhang H. Simulating the effects of atrial fibrillation induced electrical remodeling: a comprehensive simulation study. Conf Proc IEEE Eng Med Biol Soc. 2008;2008:593–596. doi: 10.1109/IEMBS.2008.4649222. [DOI] [PubMed] [Google Scholar]
- Kharche S, Garratt CJ, Boyett MR, Inada S, Holden AV, Hancox JC, Zhang H. Atrial proarrhythmia due to increased inward rectifier current (IK1) arising from KCNJ2 mutation – a simulation study. Prog Biophys Mol Biol. 2008a;98:186–197. doi: 10.1016/j.pbiomolbio.2008.10.010. [DOI] [PubMed] [Google Scholar]
- Kharche S, Seemann G, Leng J, Holden AV, Garratt CJ, Zhang H. Proceedings of the 4th International Conference on Functional Imaging and Modeling of the Heart. Springer-Verlag, Berlin, Heidelberg: 2007. Scroll waves in 3D virtual human atria: a computational study; pp. 129–138. [Google Scholar]
- Kharche S, Seemann G, Margetts L, Leng J, Holden AV, Zhang H. Simulation of clinical electrophysiology in 3D human atria: a high-performance computing and high-performance visualization application. Concurrency and Computation: Practice and Experience. 2008b;20:1317–1328. [Google Scholar]
- Khoo CW, Lip GYH. Burden of atrial fibrillation. Curr Med Res Opin. 2009;25:1261–1263. doi: 10.1185/03007990902910443. [DOI] [PubMed] [Google Scholar]
- Kim B-S, Kim Y-H, Hwang G-S, Pak H-N, Lee SC, Shim WJ, Oh DJ, Ro YM. Action potential duration restitution kinetics in human atrial fibrillation. J Am Coll Cardiol. 2002;39:1329–1336. doi: 10.1016/s0735-1097(02)01760-6. [DOI] [PubMed] [Google Scholar]
- Leon LJ, Roberge FA, Vinet A. Simulation of two-dimensional anisotropic cardiac reentry: effects of the wavelength on the reentry characteristics. Ann Biomed Eng. 1994;22:592–609. doi: 10.1007/BF02368286. [DOI] [PubMed] [Google Scholar]
- Liu P, Guo J-H, Zhang H-C, Wang M-X, Li X-B, Zhang P, Yi Z, Sun J-L. Vagal effects on the occurrence of focal atrial fibrillation originating from the pulmonary veins. Circ J. 2009;73:48–54. doi: 10.1253/circj.cj-08-0513. [DOI] [PubMed] [Google Scholar]
- Lundby A, Ravn LS, Svendsen JH, Olesen S-P, Schmitt N. KCNQ1 mutation Q147R is associated with atrial fibrillation and prolonged QT interval. Heart Rhythm. 2007;4:1532–1541. doi: 10.1016/j.hrthm.2007.07.022. [DOI] [PubMed] [Google Scholar]
- Moreno JD, Zhu Zi, Yang PC, Bankston JR, Jeng MT, Kang C, Wang L, Bayer JD, Christini DJ, Trayanova NA, Ripplinger CM, Kass RS, Clancy CE. A computational model to predict the effects of class I anti-arrhythmic drugs on ventricular rhythms. Sci Transi Med. 2011;3:98ra83. doi: 10.1126/scitranslmed.3002588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash MP, Panfilov AV. Electromechanical model of excitable tissue to study reentrant cardiac arrhythmias. Prog Biophys Mol Biol. 2004;85:501–522. doi: 10.1016/j.pbiomolbio.2004.01.016. [DOI] [PubMed] [Google Scholar]
- Nattel S, Li D, Yue L. Basic mechanisms of atrial fibrillation – very new insights into very old ideas. Annu Rev Physiol. 2000;62:51–77. doi: 10.1146/annurev.physiol.62.1.51. [DOI] [PubMed] [Google Scholar]
- Nattel S. New ideas about atrial fibrillation 50 years on. Nature. 2002;415:219–26. doi: 10.1038/415219a. [DOI] [PubMed] [Google Scholar]
- Novo G, Mansueto P, La Franca ML, Di Leo R, Di Rosa S, Fazio G, Mansueto S, Ferrara F, Novo S. Risk factors, atrial fibrillation and thromboembolic events. Int Angiol. 2008;27:433–438. [PubMed] [Google Scholar]
- Olson TM, Alekseev AE, Liu XK, Park S, Zingman LV, Bienengraeber M, Sattiraju S, Ballew JD, Jahangir A, Terzic A. Kv1.5 channelopathy due to KCNA5 loss-of-function mutation causes human atrial fibrillation. Hum Mol Genet. 2006;15:2185–2191. doi: 10.1093/hmg/ddl143. [DOI] [PubMed] [Google Scholar]
- Olson TM, Michels VV, Ballew JD, Reyna SP, Karst ML, Herron KJ, Horton SC, Rodeheffer RJ, Anderson JL. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. 2005;293:447–454. doi: 10.1001/jama.293.4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osaka T, Itoh A, Kodama I. Action potential remodeling in the human right atrium with chronic lone atrial fibrillation. Pacing Clin Electrophysiol. 2000;23:960–965. doi: 10.1111/j.1540-8159.2000.tb00881.x. [DOI] [PubMed] [Google Scholar]
- Otway R, Vandenberg JI, Guo G, Varghese A, Castro ML, Liu J, Zhao J, Bursill JA, Wyse KR, Crotty H, Baddeley O, Walker B, Kuchar D, Thorburn C, Fatkin D. Stretch-sensitive KCNQ1 mutation: A link between genetic and environmental factors in the pathogenesis of atrial fibrillation? J Am Coll Cardiol. 2007;49:578–586. doi: 10.1016/j.jacc.2006.09.044. [DOI] [PubMed] [Google Scholar]
- Pandit SV, Berenfeld O, Anumonwo JMB, Zaritski RM, Kneller J, Nattel S, Jalife J. Ionic determinants of functional reentry in a 2-D model of human atrial cells during simulated chronic atrial fibrillation. Biophys J. 2005;88:3806–3821. doi: 10.1529/biophysj.105.060459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Z, Weiss JN, Garfinkel A. Cardiac electrical restitution properties and stability of reentrant spiral waves: a simulation study. Am J Physiol Heart Circ Physiol. 1999;276:H269–H283. doi: 10.1152/ajpheart.1999.276.1.H269. [DOI] [PubMed] [Google Scholar]
- Quan W, Rudy Y. Unidirectional block and reentry of cardiac excitation: a model study. Circ Res. 1990;66:367–382. doi: 10.1161/01.res.66.2.367. [DOI] [PubMed] [Google Scholar]
- Ravens U, Cerbai E. Role of potassium currents in cardiac arrhythmias. Europace. 2008;10:1133–1137. doi: 10.1093/europace/eun193. [DOI] [PubMed] [Google Scholar]
- Ravn LS, Aizawa Y, Pollevick GD, Hofman-Bang J, Cordeiro JM, Dixen U, Jensen G, Wu Y, Burashnikov E, Haunso S, Guerchicoff A, Hu D, Svendsen JH, Christiansen M, Antzelevitch C. Gain of function in IKs secondary to a mutation in KCNE5 associated with atrial fibrillation. Heart Rhythm. 2008;5:427–435. doi: 10.1016/j.hrthm.2007.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restier L, Cheng L, Sanguinetti MC. Mechanisms by which atrial fibrillation-associated mutations in the S1 domain of KCNQ1 slow deactivation of IKs channels. J Physiol. 2008;586:4179–4191. doi: 10.1113/jphysiol.2008.157511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanoski CA. Clinical, economic, and quality of life impact of atrial fibrillation. J Manag Care Pharm. 2009;15:S4–S9. doi: 10.18553/jmcp.2009.15.s6-b.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seemann G, Höper C, Sachse FB, Dössel O, Holden AV, Zhang H. Heterogeneous three-dimensional anatomical and electrophysiological model of human atria. Philos Transact A Math Phys Eng Sci. 2006;364:1465–1481. doi: 10.1098/rsta.2006.1781. [DOI] [PubMed] [Google Scholar]
- Shaw RM, Rudy Y. The vulnerable window for unidirectional block in cardiac tissue: characterization and dependence on membrane excitability and intercellular coupling. J Cardiovasc Electrophysiol. 1995;6:115–131. doi: 10.1111/j.1540-8167.1995.tb00763.x. [DOI] [PubMed] [Google Scholar]
- Shiroshita-Takeshita A, Brundel BJJM, Nattel S. Atrial fibrillation: basic mechanisms, remodeling and triggers. J Interv Card Electrophysiol. 2005;13:181–193. doi: 10.1007/s10840-005-2362-y. [DOI] [PubMed] [Google Scholar]
- Tanabe Y, Kawamura Y, Sakamoto N, Sato N, Kikuchi K, Hasebe N. Blood pressure control and the reduction of left atrial overload is essential for controlling atrial fibrillation. Int Heart J. 2009;50:445–456. doi: 10.1536/ihj.50.445. [DOI] [PubMed] [Google Scholar]
- Turley AJ, Murray S, Thambyrajah J. Pre-excited atrial fibrillation triggered by intravenous adenosine: a commonly used drug with potentially life-threatening adverse effects. Emerg Med J. 2008;25:46–48. doi: 10.1136/emj.2007.051227. [DOI] [PubMed] [Google Scholar]
- van der Velden HM, Ausma J, Rook MB, Hellemons AJ, van Veen TA, Allessie MA, Jongsma HJ. Gap junctional remodeling in relation to stabilization of atrial fibrillation in the goat. Cardiovasc Res. 2000;46:476–486. doi: 10.1016/s0008-6363(00)00026-2. [DOI] [PubMed] [Google Scholar]
- Voigt N, Maguy A, Yeh Y-H, Qi X, Ravens U, Dobrev D, Nattel S. Changes in IK, ACh single-channel activity with atrial tachycardia remodelling in canine atrial cardiomyocytes. Cardiovasc Res. 2008;77:35–43. doi: 10.1093/cvr/cvm051. [DOI] [PubMed] [Google Scholar]
- Watanabe I, Masaki R, Ohkubo K, Okumura Y, Yamada T, Oshikawa N, Saito S, Ozawa Y, Kanmatsuse K. Rate-dependent changes in atrial action potential duration after short-duration rapid atrial pacing in humans. Circ J. 2002;66:874–875. doi: 10.1253/circj.66.874. [DOI] [PubMed] [Google Scholar]
- Workman AJ, Kane KA, Rankin AC. The contribution of ionic currents to changes in refractoriness of human atrial myocytes associated with chronic atrial fibrillation. Cardiovasc Res. 2001;52:226–235. doi: 10.1016/s0008-6363(01)00380-7. [DOI] [PubMed] [Google Scholar]
- Workman AJ, Kane KA, Rankin AC. Cellular bases for human atrial fibrillation. Heart Rhythm. 2008;5:S1–S6. doi: 10.1016/j.hrthm.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia M, Xia M, Jin Q, Bendahhou S, He Y, Larroque MM, Chen Y, Zhou Q, Yang Y, Liu Y, Liu B, Zhu Q, Zhou Y, Lin J, Liang B, Li L, Dong X, Pan Z, Wang R, Wan H, Qiu W, Eurlings P, Barhanin J, Chen Y. A Kir2.1 gain-of-function mutation underlies familial atrial fibrillation. Biochem Biophys Res Commun. 2005;332:1012–1019. doi: 10.1016/j.bbrc.2005.05.054. [DOI] [PubMed] [Google Scholar]
- Yang T, Snyders DJ, Roden DM. Rapid inactivation determines the rectification and [K+]o dependence of the rapid component of the delayed rectifier K+ current in cardiac cells. Circ Res. 1997;80:782–789. doi: 10.1161/01.res.80.6.782. [DOI] [PubMed] [Google Scholar]
- Yang Y, Yang Y, Liu Y, Dong X, Kuang Y, Lin J, Su X, Peng L, Jin Q, He Y, Liu B, Pan Z, Li J, Zhu Q, Lin X, Zhou Q, Pan Q, Eurlings PM, Fei J, Wang Z, Chen YH. Human KCNQ1 S140G mutation is associated with atrioventricular blocks. Heart Rhythm. 2007;4:611–618. doi: 10.1016/j.hrthm.2007.01.029. [DOI] [PubMed] [Google Scholar]
- Yang Y, Yang Y, Xia M, Jin Q, Bendahhou S, Shi J, Chen Y, Liang B, Lin J, Liu Y, Liu B, Zhou Q, Zhang D, Wang R, Ma N, Su X, Niu K, Pei Y, Xu W, Chen Z, Wan H, Cui J, Barhanin J, Chen Y. Identification of a KCNE2 gain-of-function mutation in patients with familial atrial fibrillation. Am J Hum Genet. 2004;75:899–905. doi: 10.1086/425342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Garratt CJ, Kharche S, Holden AV. Remodelling of cellular excitation (reaction) and intercellular coupling (diffusion) by chronic atrial fibrillation represented by a reaction-diffusion system. Physica D Nonlinear Phenomena. 2009;238:976–983. [Google Scholar]
- Zhang H, Garratt CJ, Zhu J, Holden AV. Role of up-regulation of IK1 in action potential shortening associated with atrial fibrillation in humans. Cardiovasc Res. 2005;66:493–502. doi: 10.1016/j.cardiores.2005.01.020. [DOI] [PubMed] [Google Scholar]
- Zhang H, Kharche S, Holden AV, Hancox JC. Repolarisation and vulnerability to re-entry in the human heart with short QT syndrome arising from KCNQ1 mutation – a simulation study. Prog Biophys Mol Biol. 2008a;96:112–131. doi: 10.1016/j.pbiomolbio.2007.07.020. [DOI] [PubMed] [Google Scholar]
- Zhang S, Yin K, Ren X, Wang P, Zhang S, Cheng L, Yang J, Liu JY, Liu M, Wang QK. Identification of a novel KCNQ1 mutation associated with both Jervell and Lange-Nielsen and Romano-Ward forms of long QT syndrome in a Chinese family. BMC Med Genet. 2008b;9:24. doi: 10.1186/1471-2350-9-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou R, Kneller J, Leon LJ, Nattel S. Substrate size as a determinant of fibrillatory activity maintenance in a mathematical model of canine atrium. Am J Physiol Heart Circ Physiol. 2005;289:H1002–H1012. doi: 10.1152/ajpheart.00252.2005. [DOI] [PubMed] [Google Scholar]