

Abstract
Intervertebral disc degeneration (IDD) is a common and debilitating disorder that results in reduced flexibility of the spine, pain, and reduced mobility. Risk factors for IDD include age, genetic predisposition, injury, and other environmental factors such as smoking. Loss of proteoglycans (PGs) contributes to IDD with advancing age. Currently there is a lack of a model for rapid investigation of disc aging and evaluation of therapeutic interventions. Here we examined progression of disc aging in a murine model of a human progeroid syndrome caused by deficiency of the DNA repair endonuclease, ERCC1–XPF (Ercc1−/Δ mice). The ERCC1-deficient mice showed loss of disc height and degenerative structural changes in their vertebral bodies similar to those reported for old rodents. Compared to their wild-type littermates, Ercc1−/Δ mice also exhibit other age-related IDD characteristics, including premature loss of disc PG, reduced matrix PG synthesis, and enhanced apoptosis and cell senescence. Finally, the onset of age-associated disc pathologies was further accelerated in Ercc1−/Δ mice following chronic treatment with the chemotherapeutic agent mechlorethamine. These results demonstrate that Ercc1−/Δ mice represent an accurate and rapid model of disc aging and provide novel evidence that DNA damage negatively impacts PG synthesis.
Keywords: intervertebral disc degeneration, aging, DNA repair, proteoglycan, mouse models
Intervertebral disc degeneration (IDD) is the root cause of many musculoskeletal disorders of the spine, leading to incalculable distress as well as tremendous economic loss due to its impact on work performance.1 The etiology of IDD is complex and still not well understood. Thus, current treatment options often address symptoms with little understanding of the mechanism of action. Although the efficacy varies from case to case, surgery and palliative interventions are nevertheless still the most effective available treatment modalities in spine care.
Genetic predisposition,2 excessive load,3 and cigarette smoking4 are important contributors to IDD but may only affect a subset of the IDD patient population. Aging, on the other hand, is a ubiquitous contributor to IDD in elderly patients. The incidence of disc degeneration markedly increase with age.5 Moreover, aging inevitably brings about the loss of disc matrix proteoglycans (PGs), a class of negatively charged glycosylated proteins that are critical for counteracting mechanical compressive forces to the spine.6–9 Because IDD is also invariably associated with loss of disc PG, these observations further emphasize the contribution of aging to IDD.
Understanding the complex etiology of IDD requires animal models of different contributive processes. Models to study how genetic makeup10 or injury11–14 contributes to IDD have been established. In addition, several models are presently available to evaluate the contribution of aging to IDD, but each has its own limitation. The sand rat15 and chondrodystrophoid16 dog are spontaneous models of age-related IDD, but they have incomplete disease penetrance and undefined genetic causes. Rabbit aging model17 requires an extended length of time and is restricted by limited availability of commercially research reagents (e.g., antibodies). The study using patient samples, on the other hand, is limited by the dearth of specimens from normal controls. Furthermore, there is tremendous variability in the genetic and environmental exposures between patients. This underscores the need for a rapid animal model of disc aging in which genetic and environmental variables can be controlled.
Mice offer several advantages as a model for human disc aging and degeneration. Mouse lumbar discs were recently demonstrated to be proportionally and geometrically most similar to human discs among all of the animal models tested.18 Mouse lumbar motion segments also exhibit mechanical properties such as compression and torsion stiffness similar to those of humans,19 providing further validation for mouse disc as a mechanical model of the human disc. Other obvious benefits of using mice include low cost, availability of reagents (e.g., antibodies), genomic database, and ease of genetic and surgical manipulation. Hence the availability of a murine model of the aging contribution to disc degeneration would be of tremendous benefit for studying cellular and molecular mechanisms of age-related IDD and for rapidly evaluating therapeutic interventions aimed at preventing or treating changes associated with disc aging.
Deficiency of the DNA repair nuclease ERCC1–XPF causes a dramatic progeroid syndrome, or disease of accelerated aging, affecting the epidermal, neurological, renal, hepatobiliary, musculoskeletal, hematopoietic, and endocrine systems.20 Similarly, genetic deletion of either Ercc1 or Xpf in the mouse causes dramatically accelerated aging.21,22 While progeria (accelerated aging) is not necessarily identical to natural aging, there are however a remarkable number of important similarities between the two. For example, the genome-wide expression profile of multiple tissues of ERCC1-deficient mice has a highly significant correlation with that of naturally aged mice.20,23 Similarly, the histopathology observed in multiple tissues of ERCC1-deficient mice mimics that of old wild-type mice. ERCC1-deficient mice lose functional stem cells, like aged mice.24 This has led to the conclusion that these mice and other strains that age rapidly due to defects in genome maintenance mechanisms are excellent models to probe the underlying mechanisms of age-related pathologies.25,26
In this study, we asked if ERCC1-deficient mice have early onset age-associated changes in the disc. We focused on the Ercc1−/Δ strain, which express 10% of the normal endogenous level of ERCC1–XPF and have a maximum lifespan of 7 months compared to 2–3 years in wild-type mice22,27 (Niedernhofer, submitted). We discovered that the ERCC1-deficient mice indeed have premature onset of many key features of normal disc aging. These mice represent a new and rapid murine model of disc aging that reveal novel information about a type of cellular damage that can promote disc degeneration.
MATERIALS AND METHODS
Isolation of Mouse Intervertebral Discs and Nucleus Pulposus (NP) Tissue
The experiments involving mice were approved by the University of Pittsburgh Institutional Animal Care and Use Committee. Ercc1−/Δ and Ercc1−/− were bred and genotyped by PCR as previously described.20,27 The spines were isolated from euthanized mice and dissected with the aid of a 5× magnifier, and entire intervertebral discs (IVDs) were removed en bloc from the surrounding vertebral bodies by creating an incision along the endplates. To harvest NP tissue, an axial cut was made on the disc side of the endplate to expose the disc center, followed by gentle aspiration of the NP tissue using a P-10 pipette tip under a dissecting microscope (20–40× magnification, Nikon SMZ645).
Histological Staining
Isolated spines were decalcified and embedded in paraffin (Tissue Tek processor and Leica embedder). Seven micrometer sections were stained with either hematoxylin and eosin (H&E) or safranin O and fast green dyes (Fisher Scientific, Pittsburgh, PA) by standard procedure and photographed under 40–200× magnification (Nikon Eclipse Ts100).
Quantitative Immunofluorescence
Lumbar discs from three mice per group (20- to 23-week-old Ercc1−/Δ and Wt littermate and 2- to 2.5-year-old Wt mice) were isolated. Five micrometer frozen sections were washed and blocked with 50 μg/ml purified donkey IgG at 25°C for 30 min, followed by a 60 min incubation with the primary antibody (rabbit polyclonal IgG anti-aggrecan, amino acids 1177–1326 of mouse aggrecan epitope, Millipore, Temecula, CA, ab1031). After washing, the sections were incubated for 60 min in a mixture of fluorescent-labeled secondary antibody (Alexa Fluor 488 conjugated donkey anti-rabbit IgG(H + L)) and Alexa Fluor 647 Phalloidin1 to stain cytoplasmic actin, then washed and mounted in Prolong Gold anti-fade reagent with DAPI (Invitrogen, Carlsbad, CA, P36935) to stain for nuclei. Quantitative fluorescent confocal microscopy (fluoview1000) was used to measure the number and distribution of a variety of fluorescently labeled cells and extracellular matrix molecules. At least five random fields in five sections from each tissue sample were imaged at 40× and the quantitation of the aggrecan fluorescent signal was done using Metamorph software (Universal Imaging, West Chester, PA). Average values from 15 fields (5 field/NP × 3 mice) were calculated with 1 SE.
1,9-Dimethylmethylene Blue (DMB) Colorimetric Assay for Sulfated Glycosaminoglycans (GAGs)
Three Ercc1−/Δ and three wild-type littermate mice (20- to 23-week old) were sacrificed for this assay. NP tissue was isolated and pooled from six lumbar IVDs of each mouse. The pooled tissue sample was digested using papain at 60°C for 2 h. GAG content was measured in duplicate by DMB procedure28 using chondroitin-6-sulfate (Sigma, Milwaukee, WI, C-8529) as a standard. The DNA concentration of each sample was measured using the PicoGreen assay (Molecular Probes, Sunnyvale, CA) and used to normalize the GAG values. Average values from six reaction samples (two duplicates × three mice per group) were calculated with 1 SE.
Quantitation of PG Synthesis
Disc organ cultures were performed using isolated functional spine units (FSU) each consisting of vertebra, disc, vertebra without endplates at the two ends of each FSU. Four lumbar FSUs were cultured in complete growth medium (F-12/D-MEM containing 10% FCS, 1% PS, and 25 μg/ml L-ascorbic acid) for 2 days to equilibrate after the trauma of surgical dissection, followed by 3-day incubation with 35S-sulfate (20 μCi/ml). Individual discs were dissected from the FSUs under a 5× magnifier, ground using a micropestle and 1.5 ml microfuge tube (USA Scientific, Ocala, FL) in 0.1 ml homogenizing buffer (20 mM Tris–HCl, 200 mM NaCl, 100 mM glycine, 0.1% Triton X-100, 50 μM DTT, 0.1 mg/ml soybean trypsin inhibitor) with 4 M guanidine–HCl and extracted for 2 days with rotation at 4°C. Incorporated 35S-sulfate was separated from free sulfate using size exclusion PD-10 columns, and radioactivity in the fixed fraction quantified by liquid scintillation counting as previously described.29 The rate of PG synthesis was calculated as the pmoles of sulfate incorporated per μg DNA. Average values from six measurements (duplicates × three mice per group) are shown ± 1 SE.
Radiographic Analysis of Intervertebral Disc Height
X-ray radiographs were taken of 3-week-old Ercc1−/− mutant and wt littermate mice and 2-year-old wt mice as previously described.20 In addition, three-dimensional reconstruction of the lumbar vertebrae was performed using microcomputed tomography of the lumbar spines isolated from 20-week-old Ercc1−/Δ mice and their wt littermates as well as 2-year-old wt mice. Images were acquired using a VivaCT 40 (Scanco Medical, Bassersdorf, Switzerland) at 15-μm isotropic voxel size resolution, 55 kVp of energy, and 145 μA of current. The relative heights of the lumbar IVDs were measured from the radiographs and are reported as the percent of the length of the adjacent vertebral body plus the disc thickness.30 For each disc height measurement, magnified (10–20×) radiograph sections were divided into four equal quarters by three lines (Fig. 1A). Percent disc height of were calculated as an average of the three measurements for each disc. For each mouse group, the percent disc height was calculated as the average of nine lumbar discs, three discs (L2–L5) from each of the three mice, with 1 SE. Calculations made by three researchers (inter-observer variability) yielded consistent results, and nonoverlapping of standard error between ERCC1-deficient mice and their Wt littermates suggest that the difference in their relative disc heights is significant.
Figure 1.

Intervertebral disc height of aged and young wild-type, and progeroid ERCC1-deficient mice. (A) The percent disc height was calculated as the ratio of the average of disc height to sum of the length of the vertebrae and disc height, to correct for differences in spine length due to the various size of the different animals. (B) Radiographs of the lumbar vertebra of a 3-week-old wild-type mouse (top), its Ercc1−/− littermate (middle), and a 2-year-old wild-type mouse (bottom). (C) The percent of lumbar disc heights as determined from measurements made from X-ray (top) or micro-CT (bottom). Data reported were average ± standard error of three discs from at least three mice per group (micro-CT).
Measuring Apoptosis and Cell Senescence
Frozen sections of discs were treated with the TUNEL reagents (Roche, Branchburg, NJ) to detect apoptotic cells as described by the manufacturer and counterstained with the nuclear stain Hoestch to detect total cells in the specimens. Immunohistochemistry was used to localize the senescence marker P16INK4a in disc tissue using tonsil tissue as a positive control. Briefly, slides containing 4 μm frozen sections of discs were fixed in cold acetone for 10 min, washed 3× in PBS, and incubated in Vector M.O.M (Vector Lab, Burlingame, CA) for 1 h to eliminate background binding mouse primary monoclonal antibodies on mouse tissues. Sections were incubated overnight at 4°C with mouse monoclonal primary antibody against p16INK4a (Santa Cruz Biotech, Santa Cruz, CA; 1:50 dilution). After washing with PBS, sections were incubated with biotinylated horse anti-mouse serum (1:200; Vector Lab) for 30 min at room temperature, followed by biotin detection for 30 min using ABC/horse radish peroxidase (ABC Elite; Vector Lab) and AEC chromogen substrate (Sky Teck labs, Mississauga, Ontario, Canada) for 10 min at RT. Sections were counterstained with aqueous hematoxylin and blue Scott’s T H2O.
Quantitation of Matrix Gene Expression
Total RNA was purified from NP tissue using the RNeasy Mini kit (Qiagen, Germantown, MD). The RNA was analyzed in duplicate reactions by real-time RT-PCR (iCycler IQ4, Bio-Rad, Hercules, CA) to determine aggrecan and versican mRNA levels.
The cycle threshold (Ct) values were obtained and normalized to the housekeeping gene GAPDH. The ΔΔCt method13 was used to calculate the relative mRNA levels of each target gene between Ercc1−/Δ mice and wild-type littermates. Average values from six measurements (two duplicate × three mice per group) are shown ± 1 SE.
Treatment of Mice with Mechlorethamine
Ercc1−/Δ mice (n = 6) and their wild-type littermates were chronically exposed to genotoxic stress by administration of a subtoxic dose of the chemotherapeutic agent mechlorethamine (MEC).31 Mice were injected subcutaneously with 8 μg/kg MEC once per week for 6 weeks, beginning at 8 weeks of age. At 20 weeks of age they were euthanized and tissues were collected for analysis.
| Gene | Forward (5′ → 3′) | Reverse (5′ → 3′) |
|---|---|---|
| Aggrecan | ATACCCCATCCACACGCCCCG | GCGAAGCAGTACACATCATAGG |
| Versican | AAGGAGAAGTTCGAGCAGCAACAG | AACCTTCCCAGGTAGCCAAATCAC |
| GAPDH | GGCAAATTCAACGGCACAGTCAAG | AAGACACCAGTAGACTCCACGACA |
RESULTS
ERCC1-Deficient Mice Display Age-Related Degenerative Changes in Their Spines
We measured the disc height of lumbar discs in ERCC1-deficient and old wild-type mice by micro-CT and X-ray. The data are reported as the percent disc height, which is defined as the ratio of the disc height to the total vertebra and disc height to correct for the differences in the size of the mice of various ages and genotypes (Fig. 1A). Disc height, as measured by micro-CT, was reduced 20–30% in 20-week-old Ercc1−/Δ mice relative to their wild-type littermates (Fig. 1). This is equivalent to the loss of disc height observed with natural aging in 2-year-old mice (Fig. 1). The percent disc height of 3-week-old Ercc1−/− mice, as measured by X-ray, was also significantly reduced and comparable to old wild-type mice (Fig. 1). Therefore, there is premature loss of IVD height in ERCC1-deficient mice, a hallmark feature of aged and degenerated discs.32 In addition, the Ercc1−/Δ mice show kyphosis, a pathologic convex curving of the thoracic spine due to structural abnormalities, and other degenerative changes in their vertebral bodies, including loss of bone mineralization and increased porosity (Niedernhofer, submitted), similar to those observed in aged rodents.33
Ercc1−/Δ Mice Display Age-Dependent Loss of Matrix PGs in Their Intervertebral Discs
A second hallmark feature of disc aging and disc degeneration is progressive loss of matrix PGs. NP from 3-week-old Ercc1−/Δ mice were gelatinous and similar to those of wild-type littermates (Fig. 2A). However, by 8 weeks of age, the NP of Ercc1−/Δ mice were noticeably less gelatinous and more fibrotic compared to those of wild-type littermates (Fig. 2A). The difference between the NP of Ercc1−/Δ mice and littermate controls became more exaggerated at 20 weeks of age (Fig. 2A). The structural changes observed in the Ercc1−/Δ mice were similar to those observed with natural aging in mice >2 years old (Fig. 2A).
Figure 2.

Progressive, spontaneous loss of intervertebral disc hydration and PGs with age. (A) Gross images of axially dissected discs from Ercc1−/Δ mice and wild-type littermates at 3, 8, and 20 weeks of age, as well as from old wild-type mice (27 months). (B) Safranin O and fast green staining of discs of 7- and 20-week-old Ercc1−/Δ mice and their Wt littermates and natural aged Wt mouse disc (27 months). (C) Quantitation of NP GAG. GAG levels from NP tissues of 18–20 weeks old Ercc1−/Δ mice were measured using 1,9-dimethylmethylene assay against GAG standards (chondroitin-6-sulfate) and normalized to total DNA content. Values are shown as averages from six independent experiments ± standard error. Safranin N/A: not available since Ercc1−/Δ mice die by 6–7 months. NP, nucleus pulposus; AF, annulus fibrosis; EP, endplate.
Safranin O staining of sulfated PGs revealed a substantial reduction of PGs in the discs of Ercc1−/Δ mice compared to those from wild-type littermates (Fig. 2B). The difference was already apparent at 7 weeks of age and became more pronounced by 20 weeks (Fig. 2B). Reduced PGs in Ercc1−/Δ mice was specific for the IVD; the vertebral endplates were largely unaffected. Reduced NP PGs were also observed in discs of naturally aged mice (Fig. 2B). Quantitative 1,9-dimethylmethylene blue (DMB) assay measuring total NP sulfated glycosaminoglycans (GAGs) of PGs produced similar results (Fig. 2B). The GAG content of the NP was reduced in 20-week-old Ercc1−/Δ mice (3 ± 1 μg GAG/ng DNA) compared to 20-week-old wild-type mice (5.1 ± 1.2 μg GAG/ng DNA).
To further analyze PGs in the discs of ERCC1-deficient mice, we measured the level of aggrecan, a major PG constituent responsible for the osmotic turgidity of the disc,6 by quantitative immunofluorescence (Fig. 3A, see the Materials and Methods Section). Aggrecan was significantly reduced in progeroid Ercc1−/Δ mice at 18–20 weeks of age, compared to their wild-type littermates, but only slightly reduced at 3–5 weeks of age when the mice are still healthy and show no signs of aging (Fig. 3A). The extent of reduction in aggrecan was similar to that seen in wild-type mice >2 years of age. Together, these data demonstrate progressive, accelerated loss of disc NP structure and matrix PGs in the ERCC1-deficient mice similar to that which occurs with natural aging.
Figure 3.
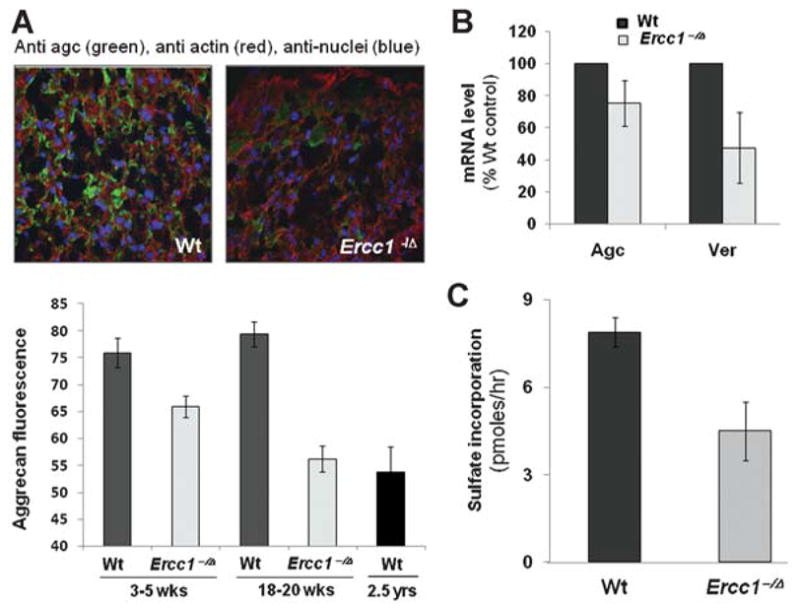
Decreased PG production with aging. (A) Axial sections of mouse discs were immunostained for extracellular aggrecan (green) and counterstained with phalloidin for cytoplasmic actin (red) and DAPI for cellular nuclei (blue). Average values of aggrecan (agc) fluorescence from at least 15 random fields within the NP of three mice per mouse group are shown with 1 SE. (B) RT-PCR quantitation of aggrecan and versican mRNA in discs of 20-week-old mice. (C) Incorporation of 35S-sulfate into proteins by intervertebral discs in organotypic cultures as a measure of PG synthesis. For B and C, average values from six measurements (two duplicate × three mice per group) are shown ± 1 SE.
Intervertebral Discs of Ercc1−/Δ Mice Exhibit Decreased Capacity for Matrix Protein Synthesis
Reduced levels of PGs in the discs of Ercc1−/Δ mice could be due to decreased synthesis of new PGs. Consistent with this, mRNA of aggrecan as well as versican, another major NP PG, were reduced by 25% and 50%, respectively, in Ercc1−/Δ mice compared to wild-type littermates at 20 weeks of age (Fig. 3B). We also directly measured PG protein synthesis in the discs by quantifying the level of 35S incorporation. Discs from 20-week-old Ercc1−/Δ mice incorporated significantly less sulfate into protein than wild-type littermates (Fig. 3C). These data demonstrate that progeroid Ercc1−/Δ mice have impaired PG synthesis, which contributes to their reduced disc PG content.
Ercc1−/Δ Mice Display Higher Level of Apoptosis and Cell Senescence in Their Intervertebral Discs
The most obvious cause of decreased PG synthesis with aging is if functional cells are lost. Thus we measured apoptotic cells by TUNEL assay and senescent cells by immunodetection of P16INK4a. More positively stained cells were detected in the Ercc1−/Δ mice compared to wild-type littermates for both assays (Fig. 4). Furthermore, examination of H&E-stained sections of the disc revealed decreased cellularity in progeroid Ercc1−/Δ mice and naturally aged mice relative to young wild-type mice (Fig. 4). Decreased cellularity was particularly prominent in the endplate region, a site previously suggested as an original source of chondrocyte-like cells for NP.34
Figure 4.
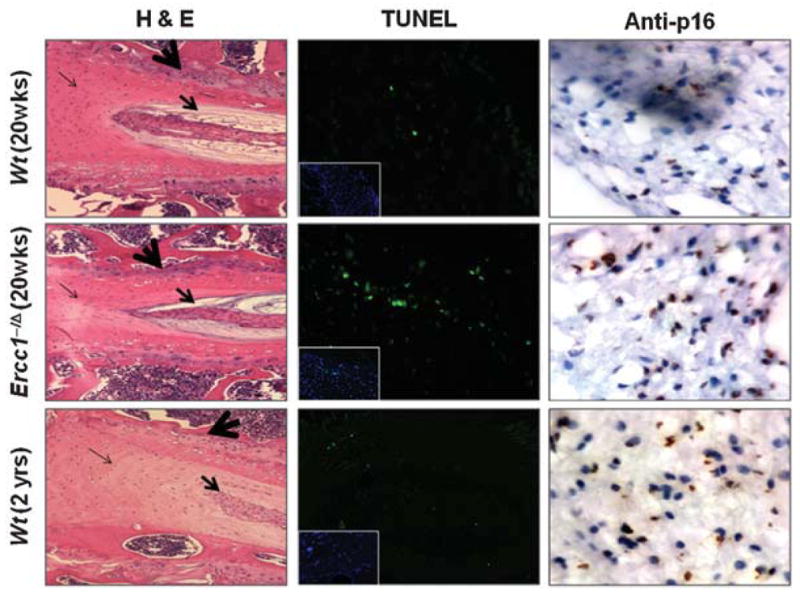
Decreased cellularity and enhanced apoptosis and cell senescence in intervertebral discs of aged and progeroid mice. Representative images of hematoxylin and eosin (H&E) stained discs from 20-week-old Ercc1−/Δ and wild-type littermates as well as 2-year-old wild-type mice (left). The big arrow marks the vertebral endplate, the medium-sized arrow indicates the nucleus pulposus, the fine arrow marks the annulus fibrosis. TUNEL assay to identify apoptotic cells (green) in disc tissue (middle). Insets, nuclear DAPI stain revealing tissue cellularity. Immunohistochemical detection of P16INK4a, a cell senescence marker, to distinguish senesced (brown) from nonsenesced (blue) cells (right).
Genotoxic Stress Further Accelerates PG Loss
ERCC1–XPF deficient mice and humans age rapidly as a consequence of DNA repair defect, implying that DNA damage, when not repaired, can promote age-related degeneration.23 Thus we asked if DNA damage can promote PG loss in IVDs. Ercc1−/Δ mice and wild-type littermates were chronically exposed to a subtoxic dose of the chemotherapeutic agent mechlorethamine, which is a DNA damaging agent, for 6 weeks beginning at 8 weeks of age. At 20 weeks of age, the mice were euthanized and tissues isolated for analysis. Vertebral sections were stained with safranin O to detect PGs. PG was substantially reduced in the discs and vertebral endplates of all animals exposed to mechlorethamine (Fig. 5 vs. Fig. 2). These data support the conclusion that DNA damage is at least one type of cellular damage that can drive age-related disc degenerative changes.
Figure 5.
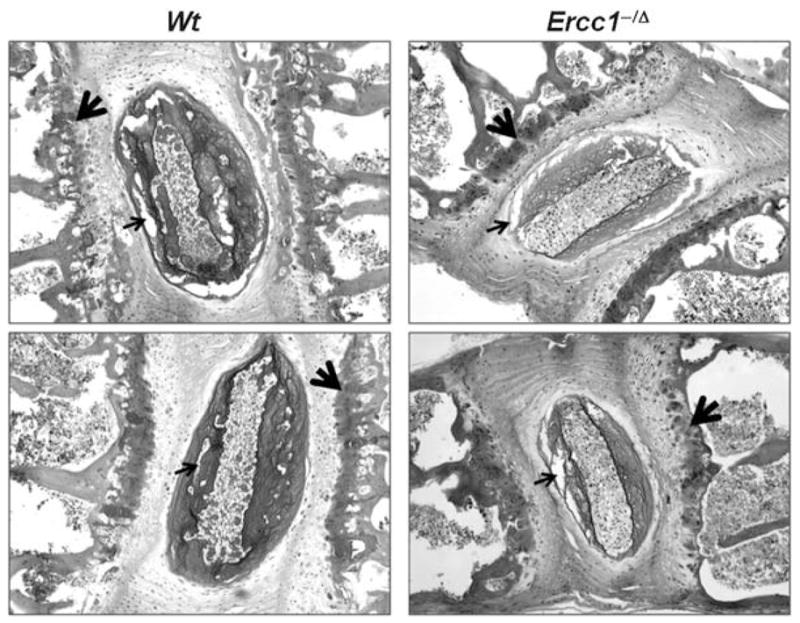
Genotoxic stress reduces disc PGs. Representative images of safranin O/fast green stained discs from 20-week-old Ercc1−/Δ and wild-type littermates treated with subtoxic dose of the chemotherapeutic agent mechlorethamine (MEC, see Materials and Methods). MEC exposure resulted in decreased safranin O staining and cellularity in cartilage end-plate (big arrow) and large unstained gaps in nucleus pulposus (medium-sized arrow).
DISCUSSION
Animal models of human diseases are vital for research aimed at identifying disease mechanism and therapeutic targets for disease prevention and treatment. Modeling age-related contribution to degenerative diseases is particularly challenging but necessary since studies in humans are implausible due to the length of the studies and the sample size necessary to overcome genetic and environmental variables. Thus an animal model of age-related contribution to IDD, in which genetic and environmental variables can be controlled, would be an extremely valuable tool for studying one important component of the etiology of IDD, identifying biomarkers of disease progression and testing therapies to prevent disease progression. In this study we establish that Ercc1−/Δ mice, which mimic a human progeroid syndrome, spontaneously develop several progressive degenerative changes associated with aging within 20 weeks.
An important hallmark of IDD is age-dependent progressive loss of disc matrix PGs, typically accompanied by reduced ability of the tissue to resist mechanical stress, resulting in compromised disc architecture and function.8 A recent longitudinal study of rabbits demonstrated a gradual decrease of disc magnetic resonance imaging (MRI) signal, suggesting loss of PGs with age.17 Other studies also reported age-dependent decline in total PG content in both the annulus fibrosis (AF) and NP of human discs.8,35 Indeed, disc PG loss appears to be universal in all models of IDD examined, including bovine, canine, ovine, and others.17,36 Deciphering the molecular mechanism behind age-dependent loss of disc matrix PGs is therefore critical to elucidate the contribution of IDD and designing rational strategies for this disease prevention and treatment.
Ercc1−/Δ mice exhibit significant matrix PG loss by 20 weeks of age, as revealed by safranin O staining of PGs, DMB colorimetric quantitation of GAGs, and immunofluorescence detection of aggrecan (Figs. 2 and 3). PG loss in Ercc1−/Δ mice is also progressive, beginning by 8 weeks of age, and might be responsible for the compromised disc structure and height in the Ercc1−/Δ mice, as it does with natural aging in both mice and humans (Figs. 1 and 2). Based on safranin O staining, it appears that PG loss is specific to NP as there was no reduced staining of disc endplate of Ercc1−/Δ mice. PG loss at the endplate is only evident when these mice were treated with MEC (Fig. 5), suggesting that NP is more susceptible to PG loss due to aging-related DNA damage than the endplate. These results establish the Ercc1−/Δ mice as a rapid model to study potential mechanisms of age-related disc PG loss.
It is well established that disc matrix protein production declines with age in mammals. PG gene expression and the rate of matrix PG synthesis decrease with age in human IVDs.8,37 Indeed, PG synthesis is also reduced in the discs of these progeroid mice compared to normal adult mice, as demonstrated by decreased mRNA of key structural PG genes (Fig. 3B) and reduced incorporation of 35S-sulfate incorporation into proteins by discs using organotypic culture (Fig. 3C). Thus Ercc1−/Δ mice rapidly lose disc PGs through a similar mechanism as naturally aged animals.
Reduced disc matrix PG synthesis in Ercc1−/Δ mice might be due, in part, to increased cell senescence and apoptosis (Fig. 4), both of which are well-demonstrated consequences of cellular response to DNA damage.38,39 It is interesting to note that apoptosis is mostly found in the AF of these mice while cell senescence, as probed by the senescence marker P16INK4a, occurs primarily in the NP. Since PG loss is most dramatic in the NP of Ercc1−/Δ mice, this suggests that PG loss in the NP might be related to increased cell senescence rather than apoptosis. Elevated level of disc cell senescence in the progeroid Ercc1−/Δ mice is consistent with previously reported correlation between the level of cell senescence and disc aging and disc degeneration.8,40–42
Discs of progeroid Ercc1−/Δ mice manifest a number of age-associated IDD characteristics. However, they show no evidence of annular fissures, osteophyte formation, or severe disc fibrosis and collapse typically observed in clinical human IDD. Thus Ercc1−/Δ mice represent a model of accelerated disc aging rather than the classical disc degeneration. In addition, the aging characteristics of progeroid Ercc1−/Δ mouse discs might not completely mimic natural aging, and thus extrapolation to normal human disc aging using this model requires experimental validation in humans. Moreover, extrapolation of disc research from mice to humans needs to be done with the awareness of other limitations, including differences in quadruped versus biped biomechanics, nutrient diffusion due to disparity in disc size, and the persistent presence of notorchordal cells in mouse discs.43,44
Nevertheless, Ercc1−/Δ mice are an extremely useful model to study the mechanism of one important aspect of clinical disc degeneration, that is, age-related disc PG loss, cell senescence and apoptosis which are also found in natural aging. Using these mice, we provide novel evidence that DNA damage can accelerate loss of PG, a key feature of aged discs. Chronic exposure of mice to a chemotherapeutic agent decreased disc PGs in adult wild-type and Ercc1−/Δ mice (Fig. 5 vs. Fig. 2). The effect was exaggerated in the DNA repair deficient Ercc1−/Δ mice, further implicating DNA damage as a potential contributor. In summary, herein, we report the characterization and early utilization of a novel and rapid murine model of age-related degenerative changes associated with IDD. This model is useful not only to probe the mechanisms of disc aging but also to rapidly screen therapeutics for age-associated contribution to IDD.
Acknowledgments
The contributions from the following individuals are gratefully acknowledged. Studer R, Sowa G, Lee J, Robbins P, Niedernhofer L, and Kang J (experimental design, data interpretation, intellectual inputs, and manuscript preparation), Taylor L, Loppini M, Alber S, Watkins S (histology and immunohistology), Usas A, Huard J (radiography), Robison A, Hyoung-Yeon S (breeding, MEC treatment, and dissection of mice), Coehlo P, Bentley D, Wang D (matrix gene expression and synthesis). The authors also thank Kevin Bell and Helga Georgescu for their technical assistance as well as Lou Duerring for her administrative assistance. This work was supported in part by the Albert B. Ferguson, Jr. M.D. Orthopaedic Fund of the Pittsburgh Foundation, The Orthopaedic Research and Education Foundation Grant to Joon Lee, The AOSpine North America Young Investigator Research Grant to Nam Vo. Laura Niedernhofer and Andria Robinson are supported by NIEHS (ES016114) and the Ellison Medical Foundation (AG-NS-0303-05).
References
- 1.Borenstein D. Epidemiology, etiology, diagnostic evaluation, and treatment of low back pain. Curr Opin Rheumatol. 1992;4:226–232. doi: 10.1097/00002281-199204000-00016. [DOI] [PubMed] [Google Scholar]
- 2.Battie MC, Videman T, Gibson LE, et al. 1995 Volvo Award in clinical sciences. Determinants of lumbar disc degeneration. A study relating lifetime exposures and magnetic resonance imaging findings in identical twins. Spine (Phila PA 1976) 1995;20:2601–2612. [PubMed] [Google Scholar]
- 3.Adams MA, Roughley PJ. What is intervertebral disc degeneration, and what causes it? Spine (Phila PA 1976) 2006;31:2151–2161. doi: 10.1097/01.brs.0000231761.73859.2c. [DOI] [PubMed] [Google Scholar]
- 4.Videman T, Battie MC, Parent E, et al. Progression and determinants of quantitative magnetic resonance imaging measures of lumbar disc degeneration: A five-year follow-up of adult male monozygotic twins. Spine (Phila PA 1976) 2008;33:1484–1490. doi: 10.1097/BRS.0b013e3181753bb1. [DOI] [PubMed] [Google Scholar]
- 5.Miller JA, Schmatz C, Schultz AB. Lumbar disc degeneration: Correlation with age, sex, and spine level in 600 autopsy specimens. Spine. 1988;13:173–178. [PubMed] [Google Scholar]
- 6.Roughley PJ, Alini M, Antoniou J. The role of proteoglycans in aging, degeneration and repair of the intervertebral disc. Biochem Soc Trans. 2002;30:869–874. doi: 10.1042/bst0300869. [DOI] [PubMed] [Google Scholar]
- 7.Antoniou J, Steffen T, Nelson F, et al. The human lumbar intervertebral disc: Evidence for changes in the biosynthesis and denaturation of the extracellular matrix with growth, maturation, ageing, and degeneration. J Clin Invest. 1996;98:996–1003. doi: 10.1172/JCI118884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singh K, Masuda K, Thonar EJ, et al. Age-related changes in the extracellular matrix of nucleus pulposus and anulus fibrosus of human intervertebral disc. Spine (Phila PA 1976) 2009;34:10–16. doi: 10.1097/BRS.0b013e31818e5ddd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buckwalter JA, Roughley PJ, Rosenberg LC. Age-related changes in cartilage proteoglycans: Quantitative electron microscopic studies. Microsc Res Tech. 1994;28:398–408. doi: 10.1002/jemt.1070280506. [DOI] [PubMed] [Google Scholar]
- 10.Watanabe H, Nakata K, Kimata K, et al. Dwarfism and age-associated spinal degeneration of heterozygote cmd mice defective in aggrecan. Proc Natl Acad Sci USA. 1997;94:6943–6947. doi: 10.1073/pnas.94.13.6943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lotz JC, Colliou OK, Chin JR, et al. Compression-induced degeneration of the intervertebral disc: An in vivo mouse model and finite-element study. Spine (Phila PA 1976) 1998;23:2493–2506. doi: 10.1097/00007632-199812010-00004. [DOI] [PubMed] [Google Scholar]
- 12.Alini M, Eisenstein S, Ito K, et al. Are animal models useful for studying human disc disorders/degeneration? Eur Spine J. 2008;17:2–19. doi: 10.1007/s00586-007-0414-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sobajima S, Kompel KF, Kim JS, et al. A slowly progressive and reproducible animal model of intervertebral disc degeneration characterized by MRI, X-ray, and histology. Spine. 2005;30:15–24. doi: 10.1097/01.brs.0000148048.15348.9b. [DOI] [PubMed] [Google Scholar]
- 14.Yang F, Leung VY, Luk KD, et al. Injury-induced sequential transformation of notochordal nucleus pulposus to chondrogenic and fibrocartilaginous phenotype in the mouse. J Pathol. 2009;218:113–121. doi: 10.1002/path.2519. [DOI] [PubMed] [Google Scholar]
- 15.Gruber HE, Gordon B, Williams C, et al. Vertebral end-plate and disc changes in the aging sand rat lumbar spine: Cross-sectional analyses of a large male and female population. Spine. 2007;32:2529–2536. doi: 10.1097/BRS.0b013e318158cd69. [DOI] [PubMed] [Google Scholar]
- 16.Cappello R, Bird JL, Pfeiffer D, et al. Notochordal cell produce and assemble extracellular matrix in a distinct manner, which may be responsible for the maintenance of healthy nucleus pulposus. Spine (Phila PA 1976) 2006;31:873–882. doi: 10.1097/01.brs.0000209302.00820.fd. discussion 883. [DOI] [PubMed] [Google Scholar]
- 17.Sowa G, Valada G, Studer R, et al. Characterization of intervertebral disc aging: Longitudinal analysis of a rabbit model by magnetic resonance imaging, histology, and gene expression. Spine (Phila PA 1976) 2008;33:1821–1828. doi: 10.1097/BRS.0b013e31817e2ce3. [DOI] [PubMed] [Google Scholar]
- 18.O’Connell GD, Vresilovic EJ, Elliott DM. Comparison of animals used in disc research to human lumbar disc geometry. Spine. 2007;32:328–333. doi: 10.1097/01.brs.0000253961.40910.c1. [DOI] [PubMed] [Google Scholar]
- 19.Elliott DM, Sarver JJ. Young investigator award winner: Validation of the mouse and rat disc as mechanical models of the human lumbar disc. Spine. 2004;29:713–722. doi: 10.1097/01.brs.0000116982.19331.ea. [DOI] [PubMed] [Google Scholar]
- 20.Niedernhofer LJ, Garinis GA, Raams A, et al. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature. 2006;444:1038–1043. doi: 10.1038/nature05456. [DOI] [PubMed] [Google Scholar]
- 21.McWhir J, Selfridge J, Harrison DJ, et al. Mice with DNA repair gene (ERCC-1) deficiency have elevated levels of p53, liver nuclear abnormalities and die before weaning. Nat Genet. 1993;5:217–224. doi: 10.1038/ng1193-217. [DOI] [PubMed] [Google Scholar]
- 22.Weeda G, Denker I, de Wit J, et al. Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr Biol. 1997;7:427–439. doi: 10.1016/s0960-9822(06)00190-4. [DOI] [PubMed] [Google Scholar]
- 23.Schumacher B, Van der Pluijm I, Moorhouse MJ, et al. Delayed and accelerated aging share common longevity assurance mechanisms. PLoS Genet. 2008;4:e1000161. doi: 10.1371/journal.pgen.1000161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prasher JM, Lalai As, Heijmans C, et al. Reduced hematopoietic reserves in DNA interstrand crosslink repair-deficient Ercc1−/− mice. EMBO J. 2005;24:861–871. doi: 10.1038/sj.emboj.7600542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schumacher B, Hoeijmakers JH, Garinis GA. Sealing the gap between nuclear DNA damage and longevity. Mol Cell Endocrinol. 2009;299:112–117. doi: 10.1016/j.mce.2008.10.031. [DOI] [PubMed] [Google Scholar]
- 26.Garinis GA, van der Horst GT, Vijg J, et al. DNA damage and ageing: New-age ideas for an age-old problem. Nat Cell Biol. 2008;10:1241–1247. doi: 10.1038/ncb1108-1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ahmad A, Robinson AR, Duensing A, et al. ERCC1-XPF endonuclease facilitates DNA double-strand break repair. Mol Cell Biol. 2008;28:5082–5092. doi: 10.1128/MCB.00293-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Farndale RW, Buttle DJ, Barrett AJ. Improved quantitation and discrimination of sulphated glycosaminoglycans by use of dimethylmethylene blue. Biochim Biophys Acta. 1986;883:173–177. doi: 10.1016/0304-4165(86)90306-5. [DOI] [PubMed] [Google Scholar]
- 29.Studer RK, Aboka AM, Gilbertson LG, et al. p38 MAPK inhibition in nucleus pulposus cells: A potential target for treating intervertebral disc degeneration. Spine. 2007;32:2827–2833. doi: 10.1097/BRS.0b013e31815b757a. [DOI] [PubMed] [Google Scholar]
- 30.Lu DS, Shono Y, Oda I, et al. Effects of chondroitinase ABC and chymopapain on spinal motion segment biomechanics. An in vivo biomechanical, radiologic, and histologic canine study. Spine (Phila PA 1976) 1997;22:1828–1834. doi: 10.1097/00007632-199708150-00006. discussion 1834–1835. [DOI] [PubMed] [Google Scholar]
- 31.Sanderson BJ, Shield AJ. Mutagenic damage to mammalian cells by therapeutic alkylating agents. Mutat Res. 1996;355:41–57. doi: 10.1016/0027-5107(96)00021-8. [DOI] [PubMed] [Google Scholar]
- 32.Imai Y, Okuma M, Ari HS, et al. Restoration of disc height loss by recombinant human osteogenic protein-1 injection into intervertebral discs undergoing degeneration induced by an intradiscal injection of chondroitinase ABC. Spine. 2007;32:1197–1205. doi: 10.1097/BRS.0b013e3180574d26. [DOI] [PubMed] [Google Scholar]
- 33.Gruber HE, Ashraf N, Kilburn J, et al. Vertebral endplate architecture and vascularization: Application of microcomputerized tomography, a vascular tracer, and immunocytochemistry in analyses of disc degeneration in the aging sand rat. Spine. 2005;30:2593–2600. doi: 10.1097/01.brs.0000187877.30149.83. [DOI] [PubMed] [Google Scholar]
- 34.Kim KW, Lim TH, Kim JG, et al. The origin of chondrocytes in the nucleus pulposus and histologic findings associated with the transition of a notochordal nucleus pulposus to a fibrocartilaginous nucleus pulposus in intact rabbit intervertebral discs. Spine. 2003;28:982–990. doi: 10.1097/01.BRS.0000061986.03886.4F. [DOI] [PubMed] [Google Scholar]
- 35.Gower WE, Pedrini V. Age-related variations in proteinpolysaccharides from human nucleus pulposus, annulus fibrosus, and costal cartilage. J Bone Joint Surg Am. 1969;51:1154–1162. [PubMed] [Google Scholar]
- 36.Melrose J, Smith SM, Fuller ES, et al. Biglycan and fibromodulin fragmentation correlates with temporal and spatial annular remodelling in experimentally injured ovine intervertebral discs. Eur Spine J. 2007;16:2193–2205. doi: 10.1007/s00586-007-0497-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boos N, Weissbach S, Rohrbach H, et al. Classification of age-related changes in lumbar intervertebral discs: 2002 Volvo Award in basic science. Spine (Phila PA 1976) 2002;27:2631–2644. doi: 10.1097/00007632-200212010-00002. [DOI] [PubMed] [Google Scholar]
- 38.Vazquez A, Bond EE, Levine AJ, et al. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov. 2008;7:979–987. doi: 10.1038/nrd2656. [DOI] [PubMed] [Google Scholar]
- 39.Niedernhofer LJ, Robbins PD. Signaling mechanisms involved in the response to genotoxic stress and regulating lifespan. Int J Biochem Cell Biol. 2008;40:176–180. doi: 10.1016/j.biocel.2007.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Le Maitre CL, Freemont AJ, Hoyland JA. Accelerated cellular senescence in degenerate intervertebral discs: A possible role in the pathogenesis of intervertebral disc degeneration. Arthritis Res Ther. 2007;9:R45. doi: 10.1186/ar2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gruber HE, Ingram JA, Norton HJ, et al. Senescence in cells of the aging and degenerating intervertebral disc: Immunolocalization of senescence-associated beta-galactosidase in human and sand rat discs. Spine. 2007;32:321–327. doi: 10.1097/01.brs.0000253960.57051.de. [DOI] [PubMed] [Google Scholar]
- 42.Zhao CQ, Wang LM, Jiang LS, et al. The cell biology of intervertebral disc aging and degeneration. Ageing Res Rev. 2007;6:247–261. doi: 10.1016/j.arr.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 43.Braund KG, Ghosh P, Taylor TK, et al. Morphological studies of the canine intervertebral disc. The assignment of the beagle to the achondroplastic classification. Res Vet Sci. 1975;19:167–172. [PubMed] [Google Scholar]
- 44.Higuchi M, Abe K, Kaneda K. Changes in the nucleus pulposus of the intervertebral disc in bipedal mice. A light and electron microscopic study. Clin Orthop Relat Res. 1983;175:251–257. [PubMed] [Google Scholar]