

Abstract
Neurofibromatosis type 1 (NF1) is the most common monogenic disorder in which individuals manifest CNS abnormalities. Affected individuals develop glial neoplasms (optic gliomas, malignant astrocytomas) and neuronal dysfunction (learning disabilities, attention deficits). Nf1 genetically engineered mouse models have revealed the molecular and cellular underpinnings of gliomagenesis, attention deficit, and learning problems with relevance to basic neurobiology. Using NF1 as a model system, these studies have revealed critical roles for the NF1 gene in non-neoplastic cells in the tumor microenvironment, the importance of brain region heterogeneity, novel mechanisms of glial growth regulation, the neurochemical bases for attention deficit and learning abnormalities, and new insights into neural stem cell function. Here we review recent studies, presented at a symposium at the 2012 Society for Neuroscience annual meeting, that highlight unexpected cell biology insights into RAS and cAMP pathway effects on neural progenitor signaling, neuronal function, and oligodendrocyte lineage differentiation.
Neurofibromatosis type 1 (NF1) is a common inherited tumor predisposition syndrome affecting 1 in 3000 individuals worldwide (Friedman et al., 1999). As such, children and adults are prone to the development of benign and malignant tumors of the peripheral nervous system (PNS) and CNS. Within the CNS, children and adults affected with NF1 manifest cognitive disabilities, behavioral issues, and motor delays, which negatively impact on scholastic achievement (Hyman et al., 2005, 2006; Isenberg et al., 2012; Soucy et al., 2012). Moreover, 15–20% of children with NF1 will develop a low-grade glioma of the optic pathway, which, when symptomatic, can cause reduced vision (Listernick et al., 1994, 1997). In addition, adults with NF1 harbor a 50- to 100-fold increased risk of high-grade (malignant) glioma development (Rasmussen et al., 2001; Gutmann et al., 2002). While uncommon, these cancers when encountered are frequently fatal and unresponsive to conventional therapies.
As a tumor predisposition syndrome, NF1 is a dominantly inherited genetic condition, where all affected individuals harbor a germline NF1 gene mutation. In this regard, children with NF1 start life with one nonfunctional and one functional NF1 gene in every cell in their body. Reduced NF1 gene expression is sufficient for some NF1-associated CNS clinical features (learning, memory, and attention deficits), whereas tumor formation requires complete loss of NF1 gene function in the neoplastic cells.
The NF1 gene resides on chromosome 17q11.2 in humans (11 B4–5 in the mouse) and encodes a 220–250 kDa cytoplasmic protein. Neurofibromin contains a 300 aa RAS GTPase activating protein (GAP) domain (Xu et al., 1990), which functions to convert RAS from its active GTP-bound form to its inactive GDP-bound form. Loss of neurofibromin expression (as observed in human NF1-associated tumors) results in increased RAS activity and cell growth (Basu et al., 1992: DeClue et al., 1992; Bollag et al., 1996). Consistent with increased RAS pathway activity in NF1-deficient tumor cells, high levels of activated RAF/MEK and AKT/mammalian target of rapamycin (mTOR) are observed (Dasgupta et al., 2005b; Johannessen et al., 2005) (Fig. 1). In addition, neurofibromin also positively controls adenylyl cyclase (AC) activity, and intracellular cAMP levels (Tong et al., 2002; Dasgupta et al., 2003). Reduced or absent Nf1 gene expression in astrocytes and neurons is associated with lower cAMP levels. While the mechanism underlying neurofibromin cAMP regulation is currently incompletely elucidated, both Ras-dependent and Ras-independent modes have been implicated (Hannan et al., 2006; Brown et al., 2010b).
Figure 1.
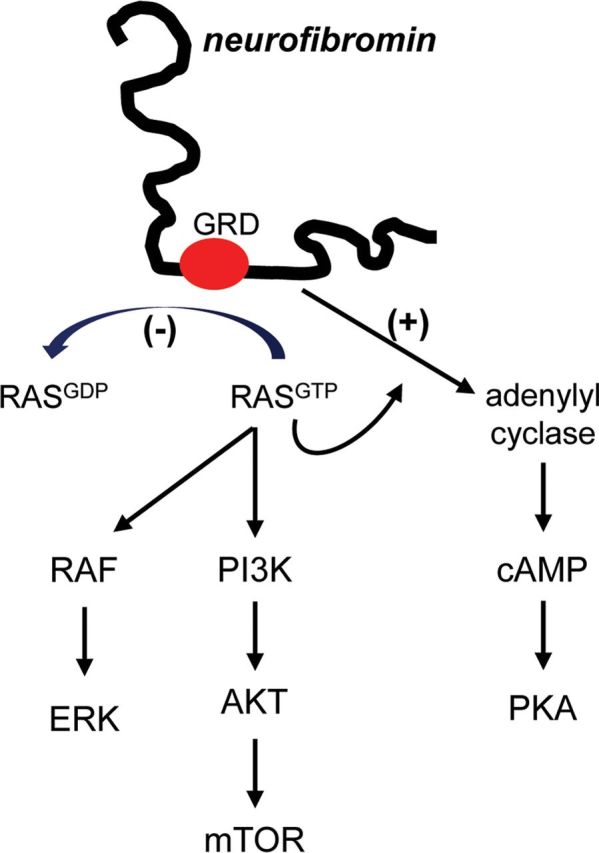
Neurofibromin signaling pathway regulation. The NF1 gene product, neurofibromin, functions as both a negative regulator of RAS activity (RAS GAP) and as a positive regulator of AC activity. Impaired neurofibromin function leads to increased RAS activation, resulting in high levels of RAF/MEK and AKT/mTOR signaling. Similarly, reduced neurofibromin function is associated with decreased cAMP levels and reduced protein kinase A (PKA) activity. GRD, GAP-related domain.
Over the past decade, researchers have developed numerous robust and accurate models of NF1-associated CNS abnormalities (Table 1). These Nf1 mutant mouse strains have helped elucidate the molecular and cellular pathogenesis of NF1-associated clinical features as well as served as tractable platforms for drug discovery and preclinical therapeutic evaluation before clinical trials in humans.
Table 1.
Nf1 genetically engineered mouse models of CNS dysfunction
| GEM strain | Description | CNS abnormality | References |
|---|---|---|---|
| Nf1+/− mice | Mice with reduced Nf1 gene expression throughout | Impaired learning, memory, and attention; increased astrogliosis | Jacks et al., 1994; Silva et al., 1997; Gutmann et al., 1999; Rizvi et al., 1999 |
| Nf1+/−Syn mice | Mice with reduced Nf1 gene expression in hippocampal neurons | Impaired learning, memory, and attention | Cui et al., 2008 |
| Nf1+/−GFAPCKO mice | Nf1+/− mice with complete Nf1 loss in neuroglial progenitors | Impaired learning, memory, and attention; optic glioma formation | Brown et al., 2010a; Bajenaru et al., 2003 |
| Nf1+/−; KRasGFAP mice | Nf1+/− mice with KRas activation in neuroglial progenitors | Optic glioma formation | Dasgupta et al., 2005a |
| Nf1;p53GFAPCKO mice | Mice with Nf1 and p53 inactivation in neuroglial progenitors | High-grade glioma formation | Zhu et al., 2005b |
| Nf1;p53; PtenGFAPCKO mice | Mice with Nf1,p53, and Pten inactivation in neuroglial progenitors | GBM formation | Kwon et al., 2008 |
| Nf1;p53; PtennestinCKO mice | Mice with Nf1,p53, and Pten inactivation in neural stem cells | GBM formation | Chen et al., 2012 |
Brain abnormalities in NF1
Remarkably little is known about structural brain abnormalities in individuals with NF1. In ∼50% of autopsied brains, disordered cortical architecture has been reported, with random orientation of neurons, focal heterotopic neurons, proliferation of glial cells to form well defined gliofibrillary nodules, and hyperplastic gliosis (Rosman and Pearce, 1967). Glial fibrillary acidic protein (GFAP) expression is increased, supporting the existence of reactive astrogliosis (Nordlund et al., 1995). Astrogliosis has also been observed in Nf1+/− mouse brains (Gutmann et al., 1999; Rizvi et al., 1999).
Magnetic resonance imaging (MRI) has revealed focal areas of high signal intensity on T2-weighted images of brains of children with NF1. Autopsy analysis of the brain regions corresponding to areas of high T2 signal intensity on MRI performed before death in two children concluded that the high-signal intensity lesions on MRI represented regions containing increased fluid within the myelin associated with hyperplastic or dysplastic glial cell proliferation (DiPaolo et al., 1995).
Based on these collective findings, it was proposed that early metabolic abnormalities in these brains leads to focal edema and vacuolization of myelin, which may be visible as T2 hyperintensities, followed by destruction of neurons, and ultimately to regression of visible lesions. Further support for a widespread myelin disorder model derives from water diffusion MRI, which identified significant increases in brain water diffusion in NF1 patient brains (Eastwood et al., 2001; Alkan et al., 2005; Zamboni et al., 2007; van Engelen et al., 2008; Ferraz-Filho et al., 2012). When T2 hyperintense lesions were specifically sampled, greater differences were observed, underscoring the notion that these hyperintense MRI abnormalities represent focal patches indicative of abnormal myelin.
Macrocephaly is another common feature of children with NF1 (Friedman and Birch, 1997; Friedman et al., 1999). Overall, macrocephalic subjects showed significant increases in white matter volumes (Steen et al., 2001; Cutting et al., 2002), with increased gray matter volumes variably observed. One heavily myelinated tract, the corpus callosum, has been extensively examined: children with NF1 have significantly larger corpus callosi compared with controls (Kayl et al., 2000; Moore et al., 2000), and a larger corpus callosum size is associated with reduced IQ (Pride et al., 2010). Even in adults, increased corpus callosum size is found in brains from NF1 patients (Wignall et al., 2010). Together, these findings provide evidence for more pervasive myelin dysfunction in the brains of individuals with NF1.
Neurofibromin, the NF1 gene product, is widely expressed in the CNS during early development. By mid-gestation, newly postmitotic neurons show increased levels of neurofibromin compared with less differentiated neuroepithelial cells (Daston and Ratner, 1992; Huynh et al., 1994). In the adult CNS, neurofibromin is strongly expressed in subsets of neurons, including projection neurons (Nordlund et al., 1993). Oligodendrocytes also express high levels of neurofibromin (Daston and Ratner, 1992; Daston et al., 1992). In contrast, astrocytes lack detectable levels of neurofibromin expression in situ (Daston and Ratner, 1992; Daston et al., 1992; Nordlund et al., 1993, 1995), but can be induced after injury (Hewett et al., 1995).
Direct evidence that oligodendrocyte lineage cells can be affected by loss of NF1 expression derives from several studies in mice: following bi-allelic Nf1 gene inactivation in neuroglial progenitor cells, oligodendrocyte precursors are increased in number in vitro and in vivo (Bennett et al., 2003; Hegedus et al., 2007). Consistent with an important role for oligodendrocytes in NF1 biology, Nf1 inactivation in the brain results in increased numbers of NG2+ glial cells in the mouse brain (Hegedus et al., 2007) and altered neuron–glial specification (Wang et al., 2012). Current studies are aimed at testing how loss of Nf1 expression affects oligodendrocyte differentiation and function.
Low-grade gliomas in NF1
Gliomas arising in the context of NF1 are largely restricted to the optic pathway (nerve, chiasm, and radiations) and brainstem of children typically younger than 7 years of age (Listernick et al., 1994, 1997; Guillamo et al., 2003). Importantly, some optic gliomas can lead to progressive visual loss, while other radiographically similar ones may be completely asymptomatic (normal vision). What determines this exquisite regional and spatial pattern of gliomagenesis and the variable effects on vision in children with NF1 are currently unknown; however, potential contributors include the timing of NF1 gene inactivation (cell of origin), the role of non-neoplastic cells in the tumor microenvironment, and the response of associated neurons.
Timing of Nf1 gene inactivation
Recent studies have revealed that Nf1 loss must occur in a specific progenitor cell during a restricted window of mouse development (Lee et al., 2012). While Nf1 loss in neuroglial progenitors before embryonic day 16.5 results in optic glioma formation in Nf1+/− mice, postnatal biallelic Nf1 loss in GFAP+ astrocytes is not sufficient for gliomagenesis. Interestingly, these Nf1 mouse optic gliomas appear to arise from progenitor cells in the third ventricle, rather than the lateral ventricle where many adult gliomas derive (Alcantara Llaguno et al., 2009). Studies are currently underway to define the role of more restricted progenitors, including NG2+ glia in Nf1 optic gliomagenesis as well as to define the unique properties of stem cells in the third ventricle (Dahiya et al., 2011).
Stromal influences on gliomagenesis
Studies using Nf1 genetically engineered mouse strains have also revealed that the location of gliomas in NF1 results from a combination of susceptible progenitor cells coupled with a permissive local microenvironment (stroma). In this regard, Nf1 loss in neuroglial progenitors is insufficient for gliomagenesis unless coupled with reduced Nf1 expression in non-neoplastic (Nf1+/−) cells in the brain (Bajenaru et al., 2002, 2003; Zhu et al., 2005b). It is curious that while Nf1+/− stromal cells and Nf1-deficient progenitors exist throughout the CNS of these mice, gliomas are restricted to the optic nerve and chiasm. Further examination of potential progenitor cells [neural stem cells (NSCs)] has demonstrated that not all NSC populations increase their proliferation and glial differentiation following Nf1 gene inactivation. Whereas Nf1−/− brainstem and third ventricle NSCs exhibit increased proliferation and glial differentiation relative to wild-type controls, Nf1−/− neocortical and lateral ventricle NSCs are nearly indistinguishable from wild-type NSCs (Lee et al., 2010). This differential response to Nf1 loss results from differential expression of one signaling component of the RAS/mTOR growth control pathway, leading to increased AKT activation in Nf1−/− brainstem, but not neocortical, NSCs.
The instructive role of the local microenvironment (stroma) is additionally underscored by studies focused on microglia. Resident microglia are abundant in human low-grade gliomas, and comprise 35–50% of the total number of cells in these tumors (Simmons et al., 2011). Using several Nf1 genetically engineered mouse strains, pharmacologic and genetic silencing of microglia function impairs optic glioma growth (Daginakatte and Gutmann, 2007; Daginakatte et al., 2008; Simmons et al., 2011). One of the stromal factors that facilitates gliomagenesis and continued growth is the chemokine CXCL12 (stroma-derived factor-1α) (Warrington et al., 2007), such that forced expression of CXCL12 in the brains of Nf1 optic glioma mice results in ectopic glioma-like formation (Sun et al., 2010). CXCL12 acting through its receptor, CXCR4, regulates glial cell survival in a cAMP-dependent manner: Restoring cAMP levels in Nf1 optic glioma strains reduces tumor growth (Warrington et al., 2010). Current work is focused on identifying the spectrum of microglia-produced growth and survival factors that drive and maintain glioma growth as potential targets for stroma-directed therapeutic drug design.
Response of associated neurons
There is considerable variability in the clinical outcome of children with NF1-associated optic glioma. While some children with progressive visual loss may have no further clinical deterioration following treatment, other optic gliomas may cause continued vision loss with little response to chemotherapy. What determines these divergent clinical responses is currently unclear, but may reflect vulnerability of Nf1+/− CNS neurons to neuronal cell death. Analysis of CNS neurons from the hippocampus, retina, and striatum of Nf1+/− mice have shown reduced neurite lengths, growth cone areas, and survival relative to wild-type littermates (Brown et al., 2010b), which are not further worsened by total Nf1 loss (Brown et al., 2012). This is in striking contrast to Nf1+/− PNS sensory and sympathetic neurons, which exhibit little change in neurite integrity or apoptosis. Moreover, Nf1−/− PNS neurons have longer processes and inappropriate cell survival following neurotrophin withdrawal (Vogel et al., 1995). On the molecular level, Nf1−/− PNS neuron survival is driven by RAS/AKT hyperactivation, whereas Nf1+/− CNS neuronal morphology reflects attenuated cAMP generation. The relationship between the specific germline NF1 gene mutation in individuals with NF1 and resulting neuronal function in the brain is presently being explored to identify genetic and potential genomic modifiers of disease progression and outcome.
Models of malignant glioma and therapeutic implications
While optic gliomas are the most common CNS tumor found in children with NF1, malignant gliomas can develop in all areas of the brains of adults with NF1 (Ilgren et al., 1985; Rasmussen et al., 2001; Gutmann et al., 2002).
Molecular analysis of high-grade glioma
While the prevalence of NF1 mutation in idiopathic glioma was initially underappreciated, data from The Cancer Genome Atlas study revealed that NF1 mutation was one of the most frequently occurring mutations found in glioblastoma (GBM), along with mutations in the TP53, PTEN, EGFR, RB, and CDKN2A/2B genes (Cancer Genome Atlas Research Network, 2008). Although these genes were well known in other cancers, this type of comprehensive analysis revealed new information about the prevalence of particular mutations in GBM and increased our understanding of the complex genetic landscape of this deadly high-grade brain tumor.
Mouse high-grade glioma models
These large-scale genome-wide sequencing studies have provided a wealth of information regarding the molecular events that take place in a particular tumor type, and offer potentially critical diagnostic and prognostic value. However, while studies using human tumor samples and cancer cell lines have provided important molecular insights, these data represent only a “snapshot” of the oncogenic process. To better understand the natural history of a tumor's development, researchers have turned to genetically engineered mouse models (GEMMs). The development of GEMMs for the study of tumorigenesis has allowed investigators to study tumors that have high fidelity to their human counterparts in an endogenous setting, as they develop within their natural macroenvironments and microenvironments. Moreover, these models recapitulate the cellular heterogeneity of the tumors typically seen in human disease physiology and, in models with 100% penetrance, allow for investigation into the very earliest molecular and cellular alterations. Thus, they have provided researchers with invaluable resources for studying the underlying mechanisms involved in tumor initiation and progression.
A number of mouse models of high-grade glioma have been developed, including mice with genetic alterations of EGFR, PDGF, Bmi1, Arf, and many other genes, either alone or in combination (Holland et al., 1998; Uhrbom et al., 1998; Bruggeman et al., 2007; Chen et al., 2012b). The Parada Laboratory developed mouse models of GBM, wherein two or three of the most commonly mutated genes found in human GBM—Nf1, p53, and Pten (Cancer Genome Atlas Research Network, 2008)—are conditionally mutated. These mice developed tumors with 100% penetrance, and the tumors faithfully recapitulated the human tumors, both histologically and molecularly, thus making them valid models for studying the natural history of tumor development. Using these models, they found that mutation in p53 must precede that of Nf1 for glioma to develop (Zhu et al., 2005a), which could account for the fact that in NF1 patients, gliomas are less frequently observed than other types of neoplasms. They also showed that while Nf1;p53 mutant mice developed tumors with 100% penetrance, mice with the additional Pten mutation had shortened survival time, with higher grade tumors that developed faster (Kwon et al., 2008). Examination of the presymptomatic mice allowed analysis of the earliest events in tumor development. Pulse chase experiments, as well as other forms of analysis, revealed that the subventricular zone—a primary niche of adult neural stem cells—exhibited early aberrant cellularity, proliferation, and migration of neural stem cells into the brain parenchyma. These studies led to the proposal that stem/progenitor cells are the cell of origin of malignant glioma (Zhu et al., 2005; Kwon et al., 2008; Alcantara Llaguno et al., 2009). Thus, GEMMs can provide important information about the earliest neoplastic events. Additionally, they are invaluable for the preclinical testing of putative drugs that target a particular oncogenic pathway.
Cell of origin and therapeutic implications
One topic of intense study in the cancer field is the “cell of origin.” Identifying the cell population that gives rise to and propagates a particular tumor type has important implications for the design of effective treatment strategies. One long-held theory is that tumors arise from differentiated cells that, after accumulating mutations, de-differentiate and acquire tumorigenic potential, including uncontrolled growth, stem-like properties, and self-renewal ability. One corollary of this theory is that a relatively large number of cells have tumorigenic potential. In the case of glioma, it was postulated that glial cells, in particular astrocytes, were the source of the tumors. Definitive evidence to support this theory, however, has been lacking.
Recently, the idea of “cancer stem cells” as the source of certain tumor types (Clarke et al., 2006) has gained considerable traction. Evidence for the existence of cancer stem cells has been provided for a number of solid tumors, including GBM (Singh et al., 2004). While standard methods for studying cancer stem cells have been extremely informative, they also have significant limitations. In the endogenous tumor setting, cancer stem cells are thought to be relatively quiescent and therefore not susceptible to the current chemotherapies and radiotherapies that target rapidly dividing cells, making them a likely candidate for the source of tumor recurrence following treatment. Therapies designed to target this particular cell population might therefore greatly improve prognosis for patients with therapy-resistant cancers such as glioma, making identification and analysis of these cells critical.
Using two different methods (genetically and by stereotactic injection), the Parada Laboratory previously demonstrated that neural stem cells in the SVZ can give rise to tumors upon conditional deletion of the Nf1/p53/Pten tumor suppressor genes (Alcantara Llaguno et al., 2009). Additionally, they have found that eliminating this population of cells in their mouse glioma model reduced tumor size and infiltration, and extended survival (Chen et al., 2012a). As the behavior of cancer stem cells is affected by their microenvironment, they sought to study the effect of eliminating this mutant cell population in the endogenous (in vivo) tumor setting by intercrossing mice carrying a Nestin-ΔTK-IRES-GFP transgene with Nf1;p53;Pten mutant mice. The Nestin-ΔTK-IRES-GFP transgene is expressed in quiescent neural stem cells of the subventricular zone and also labels a subset of glioma cells in the mutant mice. It does not, however, label the highly proliferative Ki67+ population of tumor cells. The mice were subjected to a dual regimen of temozolomide (TMZ), which kills rapidly dividing cells, and gancyclovir, which will kill the relatively quiescent Nestin-ΔTK-IRES-GFP-expressing cells once they enter the cell cycle. Such studies provide evidence that TMZ specifically targets the amplifying tumor-dividing population, but not the quiescent transgene-positive cells. Moreover, these data indicate that tumor regrowth after eradication of the proliferative cell pool is mediated by the quiescent endogenous tumor population. These data lend considerable support to the cancer stem cell hypothesis for malignant glioma and have important implications for the design of therapeutic strategies.
Understanding the learning deficits associated with NF1: mechanisms for the spatial learning, attention deficits, and working memory impairments associated with NF1
Behavioral analyses of mice heterozygous for a null mutation in the Nf1 gene (Nf1+/−) (Jacks et al., 1994) revealed a pattern of deficits in specific cognitive domains, including executive, attention, and spatial impairments, that is similar to that observed in individuals with NF1 (Costa et al., 2002; Cui et al., 2008; Shilyansky et al., 2010). For example, Nf1+/− mice required extended training to learn the position of the hidden platform in the Morris water maze (Silva et al., 1997). A pilot study on children with NF1 revealed a similar pattern of spatial learning deficits in a virtual Morris maze (Ullrich et al., 2010). As seen in Nf1+/− mice, children with NF1 needed additional training to reach the same level of performance as unaffected children. Importantly, in both mice and children with NF1, the behavioral and cognitive deficits described do not reflect generalized impairments in brain function. Nf1+/− mice show clear deficits in spatial learning and contextual conditioning (both hippocampal-dependent tasks), but are completely normal in visual learning and in auditory conditioning (hippocampal-independent tasks) (Silva et al., 1997; Costa et al., 2002; Cui et al., 2008).
The increase in RAS/ERK signaling observed in the brains of Nf1+/− mice leads to increased phosphorylation of synapsin I (Cui et al., 2008), a presynaptic molecule that regulates neurotransmitter release. In turn, this causes enhanced GABA release in hippocampal, prefrontal, and striatal circuits (Costa et al., 2002; Li et al., 2005; Cui et al., 2008; Shilyansky et al., 2010). This enhanced GABA-mediated inhibition accounts for the deficits in hippocampal long-term potentiation (LTP) observed in Nf1+/− mice. Hippocampal LTP is a key mechanism underlying spatial learning and memory (Lee and Silva 2009). Cre-mediated and cell type-specific Nf1 gene deletions confirmed the importance of changes in GABA inhibition: while Nf1 heterozygosity in inhibitory neurons leads to deficits in LTP and spatial learning, reduced neurofibromin expression in either astrocytes or excitatory neurons does not (Cui et al., 2008).
Working memory, which, like attention, depends on intact prefrontal circuitry, is also impaired in both Nf1+/− mice and in individuals with NF1 (Shilyansky et al., 2010). Functional imaging studies showed that the working memory impairments of NF1 subjects correlated with hypoactivation in the prefrontal cortex, which may reflect increased GABA-mediated inhibition in prefrontal cortical circuits of Nf1+/− mice (Shilyansky et al., 2010). Remarkably, a dose of a GABA receptor inhibitor (picrotoxin), which caused deficits in working memory in control mice, rescued the working memory deficits of Nf1+/− mice, a result consistent with the hypothesis that increased inhibition is at the root of the working memory deficits associated with NF1.
Increases in RAS/ERK signaling in Nf1+/− mice have been implicated in the working memory, attention, and spatial learning deficits of these mice (Costa et al., 2002; Li et al., 2005; Cui et al., 2008; Shilyansky et al., 2010). Genetic and pharmacological manipulations that target the RAS/ERK signaling pathway were shown to rescue the physiological and behavioral deficits of Nf1+/− mice. Importantly, pharmacological manipulations that impair the isoprenylation of RAS (statins, farnesyl transferase inhibitors), and therefore decrease the levels of RAS/ERK signaling, also rescue key electrophysiological and behavioral phenotypes of Nf1+/− mice. Indeed, at concentrations that do not affect signaling, physiology, or behavior of wild-type controls, statins reverse the signaling, electrophysiological, attention, and spatial learning deficits of Nf1+/− mice (Li et al., 2005). Prompted by these findings, clinical studies are currently underway to test the efficacy of statins as a treatment for the behavioral and cognitive deficits in individuals with NF1 (Krab et al., 2008; Acosta et al., 2011; Chabernaud et al., 2012).
Similar to individuals with NF1, Nf1 mutant mice also show attention deficits (Li et al., 2005; Brown et al., 2010a). These deficits are thought to be key contributors to academic and social problems in children with NF1 (Mautner et al., 2002; Hyman et al., 2005; Li et al., 2005). Using an additional Nf1 GEM strain to study attention, in which the Nf1+/− mutation is combined with Cre-driven homozygous Nf1 gene deletion in GFAP-expressing cells (Nf1 OPG mouse), it was found that reduced striatal dopamine was responsible for the observed attention deficits (Brown et al., 2010a). Treatment with methylphenidate (but not with drugs that affect RAS) reversed the attention deficits of these Nf1 OPG mutants (Brown et al., 2011), suggesting that defects in brain catecholamine homeostasis contribute to the attention deficits observed. These results suggest that, in addition to drugs that affect RAS/ERK signaling, drugs that manipulate dopaminergic function could also be used to treat the cognitive deficits associated with NF1.
Treatments and future directions
With the availability of genetically engineered mouse models for NF1-associated CNS pathology, it now becomes possible to envision a pipeline in which fundamental basic science discoveries lead to the identification of new cellular and molecular targets for therapeutic drug design, culminating in preclinical evaluation before testing in patients with NF1. First, the success of Nf1 mouse model implementation has already resulted in the clinical evaluation of lovastatin in children with NF1-associated learning deficits (Li et al., 2005) and rapamycin analogs for the treatment of glioma (Hegedus et al., 2008). Second, mouse models afford an opportunity to envision specific features of NF1 as distinct diseases defined by the timing of NF1 gene inactivation or the particular cell of origin. Similar to other cancers, the identification of molecular or cellular subtypes of NF1-associated nervous system tumors or learning/behavioral problems may result in more individualized treatments with a higher likelihood of success. Third, as we further exploit these powerful preclinical models, additional cellular and molecular targets may emerge as candidates for future therapeutic drug design. In this regard, one could envision more effective therapies resulting from the combined use of targeted inhibition of multiple growth control pathways regulated by neurofibromin in the neoplastic cell (NF1-deficient neuroglial precursor) or dual targeting of non-neoplastic (microglia) and neoplastic cells within NF1-associated CNS tumors.
Footnotes
This work was supported by National Cancer Institute Grant CA141549-01, National Institutes of Health Grant NS065547, and the Department of Defense (to D.H.G.).
The authors declare no competing financial interests.
References
- Acosta MT, Kardel PG, Walsh KS, Rosenbaum KN, Gioia GA, Packer RJ. Lovastatin as treatment for neurocognitive deficits in neurofibromatosis type 1: phase I study. Pediatr Neurol. 2011;45:241–245. doi: 10.1016/j.pediatrneurol.2011.06.016. [DOI] [PubMed] [Google Scholar]
- Alcantara Llaguno S, Chen J, Kwon CH, Jackson EL, Li Y, Burns DK, Alvarez-Buylla A, Parada LF. Malignant astrocytomas originate from neural stem/progenitor cells in a somatic tumor suppressor mouse model. Cancer Cell. 2009;15:45–56. doi: 10.1016/j.ccr.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkan A, Sigirci A, Kutlu R, Ozcan H, Erdem G, Aslan M, Ates O, Yakinci C, Egri M. Neurofibromatosis type 1: diffusion weighted imaging findings of brain. Eur J Radiol. 2005;56:229–234. doi: 10.1016/j.ejrad.2005.05.008. [DOI] [PubMed] [Google Scholar]
- Bajenaru ML, Zhu Y, Hedrick NM, Donahoe J, Parada LF, Gutmann DH. Astrocyte-specific inactivation of the neurofibromatosis 1 gene (NF1) is insufficient for astrocytoma formation. Mol Cell Biol. 2002;22:5100–5113. doi: 10.1128/MCB.22.14.5100-5113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajenaru ML, Hernandez MR, Perry A, Zhu Y, Parada LF, Garbow JR, Gutmann DH. Optic nerve glioma in mice requires astrocyte Nf1 gene inactivation and Nf1 brain heterozygosity. Cancer Res. 2003;63:8573–8577. [PubMed] [Google Scholar]
- Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J. Aberrant regulation of ras proteins in malignant tumours cells from type 1 neurofibromatosis patients. Nature. 1992;356:713–715. doi: 10.1038/356713a0. [DOI] [PubMed] [Google Scholar]
- Bennett MR, Rizvi TA, Karyala S, McKinnon RD, Ratner N. Aberrant growth and differentiation of CNS glial progenitors in neurofibromatosis type 1 mutants. J Neurosci. 2003;23:7207–7217. doi: 10.1523/JNEUROSCI.23-18-07207.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollag G, Clapp DW, Shih S, Adler F, Zhang YY, Thompson P, Lange BJ, Freedman MH, McCormick F, Jacks T, Shannon K. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat Genet. 1996;12:144–148. doi: 10.1038/ng0296-144. [DOI] [PubMed] [Google Scholar]
- Brown JA, Emnett RJ, White CR, Yuede CM, Conyers SB, O'Malley KL, Wozniak DF, Gutmann DH. Reduced striatal dopamine underlies the attention system dysfunction in neurofibromatosis-1 mutant mice. Hum Mol Genet. 2010a;19:4515–4528. doi: 10.1093/hmg/ddq382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JA, Gianino SM, Gutmann DH. Defective cAMP generation underlies the sensitivity of CNS neurons to neurofibromatosis-1 heterozygosity. J Neurosci. 2010b;30:5579–5589. doi: 10.1523/JNEUROSCI.3994-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JA, Xu J, Diggs-Andrews KA, Wozniak DF, Mach RH, Gutmann DH. PET imaging for attention deficit preclinical drug testing in neurofibromatosis-1 mice. Exp Neurol. 2011;232:333–338. doi: 10.1016/j.expneurol.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JA, Diggs-Andrews KA, Gianino SM, Gutmann DH. Neurofibromatosis-1 heterozygosity impairs CNS neuronal morphology in a cAMP/PKA/ROCK-dependent manner. Mol Cell Neurosci. 2012;49:13–22. doi: 10.1016/j.mcn.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruggeman SW, Hulsman D, Tanger E, Buckle T, Blom M, Zevenhoven J, van Tellingen O, van Lohuizen M. Bmi1 controls tumor development in an Ink4a/Arf-independent manner in a mouse model for glioma. Cancer Cell. 2007;12:328–341. doi: 10.1016/j.ccr.2007.08.032. [DOI] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabernaud C, Mennes M, Kardel PG, Gaillard WD, Kalbfleisch ML, Vanmeter JW, Packer RJ, Milham MP, Castellanos FX, Acosta MT. Lovastatin regulates brain spontaneous low-frequency brain activity in Neurofibromatosis type 1. Neurosci Lett. 2012;515:28–33. doi: 10.1016/j.neulet.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Li Y, Yu T-S, McKay RM, Burns DK, Kernie SG, Parada LF. A restricted cell population propagates glioblastoma growth following chemotherapy. Nature. 2012a doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, McKay RM, Parada LF. Malignant glioma: lessons from genomics, mouse models, and stem cells. Cell. 2012b;149:36–47. doi: 10.1016/j.cell.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer stem cells—perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. 2006;66:9339–9344. doi: 10.1158/0008-5472.CAN-06-3126. [DOI] [PubMed] [Google Scholar]
- Costa RM, Federov NB, Kogan JH, Murphy GG, Stern J, Ohno M, Kucherlapati R, Jacks T, Silva AJ. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature. 2002;415:526–530. doi: 10.1038/nature711. [DOI] [PubMed] [Google Scholar]
- Cui Y, Costa RM, Murphy GG, Elgersma Y, Zhu Y, Gutmann DH, Parada LF, Mody I, Silva AJ. Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell. 2008;135:549–560. doi: 10.1016/j.cell.2008.09.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutting LE, Cooper KL, Koth CW, Mostofsky SH, Kates WR, Denckla MB, Kaufmann WE. Megalencephaly in NF1: predominantly white matter contribution and mitigation by ADHD. Neurology. 2002;59:1388–1394. doi: 10.1212/01.wnl.0000032370.68306.8a. [DOI] [PubMed] [Google Scholar]
- Daginakatte GC, Gutmann DH. Neurofibromatosis-1 (Nf1) heterozygous brain microglia elaborate paracrine factors that promote Nf1-deficient astrocyte and glioma growth. Hum Mol Genet. 2007;16:1098–1112. doi: 10.1093/hmg/ddm059. [DOI] [PubMed] [Google Scholar]
- Daginakatte GC, Gianino SM, Zhao NW, Parsadanian AS, Gutmann DH. Increased JNK signaling in Neurofibromatosis-1 (Nf1) heterozygous microglia drives microglia activation and promotes optic glioma proliferation. Cancer Res. 2008;68:10358–10366. doi: 10.1158/0008-5472.CAN-08-2506. [DOI] [PubMed] [Google Scholar]
- Dahiya S, Lee da Y, Gutmann DH. Comparative characterization of the human and mouse third ventricle germinal zones. J Neuropathol Exp Neurol. 2011;70:622–633. doi: 10.1097/NEN.0b013e31822200aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta B, Dugan LL, Gutmann DH. The neurofibromatosis 1 gene product neurofibromin regulates pituitary adenylate cyclase-activating polypeptide-mediated signaling in astrocytes. J Neurosci. 2003;23:8949–8954. doi: 10.1523/JNEUROSCI.23-26-08949.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta B, Li W, Perry A, Gutmann DH. Glioma formation in neurofibromatosis 1 reflects preferential activation of K-RAS in astrocytes. Cancer Res. 2005a;65:236–245. [PubMed] [Google Scholar]
- Dasgupta B, Yi Y, Chen DY, Weber JD, Gutmann DH. Proteomic analysis reveals hyperactivation of the mTOR pathway in NF1-associated human and mouse brain tumors. Cancer Res. 2005b;65:2755–2760. doi: 10.1158/0008-5472.CAN-04-4058. [DOI] [PubMed] [Google Scholar]
- Daston MM, Ratner N. Neurofibromin, a predominantly neuronal GTPase activating protein in the adult, is ubiquitously expressed during development. Dev Dyn. 1992;195:216–226. doi: 10.1002/aja.1001950307. [DOI] [PubMed] [Google Scholar]
- Daston MM, Scrable H, Nordlund M, Sturbaum AK, Nissen LM, Ratner N. The Protein Product of the Neurofibromatosis Type 1 Gene is Expressed at Highest Abundance in Neurons, Schwann Cells, and Oligodendrocytes. Neuron. 1992;8:415–428. doi: 10.1016/0896-6273(92)90270-n. [DOI] [PubMed] [Google Scholar]
- DeClue JE, Papageorge AG, Fletcher JA, Diehl SR, Ratner N, Vass WC, Lowy DR. Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell. 1992;69:265–273. doi: 10.1016/0092-8674(92)90407-4. [DOI] [PubMed] [Google Scholar]
- DiPaolo DP, Zimmerman RA, Rorke LB, Zackai EH, Bilaniuk LT, Yachnis AT. Neurofibromatosis type 1: pathologic substrate of high-signal intensity foci in the brain. Radiology. 1995;195:721–724. doi: 10.1148/radiology.195.3.7754001. [DOI] [PubMed] [Google Scholar]
- Eastwood JD, Fiorella DJ, MacFall JF, Delong DM, Provenzale JM, Greenwood RS. Increased brain apparent diffusion coefficient in children with neurofibromatosis type 1. Radiology. 2001;219:354–358. doi: 10.1148/radiology.219.2.r01ap25354. [DOI] [PubMed] [Google Scholar]
- Ferraz-Filho JR, José da Rocha A, Muniz MP, Souza AS, Goloni-Bertollo EM, Pavarino-Bertelli EC. Unidentified bright objects in neurofibromatosis type 1: conventional MRI in the follow-up and correlation of microstructural lesions on diffusion tensor images. Eur J Paediatr Neurol. 2012;16:42–47. doi: 10.1016/j.ejpn.2011.10.002. [DOI] [PubMed] [Google Scholar]
- Friedman JM, Birch PH. Type 1 neurofibromatosis: a descriptive analysis of the disorder in 1,728 patients. Am J Med Genet. 1997;70:138–143. doi: 10.1002/(sici)1096-8628(19970516)70:2<138::aid-ajmg7>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Friedman JM, Gutmann DH, MacCollin M, Riccardi VM. Neurofibromatosis: Phenotype, Natural History, and Pathogenesis. Baltimore: Johns Hopkins University; 1999. [Google Scholar]
- Guillamo JS, Créange A, Kalifa C, Grill J, Rodriguez D, Doz F, Barbarot S, Zerah M, Sanson M, Bastuji-Garin S, Wolkenstein P. Prognostic factors of CNS tumours in Neurofibromatosis 1 (NF1): a retrospective study of 104 patients. Brain. 2003;126:152–160. doi: 10.1093/brain/awg016. [DOI] [PubMed] [Google Scholar]
- Gutmann DH, Loehr A, Zhang Y, Kim J, Henkemeyer M, Cashen A. Haploinsufficiency for the neurofibromatosis 1 (NF1) tumor suppressor results in increased astrocyte proliferation. Oncogene. 1999;18:4450–4459. doi: 10.1038/sj.onc.1202829. [DOI] [PubMed] [Google Scholar]
- Gutmann DH, Rasmussen SA, Wolkenstein P, MacCollin MM, Guha A, Inskip PD, North KN, Poyhonen M, Birch PH, Friedman JM. Gliomas presenting after age 10 in individuals with neurofibromatosis type 1 (NF1) Neurology. 2002;59:759–761. doi: 10.1212/wnl.59.5.759. [DOI] [PubMed] [Google Scholar]
- Hannan F, Ho I, Tong JJ, Zhu Y, Nurnberg P, Zhong Y. Effect of neurofibromatosis type I mutations on a novel pathway for adenylyl cyclase activation requiring neurofibromin and Ras. Hum Mol Genet. 2006;15:1087–1098. doi: 10.1093/hmg/ddl023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus B, Dasgupta B, Shin JE, Emnett RJ, Hart-Mahon EK, Elghazi L, Bernal-Mizrachi E, Gutmann DH. Neurofibromatosis-1 regulates neuronal and glial cell differentiation from neuroglial progenitors in vivo by both cAMP- and Ras-dependent mechanisms. Cell Stem Cell. 2007;1:443–457. doi: 10.1016/j.stem.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Hegedus B, Banerjee D, Yeh TH, Rothermich S, Perry A, Rubin JB, Garbow JR, Gutmann DH. Preclinical cancer therapy in a mouse model of neurofibromatosis-1 optic glioma. Cancer Res. 2008;68:1520–1528. doi: 10.1158/0008-5472.CAN-07-5916. [DOI] [PubMed] [Google Scholar]
- Hewett SJ, Choi DW, Gutmann DH. Expression of the neurofibromatosis 1 (NF1) gene in reactive astrocytes in vitro. Neuroreport. 1995;6:1565–1568. doi: 10.1097/00001756-199507310-00025. [DOI] [PubMed] [Google Scholar]
- Holland EC, Hively WP, DePinho RA, Varmus HE. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev. 1998;12:3675–3685. doi: 10.1101/gad.12.23.3675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh DP, Nechiporuk T, Pulst SM. Differential expression and tissue distribution of type I and type II neurofibromins during mouse fetal development. Dev Biol. 1994;161:538–551. doi: 10.1006/dbio.1994.1052. [DOI] [PubMed] [Google Scholar]
- Hyman SL, Shores A, North KN. The nature and frequency of cognitive deficits in children with neurofibromatosis type 1. Neurology. 2005;65:1037–1044. doi: 10.1212/01.wnl.0000179303.72345.ce. [DOI] [PubMed] [Google Scholar]
- Hyman SL, Arthur Shores E, North KN. Learning disabilities in children with neurofibromatosis type 1: subtypes, cognitive profile, and attention-deficit-hyperactivity disorder. Dev Med Child Neurol. 2006;48:973–977. doi: 10.1017/S0012162206002131. [DOI] [PubMed] [Google Scholar]
- Ilgren EB, Kinnier-Wilson LM, Stiller CA. Gliomas in neurofibromatosis: a series of 89 cases with evidence for enhanced malignancy in associated cerebellar astrocytomas. Pathol Ann. 1985;20:331–358. [PubMed] [Google Scholar]
- Isenberg JC, Templer A, Gao F, Titus JB, Gutmann DH. Attention skills in children with neurofibromatosis type 1. J Child Neurol. 2012 doi: 10.1177/0883073812439435. [DOI] [PubMed] [Google Scholar]
- Jacks T, Shih TS, Schmitt EM, Bronson RT, Bernards A, Weinberg RA. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat Genet. 1994;7:353–361. doi: 10.1038/ng0794-353. [DOI] [PubMed] [Google Scholar]
- Johannessen CM, Reczek EE, James MF, Brems H, Legius E, Cichowski K. The NF1 tumor suppressor critically regulates TSC2 and mTOR. Proc Natl Acad Sci U S A. 2005;102:8573–8578. doi: 10.1073/pnas.0503224102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayl AE, Moore BD, 3rd, Slopis JM, Jackson EF, Leeds NE. Quantitative morphology of the corpus callosum in children with neurofibromatosis and attention-deficit hyperactivity disorder. J Child Neurol. 2000;15:90–96. doi: 10.1177/088307380001500206. [DOI] [PubMed] [Google Scholar]
- Krab LC, de Goede-Bolder A, Aarsen FK, Pluijm SM, Bouman MJ, van der Geest JN, Lequin M, Catsman CE, Arts WF, Kushner SA, Silva AJ, de Zeeuw CI, Moll HA, Elgersma Y. Effect of simvastatin on cognitive functioning in children with neurofibromatosis type 1: a randomized controlled trial. JAMA. 2008;300:287–294. doi: 10.1001/jama.300.3.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon CH, Zhao D, Chen J, Alcantara S, Li Y, Burns DK, Mason RP, Lee EY, Wu H, Parada LF. Pten haploinsufficiency accelerates formation of high grade astrocytomas. Cancer Res. 2008;68:3286–3294. doi: 10.1158/0008-5472.CAN-07-6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee da Y, Yeh TH, Emnett RJ, White CR, Gutmann DH. Neurofibromatosis-1 regulates neuroglial progenitor proliferation and glial differentiation in a brain region-specific manner. Genes Dev. 2010;24:2317–2329. doi: 10.1101/gad.1957110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee da Y, Gianino SM, Gutmann DH. Innate neural stem cell heterogeneity determines the patterning of glioma formation in children. Cancer Cell. 2012;22:131–138. doi: 10.1016/j.ccr.2012.05.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Silva AJ. The molecular and cellular biology of enhanced cognition. Nat Rev Neurosci. 2009;10:126–140. doi: 10.1038/nrn2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Cui Y, Kushner SA, Brown RA, Jentsch JD, Frankland PW, Cannon TD, Silva AJ. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr Biol. 2005;15:1961–1967. doi: 10.1016/j.cub.2005.09.043. [DOI] [PubMed] [Google Scholar]
- Listernick R, Charrow J, Greenwald M, Mets M. Natural history of optic pathway tumors in children with neurofibromatosis type 1: a longitudinal study. J Pediatr. 1994;125:63–66. doi: 10.1016/s0022-3476(94)70122-9. [DOI] [PubMed] [Google Scholar]
- Listernick R, Louis DN, Packer RJ, Gutmann DH. Optic pathway gliomas in children with neurofibromatosis 1: consensus statement from the NF1 optic pathway glioma task force. Ann Neurol. 1997;41:143–149. doi: 10.1002/ana.410410204. [DOI] [PubMed] [Google Scholar]
- Mautner VF, Kluwe L, Thakker SD, Leark RA. Treatment of ADHD in neurofibromatosis type 1. Dev Med Child Neurol. 2002;44:164–170. doi: 10.1017/s0012162201001876. [DOI] [PubMed] [Google Scholar]
- Moore BD, 3rd, Slopis JM, Jackson EF, De Winter AE, Leeds NE. Brain volume in children with neurofibromatosis type 1: relation to neuropsychological status. Neurology. 2000;54:914–920. doi: 10.1212/wnl.54.4.914. [DOI] [PubMed] [Google Scholar]
- Nordlund ML, Rizvi TA, Brannan CI, Ratner N. Neurofibromin expression and astrogliosis in neurofibromatosis (type 1) brains. J Neuropathol Exp Neurol. 1995;54:588–600. doi: 10.1097/00005072-199507000-00013. [DOI] [PubMed] [Google Scholar]
- Nordlund M, Gu X, Shipley MT, Ratner N. Neurofibromin is enriched in the endoplasmic reticulum of CNS neurons. J Neurosci. 1993;13:1588–1600. doi: 10.1523/JNEUROSCI.13-04-01588.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pride N, Payne JM, Webster R, Shores EA, Rae C, North KN. Corpus callosum morphology and its relationship to cognitive function in neurofibromatosis type 1. J Child Neurol. 2010;25:834–841. doi: 10.1177/0883073809350723. [DOI] [PubMed] [Google Scholar]
- Rasmussen SA, Yang Q, Friedman JM. Mortality in neurofibromatosis 1: an analysis using U.S. death certificates. Am J Hum Genet. 2001;68:1110–1118. doi: 10.1086/320121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi TA, Akunuru S, de Courten-Myers G, Switzer RC, 3rd, Nordlund ML, Ratner N. Region-specific astrogliosis in brains of mice heterozygous for mutations in the neurofibromatosis type 1 (Nf1) tumor suppressor. Brain Res. 1999;816:111–123. doi: 10.1016/s0006-8993(98)01133-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosman NP, Pearce J. The brain in multiple neurofibromatosis (von Recklinghausen's disease): a suggested neuropathological basis for the associated mental defect. Brain. 1967;90:829–838. doi: 10.1093/brain/90.4.829. [DOI] [PubMed] [Google Scholar]
- Shilyansky C, Karlsgodt KH, Cummings DM, Sidiropoulou K, Hardt M, James AS, Ehninger D, Bearden CE, Poirazi P, Jentsch JD, Cannon TD, Levine MS, Silva AJ. Neurofibromin regulates corticostriatal inhibitory networks during working memory performance. Proc Natl Acad Sci U S A. 2010;107:13141–13146. doi: 10.1073/pnas.1004829107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AJ, Frankland PW, Marowitz Z, Friedman E, Laszlo GS, Cioffi D, Jacks T, Bourtchuladze R, Lazlo G. A mouse model for the learning and memory deficits associated with neurofibromatosis type I. Nat Genet. 1997;15:281–284. doi: 10.1038/ng0397-281. [DOI] [PubMed] [Google Scholar]
- Simmons GW, Pong WW, Emnett RJ, White CR, Gianino SM, Rodriguez FJ, Gutmann DH. Neurofibromatosis-1 heterozygosity increases microglia in a spatially- and temporally-restricted pattern relevant to mouse optic glioma formation and growth. J Neuropathol Exp Neurol. 2011;70:51–62. doi: 10.1097/NEN.0b013e3182032d37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- Soucy EA, Gao F, Gutmann DH, Dunn CM. Developmental delays in children with neurofibromatosis type 1. J Child Neurol. 2012;27:641–644. doi: 10.1177/0883073811423974. [DOI] [PubMed] [Google Scholar]
- Steen RG, Taylor JS, Langston JW, Glass JO, Brewer VR, Reddick WE, Mages R, Pivnick EK. Prospective evaluation of the brain in asymptomatic children with neurofibromatosis type 1: relationship of macrocephaly to T1 relaxation changes and structural brain abnormalities. AJNR Am J Neuroradiol. 2001;22:810–817. [PMC free article] [PubMed] [Google Scholar]
- Sun T, Gianino SM, Jackson E, Piwnica-Worms D, Gutmann DH, Rubin JB. CXCL12 alone is insufficient for gliomagenesis in Nf1 mutant mice. J Neuroimmunol. 2010;224:108–113. doi: 10.1016/j.jneuroim.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong J, Hannan F, Zhu Y, Bernards A, Zhong Y. Neurofibromin regulates G protein-stimulated adenylyl cyclase activity. Nat Neurosci. 2002;5:95–96. doi: 10.1038/nn792. [DOI] [PubMed] [Google Scholar]
- Uhrbom L, Hesselager G, Nistér M, Westermark B. Induction of brain tumors in mice using a recombinant platelet-derived growth factor B-chain retrovirus. Cancer Res. 1998;58:5275–5279. [PubMed] [Google Scholar]
- Ullrich NJ, Ayr L, Leaffer E, Irons MB, Rey-Casserly C. Pilot study of a novel computerized task to assess spatial learning in children and adolescents with neurofibromatosis type 1. J Child Neurol. 2010;25:1195–1202. doi: 10.1177/0883073809358454. [DOI] [PubMed] [Google Scholar]
- van Engelen SJ, Krab LC, Moll HA, de Goede-Bolder A, Pluijm SM, Catsman-Berrevoets CE, Elgersma Y, Lequin MH. Quantitative differentiation between healthy and disordered brain matter in patients with neurofibromatosis type I using diffusion tensor imaging. AJNR Am J Neuroradiol. 2008;29:816–822. doi: 10.3174/ajnr.A0921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel KS, Brannan CI, Jenkins NA, Copeland NG, Parada LF. Loss of neurofibromin results in neurotrophin-independent survival of embryonic sensory and sympathetic neurons. Cell. 1995;82:733–742. doi: 10.1016/0092-8674(95)90470-0. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kim E, Wang X, Novitch BG, Yoshikawa K, Chang LS, Zhu Y. RAS/ERK inhibition rescues fate-specification defects of Nf1-deficient neural progenitors and brain abnormalities. Cell. 2012;150:816–830. doi: 10.1016/j.cell.2012.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warrington NM, Woerner BM, Daginakatte GC, Dasgupta B, Perry A, Gutmann DH, Rubin JB. Spatiotemporal differences in CXCL12 expression and cyclic AMP underlie the unique pattern of optic glioma growth in neurofibromatosis type 1. Cancer Res. 2007;67:8588–8595. doi: 10.1158/0008-5472.CAN-06-2220. [DOI] [PubMed] [Google Scholar]
- Warrington NM, Gianino SM, Jackson E, Goldhoff P, Garbow JR, Piwnica-Worms D, Gutmann DH, Rubin JB. Cyclic AMP suppression is sufficient to induce gliomagenesis in a mouse model of neurofibromatosis-1. Cancer Res. 2010;70:5717–5727. doi: 10.1158/0008-5472.CAN-09-3769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wignall EL, Griffiths PD, Papadakis NG, Wilkinson ID, Wallis LI, Bandmann O, Cowell PE, Hoggard N. Corpus callosum morphology and microstructure assessed using structural MR imaging and diffusion tensor imaging: initial findings in adults with neurofibromatosis type 1. AJNR Am J Neuroradiol. 2010;31:856–861. doi: 10.3174/ajnr.A2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu GF, O'Connell P, Viskochil D, Cawthon R, Robertson M, Culver M, Dunn D, Stevens J, Gesteland R, White R. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell. 1990;62:599–608. doi: 10.1016/0092-8674(90)90024-9. [DOI] [PubMed] [Google Scholar]
- Zamboni SL, Loenneker T, Boltshauser E, Martin E, Il'yasov KA. Contribution of diffusion tensor MR imaging in detecting cerebral microstructural changes in adults with neurofibromatosis type 1. AJNR Am J Neuroradiol. 2007;28:773–776. [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Guignard F, Zhao D, Liu L, Burns DK, Mason RP, Messing A, Parada LF. Early inactivation of p53 tumor suppressor gene cooperating with NF1 loss induces malignant astrocytoma. Cancer Cell. 2005a;8:119–130. doi: 10.1016/j.ccr.2005.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Harada T, Liu L, Lush ME, Guignard F, Harada C, Burns DK, Bajenaru ML, Gutmann DH, Parada LF. Inactivation of NF1 in CNS causes increased glial progenitor proliferation and optic glioma formation. Development. 2005b;132:5577–5588. doi: 10.1242/dev.02162. [DOI] [PMC free article] [PubMed] [Google Scholar]