

Abstract
Lower brain glucose metabolism is present before the onset of clinically-measurable cognitive decline in two groups of people at risk of Alzheimer’s disease (AD) - carriers of apoE4, and in those with a maternal family history of AD. Supported by emerging evidence from in vitro and animal studies, these reports suggest that brain hypometabolism may precede and contribute to the neuropathological cascade leading cognitive decline in AD. The reason for brain hypometabolism is unclear but may include defects in glucose transport at the blood-brain barrier, glycolysis, and/or mitochondrial function. Methodological issues presently preclude knowing with certainty whether or not aging in the absence of cognitive impairment is necessarily associated with lower brain glucose metabolism. Nevertheless, aging appears to increase the risk of deteriorating systemic control of glucose utilization which, in turn, may increase the risk of declining brain glucose uptake, at least in some regions. A contributing role of deteriorating glucose availability to or metabolism by the brain in AD does not exclude the opposite effect, i.e. that neurodegenerative processes in AD further decrease brain glucose metabolism because of reduced synaptic functionality and, hence, reduced energy needs, thereby completing a vicious cycle. Strategies to reduce the risk of AD by breaking this cycle should aim to – (i) improve insulin sensitivity by improving systemic glucose utilization, or (ii) bypass deteriorating brain glucose metabolism using approaches that safely induce mild, sustainable ketonemia.
Keywords: Glucose, ketones, brain, aging, Alzheimer’s disease, PET, insulin, cognition, mitochondria
1.0 INTRODUCTION
Alzheimer’s disease (AD) is the product of slow, progressive degenerative changes that develop in the adult brain but that remain asymptomatic for a considerable time before cognitive decline becomes clinically evident. The challenge is to identify early markers of this degenerative process before it advances to the clinical stage because, at that point, most experts agree it is too late to correct the existing damage or prevent further cognitive deterioration [1]. Progress in understanding changes in brain energy metabolism during aging and AD has grown rapidly in the past three decades to the extent that it is now widely acknowledged that brain hypometabolism accompanies AD and is regionally heterogeneous. However, most view this hypometabolism as being an intermediate stage in the cellular and functional degeneration, i.e. that lower brain functionality requires less energy substrate [2]. We present here the concept that factors impeding optimal glucose utilization can contribute to or precipitate the neuropathology that becomes AD, i.e. that brain hypometabolism may be a critical part of the clinically asymptomatic early development of AD.
There is an emerging body of evidence showing that significantly lower brain glucose metabolism can be present well in advance of the onset of clinically measurable cognitive decline in AD. This evidence comes from various clinical and experimental models including studies of family history and genetic susceptibility to AD, post-mortem brain analysis, and from in vitro and animal models. For instance, in carriers of the ε4 allele of apolipoprotein (apo) E, small areas of cortical hypometabolism are present decades before the normal clinical onset of AD, making this the earliest marker thus far identified in individuals genetically at risk of AD. Therefore, a key issue is to establish whether brain hypometabolism could contribute to development and/or progression of AD, or whether these metabolic changes in the brain are predominantly a consequence of even earlier neurodegenerative processes that reduce the demand for glucose in the affected brain areas; is brain hypometabolism primary or secondary in AD?
A second issue is to establish whether brain hypometabolism in AD involves impaired brain utilization of energy substrates in general (as hypometabolism implies) or whether brain hypometabolism is actually a problem more or less specific to glucose. With 18F-fluorodeoxyglucose (FDG) as the only PET tracer validated for studies of brain metabolism, this important question has not yet been answered. Ketone bodies (ketones) are a key physiological replacement fuel preserving brain function during periods of low glucose availability, and the brain has a transport system for ketones independent of glucose transport. The recent development of 11C-acetoacetate as a ketone tracer for PET studies opens a new window to compare brain metabolism of glucose and ketones in the same individual. If brain ketone metabolism is not lower in AD or is less affected than glucose, one potential strategy to improve brain fuel availability and reduce the risk of AD that has already been targeted in clinical studies would be to develop a way to safely and reliably provide the brain with ketones as an alternative fuel to glucose.
The reason that brain glucose metabolism could decline before the clinical onset of AD is that the elderly commonly have deteriorating systemic glucose metabolism ranging from chronic, mild glucose intolerance to type 2 diabetes. If chronic, even mild systemic glucose dysregulation may gradually strain the normally finely tuned balance between brain glucose uptake and brain function. This imbalance appears to have relatively early onset in those with a genetic predisposition to or maternal family history of AD. In the meantime, outside the brain, mild hyperglycemia and/or hyperinsulinemia also prevent the physiological ketone response that would normally replace brain low glucose availability. We speculate that this situation of brain glucose insufficiency and inadequate ketone response puts high energy consuming areas of the brain in mild but chronic energy deficit. Difficulty acquiring its main fuel (glucose) and its preferred back up fuel (ketones) may force the brain to rely on gluconeogenesis as a third but insufficient option to acquire glucose. Over time, some brain regions, presumably including the hippocampus, are increasingly at risk of chronic fuel deprivation and gradually become fatigued which, in turn, permits the neuropathological changes leading to AD.
As proposed previously [3–6], we support the concept that regional brain hypometabolism contributes to the neuropathology that precipitates clinical symptoms of AD. Brain hypometabolism can also be exacerbated secondary to advancing neuropathology, but it can also contribute to the development of AD when present before the neuropathological changes begin. We extend this concept by proposing that brain hypometabolism affects glucose more than ketones. Whether it is feasible to reduce the risk of AD by correcting, preventing or bypassing a deterioration in brain glucose metabolism prior to the onset of neuropathology and cognitive decline is currently emerging as an area of considerable research interest.
2.0 BRAIN ENERGY METABOLISM
2.1 Energy requirements of the human brain
The brain, heart, liver, and kidneys consume about 60% of the body’s energy intake thereby dominating resting human energy metabolism. The heart and kidneys are metabolically more active than the brain but, being larger, the brain takes a higher proportion of the body’s total energy needs, i.e. about 20–23% of the body’s total energy requirement despite representing only 2.0–2.3% of adult body weight [7,8]. Three major parameters linked to brain energy metabolism – cerebral blood flow, oxygen consumption and glucose metabolism - can all be measured independently in humans using minimally invasive techniques.
In vitro and in vivo studies both show that most of the glucose consumed by the brain is used to maintain pre-synaptic and post-synaptic ion gradients required for glutamate neurotransmission, with the remainder used to maintain the resting potential of neurons [9,10]. Neurotransmitter signaling requires trans-membrane lipid asymmetries and constant phospholipid remodeling which may represent as much as 26% of the net energy uptake of the brain [11]. In the awake but unstimulated brain, basal energy consumption is high, so the stimulation-induced increase in the brain’s net energy consumption is actually relatively small and usually localized according to the stimulus.
Under normal conditions, glucose entering the brain is completely oxidized to CO2 and water. However, the brain does contain some glycogen which may play an active role in normal brain function [12]. Some pyruvate produced during glycolysis is converted to lactate but whether neurons consume exclusively glucose or can also use exogenous lactate or pyruvate is unclear. This uncertainty is due in part to different definitions of neuronal activity, but also to the exclusion in some studies of the supporting contribution by astrocytes to neuronal activity. Coupling of neuron and astrocyte function is an important component of the energy cost of brain function but this, too, is not always included in the modeling [13,14]. Brain activation involves inhibitory and excitatory pathways, both of which consume energy, so examining the energy requirements of one type of neuron in isolation may give an incomplete picture of the whole [9,10]. Local brain activation normally determines local cerebral blood flow so it is the integrity of oxygen and fuel transport from the capillary to the neuron, termed neurovascular coupling, which coordinates the metabolic response to produce/replace ATP during brain activation [15].
2.2 Brain glucose uptake
Glucose transporters (GLUTs) are responsible for brain glucose uptake in a process that can be compartmentalized into three steps: (i) Transport across the endothelium of the blood-brain barrier via a 55 kDa isoform of GLUT1. (ii) Transport into astrocytes by a second isoform of GLUT1 with a molecular weight of 45 kDa. (iii) Transfer into neurons via GLUT3. Net glucose uptake by the brain is linked to GLUT expression and activity but, most importantly, to glucose concentrations on both sides of the blood-brain barrier [16]. Hence, with brain activation, lower intracellular ATP and glucose concentrations rapidly stimulate brain glucose uptake. Still, the role of glucose transport itself, i.e. GLUT activity, in regulating neuronal activity is somewhat controversial. Some consider it to be less rate-limiting for brain function than glucose phosphorylation because GLUT1 is believed to normally operate at less than half its capacity [17]. Genetic forms of GLUT1 deficiency in humans in which GLUT expression is markedly decreased or totally absent cause lower brain glucose uptake and marked developmental neurological deficit in neonates. Hence, at what point partial reduction or impairment in glucose transport itself limits brain function is still unclear [18].
2.3 Measuring brain glucose metabolism
Brain glucose metabolism is normally synonymous with brain glucose uptake, so the former term will be used here rather than analogous terms such as glucose ‘uptake’, ‘utilization’ or ‘consumption’. For many years, glucose metabolism by the human brain was determined from the arterio-venous difference in blood glucose concentration across the brain multiplied by cerebral blood flow [19–22]. These studies led to broad agreement that the cerebral metabolic rate of glucose (CMRg) of the healthy, adult human brain is 6–7 mg/100 g/min or ~31 μmol/100 g/min, an amount equivalent to 120–130 g glucose/d for the whole brain [13,22].
Nowadays, the most common approach to studying brain metabolism is by PET using the tracer - FDG. In vivo 1H-nuclear magnetic resonance spectroscopy is less widely available but can also be used to calculate brain metabolism in humans [10,13,23]. For brain metabolism studies using PET, FDG is the tracer of choice because it simulates a combination of both glucose transport and subsequent phosphorylation [24]. FDG is transported into tissues including the brain at almost the same rate as glucose itself. Like glucose, FDG can be phosphorylated by the first glycolytic enzyme (hexokinase). Unlike glucose, FDG cannot be further metabolized to fructose-6-phosphate by glucose-phosphate-isomerase, so FDG remains trapped in the tissue as FDG 6-phosphate. Hence, FDG uptake represents glucose uptake but without subsequent metabolism towards CO2.
PET has several useful attributes for human diagnostic or metabolic studies [25]: (i) Although the tracer molecules need to be radioactive to be detected by PET, the radioisotopes of choice (commonly 18F or 11C) are short-lived, safe within the dose ranges permitted, and are now widely accepted for a variety of human experimental and diagnostic studies. (ii) PET is minimally invasive requiring only very low amounts of tracer on the order of 10−12-10−9 M. (iii) Dynamic tissue uptake and washout of the tracer are measurable in real time. (iv) Various treatments or disease conditions affecting the tracer’s metabolism can be studied. Relative to magnetic resonance imaging (MRI) or computed tomography (CT), which are used to examine tissue structure, PET has relatively poor spatial resolution, a limitation that can be overcome by combining the two imaging approaches, i.e. PET-CT or PET-MRI, thereby making it possible to measure regional CMRg in specific brain areas based on volumes as small as 1 cm3, i.e. the hippocampus.
Tracer kinetics is used to model the movement of a labeled molecule between compartments which are separated by membranes or metabolic reactions. Dynamic PET permits measurement of the change in concentration of a radiotracer in tissue over time, which is more informative than the simple measurement of substrate or tracer concentrations [26]. Dynamic PET image acquisition permits modeling of regional FDG uptake and determination of phosphorylation rates [27]. The common modeling techniques include region-of-interest analysis and spatial parametric mapping from voxel-based analysis [28]. A three compartment model (blood, interstitial matrix, neuron) is widely used to derive CMRg from FDG uptake data [29]. The calculation of CMRg requires a standardized correction factor, the lumped constant, which corrects FDG uptake to glucose uptake because GLUT favors FDG but hexokinase activity favors glucose [30–33]. Measuring CMRg requires the input function, or rate of arrival of the tracer at the target tissue. When the input function is not available, a relative term, the standardized uptake value can be used to semi-quantitatively express brain glucose uptake [34]. To minimize inter-individual variability, both absolute and relative measures of brain glucose uptake can be corrected against a reference brain region. In the aging or AD brain, the reference region is frequently the cerebellum.
2.4 Brain glucose metabolism in Alzheimer’s disease
AD is a neurodegenerative disorder that results in progressive loss of memory, declining cognitive function, disorientation and behavioral changes. Aging is the main risk factor for AD, with prevalence roughly doubling every five years above 65 y old [35], and affecting more than 60% of individuals above 95 y old [36]. The post-mortem AD brain is characterized by the accumulation of β-amyloid plaques in the intercellular brain parenchyma and by intracellular neurofibrillary tangles caused by hyperphosphorylation of tau protein [1,37,38]. There are two types of AD – familial or early onset and sporadic or late onset. The familial form is rarer and represents about 5% of all AD. It tends to occur before 65 y old, has different genetic predisposition, and is inherited in an autosomal dominant fashion. Besides the different age at onset, the clinical presentation and profile of cognitive deficit does not differ significantly between the familial and sporadic types [39]. There are other forms of dementia besides AD including fronto-temporal and vascular dementias but AD is the focus here.
Significant β-amyloid plaque deposition clearly occurs prior to memory loss and cognitive decline. Neverhteless, cognitive function can be normal in the presence of some β-amyloid deposition [40], soβ-amyloid plaques alone appear to be insufficient to cause dementia in the more frequent sporadic form of AD. Indeed, large scale studies, including the AD Neuroimaging Initiative, now show that β –amyloid deposition occurs slowly before cognitive symptoms become apparent, while neurodegeneration as measured by progressive brain atrophy accelerates later in the disease process, with the latter being more closely related to cognitive decline in AD [2,41].
PET studies have long pointed to lower brain glucose metabolism in AD (Table 1). Recent reviews describe the utility of CMRg for differential diagnosis of AD compared to other measures of cognitive decline in the elderly, particularly fronto-temporal dementia [42–44]. An overview of the now extensive literature on brain metabolism shows that global CMRg is ~20–25% lower in AD, with a more marked difference in some regions (Table 1). In AD, the earliest difference in CMRg is probably in the hippocampus, which is intimately involved in memory processing. CMRg of the hippocampus is not yet commonly reported because it is relatively small, making this a technically difficult measurement requiring PET-MRI or PET-CT [44]. After the hippocampus, lower CMRg in AD is seen most commonly in the posterior cingulate, temporal and parietal lobes and, later on, in the frontal lobes [45]. The medial temporal lobe includes the hippocampus and entorhinal cortex, so lower CMRg in these regions clearly seems linked to the earliest cognitive deficit in AD – impaired episodic memory. Although the same brain regions are affected in sporadic and familial AD, the posterior cingulate cortex, the parahippocampal gyrus and the occipital cortex may be more severely affected in familial compared to sporadic AD [2,46,47].
TABLE 1.
An overview of PET studies reporting cerebral metabolic rate for glucose (CMRg) in mild cognitive impairment (MCI) and Alzheimer’s disease (AD). Owing to the different study designs, it is difficult to derive the mean difference in CMRg in AD versus controls but it is on the order of 25%, and somewhat less in MCI.
| Reference | n in each group | Atrophy correction | CMRga |
|---|---|---|---|
| Frackowiak et al (1981)208 | n = 14 HC n = 13 AD n = 9 VD |
No | 28% ↓ in grey matter 23% ↓ in white matter for AD & VD |
| Benson et al (1983)209 | n = 16 HC n = 8 AD |
No | ↓ 49% for AD |
| Cutler et al (1985)210 | n = 25 HC n = 5 mild to moderate AD n = 2 severe AD |
No | 25–50% ↓ for severe AD in frontal, parietal, temporal, occipital lobes compared to mild-mod AD & HC |
| Rapoport et al (1986)211 | n = 10 HC n = 47 mild to moderate AD |
No | 18–31% ↓ for AD |
| Chawluk et al (1987)212 | n = 17 HC n = 24 AD |
Yes | 8% ↓ for AD |
| Alavi et al (1993)213 | n = 17 HC n = 20 AD |
Yes | 11 % ↓ for AD |
| Mielke et al (1994)214 | n = 13 HC n = 20 AD |
No | ↓ for AD in temporoparietal and occipital cortex |
| Metzler et al (1996)215 | n = 10 HC n = 8 AD |
Yes | ↓ for AD in frontal, temporal and posterior parietal lobes |
| Hock et al (1997)52 | n = 17 HC n = 19 AD |
No | ↓ for AD in parietal lobe |
| Ibanez et al (1998)48 | n = 19 HC n = 5 mild AD n = 6 moderate AD n = 4 severe AD |
Yes | ↓ for AD in precuneaus and posterior cingulate cortex |
| Mosconi et al (2005)50 | n = 11 HC n = 13 MCI n = 12 AD |
Yes | 18% ↓ for MCI 27% ↓ for AD in hippocampus compared with HC |
| Minoshima et al (2007)216 | n = 22 HC n = 8 AD |
Yes | 20–21% ↓ for AD in posterior cingulate and cinguloparietal transitional area |
| Li et al (2008)37 | n = 7 HC n = 13 aMCI n = 17 AD |
No | in AD; gray matter (11% ↓), hippocampus (30% ↓), inferior parietal lobe (15% ↓), middle frontal gyrus (12% ↓), posterior cingulate (17% ↓) in aMCI; hippocampus (14% ↓) and inferior parietal lobe (11% ↓) |
| Del Sole et al (2008)58 | n = 7 HC n = 16 aMCI n = 14 AD |
No | ↓ for AD in posterior cingulate, precuneaus, & parietal & temporal lobes ↓ for aMCI in posterior cingulate |
| Ishii et al (2009)66 | n = 42 MCI who converted to AD within 5 y | No | ↓ for AD in right cingulate, left inferior lobe & left temporal gyrus |
HC – healthy controls (commonly but not always well matched for age, education). Comorbidities, i.e. hypertension, not always excluded or controlled for, especially in earlier studies.
MCI – mild cognitive impairment, type not specified
aMCI – amnestic MCI
AD – Alzheimer’s disease, usually defined as 〈probable〉 AD, Most common criteria are those of the National Institute of Neurological and Communicable Diseases and Stroke, Alzheimer’s Disease and Related Dementias Association (NINCDS-ADRDA)
CMRg – cerebral metabolic rate for glucose
VD – vascular dementia
difference versus healthy controls
Different methods of PET data expression can have a significant impact on interpreting CMRg values in AD. Several studies reported regional CMRg relative to the cerebellum because CMRg in the cerebellum is relatively unaffected in AD compared to other brain regions and so can be used as an ‘internal reference’ to correct for inter-individual variation [48–50]. The brain also atrophies with age at about 1.6%/decade after the age of 30 y old (Figure 1), which also impacts on the calculation of glucose metabolism in the whole brain (global CMRg) versus regional CMRg. Correction of CMRg for brain atrophy with age is common but is not always performed (Table 1). Hence, in relation to healthy age-matched controls, and after correction for age-associated brain atrophy, there is broad agreement that CMRg in AD is reduced by ~25%. PET methodology using FDG has dominated research on brain metabolism in AD, but complementary approaches including direct measurement of cerebral blood flow [51,52] and ex vivo work on isolated microvessels from AD brain [53,54] corroborate the in vivo results obtained with brain PET.
Figure 1.
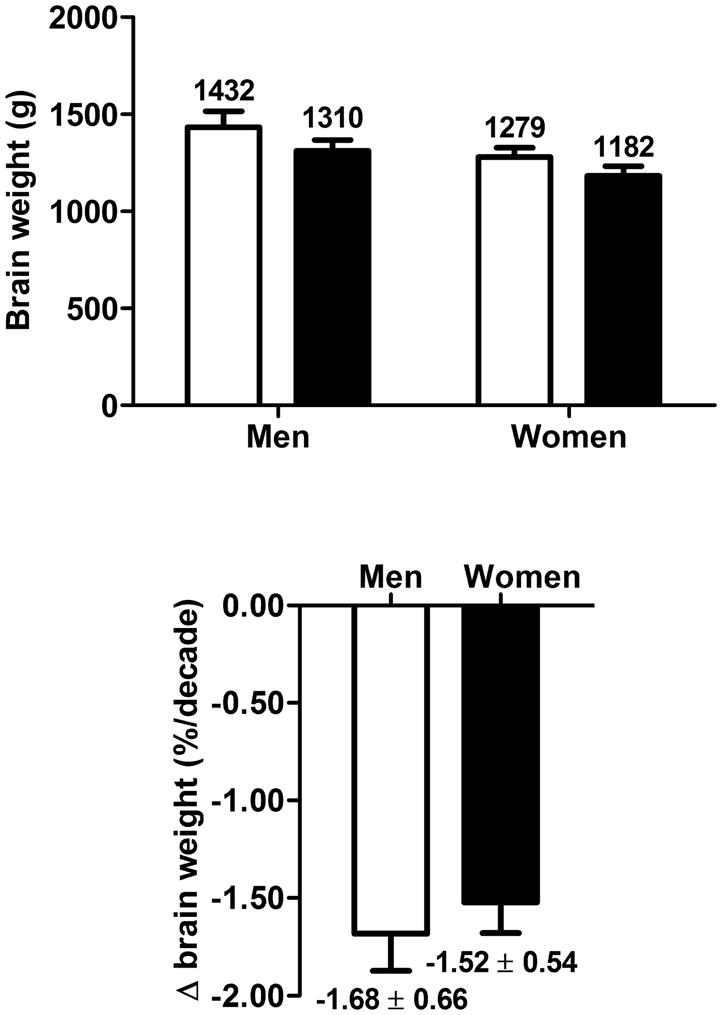
Lower overall brain volume with age modified from [206]. Based on these data, between about 30 y old (white bars) and 70 y old (black bars), the overall rate of decrease is about 1.6%/decade, or 6–7%, with no difference between men and women.
2.5 Brain glucose metabolism in mild cognitive impairment
An intermediate clinical state between healthy aging and dementia has long been recognized and is most commonly known as mild cognitive impairment (MCI). MCI includes both a complaint of cognitive decline and demonstration of objective deficits on cognitive testing, yet generally preserved functioning otherwise [55]. There are several conceptual frameworks to describe MCI [56]. Thus, when cognitive changes and memory loss are present but do not yet significantly affect social function or activities of daily living in the elderly, they are generally referred to MCI. When instrumental activities of daily living are impeded, conversion (or progression) to dementia is increasingly likely to have occurred [1,57]. Amnestic MCI (aMCI) is the form of MCI in which memory loss is the principle deficit, and is considered to be the form of cognitive decline most likely to progress to AD [58,59]. Despite ongoing efforts to establish criteria that better define MCI, it doesn’t yet have a consensus definition, so it includes a heterogeneous group of patients, some of whom may well be in the earliest detectable phase of a degenerative dementia such as AD. Consensus on definitions is emerging [60,61] and brain metabolism is being studied [62,63] in pre-clinical or prodromal forms of AD.
In the relatively few studies of brain glucose metabolism in MCI, global CMRg is lower than in controls but the difference is less than in moderate to severe AD (Table 1). Changes in CMRg are regionally heterogeneous depending on the type of MCI [64]. The pattern of change in CMRg in multi-domain MCI tend to resemble that of AD, whereas in aMCI, CMRg deficits appear to be limited to the hippocampus and parahippocampus [64]. MCI patients with normal CMRg or lower CMRg only in the hippocampus and parahippocampus are at lower risk of developing AD as compared to those with lower CMRg in broader areas, including the posterior cingulate and temporo-parietal association cortices, similar to AD. Longitudinal studies show that as MCI progresses to AD, CMRg declines in the cingulate, inferior parietal lobes and temporal lobes [65–67].
2.6 Brain glucose metabolism during aging
Our assessment of the current literature on CMRg during aging is that there is a dichotomy of results, but with no clear explanation of why that should be the case. On the one hand, we have found eight studies with a total of 359 subjects that collectively show that CMRg does not decline with age, while on the other hand, there are nine studies with a total of 573 subjects collectively demonstrating that CMRg is ~18% lower in the seventh compared to third decade of life (Table 2). There are numerous inconsistencies across these studies, regardless of whether or not an age-associated decline in CMRg was reported: (i) PET measurements of mean global CMRg have roughly doubled from ~4 mg/100 g/min in the mid-1980s to ~8 mg/100 g/min in 2002–2004. (ii) There was a wide age range of subjects defined as ‘elderly’ (55–92 y old). Most studies chose a somewhat arbitrary age range for ‘young’ versus ‘old’ subjects and grouped them such that it was a simple comparison of the two age groups, sometimes with an arbitrary, single age cut-point, eg. young <50 y old and old >50 y old. Others examined a continuum of ages, usually from the third to eighth decade of life. (iii) Control for concomitant diseases or other confounders was not always apparent so health status was often unclear. (iv) Cognitive status was not always assessed. (v) Several studies, especially the earlier ones, did not correct for aging-related brain atrophy, but after correction for brain atrophy, the aging-related decline in CMRg disappeared in two studies [68,69]. (vi) Optimal PET imaging conditions were not necessarily respected in all studies, including the need for reduced sensory input and resting conditions that minimize spurious results during the PET scan itself.
TABLE 2.
PET studies showing no difference (A) or lower (B) cerebral metabolic rate for glucose (CMRg) in the elderly with no known cognitive impairment.
| Reference | n in each group with age (y) | Atrophy correction | CMRg (mg/100 g/min) |
|---|---|---|---|
| A: NO DIFFERENCE WITH AGE | |||
| Duara et al (1983)217 | n = 21 men, 21–83 y | No | 4.3 |
| De Leon et al (1984)218 | n = 15, 26 y n = 22, 67 y |
No | 3.8 |
| Duara et al (1984)219 | n = 40 men, 21–83 y | No | 4.6 |
| Horowitz et al (1986)220 | n = 15 men, 20–32 y n = 15 men, 64–83 y |
No | 4.9 |
| Schlageter et al (1987)69 | n = 49 men, 21–83 y | Yes | 5.6 |
| Ibanez et al (2004)68 | n = 11 men, 22–24 y n = 13 men, 55–82 y |
Yes | 9.5 |
| Yanese et al (2005)221 | n = 71 men, 68 women, 24–81 y | Yes | SUV values; CMRg not given |
| Kochunov et al (2009)222 | n = 19 adults, 59–92 y | Yes | SUV values; CMRg not given |
| B: LOWER IN ELDERLY | |||
| Kety (1956)a, 223 | n = 16, 10–92 y | No | 50% ↓ >70 y, versus 10–20 y old |
| Dastur et al (1963)224 | n = 15 young men n = 26 elderlyb men |
No | young = 6.0, elderly = 4.6 (23% ↓ in elderly) |
| Kuhl et al (1982)163 | n = 40, 20–74 y old | No | young = 5.1, elderly = 3.9 (24% ↓ in elderly) |
| De Santi et al (1995)225 | n = 40, 28 y n = 31, 68 y |
No | 12–24% ↓ in frontal & temporal lobes for elderly |
| Loessner et al (1995)226 | n = 64 men, 56 women, 19–79 y | No | ↓ in frontal lobes for elderly over 6th decade |
| Moeller et al (1996)227 | n = 62 men, 68 women, 21–90 y <50 y = young >50 y = elderly |
No | 13% ↓ from 20 to 80 y; ↓ in medial frontal and frontal operculum |
| Petit-Taboué et al (1998)205 | n = 24, 20–67 y | No | 6% ↓ per decade in several cortical regions and anterior thalamus |
| Bentourkia et al (2000)228 | n = 10, 26 y n = 10, 65 y |
Yesc | young = 7.3, elderly = 6.3 (13% ↓ for elderly) |
| Willis et al (2002)191 | n = 38 men, 28 women, 20–69 y | No | young = 8.1, elderly = 6.9 (14% ↓ in elderly); ↓ in cingulate, frontal, temporal cortices from 20 to 60 y old |
| Kalpouzos et al (2009)204 | n = 21 men, 24 women, 20–83 y | Yes | ↓ in frontal cortex for elderly |
N2O method provided cerebral metabolic rate of oxygen (CMRO2)
inadequate information about criteria for state of health in elderly
no cerebral atrophy detected using MRI
Despite identifying these various confounders, they do not appear to adequately account for the dichotomy of results for CMRg during normal aging. Hence, in our opinion, it is still unclear whether or not healthy aging is truly associated with declining CMRg but this is an important issue to resolve (see Section 6.1).
3.0 FACTORS INFLUENCING BRAIN FUEL METABOLISM IN AD
Several nutritional and metabolic parameters are known to influence brain glucose metabolism, so their experimental manipulation may provide insight into whether the elderly are susceptible to brain hypometabolism and how and why brain glucose metabolism degenerates in AD. These factors include the ω3 fatty acid - docosahexaenoic acid (DHA; 22:6ω3), insulin, diabetes, dyslipidemia and mitochondrial dysfunction.
3.1 Docosahexaenoic acid
The ω3 polyunsaturated fatty acid, DHA, is now widely understood to have an important role in normal mammalian brain development. Rats made dietarily deficient in ω3 fatty acids have ~45% lower Na+/K+ pump activity in brain nerve endings [70]. More recently, rats deficient in ω3 fatty acids were shown to have lower brain glucose utilization measured by the 2-deoxyglucose method [29], as well as a reduced amount of the endothelial and astrocyte GLUT1 at the blood-brain barrier [71–72]. GLUT1 is located in endothelial cells of brain microvessels and at the end foot processes of astrocytes and is therefore at the gateway for glucose entry into the brain. Dietary supplementation with DHA increases GLUT1 expression in the rat brain endothelial cells and astrocytes [73], suggesting a positive correlation between DHA level in brain membranes and glucose transporter expression in the brain. This is confirmed by in vitro studies performed on primary cultures of rat brain endothelial cells in which glucose uptake is positively correlated to DHA levels in these cells (Pifferi et al, 2010 Neurochem. Int. [74]). ]. Expression of brain genes encoding for proteins of the mitochondrial respiratory chain was significantly increased in animals fed a diet supplemented with DHA (Kitajka et al 2004 [75]). Furthermore, in the primate, regional differences in brain DHA concentration are directly proportional to brain glucose uptake in the same regions [74].
Insufficient intake of DHA and low levels of DHA in the hippocampus may have a role in cognitive decline in the elderly and/or AD [75]. Hence, the low intake of DHA now widely but not universally reported in AD, may contribute to the evolution of cognitive decline because of its role in brain glucose transport and in other aspects of brain function and structure. This emerging role of DHA in brain energy metabolism could be linked to the early pre-symptomatic onset of brain glucose hypometabolism in AD, at least in carriers of the ε4 allele of apolipoprotein E (apoE4) [76]. Nevertheless, such an effect probably involves relatively subtle changes in DHA metabolism because plasma DHA is actually higher in the healthy elderly [77] and is widely variable in AD [75].
3.2 Insulin, diabetes, and dyslipidemia
GLUT1 in the blood brain barrier and GLUT3 in neurons are not sensitive to insulin, which is why brain glucose metabolism is so widely viewed as being independent of insulin. However, GLUT4 is insulin-responsive and is expressed particularly in brain regions involved in memory and cognition such as the hippocampus [78]. Furthermore, the insulin receptor is present in several brain regions and is linked to neuronal activity [78–81]. Brain insulin signaling may well be defective in AD [82]. Given acutely, insulin has positive effects on memory and learning but these effects seem to reverse and become detrimental during chronic insulin treatment [78,83]. Lipolysis and ketogenesis are more sensitive to insulin than tissue glucose uptake [84], so perhaps over the long term, insulin given experimentally prevents not only efficient glucose utilization but also replacement of low glucose by ketones (see Section 5.1), thereby imitating experimentally what is observed clinically in insulin resistance and type 2 diabetes.
The elderly commonly develop glucose intolerance, which may progress to the metabolic syndrome (combination of diabetes, hypertension, visceral obesity, and hyperlipidemia). In fact, type 2 diabetes is the most common metabolic disease in the elderly [85,86]. Insulin resistance is usually at or near the top of the list of known lifestyle-related factors heightening the risk of declining cognition in the elderly, [78,80,87–92]. In younger adults, obesity predisposes to the metabolic syndrome, which appears to increase the risk of degenerative changes in the brain [93]. Skeletal muscle is the main site of insulin-mediated glucose utilization in the body and so declining muscle mass (sarcopenia) in the elderly may be a factor potentially contributing to the increased risk of insulin resistance associated with aging. Whether insulin resistance adversely affects cognition directly by impairing glucose transport into certain areas of the brain or acts by glucose-independent signalling mechanisms remains to be established. Indeed, insulin resistance could impair cognition by affecting factors other than brain glucose metabolism, i.e. by mediating nitric oxide effects on blood flow [94].
In preparing tissues to utilize a blood glucose surge after a meal, insulin has long been known to inhibit lipolysis, i.e. to decrease free fatty acid release from adipose tissue triglycerides [95]. Chronically elevated insulin inhibits not only the release but also the β-oxidation of free fatty acids [96–100]. Insulin also inhibits hydroxymethylglutaryl-CoA synthase, a key step in ketone synthesis (Figure 2) [98]. Insulin resistance is commonly associated with raised plasma free fatty acids [101,102], so it is unclear whether the dominant effect of excess insulin is inhibition of lipolysis, which would lower plasma free fatty acids, or inhibition of fatty acid utilization, which would tend to raise plasma free fatty acids but inhibit ketogenesis. Since insulin resistance and cognitive decline are common in the elderly, and the former is linked to perturbed tissue utilization of glucose as well as fatty acids, it is important to establish whether impaired ketogenesis is present in insulin resistance. If so, in our view, it would appear that in insulin resistance and/or type 2 diabetes make it difficult for the brain to obtain sufficient fuel (glucose or ketones), which would eventually predispose to an increased risk of cognitive decline.
Figure 2.

The pathway of ketone synthesis. When acetyl CoA production from β-oxidized fatty acids exceeds the capacity of the tricarboxylic acid cycle, the excess acetyl CoA can condense into ketones, a process that happens predominantly but not exclusively in the liver.
3.3 Mitochondrial dysfunction in AD
Mitochondria are responsible for ATP production, so it stands to reason that if glucose metabolism is impaired in AD, it could involve down-regulated or otherwise dysfunctional mitochondria. This would also be consistent with the higher incidence of AD in those with a maternal family history of AD, in whom mitochondrial DNA mutations are observed [103]. Mutations in mitochondrial genes and some phenotypic changes affecting mitochondria underlie the concept that mitochondrial dysfunction contributes to the early stages and/or development of epilepsy [104] and various other neurodegenerative diseases [105–113]. Mitochondrial enzyme expression within neurons declines as neurofibrillary tangles are formed in AD [114–116].
Nevertheless, the link between mitochondrial dysfunction and development of AD is controversial. Mitochondrial surface area [117], enzyme markers [118] and ultrastructure [119] can be normal in AD. Mitochondrial mutational burden can be similar between AD and healthy controls, and the mutational changes that are present seem to be subtle and may lack reproducibility [108], so they remain of unknown importance.
Production of free radicals is a feature of electron transfer during normal mitochondrial function. Increased free radical release leading to peroxidative damage is also seen in the AD brain [120,121] and in epidemiological studies [122], so it is possible that disrupted mitochondrial function in AD prevents the appropriate balance between production and destruction of free radicals, leading to further neurological damage [107,110]. The location of mitochondria, whether at the synapse or elsewhere, may also be important; those at the synapse are present at higher density and have a higher metabolic responsibility for neuronal function, so their dysfunction could also contribute significantly to declining neuronal glucose requirement [113]. Whether or not mitochondrial dysfunction is a function of genetic or metabolic disturbances, clinical trials attempting to redress the energy deficit in the AD brain suggest that cognitive function can be at least transiently improved if more fuel (glucose or ketones) can be supplied to the brain (see Section 5.3).
4.0 BRAIN HYPOMETABOLISM: THE CART OR THE HORSE IN AD?
Which comes first in AD – the combination of neurodegeneration, decreased neurotransmitter production and declining neuronal function that collectively require less glucose, or the reverse – a progressive decline in some aspect of brain glucose metabolism that inhibits normal neurotransmitter production and permits less neuronal activity? Normally, CMRg and glucose uptake are essentially synonymous and are dictated by brain activity, so lower CMRg in AD has long been thought to be a consequence of lower neuronal activity due to lower functionality of brain structures that normally have high energy consumption [123]. Synaptic loss and decreased neurotransmitter production [124] have been proposed as components of an early neuropathological process that could inhibit mitochondrial enzymes, increase oxidative stress [125], and thereby initiate synaptic dysfunction that then reduces demand for glucose in brain regions affected in AD. Nevertheless, the idea is also well-supported that altered brain metabolism can predispose to neuronal damage, dysfunction and death, and lead to various acute and chronic forms of neuropathology [4,6,126]. There are several examples of how decreasing CMRg could be one of the early changes contributing to the neuropathogenesis of AD:
4.1 Tau hyperphosphorylation
Tau hyperphosphorylation appears to be the neuropathological change most closely linked to cognitive decline in AD [41]. In both in vivo and in vitro models, lower glucose availability induces hyperphosphorylation of tau protein in a site-specific manner [127]. The same form of tau O-glycosylation that regulates tau phosphorylation in mice is reduced in AD brain. Further work by the same group shows that GLUT1 and GLUT3 are reduced in the AD brain and directly correlated to lower O-glycosylation and hyperphosphorylation of tau [128]. Because the reciprocal changes in O-glycosylation and hyperphsohorylation of tau respond so rapidly to lower glucose supply in the fasted mouse model and because tau hyperphosphorylation is so sensitive to glucose and is directly linked to cognitive decline in AD, decreasing brain glucose uptake could contribute to the development of AD [127,128].
4.2 Glycolysis and pyruvate dehydrogenase activity
Lower activity of glycolytic enzymes leading to lower pyruvate synthesis, lower pyruvate dehydrogenase activity and lower acetyl CoA production have all been observed in the AD brain [3,118,129]. Pyruvate dehydrogenase complex operates in only slight excess capacity relative to demand for ATP so even its modest impairment might be critical. Furthermore, acetylcholine synthesis is acutely sensitive to brain glucose metabolism [118,129], so mildly impaired glucose availability in the synapse may be sufficient to impair cholinergic neurotransmission, which is a hallmark of AD [3,119]. Increased amino acid degradation for gluconeogenesis may partially compensate for impaired glycolysis but this pathway can supply at most 25% of brain glucose requirements [22], and is very costly in terms of muscle protein breakdown to supply the gluconeogenic amino acids. Astrocytes are capable of gluconeogenesis but endogenous to exogenous to the brain, this is still essentially a catabolic process that was never intended to meet glucose requirements for very long [4]; that was the role of ketones (see Section 5.1). However, ketones are produced in response to low systemic glucose and insulin, neither of which occurs very often in the elderly. Hence, disrupted glycolysis and lower pyruvate dehydrogenase complex activity may precede mitochondrial dysfunction and then contribute to increased oxidative stress and molecular changes affecting protein structure that precipitate declining memory and cognition [4].
4.3 Brain microvasculature
The etiology of AD appears to implicate structural and peroxidative changes not only in neurons [121,130] but also in the endothelial cells of capillaries that constitute the brain’s microvasculature and the blood-brain barrier [131–133]. The auditory cortex and inferior colliculus are the brain areas with the highest energy requirement [25] and, interestingly, both have the highest capillary density [134]; thus regional variations the density of the brain’s microvasculature are roughly proportional to the energy requirements of these regions.
Endothelial cells of brain capillaries represents <1% of brain weight but these cells are estimated to transport ten times their weight in glucose/min [135]. This phenomenal work capacity is accomplished by a mitochondrial density about five times that of skeletal muscle [136]. If mutations in mitochondria can increase the risk of AD (see Section 3.3), this may have an even greater impact on mitochondria in the blood-brain barrier than in brain cells per se. Type 2 diabetes causes microvascular damage and enhances the risk of both AD and vascular dementia [137], possibly by impairing blood brain barrier function. Since the brain’s microvasculature is the exclusive gateway for glucose to the brain, could subtle microvascular changes in those predisposed to AD disrupt the normally fine-tuned equilibrium between supply and demand for glucose (and oxygen) such that glucose transport into astrocytes and neurons no longer efficiently meets their energy needs, leading to synaptic drop-out and neuronal dysfunction?
4.4 Brain hypometabolic changes pre-date cognitive decline
In offspring with a family history of AD, lower CMRg can occur decades before appearance of cognitive symptoms [45,50,67]. In the case of asymptomatic offspring with a family history of AD, maternal but not paternal family history is associated with lower CMRg (Table 4). Carriers of apoE4 have the highest known genetic risk of AD and have lower CMRg before even the AD-associated decline in hippocampal volume or overall brain volume, both of which are also observed well before cognition symptoms of AD typically develop [2,62,138–140]. In carriers of apoE4, small areas of lower brain glucose metabolism are observed at an age as young as 30 y old, e.g. 30–40 y before clinical onset (Table 3). Indeed, we see an inverse relationship between CMRg in several brain regions and fasting plasma glucose, so brain metabolism seems to be sensitive to even mild elevations disturbances in systemic glucose control even if no clinical symptoms of cognitive decline are observed (Nugent et al, unpublished observations). Compared to non-carriers, apoE4 carriers also have altered ω3 fatty acid metabolism [76] and higher measures of oxidative stress in the brain [130], both of which may contribute to a higher risk of an early onset of brain hypometabolism. If brain hypometabolism can be present before clinical symptoms are apparent, this doesn’t prove that hypometabolism is the earliest event in AD. However, to the best of our knowledge, hypometabolism is currently the earliest measurable abnormality in the brain that is connected to AD so its features and the reasons for it should shed light on the etiology of AD.
TABLE 4.
PET studies showing lower cerebral metabolic rate of glucose (CMRg) in clinically asymptomatic subjects with a family history of Alzheimer’s disease.
| Reference | n in each group | CMRga |
|---|---|---|
| Kennedy et al (1995)233 | n = 16 HC n = 24 aFH-AD n = 18 sFH-VD |
14% ↓ in aFH-AD versus HC 34% ↓ in sFH-AD versus HC |
| Mosconi et al (2006)b, 234 | n = 7 HC n = 7 FH-AD |
18% ↓ in inferior parietal lobe for FH-AD |
| Mosconi et al (2007)b, 45 | n = 25 FH-AD n = 8 pFH-AD n = 16 mFH-AD |
10–15% ↓ in mFH-AD compared with other groups |
| Mosconi et al (2009)64 | n = 37 FH-AD n = 9 pFH-AD n = 20 mFH-AD |
↓ in mFH-AD compared with other groups |
HC – healthy controls
AD – Alzheimer’s disease, usually defined as 〈probable〉 AD, and most commonly by National Institute of Neurological and Communicable Diseases and Stroke, Alzheimer’s Disease and Related Dementias Association (NINCDS-ADRDA) criteria
FH-AD – family history of AD and, on average, mean of 13 y pre-symptomatic
aFH-AD – asymptomatic FH-AD
sFH-AD symptomatic FH-AD
pFH-AD – paternal family history of AD but cognitively normal
mFH-AD – maternal family history of AD but cognitively normal
difference versus healthy controls
corrected for brain atrophy
TABLE 3.
PET studies showing lower cerebral metabolic rate for glucose (CMRg) in carriers of apolipoprotein E4.
| Reference | n in each group | CMRg |
|---|---|---|
| Reiman et al (1996)138 | n = 11 E4+ homozygous n = 22 E4− |
↓ in posterior cingulate, parietal, temporal, & prefrontal regions of E4+ |
| Reiman et al (1998)140 | n = 11 E4+ homozygous n = 22 E4− |
↓ in posterior cingulate, parietal, temporal, & prefrontal regions of E4+ |
| De Leon et al (2001)229 | n = 48 HC of whom n = 12 → MCI, and n = 1 → AD |
↓ in entorhinal cortex predicted conversion of HC to MCI ↓ in hippocampus and temporal cortex in MCI compared with HC at follow-up ↓ in temporal cortex of E4+ |
| Mosconi et al (2004)230 | n = 37 MCI n = 16 E4+ n = 21 E4−, of whom n = 8 → AD |
↓ in inferior parietal cortex for converters to AD ↓ in temporoparietal & posterior cingulate cortex for E4+ further ↓ in anterior cingulate & inferior frontal cortex for E4+ MCI converters |
| Reiman (2004)139 | n = 12 E4+ heterozygous n = 15 E4− |
↓ in posterior cingulate, parietal, temporal, & prefrontal cortex for E4+ compared to HC |
| Reiman (2005)62 | n = 78 E4− n = 26 E4+ homozygous n = 46 E4+ heterozygous |
↓ in posterior cingulate, precuneaus, parietotemporal, & frontal regions; negative correlations between CMRg & gene dose of E4+ allele |
| Rimajova et al (2008)231 | n = 30 E4+ | ↓ in anterior & posterior cingulate cortex, temporal association cortex for E4+ |
| Langbaum et al (2009)232 | n = 82 HC n = 142 aMCI n = 74 pAD |
↓ in posterior cingulate, precuneaus, parietotemporal, frontal cortex ↓ in precuneaus and frontal cortex for HC who were E4+ ↓ in lateral temporal cortex for aMCI who were E4+ |
AD – Alzheimer’s disease
aMCI – amnestic mild cognitive impairment
E4− – no apolipoprotein E4 allele
HC – healthy controls
pAD – probable Alzheimer’s disease
→ progressed to
5.0 BRAIN HYPOMETABOLISM IN AD: SPECIFIC TO GLUCOSE OR GENERALIZED?
In the brain, glucose uptake (transport) is normally equivalent to its utilization, consumption or metabolism (see Section 2). Still, the common assumption is that brain metabolism is synonymous with glucose. While that is of course broadly true, it is also true that maintaining brain function depends on highly efficient availability of a back up fuel to occasionally replace glucose during periods of hypoglycemia. PET is an invaluable tool to study brain metabolism but such studies have been almost exclusively limited to glucose (FDG) measurements because, with rare exception, no other tracer form of a brain fuel has been available. This section describes the importance of ketones as the main replacement fuel for the brain and the need to assess the effect of aging on brain metabolism of fuels other than glucose. Clinical studies showing the potential therapeutic role of ketones in redressing brain hypometabolism are also described, the outcome of which may have an important bearing on understanding whether or not brain hypometabolism in AD can be corrected or circumvented.
5.1 Ketones: the key alternative brain fuel to glucose
When hypoglycemia develops over a period of several hours to days, i.e. during fasting or starvation, the energy requirements and normal function of the adult human brain are absolutely dependent on increased availability of two ketones – acetoacetate and β-hydroxybutyrate [13,22,25,141–144]. Whether the third ketone, acetone, contributes directly to the brain’s energy requirements is unknown but, during prolonged fasting or starvation, up to ~60% of the human brain’s energy requirements can be met by a combination of acetoacetate and β-hydroxybutyrate [141]. The brain is fully capable of converting ketones to ATP through all the necessary steps, including conversion of β-hydroxybutyrate to acetoacetate, acetoacetate to acetoacetyl CoA, and acetoacetyl CoA to acetyl CoA (Figure 2). Two observations indicate that ketone metabolism is a constitutive feature of brain function: (i) The amounts and activities of ketone-metabolizing enzymes in the brain are not changed by glucose status and always exceed the amount necessary to supply the brain’s total energy needs. (ii) During infancy, the brain has an obligatory requirement for ketones [142].
In contrast to neurons, brain capillaries can readily β-oxidize fatty acids [145] as can astrocytes [146,147]. Nevertheless, the transport of fatty acids through the blood brain barrier is too slow to make fatty acids a useful alternative to glucose for the brain [148,149]. Ketones cannot fully replace glucose as a brain fuel because, in addition to providing the carbon to replace oxaloacetate in the tricarboxylic acid cycle, glucose is still essential for the brain as the precursor to lactate, which can be exchanged with ketones when the latter are taken up by the brain’s monocarboxylic acid transporter (MCT). Several human studies show that mild, experimental ketonemia can maintain normal brain function even when plasma glucose would normally be low enough to result in acute cognitive and functional deficits (see Section 5.3).
Ketogenesis depends on a decrease in plasma insulin provoked by lower plasma glucose. When insulin decreases, its blockade of free fatty acid release from adipose tissue is overcome and free fatty acids are liberated into the plasma. These long chain fatty acids are transported to the liver and other organs where they are converted (β-oxidized) to acetyl CoA. Once a threshold concentration of acetyl CoA is reached in the liver, through a series of enzyme-catalyzed steps, it is then condensed into ketones (Figure 2). When ketone production is stimulated by low plasma glucose and insulin, maximal ketone production does not induce ketoacidosis because it is autoregulated through insulinotropic and antilypolytic feedback [60]. Hence, the brain is always prepared to burn ketones as soon as they are available; a situation totally consistent with their physiological role as its main back-up fuel.
5.2 Brain ketone uptake
The cerebral metabolic rate of ketones (CMRk) varies directly with their blood concentration, starting at very low ketone concentrations. This has been demonstrated using various approaches in small animals [150–154], by arterio-venous difference in humans [22,148,155,156], by PET with 11C-β-hydroxybutyrate in humans [157] and 11C-acetoacetate in rats (11C-AcAc; [154]), and by 1H-nuclear magnetic resonance in humans [23]. Hence, conditions that physiologically raise plasma ketones, including fasting, starvation [158] and very high fat ketogenic diets, also raise breath acetone [159–161], brain MCT1 expression [162], and brain ketone uptake [154]. Arterio-venous difference studies suggests that CMRk is not significantly affected by aging nor in suspected cases of AD [21,156,163], but that CMRk is lower in diabetes mellitus [148]. Although much more research on CMRk in humans has been done with arterio-venous differences than by PET, both approaches show excellent agreement that CMRk uptake is directly proportional to the arterial concentration of β-hydroxybutyrate and acetoacetate. Hence, at a plasma β-hydroxybutyrate concentration of 0.3–0.5 mM, such as can be achieved during 12–24 h fasting, β-hydroxybutyrate supplies 3–5% of whole brain energy requirements (Figure 3). As plasma ketones rise, CMRk also rises such that at a β-hydroxybutyrate of about 1.5 mM, ketones provide about 18% [20], and at 6 mM, they provide about 60% of brain fuel [22].
Figure 3.

The relationship between plasma ketones (shown here only as β-hydroxybutyrate; β-HB) and cerebral metabolic rate of ketones (CMRβ-HB; left hand y-axis) over a physiological range of plasma β-HB. In the original studies, two different methods were used to calculate CMRβ-HB: (i) PET (n=5 points collected close together in the bottom left of the graph between β-HB values of 0.0–0.2 mM; ○) [207], and (ii) arterio-venous difference taking into account cerebral blood flow – (n=26; ◇ [19]), and (n=10 controls □, and n=12 presumed Alzheimer’s disease ▲) [21]. Arterio-venous difference in brain β-HB uptake was reportedly the same in Alzheimer’s disease as in the controls [21]. Combined together, the 53 data points from these three studies provide the following equation of the line for CMRβ-HB over a plasma β-HB concentration range from 0.0–1.5 mM: y = 1.677x + 0.0454 (r = 0.638; p < 0.0001). The right hand y axis shows the percentage contribution of ketones to the energy requirements of the whole human brain over a physiological range of plasma β-HB. The intercept of ~18% of brain energy requirements being met by β-HB at a plasma β-HB concentration of 1.5 mM is corroborated by two additional papers that reported arterio-venous differences across the brain to derive CMRβ-HB for higher plasma β-HB values averaging 2 and 7 mM, achieved during β-HB infusion [20] and experimental starvation [22], respectively.
Brain ketone uptake is principally controlled by the permeability of the blood-brain barrier [164]. This, in turn, is a function of MCTs that transport ketones and other small monocarboxylic acids. Several MCTs are expressed in the brain [165] and their expression responds positively to raised plasma ketones [162]. Astrocytes express MCT4, neurons express MCT2 and brain capillaries express MCT1 [166]. The ability of the brain to use ketones in place of glucose is better in humans than in other omnivores such as the rat, pig, or monkey [144]. Dogs, sheep and pigs are unable to achieve significant ketosis, even during prolonged fasting [167] perhaps because their relative brain size is not very large, thereby permitting gluconeogenesis and glycerol liberated during lipolysis to support brain function without resorting to an entirely different fuel.
5.3 PET studies of brain 11C-acetoacetate uptake
Using 1-11C-AcAc [168], we have validated a model of brain ketone uptake in the rat using PET in which, as expected from many previous studies, brain ketone uptake is proportional to plasma ketone concentration [154,169] (Figures 4, 5). These studies have led us to the first human work with 11C-AcAc and PET with the aim of answering the questions – (i) is brain uptake of ketones lower in the healthy elderly or in AD and, if not, (ii) does this open up ketone-based therapeutic opportunities to treat cognitive decline before it becomes too severe to correct? To address these two questions we have just begun working with a double tracer human PET protocol in which brain uptake of FDG is imaged immediately following the 11C-AcAc scan (Figure 4). Our preliminary results are in line with previous results [156] suggesting that if brain ketone uptake is lower in the healthy elderly, the difference is not as significant as it is for the uptake of FDG. We are currently applying this approach to assess brain 11C-AcAc and FDG uptake in AD and MCI.
Figure 4.

11C-Acetoacetate uptake into the brain of rats on a control diet (normal), ketogenic diet, or fasted 48 h (means ± SD; n = 4/group), expressed as metabolic rate for acetoacetate. The uptake was measured by PET imaging and shows that brain uptake of ketones is stimulated to an approximately equivalent extent by 48 h fasting or 10 d on a very high fat ketogenic diet [154].
Figure 5.

Brain PET images showing 11C-acetoacetate (11C-AcAc; left) and 18F-fluorodeoxyglucose (18F-FDG; right) uptake by the human brain. Note that the color scales are not the same. As shown in the experimental protocol below the images, the tracers are injected sequentially with computed tomography scans before each injection. By sequentially injecting the tracers during one experiment, this approach minimizes intra-individual variability and provides a direct comparison between the brain uptake of the two tracers. Expressed as the relative term - standardized uptake values - our preliminary, unpublished data show that the brain uptake of 11C-AcAc is somewhat lower in the healthy elderly (mean - 74 y old; n=5) than in healthy young adults (mean – 26 y old; n=5). In this study, all subjects were screened to eliminate those with cognitive deficit and symptomatic disease, and were on no medications. When summed across 18 brain regions, the aging-related difference in brain 11C-AcAc uptake was about 50% less than for 18F-FDG. These measurements have not yet been corrected for any possible effect of aging-related brain atrophy, nor have they been expressed as metabolic rates. The aim of reporting these preliminary data here is to demonstrate the feasibility of these dual tracer PET measurements and not to make a claim at this time as to differences between the two age groups.
5.4 Ketogenic supplements and brain function
The potential application of stimulating mild ketonemia to circumvent problems with brain fuel supply or function has a long clinical history, starting with epilepsy about a century ago. The beneficial effect of mild to moderate ketonemia in 30–50% of children with intractable epilepsy is nothing short of remarkable [170,171]. Despite its benefits, the very high fat diet needed to induce mild ketonemia is extremely difficult to manage, has significant side effects and, despite decades of research, the mechanism by which this treatment works is still a mystery. Nevertheless, the relatively high success rate of this clearly unorthodox treatment in what are generally considered to be hopeless cases of intractable epilepsy is a testament to the rigorous procedures used to implement and efficiently maintain mild to moderate ketogenesis for periods of 2–3 y. Equally remarkable examples of the clinical efficacy of the same very high fat ketogenic diet are the rare cases of inborn errors of metabolism involving genetic GLUT-1 deficiency [172,173] and pyruvate dehydrogenase deficiency [174,175], without which such cases would rapidly be fatal.
Acute, controlled human experiments show that ketone infusion or ketogenesis inhibits the cognitive and behavioral sequelae of acute, experimental hypoglycemia, both in healthy adults [176,177] and in those with type 1 diabetes [178]. It is generally assumed that the cognitive effects of hypoglycemia can be prevented by ketones because they seamlessly replace glucose to meet the brain’s energy requirements. However, acutely raising plasma ketones also increases cerebral blood flow in humans, an effect that may contribute to their beneficial impact on cognition during hypoglycemia [20]. Studies in humans and animal models suggest further protective effects of ketones in the brain after ischemic insult and other treatments damaging neuronal function [179–181]. Assessment of plasma ketones, breath acetone and β-hydroxybutyrate oxidation all indicate that the ketogenic response to a low carbohydrate breakfast after an overnight fast is entirely normal in the healthy elderly [182].
Almost 20 years ago, brain activation with memory tasks was shown to improve glucose metabolism in the brain regions affected in AD [183]. More recent controlled clinical trials confirm that short-term improvement can occur in cognitive tests when individuals with mild to moderate AD are provided with an exogenous source of glucose, ketones, insulin, or insulin sensitizers [78,184–188]. These clinical studies show that the affected brain regions in AD are at least partially viable and that cognition can improve when exogenous fuel supply to the brain is improved. In two of these studies, ketogenic supplements based on medium chain triglycerides were used in order to permit a relatively normal choice of meals [185,186]. Medium chain triglycerides have long been known to be ketogenic because they contain medium chain fatty acids (octanoic [8:0] and decanoic [10:0] acids), which do not require activation by CoA to enter the mitochondria [189]. The mild beneficial effects on cognition and relatively good tolerance to the doses of medium chain triglyceride used are promising, notwithstanding the possibility that carriers of apoE4 with AD derive little benefit from this treatment [185]. The explanation for the beneficial effect of mild, experimental ketonemia on cognition in AD may be as simple exchanging one brain fuel for another as occurs in fasting or starvation. It may also be due to the observation that although glycolysis may be impaired in the AD brain, cerebral metabolic rate of ketones [21] and metabolic capacity [190] to use a fuel other than glucose are both apparently normal in AD.
Our own unpublished results suggest that mild ketonemia may also increase brain glucose uptake. Whatever the explanation, the relatively rapid onset of the benefit of a ketone-inducing supplement suggests a fairly direct mode of action involving improved energy substrate supply permitting improved neuronal function in AD. This implies that if brain hypometabolism in AD can be overcome (or prevented), the clinical fallout may be reduced. Nevertheless, it remains to be seen as to whether long term metabolic and cognitive benefits of ketogenic therapies can be demonstrated in AD.
6.0 METHODOLOGICAL CONSIDERATIONS
Advances in knowledge of how brain metabolism and cognition in the elderly are interconnected will depend on further advances in methodology and the resolution of issues that confound the interpretation of existing research. Amongst the key issues to resolve in relation to AD are whether brain metabolism changes with normal aging, how PET data are expressed, and development of consensus definitions of normal aging and cognitive decline. Even in clinically well-defined subjects (normal or diseased), a general limitation of most studies of brain metabolism is their cross-sectional design which does not allow assessment of the chronology of events or magnitude of change (decrease) in a given individual [2]. Moreover, the relatively small sample sizes do not permit adjustment for confounding factors that might be related to brain glucose metabolism and explain (or mask) some differences attributed to aging or early stages of AD (Tables 1–4).
6.1 Brain metabolism during aging
It is important to resolve whether healthy aging is really associated with a significant decline in CMRg, whether globally or regionally (Table 2) [191]. If it can be shown that lower CMRg with age is really a function of the way PET data are presented or interpreted, i.e. if it is a methodological issue, then the true decline in CMRg in AD is a function of a disease process and not a function of aging per se. If a decline in CMRg with age is real, we must conclude that some degree of decline in CMRg in the elderly is inevitable, i.e. that lower CMRg is a physiological, hence, obligatory part of normal aging. The pattern of lower CMRg that has been reported for normal aging [192] is not the same as that in AD, so it seems unlikely that, even if truly present, the apparent decline in CMRg with aging is simply an early stage of the form of brain hypometabolism seen in AD.
A combination of PET imaging methods that simultaneously measures brain glucose metabolism and β-amyloid deposition could improve the diagnosis and/or prognosis of AD, especially if applied longitudinally [43,193]. This dual tracer PET approach is inherently attractive because it permits simultaneous investigation of the potential link between development of the neuropathology of AD (β-amyloid accumulation or neurotransmitter defect) and the metabolic changes, i.e. the sequence of events leading to clinical expression of the disease. We are currently applying this dual tracer approach to see whether brain uptake of acetoacetate and FDG are both altered during normal aging (Figure 4) and will soon be applying this method to AD.
Focusing uniquely on PET methods may never completely resolve this issue but other experimental approaches shed light in this question. Acetylcholine synthesis is critically dependent on glucose metabolism but is also localized to the synapse, so PET may not be able to detect a difference in glucose metabolism that would be so highly localized [119]. Other minimally invasive methods may also be applicable depending on the question. Nuclear magnetic resonance (NMR) spectroscopy is applicable to the in vivo detection of molecules relevant to brain metabolism, including glucose by 1H-NMR [23], glycogen by 31P-NMR [194], and ketones by 13C-NMR [195]. The importance of glycogen in brain metabolism is still poorly understood but is amenable to dietary manipulation [194], which may have a bearing on changes in brain glucose metabolism accompanying aging or AD.
6.2 Expression of PET data
A morphological atlas based on CT or MRI images is now essential to obtain accurate CMRg measurements of specific brain regions. CT or MRI images are also invaluable for defining gray and white matter, correction for cerebrospinal fluid volume, all of which are pertinent to determining CMRg in the aging brain. In recent years, substantial improvements in PET technology, including reconstruction algorithms, sensitivity, and spatial resolution have led to better PET image quality. Increased axial field of view in a single acquisition now allows calculation of carotid blood radioactivity used to calculate FDG entering the brain, which may eventually replace the current gold standard based on blood sampling [196]. These new developments may help account for why the global CMRg of the adult human brain was calculated in the 1980s to be about 4 mg/100 g/min but more recently are about 7–8 mg/100 g/min (Table 2), which corresponds more to the classical value obtained by arterio-venous difference [13,22]. The different ways of expressed brain metabolism (SUV, global CMRg, regional CMRg, correction against the cerebellum or for brain atrophy) ultimately affect interpreting whether the metabolic rate of individual neurons changes with age or AD, or whether the number of neurons changes but not their individual activity. Modern PET technology has significantly increased the robustness of CMRg measurements and now needs application one of the central issues raised in this review – whether normal aging is accompanied by deteriorating brain glucose metabolism and, if so, why?
6.3 Criteria for healthy aging and assessment of MCI
Both biological and methodological confounders complicate the interpretation of brain glucose metabolism data during aging, including even such fundamental parameters as the definitions of ‘elderly’ and ‘healthy’. A full description of the status quo and evolution of these definitions is not the central focus of this paper but they are very relevant to the appropriate screening and classification of subjects for brain metabolic studies. Aging is commonly accompanied by increasing glucose intolerance relative to younger adults, but whether brain glucose uptake also starts to deteriorate in cognitively intact, glucose intolerant elderly is not yet known. Glucose intolerance may therefore be a key parameter in defining the risk of deteriorating brain glucose uptake in the elderly and, hence, whether they should be considered as ‘healthy’. Still, there appears to be a distinction between AD and accelerated aging that has been the focus of attention for some time [129]. For instance, as with some but not all PET data on brain metabolism (Table 4), production of acetylcholine and metabolism of glucose to CO2 by fresh human cortex do not necessarily change with age [119].
Aside from the methodological parameters related to obtaining quantitative data for brain fuel metabolism from PET images (Section 6.2), selection of an appropriate elderly control group is also of critical importance to applying CMRg to the study of disturbed brain glucose metabolism in the AD brain. Since AD is a disease of middle to late adulthood, appropriate age-matching is essential. Amongst non-genetic risk factors, AD is also affected by lifestyle, education, physical activity, diet and social integration, so effective screening to match for these parameters is also desirable. Amongst treatable diseases, hypertension, diabetes and blood lipid abnormalities are very frequent in the elderly and are linked to a higher risk of AD [197–199], so medication-free age-matched, healthy elderly subjects are the ideal controls for AD. Carriers of apoE4 are clearly at much higher risk of AD [38] and have disturbed brain glucose metabolism as much as 30 y before the typical clinical onset of AD (Table 3), so genotyping may also help reduce variability in the control population.
Diabetes and insulin resistance are major risk factors for AD, but why this is so seems paradoxical in light of the fact that brain glucose uptake has long been considered to be largely independent of insulin. One old report suggests CMRg is lower in type 1 diabetes [148], but whether brain glucose uptake is impaired in type 2 diabetes or insulin resistance is presently unknown. However, both these conditions are highly prevalent in the elderly so it is important to establish whether they confound interpretation of brain metabolism data in the elderly, or in individuals with a family history or genetic predisposition to AD.
A group of experts from various countries has recently attempted to overcome the heterogeneity in defining MCI, and has proposed specific criteria for AD before the dementia stage [46]. These criteria have yet to be adopted widely, mainly because they are very resource-intensive. There is mounting evidence that otherwise healthy elderly, with a higher burden of β-amyloid as detected by PET with a tracer such as Pittsburgh compound B, may have a lower performance on memory tests [200]. This raises the problem of defining healthy elderly controls according to clinical or imaging criteria. The healthy elderly included in studies of brain aging undoubtedly include some with MCI or pre-clinical AD. Indeed, longitudinal studies of healthy aging show that much of the cognitive decline attributed to ‘normal’ aging is actually a pre-clinical form of MCI or AD [201]. The general tendency is to diagnose neurodegenerative dementias at the earliest possible phase with the support of ancillary tests [202], but defining normal cognitive aging and applying such a definition widely enough to make studies comparable remains a challenge.
7.0 CONCLUSION
We have presented here an overview of the published evidence suggesting that impaired brain glucose metabolism may contribute to the development of AD, a concept developed by several independent research groups over at least the past 25 years (Figure 6) [3–6,78,81,126,181,183,185]. The gradual deterioration in systemic glucose metabolism commonly accompanying aging probably helps strain the finely tuned relationship between brain glucose uptake and brain function. This relationship normally involves minimal brain glucose storage, so if glucose supply to or glycolysis within the brain chronically decreases even a small amount, this would stand to compromise brain function. Normally, low brain glucose metabolism would be a reflection of low plasma glucose, and would be seamlessly compensated for by increased ketone production, the brain uptake of which is rapid and proportional to plasma ketone level (see Section 5.2, Figure 3). The ketogenic response to a low carbohydrate breakfast is normal in the elderly as long as their glucose tolerance is close to normal [182]. However, although not extensively studied yet, the ketogenic response appears to be less efficient when glucose intolerance develops, i.e. in type 2 diabetes and insulin resistance which are well-known to predispose to AD [98,137].
Figure 6.

Schematic overview of the concept that brain hypometabolism (Phase 1) contributes to the neuropathology underlying Alzheimer’s disease (AD; Phase 2), leading to the clinical symptoms of AD (Phase 3). The neuropathology and declining functionality of the brain can further contribute to brain hypometabolism, thereby completing a vicious cycle. In Phase 1, the hypometabolism is reportedly associated with various components of glucose utilization including one or a combination of impaired glucose transport (GLUT), impaired pyruvate dehydrogenase complex (PDHC) activity, and/or impaired α-ketoglutarate dehydrogenase complex activity (KDHC). Although the focus is on hypometabolism of glucose, it is not yet clear whether brain hypometabolism in AD affects glucose specifically or whether metabolism of other brain fuels such as ketones is also impaired. Brain hypometabolism is represented here as the first phase in the etiology of AD because it is the earliest known change in the brain associated with a risk factor predisposing to AD (presence of an apo E4 allele). In Phase 2, the microvascular changes involve altered blood-brain barrier function. In the normal brain, tau hyperphosphorylation can be stimulated by acute low glucose availability (see Section 4.1), so we propose that it is a consequence of brain hypometabolism. We propose that some brain areas are more susceptible to chronic brain hypometabolism, which can lead to regionalized brain starvation in areas that cannot adequately compensate by endogeneous gluconeogenesis. Hence, regionalized starvation and gluconeogenesis are shown here as consequences of brain hypometabolism. The cause brain hypometabolism is not yet known so it is possible that components of Phase 2 (particularly microvasculature changes and/or tau hyperphosphorylation can contribute to Phase 1, thereby further increasing the chances of developing Phase 3). Phase 3 represents the clinically observable phase which starts when the brain can no longer cope with the combination of chronic hypometabolism and neuropathological changes (Phases 1 and 2). Two related strategies are predicted to be potentially able to break the cycle (or delay it) and both involve a sustained improvement in brain fuel supply (dotted circle): (i) sustained improvement in brain glucose metabolism, which is probably dependent on sustained improvement in systemic glucose metabolism, and/or (ii) glucose replacement by ketones which are the brain’s preferred alternative physiological fuel to glucose. Such strategies are only likely to be effective if they can prevent Phase 1 becoming Phase 2, i.e. interrupting the deleterious impact of brain hypometabolism on the development of neuropathology, thereby preventing clinical symptoms of AD.
Since compensatory ketogenesis seems to be less efficient as systemic and brain glucose metabolism changes with age, to get sufficient fuel, we speculate that the brain may be forced to rely on a third option - the essentially catabolic process of gluconeogenesis. Since there is already a problem getting exogenous glucose into the brain, only gluconeogenesis endogenous to the brain is likely to increase glucose supply within the brain. Gluconeogenesis is at best a stop gap solution, so in the long run the energy deficit is exacerbated and some brain regions undoubtedly become fatigued [4]. The hippocampus appears to be one of the regions most vulnerable to this ongoing fuel deficit, which becomes essentially equivalent to chronic starvation. In turn, this brings on atrophy of these vulnerable regions, and the neurodegenerative changes and functional decline associated with AD. Impaired brain glucose metabolism appears to be a particularly pernicious problem in those with a genetic predisposition to AD (apoE4) or maternal family history of AD (Tables 3, 4), and can clearly be present asymptomatically 30 or more years before memory problems appear. In that sense, reduced brain activity may initially be a protective response to lower brain metabolism analogous to lower physical activity with chronic hunger or starvation [5]. Indeed, perhaps the ability to resist metabolic fatigue is part of what is known as the brain’s cognitive reserve.
Problems with brain uptake or metabolism of glucose that may precipitate AD don’t exclude brain hypometabolism from also being secondary to, e.g. a consequence of, AD (Figure 6). We agree that oxidative stress, mitochondrial dysfunction and mismanagement of amyloid precursor protein could contribute to synaptic failure which would further decrease brain glucose metabolism [2,38]. Nevertheless, two observations in particular support the notion that the neurons affected in AD are still functional so if brain fuel metabolism could be optimized or even partially returned towards normal, the risk of further cognitive decline should diminish: (i) in AD, brain ketone uptake is apparently normal or at least less affected than is glucose [21], and (ii) there is a functional response to nutritional supplements that increase brain fuel availability, particularly ketones (see Section 5.4). Raising plasma ketones to 0.4–0.5 mM would contribute to 5–10% of the brain’s energy requirements (Figure 3), which is equivalent to the early cortical glucose deficit in those genetically at risk AD [139]. Such a mild, safe level of ketonemia is achievable with ketogenic supplements, so if implemented before symptoms develop, it seems plausible that they could diminish the risk of further metabolic deterioration and clinical onset of cognitive decline.
The longstanding debate over whether AD is a form of accelerated aging [129] may never be resolved by PET studies of CMR alone, so it is important to develop a diversified arsenal of tools to fully explore the links between brain fuel metabolism, brain activity and the development, progression and treatment of AD. AD seems not to be an inevitable consequence of aging because although aging sets the stage for AD, about 50% of those >80 y old do not develop AD [203]. Even those who report that healthy aging is accompanied by declining brain metabolism [204,205] describe a pattern of brain hypometabolism in healthy aging that is distinct from what is observed in AD. Thus, cross-sectional studies are inadequate to establish causality and longitudinal studies are ultimately needed to definitively establish what is physiological and what is pathological about brain metabolism during aging.
If deteriorating brain glucose metabolism during normal aging does contribute to the development of AD, what causes impaired glucose transport, glycolysis or other possible forms of glucose mismanagement in the brain? Therapeutic and preventive approaches focused on improving brain fuel supply suggest that synaptic loss in the early stages of AD is incomplete or reversible, such that function can at least be transiently restored (see Section 5.4). Can preventive nutrition or lifestyle strategies be used to reduce the risk of AD, like with hypertension, type 2 diabetes and common forms of hypercholesterolemia? If so, how does this occur – by improving the functionality of existing synapses, neurons and pathways, or by sprouting new neural connections? Could the efficacy of pharmacological therapies targeting neurotransmitter availability be improved if brain fuel supply were also improved in AD? Once clinically apparent, is long term cognitive improvement in AD a realistic possibility?
Acknowledgments
Financial support for the research described here that was done by our group came from the Canada Research Chairs secretariat (SCC), CIHR, NSERC, CFI, AFMNet, Université de Sherbrooke (Faculty of Medicine and Health Sciences and the Department of Medicine), the Sherbrooke Molecular Imaging Center, the Etienne-Le Bel Clinical Research Centre, and the Research Center on Aging (both FRSQ funded), and FQRNT (CFQCU program). Excellent assistance was provided by Jennifer Mercier-Tremblay, Mélanie Fortier, Julie Desgagné, Conrad Filteau, Chantal Langevin, Dr. Otman Sarrhini, and Esteban O Espinosa.
ABBREVIATIONS
- aFH-AD
asymptomatic FH-AD
- aMCI
amnestic MCI
- AcAc
acetoacetate
- AD
Alzheimer’s disease
- CMRg
cerebral metabolic rate of glucose
- CMRk
cerebral metabolic rate of ketones
- DHA
docosahexaenoic acid
- FDG
fluorodeoxyglucose
- FH-AD
family history of AD
- FH(-)AD
no family history of AD and cognitively normal
- GLUT
glucose transporter
- HC
healthy controls
- MCI
mild cognitive impairment
- MCT
monocarboxylic acid transporter
- mFH-AD
maternal family history of AD but cognitively normal
- PET
positron emission tomography
- pFH-AD
paternal family history of AD but cognitively normal
- sFH-AD
symptomatic FH-AD
Footnotes
The authors declare no conflicts.
References
- 1.Blennow K, de Leon MJ, Zetterberg H. Alzheimer’s disease. Lancet. 2006;368(9533):387–403. doi: 10.1016/S0140-6736(06)69113-7. [DOI] [PubMed] [Google Scholar]
- 2.Jack CR, Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9:119–28. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sims NR, Bowen DM, Neary D, Davison AN. Metabolic processes in Alzheimer’s disease: adenine nucleotide content and production of 14CO2 from [U-14C]glucose in vitro in human neocortex. J Neurochem. 1983;41(5):1329–34. doi: 10.1111/j.1471-4159.1983.tb00829.x. [DOI] [PubMed] [Google Scholar]
- 4.Hoyer S, Oesterreich K, Wagner O. Glucose metabolism as the site of the primary abnormality in early-onset dementia of Alzheimer type? J Neurol. 1988;235(3):143–8. doi: 10.1007/BF00314304. [DOI] [PubMed] [Google Scholar]
- 5.Heininger K. The cerebral glucose-fatty acid cycle: evolutionary roots, regulation, and (patho)physiological importance. Int Rev Neurobiol. 2002;51:103–58. doi: 10.1016/s0074-7742(02)51004-7. [DOI] [PubMed] [Google Scholar]
- 6.Blass JP. A new approach to treating Alzheimer’s disease. Ann N Y Acad Sci. 2008;1147:122–8. doi: 10.1196/annals.1427.022. [DOI] [PubMed] [Google Scholar]
- 7.Holliday MA. Metabolic rate and organ size during growth from infancy to maturity and during late gastation and early infancy. Pediatrics. 1971;47(1 Suppl 2):169. [PubMed] [Google Scholar]
- 8.Sokoloff L. Energetics of functional activation in neural tissues. Neurochem Res. 1999;24(2):321–9. doi: 10.1023/a:1022534709672. [DOI] [PubMed] [Google Scholar]
- 9.Attwell D, Iadecola C. The neural basis of functional brain imaging signals. Trends Neurosci. 2002;25(12):621–5. doi: 10.1016/s0166-2236(02)02264-6. [DOI] [PubMed] [Google Scholar]
- 10.Shulman RG, Rothman DL, Behar KL, Hyder F. Energetic basis of brain activity: implications for neuroimaging. Trends Neurosci. 2004;27(8):489–95. doi: 10.1016/j.tins.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 11.Purdon AD, Rapoport SI. Energy consumption by phospholipid metabolism in mammalian brain. New York: Springer; 2007. [DOI] [PubMed] [Google Scholar]
- 12.Oz G, Kumar A, Rao JP, Kodl CT, Chow L, Eberly LE, et al. Human brain glycogen metabolism during and after hypoglycemia. Diabetes. 2009;58(9):1978–85. doi: 10.2337/db09-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Magistretti PJ. Brain energy metabolism. In: Bryrne JH, Roberts JL, editors. An introducation to cellular and molecular neuroscience. Hong Kong: Academic Press; 2004. pp. 67–90. [Google Scholar]
- 14.Pellerin L, Bouzier-Sore AK, Aubert A, Serres S, Merle M, Costalat R, et al. Activity-dependent regulation of energy metabolism by astrocytes: an update. Glia. 2007;55(12):1251–62. doi: 10.1002/glia.20528. [DOI] [PubMed] [Google Scholar]
- 15.Girouard H, Iadecola C. Neurovascular coupling in the normal brain and in hypertension, stroke, and Alzheimer disease. J Appl Physiol. 2006;100(1):328–35. doi: 10.1152/japplphysiol.00966.2005. [DOI] [PubMed] [Google Scholar]
- 16.Duelli R, Kuschinsky W. Brain glucose transporters: relationship to local energy demand. News Physiol Sci. 2001;16:71–6. doi: 10.1152/physiologyonline.2001.16.2.71. [DOI] [PubMed] [Google Scholar]
- 17.Leybaert L, De Bock M, Van Moorhem M, Decrock E, De Vuyst E. Neurobarrier coupling in the brain: adjusting glucose entry with demand. J Neurosci Res. 2007;85(15):3213–20. doi: 10.1002/jnr.21189. [DOI] [PubMed] [Google Scholar]
- 18.Barros LF, Porras OH, Bittner CX. Why glucose transport in the brain matters for PET. Trends Neurosci. 2005;28(3):117–9. doi: 10.1016/j.tins.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 19.Gottstein VU, Held K, Muller W, Berghoff W. Utilization of ketone bodies by the human brain. In: Meyer JS, Reivich M, Lecher H, Thomas CC, editors. Research on cerebral circulation: 5th international Salzburg conference; Springfield. 1970. pp. 137–45. [Google Scholar]
- 20.Hasselbalch SG, Madsen PL, Hageman LP, Olsen KS, Justesen N, Holm S, et al. Changes in cerebral blood flow and carbohydrate metabolism during acute hyperketonemia. Am J Physiol. 1996;270(5 Pt 1):E746–51. doi: 10.1152/ajpendo.1996.270.5.E746. [DOI] [PubMed] [Google Scholar]
- 21.Lying-Tunell U, Lindblad BS, Malmlund HO, Persson B. Cerebral blood flow and metabolic rate of oxygen, glucose, lactate, pyruvate, ketone bodies and amino acids. Acta Neurol Scand. 1981;63(6):337–50. doi: 10.1111/j.1600-0404.1981.tb00788.x. [DOI] [PubMed] [Google Scholar]
- 22.Owen OE, Morgan AP, Kemp HG, Sullivan JM, Herrera MG, Cahill GF., Jr Brain metabolism during fasting. J Clin Invest. 1967;46(10):1589–95. doi: 10.1172/JCI105650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pan JW, Telang FW, Lee JH, de Graaf RA, Rothman DL, Stein DT, et al. Measurement of beta-hydroxybutyrate in acute hyperketonemia in human brain. J Neurochem. 2001;79(3):539–44. doi: 10.1046/j.1471-4159.2001.00575.x. [DOI] [PubMed] [Google Scholar]
- 24.Phelps ME, Huang SC, Hoffman EJ, Selin C, Sokoloff L, Kuhl DE. Tomographic measurement of local cerebral glucose metabolic rate in humans with (F-18)2-fluoro-2-deoxy-D-glucose: validation of method. Annals of neurology. 1979;6(5):371–88. doi: 10.1002/ana.410060502. [DOI] [PubMed] [Google Scholar]
- 25.Sokoloff L. Measurement of local cerebral glucose utilization and its relation to local functional activity in the brain. Adv Exp Med Biol. 1991;291:21–42. doi: 10.1007/978-1-4684-5931-9_4. [DOI] [PubMed] [Google Scholar]
- 26.O’Sullivan F, Muzi M, Spence AM, Mankoff DM, O’Sullivan JN, Fitzgerald N, et al. Nonparametric Residue Analysis of Dynamic PET Data With Application to Cerebral FDG Studies in Normals. J Am Stat Assoc. 2009;104(486):556–71. doi: 10.1198/jasa.2009.0021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kimura N, Yamamoto Y, Kameyama R, Hatakeyama T, Kawai N, Nishiyama Y. Diagnostic value of kinetic analysis using dynamic 18F-FDG-PET in patients with malignant primary brain tumor. Nucl Med Commun. 2009;30(8):602–9. doi: 10.1097/MNM.0b013e32832e1c7d. [DOI] [PubMed] [Google Scholar]
- 28.Gunn RN, Gunn SR, Turkheimer FE, Aston JA, Cunningham VJ. Positron emission tomography compartmental models: a basis pursuit strategy for kinetic modeling. J Cereb Blood Flow Metab. 2002;22(12):1425–39. doi: 10.1097/01.wcb.0000045042.03034.42. [DOI] [PubMed] [Google Scholar]
- 29.Sokoloff L, Reivich M, Kennedy C, Des Rosiers MH, Patlak CS, Pettigrew KD, et al. The [14C]deoxyglucose method for the measurement of local cerebral glucose utilization: theory, procedure, and normal values in the conscious and anesthetized albino rat. J Neurochem. 1977;28(5):897–916. doi: 10.1111/j.1471-4159.1977.tb10649.x. [DOI] [PubMed] [Google Scholar]
- 30.Graham MM, Muzi M, Spence AM, O’Sullivan F, Lewellen TK, Link JM, et al. The FDG lumped constant in normal human brain. J Nucl Med. 2002;43(9):1157–66. [PubMed] [Google Scholar]
- 31.Huang SC. Anatomy of SUV. Standardized uptake value. Nucl Med Biol. 2000;27(7):643–6. doi: 10.1016/s0969-8051(00)00155-4. [DOI] [PubMed] [Google Scholar]
- 32.Krohn KA, Muzi M, Spence AM. What is in a number? The FDG lumped constant in the rat brain. J Nucl Med. 2007;48(1):5–7. [PubMed] [Google Scholar]
- 33.Lear JL, Ackerman RF. Comparison of regional blood-brain transport kinetics between glucose and fluorodeoxyglucose. J Nucl Med. 1992;33(10):1819–24. [PubMed] [Google Scholar]
- 34.Thie JA. Understanding the standardized uptake value, its methods, and implications for usage. J Nucl Med. 2004;45(9):1431–4. [PubMed] [Google Scholar]
- 35.Canadian Study of Health, and Aging Working Group. Canadian Study of Health and Aging: study methods and prevalence of dementia. Can Med Assoc J. 1994;150:899–913. [PMC free article] [PubMed] [Google Scholar]
- 36.Brayne C, Gao L, Dewey M, Matthews FE. Dementia before death in ageing societies--the promise of prevention and the reality. PLoS Med. 2006;3(10):e397. doi: 10.1371/journal.pmed.0030397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Y, Rinne JO, Mosconi L, Pirraglia E, Rusinek H, DeSanti S, et al. Regional analysis of FDG and PIB-PET images in normal aging, mild cognitive impairment, and Alzheimer’s disease. European journal of nuclear medicine and molecular imaging. 2008;35(12):2169–81. doi: 10.1007/s00259-008-0833-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Querfurth HW, LaFerla FM. Alzheimer’s disease. The New England journal of medicine. 2010;362(4):329–44. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 39.Lehtovirta M, Soininen H, Helisalmi S, Mannermaa A, Helkala EL, Hartikainen P, et al. Clinical and neuropsychological characteristics in familial and sporadic Alzheimer’s disease: relation to apolipoprotein E polymorphism. Neurology. 1996;46(2):413–9. doi: 10.1212/wnl.46.2.413. [DOI] [PubMed] [Google Scholar]
- 40.Erten-Lyons D, Woltjer RL, Dodge H, Nixon R, Vorobik R, Calvert JF, et al. Factors associated with resistance to dementia despite high Alzheimer disease pathology. Neurology. 2009;72(4):354–60. doi: 10.1212/01.wnl.0000341273.18141.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jack CR, Jr, Lowe VJ, Weigand SD, Wiste HJ, Senjem ML, Knopman DS, et al. Serial PIB and MRI in normal, mild cognitive impairment and Alzheimer’s disease: implications for sequence of pathological events in Alzheimer’s disease. Brain. 2009;132(Pt 5):1355–65. doi: 10.1093/brain/awp062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Herholz K, Carter SF, Jones M. Positron emission tomography imaging in dementia. The British journal of radiology. 2007;80(Spec No 2):S160–7. doi: 10.1259/bjr/97295129. [DOI] [PubMed] [Google Scholar]
- 43.Nordberg A, Rinne JO, Kadir A, Langstrom B. The use of PET in Alzheimer disease. Nat Rev Neurol. 2010;6(2):78–87. doi: 10.1038/nrneurol.2009.217. [DOI] [PubMed] [Google Scholar]
- 44.Silverman DH, Mosconi L, Ercoli L, Chen W, Small GW. Positron emission tomography scans obtained for the evaluation of cognitive dysfunction. Seminars in nuclear medicine. 2008;38(4):251–61. doi: 10.1053/j.semnuclmed.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mosconi L, Brys M, Switalski R, Mistur R, Glodzik L, Pirraglia E, et al. Maternal family history of Alzheimer’s disease predisposes to reduced brain glucose metabolism. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(48):19067–72. doi: 10.1073/pnas.0705036104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dubois B, Feldman HH, Jacova C, Dekosky ST, Barberger-Gateau P, Cummings J, et al. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria. Lancet Neurol. 2007;6(8):734–46. doi: 10.1016/S1474-4422(07)70178-3. [DOI] [PubMed] [Google Scholar]
- 47.Mosconi L, Sorbi S, Nacmias B, De Cristofaro MT, Fayyaz M, Cellini E, et al. Brain metabolic differences between sporadic and familial Alzheimer’s disease. Neurology. 2003;61(8):1138–40. doi: 10.1212/01.wnl.0000086816.30011.75. [DOI] [PubMed] [Google Scholar]
- 48.Ibanez V, Pietrini P, Alexander GE, Furey ML, Teichberg D, Rajapakse JC, et al. Regional glucose metabolic abnormalities are not the result of atrophy in Alzheimer’s disease. Neurology. 1998;50(6):1585–93. doi: 10.1212/wnl.50.6.1585. [DOI] [PubMed] [Google Scholar]
- 49.Meltzer CC, Zubieta JK, Brandt J, Tune LE, Mayberg HS, Frost JJ. Regional hypometabolism in Alzheimer’s disease as measured by positron emission tomography after correction for effects of partial volume averaging. Neurology. 1996;47(2):454–61. doi: 10.1212/wnl.47.2.454. [DOI] [PubMed] [Google Scholar]
- 50.Mosconi L, Tsui WH, De Santi S, Li J, Rusinek H, Convit A, et al. Reduced hippocampal metabolism in MCI and AD: automated FDG-PET image analysis. Neurology. 2005;64(11):1860–7. doi: 10.1212/01.WNL.0000163856.13524.08. [DOI] [PubMed] [Google Scholar]
- 51.Crawford JG. Alzheimer’s disease risk factors as related to cerebral blood flow. Med Hypotheses. 1996;46(4):367–77. doi: 10.1016/s0306-9877(96)90189-9. [DOI] [PubMed] [Google Scholar]
- 52.Hock C, Villringer K, Muller-Spahn F, Wenzel R, Heekeren H, Schuh-Hofer S, et al. Decrease in parietal cerebral hemoglobin oxygenation during performance of a verbal fluency task in patients with Alzheimer’s disease monitored by means of near-infrared spectroscopy (NIRS)–-correlation with simultaneous rCBF-PET measurements. Brain Res. 1997;755(2):293–303. doi: 10.1016/s0006-8993(97)00122-4. [DOI] [PubMed] [Google Scholar]
- 53.Kalaria RN, Harik SI. Reduced glucose transporter at the blood-brain barrier and in cerebral cortex in Alzheimer disease. J Neurochem. 1989;53(4):1083–8. doi: 10.1111/j.1471-4159.1989.tb07399.x. [DOI] [PubMed] [Google Scholar]
- 54.Marcus DL, de Leon MJ, Goldman J, Logan J, Christman DR, Wolf AP, et al. Altered glucose metabolism in microvessels from patients with Alzheimer’s disease. Annals of neurology. 1989;26(1):91–4. doi: 10.1002/ana.410260114. [DOI] [PubMed] [Google Scholar]
- 55.Petersen RC. Clinical trials for early (pre-dementia) Alzheimer’s disease: a case for mild cognitive impairment. J Nutr Health Aging. 2010;14(4):304–5. doi: 10.1007/s12603-010-0068-z. [DOI] [PubMed] [Google Scholar]
- 56.Chertkow H, Massoud F, Nasreddine Z, Belleville S, Joanette Y, Bocti C, et al. Diagnosis and treatment of dementia: 3. Mild cognitive impairment and cognitive impairment without dementia. CMAJ. 2008;178(10):1273–85. doi: 10.1503/cmaj.070797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barberger-Gateau P, Commenges D, Gagnon M, Letenneur L, Sauvel C, Dartigues JF. Instrumental activities of daily living as a screening tool for cognitive impairment and dementia in elderly community dwellers. J Am Geriatr Soc. 1992;40:1129–34. doi: 10.1111/j.1532-5415.1992.tb01802.x. [DOI] [PubMed] [Google Scholar]
- 58.Del Sole A, Clerici F, Chiti A, Lecchi M, Mariani C, Maggiore L, et al. Individual cerebral metabolic deficits in Alzheimer’s disease and amnestic mild cognitive impairment: an FDG PET study. European journal of nuclear medicine and molecular imaging. 2008;35(7):1357–66. doi: 10.1007/s00259-008-0773-6. [DOI] [PubMed] [Google Scholar]
- 59.Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Archives of neurology. 1999;56(3):303–8. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- 60.Balasse EO, Fery F. Ketone body production and disposal: effects of fasting, diabetes, and exercise. Diabetes Metab Rev. 1989;5(3):247–70. doi: 10.1002/dmr.5610050304. [DOI] [PubMed] [Google Scholar]
- 61.Dubois B, Picard G, Sarazin M. Early detection of Alzheimer’s disease: new diagnostic criteria. Dialogues Clin Neurosci. 2009;11(2):135–9. doi: 10.31887/DCNS.2009.11.2/bdubois. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, et al. Correlations between apolipoprotein E epsilon4 gene dose and brain-imaging measurements of regional hypometabolism. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(23):8299–302. doi: 10.1073/pnas.0500579102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reiman EM, Chen K, Liu X, Bandy D, Yu M, Lee W, et al. Fibrillar amyloid-beta burden in cognitively normal people at 3 levels of genetic risk for Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(16):6820–5. doi: 10.1073/pnas.0900345106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Mosconi L, Mistur R, Switalski R, Tsui WH, Glodzik L, Li Y, et al. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. European journal of nuclear medicine and molecular imaging. 2009;36(5):811–22. doi: 10.1007/s00259-008-1039-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Drzezga A, Lautenschlager N, Siebner H, Riemenschneider M, Willoch F, Minoshima S, et al. Cerebral metabolic changes accompanying conversion of mild cognitive impairment into Alzheimer’s disease: a PET follow–up study. European journal of nuclear medicine and molecular imaging. 2003;30(8):1104–13. doi: 10.1007/s00259-003-1194-1. [DOI] [PubMed] [Google Scholar]
- 66.Ishii H, Ishikawa H, Meguro K, Tashiro M, Yamaguchi S. Decreased cortical glucose metabolism in converters from CDR 0. 5 to Alzheimer’s disease in a community: the Osaki-Tajiri Project. Int Psychogeriatr. 2009;21(1):148–56. doi: 10.1017/S1041610208008132. [DOI] [PubMed] [Google Scholar]
- 67.Mosconi L, Mistur R, Switalski R, Brys M, Glodzik L, Rich K, et al. Declining brain glucose metabolism in normal individuals with a maternal history of Alzheimer disease. Neurology. 2009;72(6):513–20. doi: 10.1212/01.wnl.0000333247.51383.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ibanez V, Pietrini P, Furey ML, Alexander GE, Millet P, Bokde AL, et al. Resting state brain glucose metabolism is not reduced in normotensive healthy men during aging, after correction for brain atrophy. Brain research bulletin. 2004;63(2):147–54. doi: 10.1016/j.brainresbull.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 69.Schlageter NL, Carson RE, Rapoport SI. Examination of blood-brain barrier permeability in dementia of the Alzheimer type with [68Ga]EDTA and positron emission tomography. J Cereb Blood Flow Metab. 1987;7(1):1–8. doi: 10.1038/jcbfm.1987.1. [DOI] [PubMed] [Google Scholar]
- 70.Bourre JM, Francois M, Youyou A, Dumont O, Piciotti M, Pascal G, et al. The effects of dietary alpha-linolenic acid on the composition of nerve membranes, enzymatic activity, amplitude of electrophysiological parameters, resistance to poisons and performance of learning tasks in rats. J Nutr. 1989;119(12):1880–92. doi: 10.1093/jn/119.12.1880. [DOI] [PubMed] [Google Scholar]
- 71.Pifferi F, Roux F, Langelier B, Alessandri JM, Vancassel S, Jouin M, et al. (n-3) polyunsaturated fatty acid deficiency reduces the expression of both isoforms of the brain glucose transporter GLUT1 in rats. J Nutr. 2005;135(9):2241–6. doi: 10.1093/jn/135.9.2241. [DOI] [PubMed] [Google Scholar]
- 72.Ximenes da Silva A, Lavialle F, Gendrot G, Guesnet P, Alessandri JM, Lavialle M. Glucose transport and utilization are altered in the brain of rats deficient in n-3 polyunsaturated fatty acids. J Neurochem. 2002;81(6):1328–37. doi: 10.1046/j.1471-4159.2002.00932.x. [DOI] [PubMed] [Google Scholar]
- 73.Pifferi F, Jouin M, Alessandri JM, Haedke U, Roux F, Perrière N, et al. n-3 Fatty acids modulate brain glucose transport in endothelial cells of the blood-brain barrier. Prostaglandins Leukot Essent Fatty Acids. 2007;77(5–6):279–86. doi: 10.1016/j.plefa.2007.10.011. [DOI] [PubMed] [Google Scholar]
- 74.Brenna JT, Diau GY. The influence of dietary docosahexaenoic acid and arachidonic acid on central nervous system polyunsaturated fatty acid composition. Prostaglandins Leukot Essent Fatty Acids. 2007;77(5–6):247–50. doi: 10.1016/j.plefa.2007.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Cunnane SC, Plourde M, Pifferi F, Begin M, Feart C, Barberger-Gateau P. Fish, docosahexaenoic acid and Alzheimer’s disease. Prog Lipid Res. 2009;48(5):239–56. doi: 10.1016/j.plipres.2009.04.001. [DOI] [PubMed] [Google Scholar]
- 76.Plourde M, Vohl MC, Vandal M, Couture P, Lemieux S, Cunnane SC. Plasma n-3 fatty acid response to an n-3 fatty acid supplement is modulated by apoE epsilon4 but not by the common PPAR-alpha L162V polymorphism in men. Br J Nutr. 2009;102(8):1121–4. doi: 10.1017/S000711450938215X. [DOI] [PubMed] [Google Scholar]
- 77.Fortier M, Tremblay-Mercier J, Plourde M, Chouinard-Watkins R, Vandal M, Pifferi F, et al. Higher plasma n-3 fatty acid status in the moderately healthy elderly in southern Quebec: Higher fish intake or aging-related change in n-3 fatty acid metabolism? Prostaglandins Leukot Essent Fatty Acids. 2010 doi: 10.1016/j.plefa.2010.02.010. [DOI] [PubMed] [Google Scholar]
- 78.Watson GS, Craft S. Modulation of memory by insulin and glucose: neuropsychological observations in Alzheimer’s disease. Eur J Pharmacol. 2004;490(1–3):97–113. doi: 10.1016/j.ejphar.2004.02.048. [DOI] [PubMed] [Google Scholar]
- 79.Hoyer S. The aging brain. Changes in the neuronal insulin/insulin receptor signal transduction cascade trigger late-onset sporadic Alzheimer disease (SAD). A mini-review. J Neural Transm. 2002;109(7–8):991–1002. doi: 10.1007/s007020200082. [DOI] [PubMed] [Google Scholar]
- 80.Schubert M, Gautam D, Surjo D, Ueki K, Baudler S, Schubert D, et al. Role for neuronal insulin resistance in neurodegenerative diseases. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(9):3100–5. doi: 10.1073/pnas.0308724101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Steen E, Terry BM, Rivera EJ, Cannon JL, Neely TR, Tavares R, et al. Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease--is this type 3 diabetes? J Alzheimers Dis. 2005;7(1):63–80. doi: 10.3233/jad-2005-7107. [DOI] [PubMed] [Google Scholar]
- 82.Cole GM, Frautschy SA. The role of insulin and neurotrophic factor signaling in brain aging and Alzheimer’s Disease. Exp Gerontol. 2007;42(1–2):10–21. doi: 10.1016/j.exger.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 83.Van den Berghe G, Schoonheydt K, Becx P, Bruyninckx F, Wouters PJ. Insulin therapy protects the central and peripheral nervous system of intensive care patients. Neurology. 2005;64(8):1348–53. doi: 10.1212/01.WNL.0000158442.08857.FC. [DOI] [PubMed] [Google Scholar]
- 84.Soeters MR, Sauerwein HP, Faas L, Smeenge M, Duran M, Wanders RJ, et al. Effects of insulin on ketogenesis following fasting in lean and obese men. Obesity (Silver Spring) 2009;17(7):1326–31. doi: 10.1038/oby.2008.678. [DOI] [PubMed] [Google Scholar]
- 85.Elahi D, Muller DC, Egan JM, Andres R, Veldhuist J, Meneilly GS. Glucose tolerance, glucose utilization and insulin secretion in ageing. Novartis Found Symp. 2002;242:222–42. discussion 42–6. [PubMed] [Google Scholar]
- 86.Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL, et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300(5622):1140–2. doi: 10.1126/science.1082889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Bortz WM., 2nd A conceptual framework of frailty: a review. J Gerontol A Biol Sci Med Sci. 2002;57(5):M283–8. doi: 10.1093/gerona/57.5.m283. [DOI] [PubMed] [Google Scholar]
- 88.Craft S, Asthana S, Schellenberg G, Baker L, Cherrier M, Boyt AA, et al. Insulin effects on glucose metabolism, memory, and plasma amyloid precursor protein in Alzheimer’s disease differ according to apolipoprotein-E genotype. Ann N Y Acad Sci. 2000;903:222–8. doi: 10.1111/j.1749-6632.2000.tb06371.x. [DOI] [PubMed] [Google Scholar]
- 89.Guigoz Y, Vellas B, Garry PJ. Assessing the nutritional status of the elderly: The Mini Nutritional Assessment as part of the geriatric evaluation. Nutr Rev. 1996;54(1 Pt 2):S59–65. doi: 10.1111/j.1753-4887.1996.tb03793.x. [DOI] [PubMed] [Google Scholar]
- 90.Raffaitin C, Gin H, Empana JP, Helmer C, Berr C, Tzourio C, et al. Metabolic syndrome and risk for incident Alzheimer’s disease or vascular dementia: the Three-City Study. Diabetes Care. 2009;32(1):169–74. doi: 10.2337/dc08-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schmuck A, Villet A, Payen N, Alary J, Franco A, Roussel AM. Fatty acid nutriture in hospitalized elderly women. J Am Coll Nutr. 1998;17(5):448–53. doi: 10.1080/07315724.1998.10718792. [DOI] [PubMed] [Google Scholar]
- 92.Walston J, Fried LP. Frailty and the older man. Med Clin North Am. 1999;83(5):1173–94. doi: 10.1016/s0025-7125(05)70157-7. [DOI] [PubMed] [Google Scholar]
- 93.Bruce-Keller AJ, Keller JN, Morrison CD. Obesity and vulnerability of the CNS. Biochim Biophys Acta. 2009;1792(5):395–400. doi: 10.1016/j.bbadis.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Floras JS, Meneilly G. Insulin-mediated blood flow and glucose uptake. Can J Cardiol. 2001;17 (Suppl A):7A–10A. [PubMed] [Google Scholar]
- 95.Lewis GF, Carpentier A, Adeli K, Giacca A. Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes. Endocr Rev. 2002;23(2):201–29. doi: 10.1210/edrv.23.2.0461. [DOI] [PubMed] [Google Scholar]
- 96.Carpentier AC, Frisch F, Cyr D, Genereux P, Patterson BW, Giguere R, et al. On the suppression of plasma nonesterified fatty acids by insulin during enhanced intravascular lipolysis in humans. Am J Physiol Endocrinol Metab. 2005;289(5):E849–56. doi: 10.1152/ajpendo.00073.2005. [DOI] [PubMed] [Google Scholar]
- 97.Fernandez-Figares I, Shannon AE, Wray-Cahen D, Caperna TJ. The role of insulin, glucagon, dexamethasone, and leptin in the regulation of ketogenesis and glycogen storage in primary cultures of porcine hepatocytes prepared from 60 kg pigs. Domest Anim Endocrinol. 2004;27(2):125–40. doi: 10.1016/j.domaniend.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 98.Fukao T, Lopaschuk GD, Mitchell GA. Pathways and control of ketone body metabolism: on the fringe of lipid biochemistry. Prostaglandins Leukot Essent Fatty Acids. 2004;70(3):243–51. doi: 10.1016/j.plefa.2003.11.001. [DOI] [PubMed] [Google Scholar]
- 99.Jauch KW, Gunther B, Wicklmayr M, Teichmann R, Hartl W, Dietze G. Postoperative insulin resistance of lipolysis and ketogenesis using the glucose clamp technic. Infusionsther Klin Ernahr. 1984;11(5):271–4. [PubMed] [Google Scholar]
- 100.Wolfrum C, Asilmaz E, Luca E, Friedman JM, Stoffel M. Foxa2 regulates lipid metabolism and ketogenesis in the liver during fasting and in diabetes. Nature. 2004;432(7020):1027–32. doi: 10.1038/nature03047. [DOI] [PubMed] [Google Scholar]
- 101.Beylot M, Sautot G, Martin C, Cohen R, Berthezene F, Mornex R. Measurement of insulin sensitivity using the somatostatin-insulin-glucose test. Importance of the determination of plasma free fatty acids and insulin clearance. Presse Med. 1983;12(40):2517–20. [PubMed] [Google Scholar]
- 102.Carpentier A, Mittelman SD, Bergman RN, Giacca A, Lewis GF. Prolonged elevation of plasma free fatty acids impairs pancreatic beta-cell function in obese nondiabetic humans but not in individuals with type 2 diabetes. Diabetes. 2000;49(3):399–408. doi: 10.2337/diabetes.49.3.399. [DOI] [PubMed] [Google Scholar]
- 103.Schapira AH. Mitochondrial disease. Lancet. 2006;368(9529):70–82. doi: 10.1016/S0140-6736(06)68970-8. [DOI] [PubMed] [Google Scholar]
- 104.Bough KJ, Wetherington J, Hassel B, Pare JF, Gawryluk JW, Greene JG, et al. Mitochondrial biogenesis in the anticonvulsant mechanism of the ketogenic diet. Annals of neurology. 2006;60(2):223–35. doi: 10.1002/ana.20899. [DOI] [PubMed] [Google Scholar]
- 105.Aksenov MY, Tucker HM, Nair P, Aksenova MV, Butterfield DA, Estus S, et al. The expression of several mitochondrial and nuclear genes encoding the subunits of electron transport chain enzyme complexes, cytochrome c oxidase, and NADH dehydrogenase, in different brain regions in Alzheimer’s disease. Neurochem Res. 1999;24(6):767–74. doi: 10.1023/a:1020783614031. [DOI] [PubMed] [Google Scholar]
- 106.Bonilla E, Tanji K, Hirano M, Vu TH, DiMauro S, Schon EA. Mitochondrial involvement in Alzheimer’s disease. Biochim Biophys Acta. 1999;1410(2):171–82. doi: 10.1016/s0005-2728(98)00165-0. [DOI] [PubMed] [Google Scholar]
- 107.Castellani R, Hirai K, Aliev G, Drew KL, Nunomura A, Takeda A, et al. Role of mitochondrial dysfunction in Alzheimer’s disease. J Neurosci Res. 2002;70(3):357–60. doi: 10.1002/jnr.10389. [DOI] [PubMed] [Google Scholar]
- 108.Coon KD, Valla J, Szelinger S, Schneider LE, Niedzielko TL, Brown KM, et al. Quantitation of heteroplasmy of mtDNA sequence variants identified in a population of AD patients and controls by array-based resequencing. Mitochondrion. 2006;6(4):194–210. doi: 10.1016/j.mito.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 109.Gibson GE, Starkov A, Blass JP, Ratan RR, Beal MF. Cause and consequence: mitochondrial dysfunction initiates and propagates neuronal dysfunction, neuronal death and behavioral abnormalities in age-associated neurodegenerative diseases. Biochim Biophys Acta. 2010;1802(1):122–34. doi: 10.1016/j.bbadis.2009.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Heales SJ, Bolanos JP, Stewart VC, Brookes PS, Land JM, Clark JB. Nitric oxide, mitochondria and neurological disease. Biochim Biophys Acta. 1999;1410(2):215–28. doi: 10.1016/s0005-2728(98)00168-6. [DOI] [PubMed] [Google Scholar]
- 111.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–95. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 112.Studzinski CM, MacKay WA, Beckett TL, Henderson ST, Murphy MP, Sullivan PG, et al. Induction of ketosis may improve mitochondrial function and decrease steady-state amyloid-beta precursor protein (APP) levels in the aged dog. Brain Res. 2008;1226:209–17. doi: 10.1016/j.brainres.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 113.Villa RF, Gorini A, Hoyer S. Differentiated effect of ageing on the enzymes of Krebs’ cycle, electron transfer complexes and glutamate metabolism of non-synaptic and intra-synaptic mitochondria from cerebral cortex. J Neural Transm. 2006;113(11):1659–70. doi: 10.1007/s00702-006-0569-4. [DOI] [PubMed] [Google Scholar]
- 114.Hatanpaa K, Brady DR, Stoll J, Rapoport SI, Chandrasekaran K. Neuronal activity and early neurofibrillary tangles in Alzheimer’s disease. Annals of neurology. 1996;40(3):411–20. doi: 10.1002/ana.410400310. [DOI] [PubMed] [Google Scholar]
- 115.Hatanpaa K, Chandrasekaran K, Brady DR, Rapoport SI. No association between Alzheimer plaques and decreased levels of cytochrome oxidase subunit mRNA, a marker of neuronal energy metabolism. Brain Res Mol Brain Res. 1998;59(1):13–21. doi: 10.1016/s0169-328x(98)00117-x. [DOI] [PubMed] [Google Scholar]
- 116.Hatanpaa K, Isaacs KR, Shirao T, Brady DR, Rapoport SI. Loss of proteins regulating synaptic plasticity in normal aging of the human brain and in Alzheimer disease. J Neuropathol Exp Neurol. 1999;58(6):637–43. doi: 10.1097/00005072-199906000-00008. [DOI] [PubMed] [Google Scholar]
- 117.Sumpter PQ, Mann DM, Davies CA, Yates PO, Snowden JS, Neary D. An ultrastructural analysis of the effects of accumulation of neurofibrillary tangle in pyramidal neurons of the cerebral cortex in Alzheimer’s disease. Neuropathol Appl Neurobiol. 1986;12(3):305–19. doi: 10.1111/j.1365-2990.1986.tb00142.x. [DOI] [PubMed] [Google Scholar]
- 118.Sorbi S, Bird ED, Blass JP. Decreased pyruvate dehydrogenase complex activity in Huntington and Alzheimer brain. Annals of neurology. 1983;13(1):72–8. doi: 10.1002/ana.410130116. [DOI] [PubMed] [Google Scholar]
- 119.Sims NR, Bowen DM, Davison AN. 14C]acetylcholine synthesis and [14C]carbon dioxide production from [U-14C]glucose by tissue prisms from human neocortex. Biochem J. 1981;196(3):867–76. doi: 10.1042/bj1960867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.de la Monte SM. Insulin resistance and Alzheimer’s disease. BMB Rep. 2009;42(8):475– 81. doi: 10.5483/bmbrep.2009.42.8.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ramassamy C. Emerging role of polyphenolic compounds in the treatment of neurodegenerative diseases: a review of their intracellular targets. Eur J Pharmacol. 2006;545(1):51–64. doi: 10.1016/j.ejphar.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 122.Deschamps V, Barberger-Gateau P, Peuchant E, Orgogozo JM. Nutritional factors in cerebral aging and dementia: epidemiological arguments for a role of oxidative stress. Neuroepidemiology. 2001;20:7–15. doi: 10.1159/000054752. [DOI] [PubMed] [Google Scholar]
- 123.Lassen NA, Munck O, Tottey ER. Mental function and cerebral oxygen consumption in organic dementia. AMA Arch Neurol Psychiatry. 1957;77(2):126–33. doi: 10.1001/archneurpsyc.1957.02330320024003. [DOI] [PubMed] [Google Scholar]
- 124.Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, et al. Physical basis of cognitive alterations in Alzheimer’s disease: synapse loss is the major correlate of cognitive impairment. Annals of neurology. 1991;30(4):572–80. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- 125.Mosconi L, Pupi A, De Leon MJ. Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann N Y Acad Sci. 2008;1147:180–95. doi: 10.1196/annals.1427.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Freemantle E, Vandal M, Tremblay-Mercier J, Tremblay S, Blachere JC, Begin ME, et al. Omega-3 fatty acids, energy substrates, and brain function during aging. Prostaglandins Leukot Essent Fatty Acids. 2006;75(3):213–20. doi: 10.1016/j.plefa.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 127.Liu F, Iqbal K, Grundke-Iqbal I, Hart GW, Gong CX. O-GlcNAcylation regulates phosphorylation of tau: a mechanism involved in Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(29):10804–9. doi: 10.1073/pnas.0400348101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Liu Y, Liu F, Iqbal K, Grundke-Iqbal I, Gong CX. Decreased glucose transporters correlate to abnormal hyperphosphorylation of tau in Alzheimer disease. FEBS Lett. 2008;582(2):359–64. doi: 10.1016/j.febslet.2007.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bowen DM, White P, Spillane JA, Goodhardt MJ, Curzon G, Iwangoff P, et al. Accelerated ageing or selective neuronal loss as an important cause of dementia? Lancet. 1979;1(8106):11–4. doi: 10.1016/s0140-6736(79)90454-9. [DOI] [PubMed] [Google Scholar]
- 130.Ramassamy C, Averill D, Beffert U, Theroux L, Lussier-Cacan S, Cohn JS, et al. Oxidative insults are associated with apolipoprotein E genotype in Alzheimer’s disease brain. Neurobiol Dis. 2000;7(1):23–37. doi: 10.1006/nbdi.1999.0273. [DOI] [PubMed] [Google Scholar]
- 131.Aliev G, Palacios HH, Walrafen B, Lipsitt AE, Obrenovich ME, Morales L. Brain mitochondria as a primary target in the development of treatment strategies for Alzheimer disease. Int J Biochem Cell Biol. 2009;41(10):1989–2004. doi: 10.1016/j.biocel.2009.03.015. [DOI] [PubMed] [Google Scholar]
- 132.Bailey TL, Rivara CB, Rocher AB, Hof PR. The nature and effects of cortical microvascular pathology in aging and Alzheimer’s disease. Neurol Res. 2004;26(5):573–8. doi: 10.1179/016164104225016272. [DOI] [PubMed] [Google Scholar]
- 133.Grinberg LT, Thal DR. Vascular pathology in the aged human brain. Acta Neuropathol. 2010 doi: 10.1007/s00401-010-0652-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Borowsky IW, Collins RC. Metabolic anatomy of brain: a comparison of regional capillary density, glucose metabolism, and enzyme activities. J Comp Neurol. 1989;288(3):401–13. doi: 10.1002/cne.902880304. [DOI] [PubMed] [Google Scholar]
- 135.Harik SI, LaManna JC. Altered glucose metabolism in microvessels from patients with Alzheimer’s disease. Annals of neurology. 1991;29(5):573. doi: 10.1002/ana.410290521. [DOI] [PubMed] [Google Scholar]
- 136.Oldendorf WH, Cornford ME, Brown WJ. The large apparent work capability of the blood-brain barrier: a study of the mitochondrial content of capillary endothelial cells in brain and other tissues of the rat. Annals of neurology. 1977;1(5):409–17. doi: 10.1002/ana.410010502. [DOI] [PubMed] [Google Scholar]
- 137.Pasquier F, Boulogne A, Leys D, Fontaine P. Diabetes mellitus and dementia. Diabetes Metab. 2006;32(5 Pt 1):403–14. doi: 10.1016/s1262-3636(07)70298-7. [DOI] [PubMed] [Google Scholar]
- 138.Reiman EM, Caselli RJ, Yun LS, Chen K, Bandy D, Minoshima S, et al. Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. The New England journal of medicine. 1996;334(12):752–8. doi: 10.1056/NEJM199603213341202. [DOI] [PubMed] [Google Scholar]
- 139.Reiman EM, Chen K, Alexander GE, Caselli RJ, Bandy D, Osborne D, et al. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(1):284–9. doi: 10.1073/pnas.2635903100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Reiman EM, Uecker A, Caselli RJ, Lewis S, Bandy D, de Leon MJ, et al. Hippocampal volumes in cognitively normal persons at genetic risk for Alzheimer’s disease. Annals of neurology. 1998;44(2):288–91. doi: 10.1002/ana.410440226. [DOI] [PubMed] [Google Scholar]
- 141.Cahill GF., Jr Fuel metabolism in starvation. Annu Rev Nutr. 2006;26:1–22. doi: 10.1146/annurev.nutr.26.061505.111258. [DOI] [PubMed] [Google Scholar]
- 142.Cunnane SC. Origins and evolution of the Western diet: implications of iodine and seafood intakes for the human brain. Am J Clin Nutr. 2005;82(2):483. doi: 10.1093/ajcn/82.2.483. author reply 83–4. [DOI] [PubMed] [Google Scholar]
- 143.Mason GF, Petersen KF, Lebon V, Rothman DL, Shulman GI. Increased brain monocarboxylic acid transport and utilization in type 1 diabetes. Diabetes. 2006;55(4):929–34. doi: 10.2337/diabetes.55.04.06.db05-1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Robinson AM, Williamson DH. Physiological roles of ketone bodies as substrates and signals in mammalian tissues. Physiol Rev. 1980;60(1):143–87. doi: 10.1152/physrev.1980.60.1.143. [DOI] [PubMed] [Google Scholar]
- 145.Goldstein GW. Relation of potassium transport to oxidative metabolism in isolated brain capillaries. J Physiol. 1979;286:185–95. doi: 10.1113/jphysiol.1979.sp012613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Guzman M, Blazquez C. Is there an astrocyte-neuron ketone body shuttle? Trends Endocrinol Metab. 2001;12(4):169–73. doi: 10.1016/s1043-2760(00)00370-2. [DOI] [PubMed] [Google Scholar]
- 147.Melo TM, Nehlig A, Sonnewald U. Neuronal-glial interactions in rats fed a ketogenic diet. Neurochem Int. 2006;48(6–7):498–507. doi: 10.1016/j.neuint.2005.12.037. [DOI] [PubMed] [Google Scholar]
- 148.Gottstein WJ, Eachus AH, Cheney LC. A novel acylation of amino acids with S-carboxymethyl dialkyldithiocarbamates. J Org Chem. 1970;35(5):1693–4. doi: 10.1021/jo00830a103. [DOI] [PubMed] [Google Scholar]
- 149.Pardridge WM. Blood-brain barrier transport of glucose, free fatty acids, and ketone bodies. Adv Exp Med Biol. 1991;291:43–53. doi: 10.1007/978-1-4684-5931-9_5. [DOI] [PubMed] [Google Scholar]
- 150.Bates MW, Krebs HA, Williamson DH. Turnover rates of ketone bodies in normal, starved and alloxan-diabetic rats. Biochem J. 1968;110(4):655–61. doi: 10.1042/bj1100655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Daniel PM, Love ER, Moorehouse SR, Pratt OE, Wilson P. Factors influencing utilisation of ketone–bodies by brain in normal rats and rats with ketoacidosis. Lancet. 1971;2(7725):637–8. doi: 10.1016/s0140-6736(71)80073-9. [DOI] [PubMed] [Google Scholar]
- 152.Gjedde A, Crone C. Induction processes in blood–brain transfer of ketone bodies during starvation. Am J Physiol. 1975;229(5):1165–9. doi: 10.1152/ajplegacy.1975.229.5.1165. [DOI] [PubMed] [Google Scholar]
- 153.Hawkins RA, Mans AM, Davis DW. Regional ketone body utilization by rat brain in starvation and diabetes. Am J Physiol. 1986;250(2 Pt 1):E169–78. doi: 10.1152/ajpendo.1986.250.2.E169. [DOI] [PubMed] [Google Scholar]
- 154.Bentourkia M, Tremblay S, Pifferi F, Rousseau J, Lecomte R, Cunnane S. PET study of 11C-acetoacetate kinetics in rat brain during dietary treatments affecting ketosis. Am J Physiol Endocrinol Metab. 2009;296(4):E796–801. doi: 10.1152/ajpendo.90644.2008. [DOI] [PubMed] [Google Scholar]
- 155.Hasselbalch SG, Knudsen GM, Jakobsen J, Hageman LP, Holm S, Paulson OB. Blood-brain barrier permeability of glucose and ketone bodies during short-term starvation in humans. Am J Physiol. 1995;268(6 Pt 1):E1161–6. doi: 10.1152/ajpendo.1995.268.6.E1161. [DOI] [PubMed] [Google Scholar]
- 156.Lying-Tunell U, Lindblad BS, Malmlund HO, Persson B. Cerebral blood flow and metabolic rate of oxygen, glucose, lactate, pyruvate, ketone bodies and amino acids. Acta Neurol Scand. 1980;62(5):265–75. doi: 10.1111/j.1600-0404.1980.tb03035.x. [DOI] [PubMed] [Google Scholar]
- 157.Blomqvist G, Alvarsson M, Grill V, Von Heijne G, Ingvar M, Thorell JO, et al. Effect of acute hyperketonemia on the cerebral uptake of ketone bodies in nondiabetic subjects and IDDM patients. Am J Physiol Endocrinol Metab. 2002;283(1):E20–8. doi: 10.1152/ajpendo.00294.2001. [DOI] [PubMed] [Google Scholar]
- 158.Hasselbalch SG, Knudsen GM, Jakobsen J, Hageman LP, Holm S, Paulson OB. Brain metabolism during short-term starvation in humans. J Cereb Blood Flow Metab. 1994;14(1):125–31. doi: 10.1038/jcbfm.1994.17. [DOI] [PubMed] [Google Scholar]
- 159.Morris AA. Cerebral ketone body metabolism. J Inherit Metab Dis. 2005;28(2):109–21. doi: 10.1007/s10545-005-5518-0. [DOI] [PubMed] [Google Scholar]
- 160.Musa-Veloso K, Likhodii SS, Cunnane SC. Breath acetone is a reliable indicator of ketosis in adults consuming ketogenic meals. Am J Clin Nutr. 2002;76(1):65–70. doi: 10.1093/ajcn/76.1.65. [DOI] [PubMed] [Google Scholar]
- 161.Musa-Veloso K, Likhodii SS, Rarama E, Benoit S, Liu YM, Chartrand D, et al. Breath acetone predicts plasma ketone bodies in children with epilepsy on a ketogenic diet. Nutrition. 2006;22(1):1–8. doi: 10.1016/j.nut.2005.04.008. [DOI] [PubMed] [Google Scholar]
- 162.Leino RL, Gerhart DZ, Duelli R, Enerson BE, Drewes LR. Diet-induced ketosis increases monocarboxylate transporter (MCT1) levels in rat brain. Neurochem Int. 2001;38(6):519–27. doi: 10.1016/s0197-0186(00)00102-9. [DOI] [PubMed] [Google Scholar]
- 163.Kuhl DE, Metter EJ, Riege WH, Phelps ME. Effects of human aging on patterns of local cerebral glucose utilization determined by the [18F]fluorodeoxyglucose method. J Cereb Blood Flow Metab. 1982;2(2):163–71. doi: 10.1038/jcbfm.1982.15. [DOI] [PubMed] [Google Scholar]
- 164.Ruderman NB, Ross PS, Berger M, Goodman MN. Regulation of glucose and ketone- body metabolism in brain of anaesthetized rats. Biochem J. 1974;138(1):1–10. doi: 10.1042/bj1380001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Pierre K, Pellerin L. Monocarboxylate transporters in the central nervous system: distribution, regulation and function. J Neurochem. 2005;94(1):1–14. doi: 10.1111/j.1471-4159.2005.03168.x. [DOI] [PubMed] [Google Scholar]
- 166.Simpson IA, Carruthers A, Vannucci SJ. Supply and demand in cerebral energy metabolism: the role of nutrient transporters. J Cereb Blood Flow Metab. 2007;27(11):1766–91. doi: 10.1038/sj.jcbfm.9600521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wiener R, Hirsch HJ, Spitzer JJ. Cerebral extraction of ketones and their penetration into CSF in the dog. Am J Physiol. 1971;220(5):1542–6. doi: 10.1152/ajplegacy.1971.220.5.1542. [DOI] [PubMed] [Google Scholar]
- 168.Tremblay S, Ouellet R, Rodrigue S, Langlois R, Benard F, Cunnane SC. Automated synthesis of 11C–acetoacetic acid, a key alternate brain fuel to glucose. Appl Radiat Isot. 2007;65(8):934–40. doi: 10.1016/j.apradiso.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 169.Pifferi F, Tremblay S, Plourde M, Tremblay-Mercier J, Bentourkia M, Cunnane SC. Ketones and brain function: possible link to polyunsaturated fatty acids and availability of a new brain PET tracer, 11C-acetoacetate. Epilepsia. 2008;49 (Suppl 8):76–9. doi: 10.1111/j.1528-1167.2008.01842.x. [DOI] [PubMed] [Google Scholar]
- 170.Cross H, Ferrie C, Lascelles K, Livingston J, Mewasingh L. Old versus new antiepileptic drugs: the SANAD study. Lancet. 2007;370(9584):314. doi: 10.1016/S0140-6736(07)61151-9. author reply 15–6. [DOI] [PubMed] [Google Scholar]
- 171.Freeman J, Veggiotti P, Lanzi G, Tagliabue A, Perucca E. The ketogenic diet: from molecular mechanisms to clinical effects. Epilepsy Res. 2006;68(2):145–80. doi: 10.1016/j.eplepsyres.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 172.Klepper J. Impaired glucose transport into the brain: the expanding spectrum of glucose transporter type 1 deficiency syndrome. Curr Opin Neurol. 2004;17(2):193–6. doi: 10.1097/00019052-200404000-00018. [DOI] [PubMed] [Google Scholar]
- 173.Wang D, Pascual JM, Yang H, Engelstad K, Jhung S, Sun RP, et al. Glut-1 deficiency syndrome: clinical, genetic, and therapeutic aspects. Annals of neurology. 2005;57(1):111–8. doi: 10.1002/ana.20331. [DOI] [PubMed] [Google Scholar]
- 174.De Vivo DC, Leary L, Wang D. Glucose transporter 1 deficiency syndrome and other glycolytic defects. J Child Neurol. 2002;17(Suppl 3):3S15–23. discussion 3S24–5. [PubMed] [Google Scholar]
- 175.Falk RE, Cederbaum SD, Blass JP, Gibson GE, Kark RA, Carrel RE. Ketonic diet in the management of pyruvate dehydrogenase deficiency. Pediatrics. 1976;58(5):713–21. [PubMed] [Google Scholar]
- 176.Amiel SA, Archibald HR, Chusney G, Williams AJ, Gale EA. Ketone infusion lowers hormonal responses to hypoglycaemia: evidence for acute cerebral utilization of a non-glucose fuel. Clin Sci (Lond) 1991;81(2):189–94. doi: 10.1042/cs0810189. [DOI] [PubMed] [Google Scholar]
- 177.Veneman T, Mitrakou A, Mokan M, Cryer P, Gerich J. Effect of hyperketonemia and hyperlacticacidemia on symptoms, cognitive dysfunction, and counterregulatory hormone responses during hypoglycemia in normal humans. Diabetes. 1994;43(11):1311–7. doi: 10.2337/diab.43.11.1311. [DOI] [PubMed] [Google Scholar]
- 178.Page KA, Williamson A, Yu N, McNay EC, Dzuira J, McCrimmon RJ, et al. Medium-chain fatty acids improve cognitive function in intensively treated type 1 diabetic patients and support in vitro synaptic transmission during acute hypoglycemia. Diabetes. 2009;58(5):1237–44. doi: 10.2337/db08-1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Prins ML. Cerebral metabolic adaptation and ketone metabolism after brain injury. J Cereb Blood Flow Metab. 2008;28(1):1–16. doi: 10.1038/sj.jcbfm.9600543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Van der Auwera I, Wera S, Van Leuven F, Henderson ST. A ketogenic diet reduces amyloid beta 40 and 42 in a mouse model of Alzheimer’s disease. Nutr Metab (Lond) 2005;2:28. doi: 10.1186/1743-7075-2-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Veech RL. The therapeutic implications of ketone bodies: the effects of ketone bodies in pathological conditions: ketosis, ketogenic diet, redox states, insulin resistance, and mitochondrial metabolism. Prostaglandins Leukot Essent Fatty Acids. 2004;70(3):309–19. doi: 10.1016/j.plefa.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 182.Freemantle E, Vandal M, Tremblay Mercier J, Plourde M, Poirier J, Cunnane SC. Metabolic response to a ketogenic breakfast in the healthy elderly. J Nutr Health Aging. 2009;13(4):293–8. doi: 10.1007/s12603-009-0026-9. [DOI] [PubMed] [Google Scholar]
- 183.Duara R, Barker WW, Chang J, Yoshii F, Loewenstein DA, Pascal S. Viability of neocortical function shown in behavioral activation state PET studies in Alzheimer disease. J Cereb Blood Flow Metab. 1992;12(6):927–34. doi: 10.1038/jcbfm.1992.129. [DOI] [PubMed] [Google Scholar]
- 184.Greenwood CE, Winocur G. High-fat diets, insulin resistance and declining cognitive function. Neurobiology of aging. 2005;26 (Suppl 1):42–5. doi: 10.1016/j.neurobiolaging.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 185.Henderson ST, Vogel JL, Barr LJ, Garvin F, Jones JJ, Costantini LC. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: a randomized, double-blind, placebo-controlled, multicenter trial. Nutr Metab (Lond) 2009;6:31. doi: 10.1186/1743-7075-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Reger MA, Henderson ST, Hale C, Cholerton B, Baker LD, Watson GS, et al. Effects of beta-hydroxybutyrate on cognition in memory-impaired adults. Neurobiology of aging. 2004;25(3):311–4. doi: 10.1016/S0197-4580(03)00087-3. [DOI] [PubMed] [Google Scholar]
- 187.Risner ME, Saunders AM, Altman JF, Ormandy GC, Craft S, Foley IM, et al. Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics J. 2006;6(4):246–54. doi: 10.1038/sj.tpj.6500369. [DOI] [PubMed] [Google Scholar]
- 188.Roses AD, Saunders AM, Huang Y, Strum J, Weisgraber KH, Mahley RW. Complex disease-associated pharmacogenetics: drug efficacy, drug safety, and confirmation of a pathogenetic hypothesis (Alzheimer’s disease) Pharmacogenomics J. 2007;7(1):10–28. doi: 10.1038/sj.tpj.6500397. [DOI] [PubMed] [Google Scholar]
- 189.Freund G, Weinsier RL. Standardized ketosis in man following medium chain triglyceride ingestion. Metabolism. 1966;15(11):980–91. doi: 10.1016/0026-0495(66)90046-1. [DOI] [PubMed] [Google Scholar]
- 190.Sims NR, Bowen DM, Smith CC, Flack RH, Davison AN, Snowden JS, et al. Glucose metabolism and acetylcholine synthesis in relation to neuronal activity in Alzheimer’s disease. Lancet. 1980;1(8164):333–6. doi: 10.1016/s0140-6736(80)90884-3. [DOI] [PubMed] [Google Scholar]
- 191.Willis MW, Ketter TA, Kimbrell TA, George MS, Herscovitch P, Danielson AL, et al. Age, sex and laterality effects on cerebral glucose metabolism in healthy adults. Psychiatry research. 2002;114(1):23–37. doi: 10.1016/s0925-4927(01)00126-3. [DOI] [PubMed] [Google Scholar]
- 192.Kalpouzos G, Eustache F, Desgranges B. Cognitive reserve and neural networks in normal aging and Alzheimer’s disease. Psychol Neuropsychiatr Vieil. 2008;6(2):97–105. doi: 10.1684/pnv.2008.0120. [DOI] [PubMed] [Google Scholar]
- 193.Engler H, Forsberg A, Almkvist O, Blomquist G, Larsson E, Savitcheva I, et al. Two-year follow-up of amyloid deposition in patients with Alzheimer’s disease. Brain. 2006;129(Pt 11):2856–66. doi: 10.1093/brain/awl178. [DOI] [PubMed] [Google Scholar]
- 194.Bodoky G, Yang ZJ, Meguid MM, Laviano A, Szeverenyi N. Effects of fasting, intermittent feeding, or continuous parenteral nutrition on rat liver and brain energy metabolism as assessed by 31P-NMR. Physiol Behav. 1995;58(3):521–7. doi: 10.1016/0031-9384(95)00078-w. [DOI] [PubMed] [Google Scholar]
- 195.Cross TA, Pahl C, Oberhansli R, Aue WP, Keller U, Seelig J. Ketogenesis in the living rat followed by 13C NMR spectroscopy. Biochemistry. 1984;23(26):6398–402. doi: 10.1021/bi00321a018. [DOI] [PubMed] [Google Scholar]
- 196.Croteau E, Lavallee E, Labbe SM, Hubert L, Pifferi F, Rousseau JA, et al. Image-derived input function in dynamic human PET/CT: methodology and validation with 11Cacetate and 18F-fluorothioheptadecanoic acid in muscle and 18F-fluorodeoxyglucose in brain. European journal of nuclear medicine and molecular imaging. 2010 doi: 10.1007/s00259-010-1443-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Feigin V, Ratnasabapathy Y, Anderson C. Does blood pressure lowering treatment prevents dementia or cognitive decline in patients with cardiovascular and cerebrovascular disease? Journal of the neurological sciences. 2005;229–230:151–5. doi: 10.1016/j.jns.2004.11.020. [DOI] [PubMed] [Google Scholar]
- 198.Forette F, Seux ML, Staessen JA, Thijs L, Birkenhager WH, Babarskiene MR, et al. Prevention of dementia in randomised double-blind placebo-controlled Systolic Hypertension in Europe (Syst-Eur) trial. Lancet. 1998;352(9137):1347–51. doi: 10.1016/s0140-6736(98)03086-4. [DOI] [PubMed] [Google Scholar]
- 199.Patterson C, Feightner J, Garcia A, MacKnight C. General risk factors for dementia: a systematic evidence review. Alzheimers Dement. 2007;3(4):341–7. doi: 10.1016/j.jalz.2007.07.001. [DOI] [PubMed] [Google Scholar]
- 200.Pike KE, Savage G, Villemagne VL, Ng S, Moss SA, Maruff P, et al. Beta-amyloid imaging and memory in non-demented individuals: evidence for preclinical Alzheimer’s disease. Brain. 2007;130(Pt 11):2837–44. doi: 10.1093/brain/awm238. [DOI] [PubMed] [Google Scholar]
- 201.Sliwinski MJ, Hofer SM, Hall C, Buschke H, Lipton RB. Modeling memory decline in older adults: the importance of preclinical dementia, attrition, and chronological age. Psychol Aging. 2003;18(4):658–71. doi: 10.1037/0882-7974.18.4.658. [DOI] [PubMed] [Google Scholar]
- 202.Petersen RC. Early diagnosis of Alzheimer’s disease: is MCI too late? Curr Alzheimer Res. 2009;6(4):324–30. doi: 10.2174/156720509788929237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.den Dunnen WF, Brouwer WH, Bijlard E, Kamphuis J, van Linschoten K, Eggens-Meijer E, et al. No disease in the brain of a 115-year-old woman. Neurobiology of aging. 2008;29(8):1127–32. doi: 10.1016/j.neurobiolaging.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 204.Kalpouzos G, Chetelat G, Baron JC, Landeau B, Mevel K, Godeau C, et al. Voxel-based mapping of brain gray matter volume and glucose metabolism profiles in normal aging. Neurobiology of aging. 2009;30(1):112–24. doi: 10.1016/j.neurobiolaging.2007.05.019. [DOI] [PubMed] [Google Scholar]
- 205.Petit-Taboue MC, Landeau B, Desson JF, Desgranges B, Baron JC. Effects of healthy aging on the regional cerebral metabolic rate of glucose assessed with statistical parametric mapping. NeuroImage. 1998;7(3):176–84. doi: 10.1006/nimg.1997.0318. [DOI] [PubMed] [Google Scholar]
- 206.Svennerholm L, Bostrom K, Jungbjer B. Changes in weight and compositions of major membrane components of human brain during the span of adult human life of Swedes. Acta Neuropathol. 1997;94(4):345–52. doi: 10.1007/s004010050717. [DOI] [PubMed] [Google Scholar]
- 207.Blomqvist G, Thorell JO, Ingvar M, Grill V, Widen L, Stone-Elander S. Use of R-beta-[1-11C]hydroxybutyrate in PET studies of regional cerebral uptake of ketone bodies in humans. Am J Physiol. 1995;269(5 Pt 1):E948–59. doi: 10.1152/ajpendo.1995.269.5.E948. [DOI] [PubMed] [Google Scholar]
- 208.Frackowiak RS, Pozzilli C, Legg NJ, Du Boulay GH, Marshall J, Lenzi GL, et al. Regional cerebral oxygen supply and utilization in dementia. A clinical and physiological study with oxygen-15 and positron tomography. Brain. 1981;104(Pt 4):753–78. doi: 10.1093/brain/104.4.753. [DOI] [PubMed] [Google Scholar]
- 209.Benson DF, Kuhl DE, Hawkins RA, Phelps ME, Cummings JL, Tsai SY. The fluorodeoxyglucose 18F scan in Alzheimer’s disease and multi-infarct dementia. Archives of neurology. 1983;40(12):711–4. doi: 10.1001/archneur.1983.04050110029003. [DOI] [PubMed] [Google Scholar]
- 210.Cutler NR, Haxby JV, Duara R, Grady CL, Kay AD, Kessler RM, et al. Clinical history, brain metabolism, and neuropsychological function in Alzheimer’s disease. Annals of neurology. 1985;18(3):298–309. doi: 10.1002/ana.410180305. [DOI] [PubMed] [Google Scholar]
- 211.Rapoport SI. Positron emission tomography in normal aging and Alzheimer’s disease. Gerontology. 1986;32 (Suppl 1):6–13. doi: 10.1159/000212819. [DOI] [PubMed] [Google Scholar]
- 212.Chawluk JB, Alavi A, Dann R, Hurtig HI, Bais S, Kushner MJ, et al. Positron emission tomography in aging and dementia: effect of cerebral atrophy. J Nucl Med. 1987;28(4):431–7. [PubMed] [Google Scholar]
- 213.Alavi A, Newberg AB, Souder E, Berlin JA. Quantitative analysis of PET and MRI data in normal aging and Alzheimer’s disease: atrophy weighted total brain metabolism and absolute whole brain metabolism as reliable discriminators. J Nucl Med. 1993;34(10):1681–7. [PubMed] [Google Scholar]
- 214.Mielke R, Pietrzyk U, Jacobs A, Fink GR, Ichimiya A, Kessler J, et al. HMPAO SPET and FDG PET in Alzheimer’s disease and vascular dementia: comparison of perfusion and metabolic pattern. Eur J Nucl Med. 1994;21(10):1052–60. doi: 10.1007/BF00181059. [DOI] [PubMed] [Google Scholar]
- 215.Meltzer CC, Zubieta JK, Brandt J, Tune LE, Mayberg HS, Frost JJ. Regional hypometabolism in Alzheimer’s disease as measured by positron emission tomography after correction for effects of partial volume averaging. Neurology. 1996;47(2):454–61. doi: 10.1212/wnl.47.2.454. [DOI] [PubMed] [Google Scholar]
- 216.Minoshima S, Giordani B, Berent S, Frey KA, Foster NL, Kuhl DE. Metabolic reduction in the posterior cingulate cortex in very early Alzheimer’s disease. Annals of neurology. 1997;42(1):85–94. doi: 10.1002/ana.410420114. [DOI] [PubMed] [Google Scholar]
- 217.Duara R, Margolin RA, Robertson-Tchabo EA, London ED, Schwartz M, Renfrew JW, et al. Cerebral glucose utilization, as measured with positron emission tomography in 21 resting healthy men between the ages of 21 and 83 years. Brain. 1983;106 (Pt 3):761–75. doi: 10.1093/brain/106.3.761. [DOI] [PubMed] [Google Scholar]
- 218.de Leon MJ, George AE, Ferris SH, Christman DR, Fowler JS, Gentes CI, et al. Positron emission tomography and computed tomography assessments of the aging human brain. Journal of computer assisted tomography. 1984;8(1):88–94. doi: 10.1097/00004728-198402000-00017. [DOI] [PubMed] [Google Scholar]
- 219.Duara R, Grady C, Haxby J, Ingvar D, Sokoloff L, Margolin RA, et al. Human brain glucose utilization and cognitive function in relation to age. Annals of neurology. 1984;16(6):703–13. doi: 10.1002/ana.410160613. [DOI] [PubMed] [Google Scholar]
- 220.Horwitz B, Duara R, Rapoport SI. Age differences in intercorrelations between regional cerebral metabolic rates for glucose. Annals of neurology. 1986;19(1):60–7. doi: 10.1002/ana.410190111. [DOI] [PubMed] [Google Scholar]
- 221.Yanase D, Matsunari I, Yajima K, Chen W, Fujikawa A, Nishimura S, et al. Brain FDG PET study of normal aging in Japanese: effect of atrophy correction. European journal of nuclear medicine and molecular imaging. 2005;32(7):794–805. doi: 10.1007/s00259-005-1767-2. [DOI] [PubMed] [Google Scholar]
- 222.Kochunov P, Ramage AE, Lancaster JL, Robin DA, Narayana S, Coyle T, et al. Loss of cerebral white matter structural integrity tracks the gray matter metabolic decline in normal aging. NeuroImage. 2009;45(1):17–28. doi: 10.1016/j.neuroimage.2008.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Kety SS. Human cerebral blood flow and oxygen consumption as related to aging. Research publications - Association for Research in Nervous and Mental Disease. 1956;35:31–45. [PubMed] [Google Scholar]
- 224.Dastur DK, Lane MH, Hansen DB, Ketty SS, Butler RN, Perlin S, et al. Effects of aging on cerebral circulation and metabolism in man. In: Biren JE, Butler RN, Greenhouse SW, Sokoloff L, Yarrow MR, editors. Human aging: A biological and biochemical study. Washington: US Government Printing Office; 1963. pp. 59–78. [Google Scholar]
- 225.De Santi S, de Leon MJ, Convit A, Tarshish C, Rusinek H, Tsui WH, et al. Age-related changes in brain: II. Positron emission tomography of frontal and temporal lobe glucose metabolism in normal subjects. Psychiatr Q. 1995;66(4):357–70. doi: 10.1007/BF02238755. [DOI] [PubMed] [Google Scholar]
- 226.Loessner A, Alavi A, Lewandrowski KU, Mozley D, Souder E, Gur RE. Regional cerebral function determined by FDG–PET in healthy volunteers: normal patterns and changes with age. J Nucl Med. 1995;36(7):1141–9. [PubMed] [Google Scholar]
- 227.Moeller JR, Ishikawa T, Dhawan V, Spetsieris P, Mandel F, Alexander GE, et al. The metabolic topography of normal aging. J Cereb Blood Flow Metab. 1996;16(3):385–98. doi: 10.1097/00004647-199605000-00005. [DOI] [PubMed] [Google Scholar]
- 228.Bentourkia M, Bol A, Ivanoiu A, Labar D, Sibomana M, Coppens A, et al. Comparison of regional cerebral blood flow and glucose metabolism in the normal brain: effect of aging. Journal of the neurological sciences. 2000;181(1–2):19–28. doi: 10.1016/s0022-510x(00)00396-8. [DOI] [PubMed] [Google Scholar]
- 229.de Leon MJ, Convit A, Wolf OT, Tarshish CY, DeSanti S, Rusinek H, et al. Prediction of cognitive decline in normal elderly subjects with 2-[(18)F]fluoro-2-deoxy-D-glucose/poitron-emission tomography (FDG/PET) Proceedings of the National Academy of Sciences of the United States of America. 2001;98(19):10966–71. doi: 10.1073/pnas.191044198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Mosconi L, Perani D, Sorbi S, Herholz K, Nacmias B, Holthoff V, et al. MCI conversion to dementia and the APOE genotype: a prediction study with FDG-PET. Neurology. 2004;63(12):2332–40. doi: 10.1212/01.wnl.0000147469.18313.3b. [DOI] [PubMed] [Google Scholar]
- 231.Rimajova M, Lenzo NP, Wu JS, Bates KA, Campbell A, Dhaliwal SS, et al. Fluoro-2-deoxy-D-glucose (FDG)-PET in APOEepsilon4 carriers in the Australian population. J Alzheimers Dis. 2008;13(2):137–46. doi: 10.3233/jad-2008-13203. [DOI] [PubMed] [Google Scholar]
- 232.Langbaum JB, Chen K, Lee W, Reschke C, Bandy D, Fleisher AS, et al. Categorical and correlational analyses of baseline fluorodeoxyglucose positron emission tomography Images from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) NeuroImage. 2009;45(4):1107–16. doi: 10.1016/j.neuroimage.2008.12.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Kennedy AM, Frackowiak RS, Newman SK, Bloomfield PM, Seaward J, Roques P, et al. Deficits in cerebral glucose metabolism demonstrated by positron emission tomography in individuals at risk of familial Alzheimer’s disease. Neuroscience letters. 1995;186(1):17–20. doi: 10.1016/0304-3940(95)11270-7. [DOI] [PubMed] [Google Scholar]
- 234.Mosconi L, Sorbi S, de Leon MJ, Li Y, Nacmias B, Myoung PS, et al. Hypometabolism exceeds atrophy in presymptomatic early-onset familial Alzheimer’s disease. J Nucl Med. 2006;47(11):1778–86. [PubMed] [Google Scholar]