

Abstract
Misexpression and cytosolic retention of peripheral myelin protein 22 (PMP22) within Schwann cells (SCs) is associated with a genetically heterogeneous group of demyelinating peripheral neuropathies. PMP22 overproducer C22 and spontaneous mutant Trembler J (TrJ) mice display neuropathic phenotypes and affected nerves contain abnormally localized PMP22. Nutrient deprivation-induced autophagy is able to suppress the formation of PMP22 aggregates in a toxin-induced cellular model, and improve locomotor performance and myelination in TrJ mice. As a step toward therapies, we assessed whether pharmacological activation of autophagy by rapamycin (RM) could facilitate the processing of PMP22 within neuropathic SCs and enhance their capacity to myelinate peripheral axons. Exposure of mouse SCs to RM induced autophagy in a dose- and time-dependent manner and decreased the accumulation of poly-ubiquitinated substrates. The treatment of myelinating dorsal root ganglion (DRG) explant cultures from neuropathic mice with RM (25 nm) improved the processing of PMP22 and increased the abundance and length of myelin internodes, as well as the expression of myelin proteins. Notably, RM is similarly effective in both the C22 and TrJ model, signifying that the benefit overlaps among distinct genetic models of PMP22 neuropathies. Furthermore, lentivirus-mediated shRNA knockdown of the autophagy-related gene 12 (Atg12) abolished the activation of autophagy and the increase in myelin proteins, demonstrating that autophagy is critical for the observed improvement. Together, these results support the potential use of RM and other autophagy-enhancing compounds as therapeutic agents for PMP22-associated demyelinating neuropathies.
Introduction
Aggregation of misfolded proteins and their sequestration into intracellular inclusions is a problem common to diseases of the central and peripheral nervous systems (Stefani and Dobson, 2003). Charcot-Marie-Tooth disease type 1A (CMT1A) is a prevalent protein misfolding disease in humans characterized by progressive demyelination of peripheral nerves and associated neuromuscular deficits. During the last 10 years there have been many advances with the molecular diagnosis of CMT neuropathies; however, effective drug therapy is still not available (Pareyson and Marchesi, 2009). Duplications of, or point mutations within, the Peripheral Myelin Protein 22 (PMP22) gene are known to cause CMT1A (Shy et al., 2001) and other related neuropathies. PMP22 is a hydrophobic integral membrane glycoprotein that is mostly expressed by myelinating Schwann cells (SCs) (Naef and Suter, 1998). Transgenic mice based on the overexpression of the human wild-type (Wt) PMP22 termed C22, and spontaneous mutant (L16P) Trembler J (TrJ), reproduce features of the human neuropathy and provide valuable experimental models to study disease pathogenesis (Suter et al., 1992; Huxley et al., 1996).
PMP22 folds with only a modest efficiency under normal conditions (Sanders et al., 2001) and nearly 80% of the newly synthesized protein is rapidly turned over by the proteasome (Pareek et al., 1997; Notterpek et al., 1999). In response to PMP22 overproduction and the L16P mutation, excessive or defective PMP22 polypeptides are targeted for degradation by the ubiquitin-proteasome system and accumulate in cytosolic aggregates (Fortun et al., 2003, 2005, 2006). Autophagic and lysosomal components as well as heat shock proteins (HSPs) are recruited to ubiquitin-positive PMP22 aggregates in nerves of C22 and TrJ mice, likely reflecting an attempt by the cells to clear them through alternate pathways. This sequence of events decreases the amount of PMP22 protein within the SC plasma membrane, and likely contributes to the pronounced demyelinating phenotype (Huxley et al., 1996, 1998; Notterpek et al., 1997; Fortun et al., 2003, 2006).
Promising therapeutic approaches for protein misfolding disorders, such as PMP22-associated-neuropathies, include increasing the synthesis of HSPs (Muchowski and Wacker, 2005) and stimulating autophagic protein degradation (Sarkar et al., 2009). In previous studies, we demonstrated that the activation of autophagy by amino acid and serum deprivation (Fortun et al., 2003, 2007), or intermittent-fasting, suppressed the accumulation of misfolded proteins within neuropathic SCs and improved myelination in TrJ mice (Madorsky et al., 2009). Since such dramatic dietary restriction is not suitable for therapy in humans, here we asked whether pharmacological activation of autophagy within myelinating SCs could offer similar benefits. Rapamycin (RM), a macrolide antibiotic, is a widely used inhibitor of the mammalian target of rapamycin (mTOR) which induces autophagy in a variety of cell types (Sabers et al., 1995; Sarkar and Rubinsztein, 2008). In this study, we show that autophagy is a critical pathway for RM-mediated myelin improvement in neuropathic SCs.
Materials and Methods
Mouse colonies.
PMP22 overexpressor (C22) (Huxley et al., 1996) and spontaneous mutant TrJ (Suter et al., 1992) neuropathic mouse breeding colonies are housed under SPF conditions at the University of Florida, McKnight Brain Institute animal facility. The use of animals for these studies has been approved by an Institutional Animal Care and Use Committee. We used both male and female mouse embryos (E13) and pups (P6) to set up the dorsal root ganglion and Schwann cell cultures, respectively. Genomic DNA was isolated from tail biopsies of embryos and pups (<10 d old) and litters were genotyped by PCR (Huxley et al., 1996; Notterpek et al., 1997).
Primary mouse SC cultures.
SC cultures from genotyped postnatal day 6 Wt, C22 and TrJ mouse pups were prepared and maintained as described with slight modifications (Nicholson et al., 2001). Nerves were dissected and enzymatically digested over a period of 2 h. The digestion medium consisted of DMEM/F12 with GlutaMAX-I (Invitrogen), 15% FBS (Hyclone), penicillin streptomycin (Invitrogen) and an enzyme mixture of 0.03% collagenase type III (Worthington), 0.1% hyaluronidase (Sigma-Aldrich) and 1.25 U/ml dispase (Worthington). Next, the cell suspensions were washed once and resuspended in culture medium (DMEM/F12 containing 15% FBS). Cells were then plated in drops on poly-l-lysine (Sigma-Aldrich)-coated plastic Petri-dishes and allowed to adhere overnight. The next day, cells were washed with DMEM/F12 followed by addition of DMEM/F12 containing 15% FBS, 10 μm antimitotic agent cytosine β-d-arabinofuranoside (Sigma-Aldrich), to eradicate contaminating fibroblasts. After two 48 h periods of antimitotic treatments, given on alternate days, standard growth medium [DMEM/F12 containing 10% FBS, 10 μg/ml bovine pituitary extract (Biomedical Technologies), and 2.5 μm forskolin (Calbiochem)] was added and the cells were allowed to proliferate for 2 d to reach 80% confluency.
Autophagic flux measurement.
Rapamycin (RM) (Calbiochem) was dissolved in ethanol vehicle and 0.5 mm stocks were stored at −80°C. Chloroquine (CQ) (Sigma-Aldrich) was dissolved in water to a stock of 50 mm and aliquots were stored at −80°C. SCs were treated with vehicle control (Ct) or RM at a final concentration of 25 nm, either in the presence or absence of CQ for 48 h. Following this, the cultures were lysed and analyzed for microtubule-associated protein light chain 3 (MAP-LC3, from here on referred to as LC3) by Western blots. A statistically significant increase in LC3 II band intensity in RM+CQ compared with Ct+CQ samples indicates an enhanced autophagic flux and signifies that the pathway attains completion (Mizushima and Yoshimori, 2007).
Metabolic labeling and 35S-pulse studies.
SCs were treated with Ct, RM (25 nm) for 48 h or positive control cyclohexamide (Tocris Bioscience) (Chx, 100 μg/ml) for 12 h. During the last 1.5 h of incubation, the cells were starved in methionine- and cysteine-free media with or without RM for 1 h. Following this, a 30 min pulse of medium containing 0.25 mCi/ml trans-35S (ICN Biochemicals) was added. After lysis in RIPA buffer (Pareek et al., 1997), proteins were precipitated with 5% trichloroacetate and bound 35S-radioactivity was counted in a scintillation counter (LS6500, Beckman Coulter). The counts per minute per micrograms of protein (cpm/μg protein) were quantified from triplicate experiments. Simultaneously, equal amounts of proteins were separated by SDS-PAGE and the gels were dried and treated with AMPLIFY (GE Healthcare). Dried gels were exposed to Classic autoradiography film (Molecular Technologies) at −80°C. Densitometry was performed on the trans-35S-labeled protein bands using Scion Image software. Quantification was performed from triplicate experiments and statistical analysis was performed by Student's t test using GraphPad Prism.
Dorsal root ganglion (DRG) explant cultures and RM treatment paradigms.
DRG explant cultures were established from Wt, C22 and TrJ mouse embryos as described previously (Fortun et al., 2006). Cultures were exposed to RM (25 nm) for the indicated time points (see Fig. 3A), including the last 8 h before analysis by immunostaining and Western blots. SC-depleted neuronal cultures (Einheber et al., 1993) were treated for 48 h with RM (25 nm) or vehicle.
Figure 3.

Rapamycin promotes myelination by SCs. A, Schematic representation of the RM treatment paradigms. The black line indicates DIV, while the green lines represent the expression profile of myelin proteins in Wt (bold green) and neuropathic (dashed green) samples. The blue line indicates the time-scale for RM treatment in hours (h). Asterisks indicate the DIV at which the control samples Ct* and Ct** were obtained. For early RM treatment (a), starting on DIV9, or for late treatment (b) starting on DIV15, a pulse of RM (green arrows) was given for 48 h, followed by 48 h washout (wo, black arrows). This sequence was repeated and a third RM treatment (8 h) was given before analysis (arrowhead). B, C, DRG explants from Wt and C22 mice were treated with RM as indicated and whole protein lysates were analyzed for the myelin proteins, MBP and P0. The main isoforms of MBP are marked. Tubulin is shown as protein loading control. Molecular mass in kDa.
Lentiviral transduction of mouse SCs, DRG explants, and SC-depleted neuronal cultures.
Prepackaged lentiviral particles were purchased from a commercial provider (Santa Cruz Biotechnology), which either encoded a nontargeting shRNA (negative shRNA) (catalog # sc-108080), or a sequence specifically targeting mouse Atg12 (catalog # sc-72579-V). For viral infection of isolated mouse SC cultures, 48 h after the last antimitotic treatment, the cells were gently washed and the media was replaced with SC culture media containing 5 μg/ml Polybrene (Santa Cruz Biotechnology, catalog # sc-134220). The SCs were then exposed to ∼1.0 × 105 infectious units of virus per 6 cm plate of cells for 48 h. Since the lentiviral shRNA particles also encode for a puromycin resistance gene for transduction selection, the cells were then washed and grown in culture media containing 10 μg/ml puromycin dihydrochloride (Sigma, catalog # P9620) for an additional 48 h. The SCs were allowed to recover and proliferate for 1 week before treatment with RM and then processed for analyses.
For the Atg12 knock-down experiments in DRGs, negative (Neg.) or Atg12 shRNA lentiviral particles were combined with the explants from Wt, C22 or TrJ mouse embryos at the time of plating. Each individual culture was transduced with ∼7.5 × 104 infectious units of virus. Following transduction, cultures expressing either Neg. or Atg12 shRNA were subjected to the late RM treatment paradigm (see Fig. 3A). Each shRNA transduction experiment was performed at least three times from independent DRG isolations. For Atg12 knock-down of SC-depleted neurons, the DRGs were transduced as mentioned above, followed by elimination of SCs to obtain pure neuronal culture (Einheber et al., 1993). Subsequently, the neurons were treated with RM.
To monitor the lentiviral transduction efficiency and transgene expression for the duration of the experiments, we incubated additional subsets of DRG explants with lentiviral particles encoding a green fluorescence protein (GFP) reporter (catalog # sc-108084). Transduction efficiency was obtained as the ratio of the number of GFP+ to total number of cells, from eight random visual fields from three independent culture experiments.
Biochemical studies.
Control and RM-treated samples were lysed and processed for Western analyses. The processing of PMP22 was assessed by treatment of the protein lysates with PNGaseF (N) or endo-H (H) (New England Biolabs) (Pareek et al., 1997). To detect PMP22, a 1:1 mixture of two rabbit polyclonal antibodies, developed against a peptide corresponding to the second extracellular loop of the human or the rat PMP22, was used (Pareek et al., 1997; Fortun et al., 2006). For the detection of protein zero (P0) we used a previously characterized mouse monoclonal antibody (Archelos et al., 1993). All other commercially available primary antibodies are listed in supplemental Table 1, available at www.jneurosci.org as supplemental material. For the Western blots, bound primary antibodies were detected with appropriate HRP-linked secondary antibodies and visualized using the enhanced chemiluminescence detection kit (PerkinElmer Life Sciences). Films were digitally imaged using a GS-710 densitometer (Bio-Rad Laboratories) and densitometric analysis of scans from three independent experiments was performed using Scion image software. Statistical significance was determined by Student's t test using GraphPad Prism.
Immunolabeling studies.
Myelinating DRG explant cultures on glass coverslips were fixed, permeabilized and processed for immunostaining with anti-MBP antibodies (Amici et al., 2007). Bound primary antibodies were detected with Alexa Fluor 594 goat anti-rat IgGs (Invitrogen). Hoechst dye (Invitrogen) was included in the secondary antibody solution to visualize nuclei. Coverslips were mounted using the ProLong Antifade kit (Invitrogen). Samples were imaged with an Olympus 1X81-DSU spinning disk confocal microscope and were formatted for printing by using Adobe Photoshop 5.5.
Quantification of myelin internode abundance and lengths.
DRG explant cultures were subjected to the treatment paradigms described above and immunostained with an anti-MBP antibody to label internodal myelin segments (Amici et al., 2007). The abundance of myelin internodes was counted as the number of MBP+ internodal segments per 0.1 mm2 area from three independent experiments, and eight random visual fields per condition (see Fig. 3A). Internode lengths from the same experiments were measured with Spot RT software (Diagnostic Instruments). Measurements were collected from three coverslips per genotype per treatment paradigm (n = 130–260). Statistical significance was determined by Student's t test using GraphPad Prism software.
Results
Rapamycin activates autophagy in primary mouse SCs
The activation of autophagy by dietary restriction is able to suppress the formation of PMP22 aggregates and stimulate myelin formation in TrJ mice (Madorsky et al., 2009). To enhance autophagy by pharmacologic means, we exposed mouse SCs to varying doses (10–500 nm) of RM (Sarkar and Rubinsztein, 2008). After 6 h incubation, mTOR complex 1 (mTORC1) activity was monitored by the dephosphorylation of phosphorylated S6 (pS6) ribosomal protein and the activation of autophagy via the lipidation of LC3 (Fig. 1A). During autophagy, a cytosolic form of microtubule-associated protein light-chain I (LC3 I) is lipidated to form LC3 II, which is recruited to autophagosomal membranes (Yang and Klionsky, 2009). Autophagosomes fuse with lysosomes and captured intra-autophagosomal components are degraded by lysosomal hydrolases along with LC3 II (Mizushima and Yoshimori, 2007). Based on the observed response, we chose the 25 nm concentration for all subsequent studies as it was the lowest dosage that consistently activated autophagy within SCs (Fig. 1A–C). To optimize the frequency of the treatment, the activation of autophagy in SCs from Wt and C22 mice over a 2–48 h period was examined (Fig. 1B,C). In addition to the dephosphorylation of pS6, and the conversion of LC3 I to LC3 II (lipidated form), we analyzed p62, an autophagy substrate that aids the degradation of poly-ubiquitinated (pUb) proteins (Komatsu and Ichimura, 2010). As shown on the Western blots, RM exposure activates autophagy for at least 48 h in cells from both Wt (Fig. 1B) and C22 (Fig. 1C) mice, leading to a pronounced increase in the ratio of lipidated LC3 II/LC3 I and a decrease in p62. Quantification of three experiments indicate that this effect is consistent among independent primary cultures (Fig. 1B,C, right, **p < 0.01, *p < 0.05).
Figure 1.
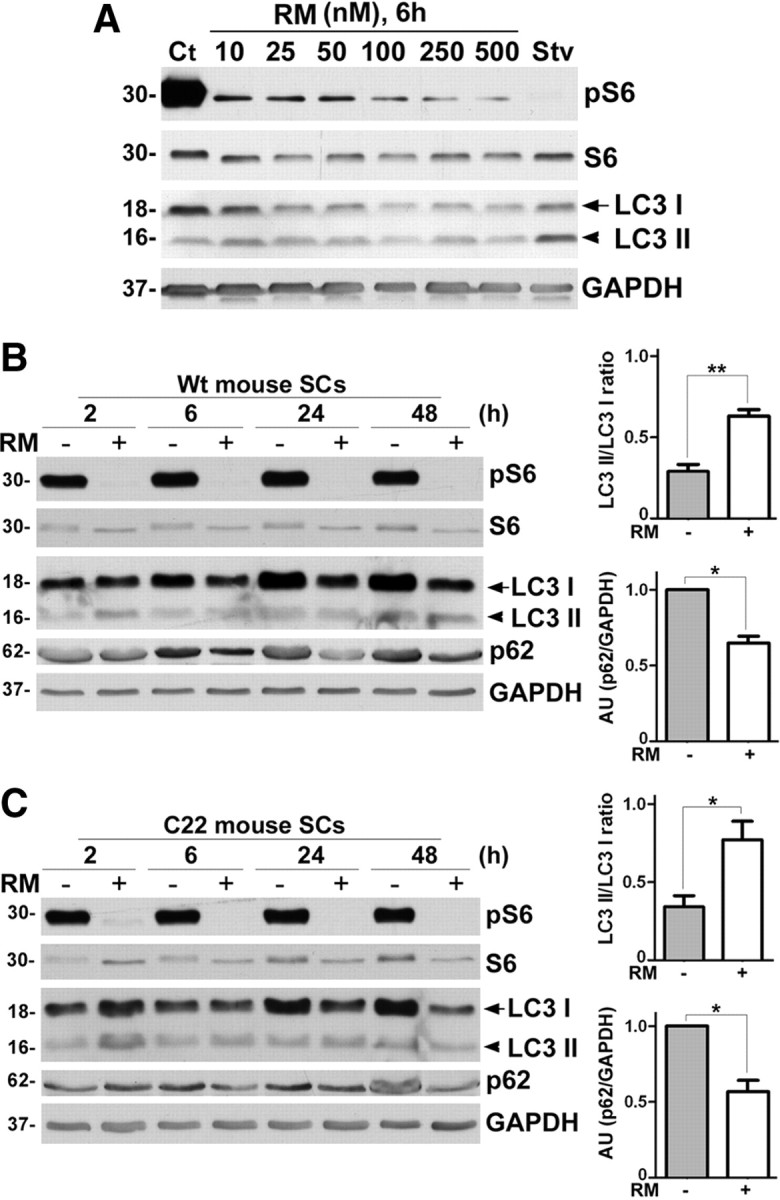
Rapamycin effectively induces autophagy in cultured mouse SCs in a dose- and time-dependent manner. A, For the RM dose–response, the expression levels of pS6, S6, LC3 I and LC3 II proteins were monitored in SCs from Wt mice via Western blots. Lysates of Ct and 4 h starvation (Stv) medium-treated cells were included as negative and positive controls, respectively. B, For the time course studies in SCs from Wt mice, the levels of pS6, S6, LC3 I, LC3 II and p62 were analyzed after treatment with Ct (−) or 25 nm RM (+) for the time points 2, 6, 24, 48 h. The ratios of LC3 II to LC3 I band intensities measured from Western blots of Wt cell lysates treated with Ct (−) or 25 nm RM (+) for 48 h are shown. (**p < 0.01, t test, mean ± SEM, n = 3). Quantification for p62 band intensities normalized to GAPDH in SCs from Wt mice treated with Ct (−) or 25 nm RM (+) for 48 h is shown (*p < 0.05, t test, mean ± SEM, n = 3). C, Time course studies in SCs from C22 mice, analyzed for the same proteins and time points as in B after treatment with Ct (−) or 25 nm RM (+).The ratios of LC3 II to LC3 I band intensities measured in SCs from C22 mice treated with Ct (−) or 25 nm RM (+) for 48 h are shown. (*p < 0.05, t test, mean ± SEM, n = 3). Quantification for p62 band intensities normalized to GAPDH in C22 samples in C at 48 h is shown (*p < 0.05, t test, mean ± SEM, n = 3). A–C, GAPDH is the protein loading control. AU, arbitrary units. Molecular mass in kDa.
Exposure of certain cell lines to RM at 100 nm concentration can lead to dephosphorylation of Akt (at Ser473) through the mTOR complex 2 (mTORC2) pathway (Sarbassov et al., 2006), and phosphorylated Akt (pAkt) (Ser473) is essential for myelination in peripheral nerves (Wakatsuki et al., 2009). Hence, we analyzed the SC lysates treated with RM (25 nm) for 2, 4, 6, 8, 24, and 48 h for pS6 and pAkt (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). The downstream substrate of mTORC1, namely pS6, is almost completely dephosphorylated in response to RM treatment at 48 h (supplemental Fig. S1A,B, available at www.jneurosci.org as supplemental material). In contrast, we found only a slight decline in the levels of pAkt over the 48 h treatment period, an effect that did not reach statistical significance (supplemental Fig. S1C, available at www.jneurosci.org as supplemental material). These data suggest that under the described conditions RM acts primarily via the mTORC1 complex within primary mouse SCs.
To assess autophagic flux, a subset of samples were cotreated with RM and chloroquine (CQ), which inhibits the degradation of LC3 II by lysosomal hydrolases (Maiuri et al., 2007; Mizushima and Yoshimori, 2007). As shown on the blot, the simultaneous exposure of SCs to RM and CQ leads to a 2.8-fold (2.88 ± 0.40) increase (**p < 0.01, t test, n = 4) in LC3 II, compared with CQ alone. In agreement with the elevated basal lysosomal activity in neuropathic samples (Notterpek et al., 1997; Fortun et al., 2006), the response of C22 SCs to CQ alone is more robust than those of Wt. Still, we detected a 2.5-fold (2.57 ± 0.49) increase (*p < 0.05, t test, n = 3) in LC3 II levels upon cotreatment. These data indicate that while the basal level of autophagy is distinct between Wt and neuropathic samples, RM enhances the response in both cell types.
To further substantiate the activation of autophagy by RM in SCs, we used lentivirus-mediated knockdown of the autophagy related gene 12 (Atg12), which is known to prevent the elongation of the autophagosome and curtail autophagy (Dreux et al., 2009). To control for any off-target effects of shRNA transduction, control Neg. lentiviral particles were used. Following Neg. or Atg12 shRNA transduction, SCs from Wt and C22 mice were treated with RM (25 nm, 48 h) (Fig. 2B). Upon analysis of the protein lysates, the steady-state levels of the covalently linked Atg12-Atg5 functional conjugates are reduced in samples transduced with Atg12 shRNA, both in the absence and presence of RM (Fig. 2B). Quantification from independent experiments shows an ∼50% reduction in Atg12-Atg5 conjugate (*p < 0.05, n = 3), which confirms efficient knock-down. We monitored the activity of RM in the transduced samples by the dephosphorylation of pS6. While the increase in LC3 II/LC3 I ratio and the decrease in p62 with RM treatment is reproducible in the Neg. shRNA-treated cells, in Atg12-deficient samples autophagy is hindered, as reflected by the accumulation of p62 and reduced conversion of LC3 I to LC3 II (Dreux et al., 2009) (Fig. 2B). The decline in LC3 II/LC3 I ratios is consistent among independent experiments (*p < 0.05, **p < 0.01, n = 3).
Figure 2.
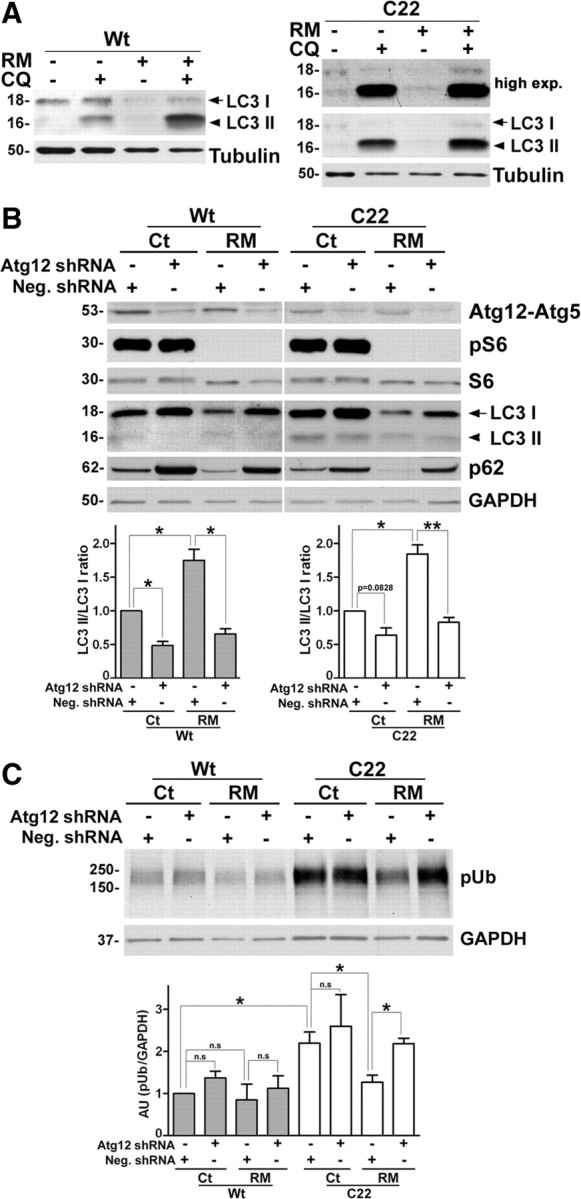
Enhancement of autophagy reduces the levels of ubiquitinated substrates. A, Wt (left) and C22 (right) mouse SCs were treated with vehicle control (−) or RM (+), without (−) or with (+) the lysosomotropic alkaline CQ. The level of autophagy markers LC3 I (arrow) and LC3 II (arrowhead) were determined by Western blots. For the C22 samples, a higher exposure (high exp.) blot for LC3 is shown to visualize LC3 I levels. B, SCs from Wt and C22 mice were exposed to lentiviral particles encoding Neg. or Atg12 shRNA followed by treatment with vehicle (−) or RM (+). The lysates were probed for Atg12-Atg5 conjugate, pS6, S6, LC3 and p62 proteins. Semiquantitative analysis for LC3 II/ LC3 I ratio in B is shown. The value of LC3 II/LC3 I in Ct samples treated with Neg. shRNA was set as 1 (p = 0.0828, *p < 0.05, **p < 0.01, t test, mean ± SEM, n = 3). C, The steady-state levels of slow-migrating pUb proteins in the same lysates in B of SCs from Wt and C22 mice are shown. Quantification of pUb/GAPDH in A. The value of pUb/GAPDH in Wt was set as 1 (*p < 0.05, **p < 0.01, t test, mean ± SEM, n = 3). B, C, GAPDH is a loading control. AU, arbitrary units. Molecular mass in kDa.
The intracellular retention of PMP22 in samples from C22 mice is associated with the accumulation of pUb proteins and impairment of proteasome activity (Fortun et al., 2006). Hence, we asked whether RM-activated autophagy could suppress the accrual of pUb substrates within the same cells that were analyzed in Figure 2B. The Atg12 knockdown in Wt and C22 samples (Fig. 2B) resulted in a slight increase in pUb proteins, which did not attain statistical significance. With RM treatment, we detected a slight reduction in slow-migrating pUb proteins in Wt samples; however, this change was more pronounced in cells from neuropathic mice (Fig. 2C). As reported previously (Fortun et al., 2006), semiquantitative analyses confirmed the accumulation of pUb proteins in Neg. shRNA-treated C22 SCs, compared with Wt (*p < 0.05, n = 3), and an ∼50% reduction by RM (*p < 0.05, n = 3). Notably, the reduction in pUb substrates by RM is attenuated in Atg12-deficient cells, an effect that is more pronounced in C22 (Fig. 2C,*p < 0.05, n = 3). As high concentration of RM treatment (200 nm) is known to have an effect on global protein synthesis (King et al., 2008), we performed 35S-pulse labeling and autoradiography (Pareek et al., 1997). Analyses of autoradiographs (supplemental Fig. S2A,C, available at www.jneurosci.org as supplemental material) and quantifications of protein bound 35S (supplemental Fig. S2B,D, available at www.jneurosci.org as supplemental material) reveal that 48 h exposure to RM reduces new protein synthesis by ∼40% in the Wt and by ∼30% in C22 SCs, which is less than the observed decrease in pUb proteins. Together, these results support the notion that RM promotes the clearance of pUb proteins from neuropathic SCs predominantly through the autophagic pathway.
Myelin production in C22 neuropathic samples is enhanced by rapamycin
DRG explant cultures from C22 neuropathic mice are impaired in their ability to make myelin (Fortun et al., 2006; Rangaraju et al., 2008), therefore serve as an ideal model to test potential therapeutic compounds for enhancing myelination. Control samples Ct* and Ct** were obtained on days in vitro (DIV) 9 and 15, respectively, either 2 or 8 d post-ascorbate, which time points coincided with the initiation of early and late RM exposures (Fig. 3A). Ascorbate is necessary for the initiation of myelination (Eldridge et al., 1987) and was included in all of our experiments. As expected from previous in vivo and in vitro studies (Huxley et al., 1996; Fortun et al., 2006), whole protein lysates of vehicle-treated neuropathic cultures harvested on DIV24 contain low levels of myelin proteins, compared with Wt (Fig. 3B,C). However, in response to RM they show an increase in the steady-state expression of both myelin basic protein (MBP) and protein zero (P0) (Fig. 3B,C). The effect is more pronounced when the cultures were analyzed after the late treatment paradigm, as seen from quantification of three independent experiments (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). These data also show that RM exposure enhanced myelin protein synthesis by Wt SCs, which is nearly significant in the late treatment paradigm.
We confirmed the positive effect of RM on myelination by immunostaining DRG explant cultures with an anti-MBP antibody (Fig. 4). Cultures from Wt mice contain numerous MBP-positive myelin internodes, while C22 explants form only a few shortened internodes (Fortun et al., 2006). Significantly, RM treatment of affected cultures is associated with a dramatic improvement in the number and length of myelin internodes, as shown on the confocal images (Fig. 4A). The characteristic, railroad-like appearance of myelin tracks is displayed in the enlargements in each of the panels (Fig. 4A, insets, 3× magnification). Quantifications of the number (Fig. 4B) and length (Fig. 4C) of myelin internodes reveal highly significant improvements in samples from neuropathic mice, after both early and late RM treatments (***p < 0.001, n = 3). Wt DRGs show a slight, but not significant increase in the number of myelin internodes with the intervention [Fig. 4B, not significant (n.s), n = 3]; however, the lengthening of internodal myelin is significant for both early and late treatment paradigms (Fig. 4C, *p < 0.05, n = 3)].
Figure 4.
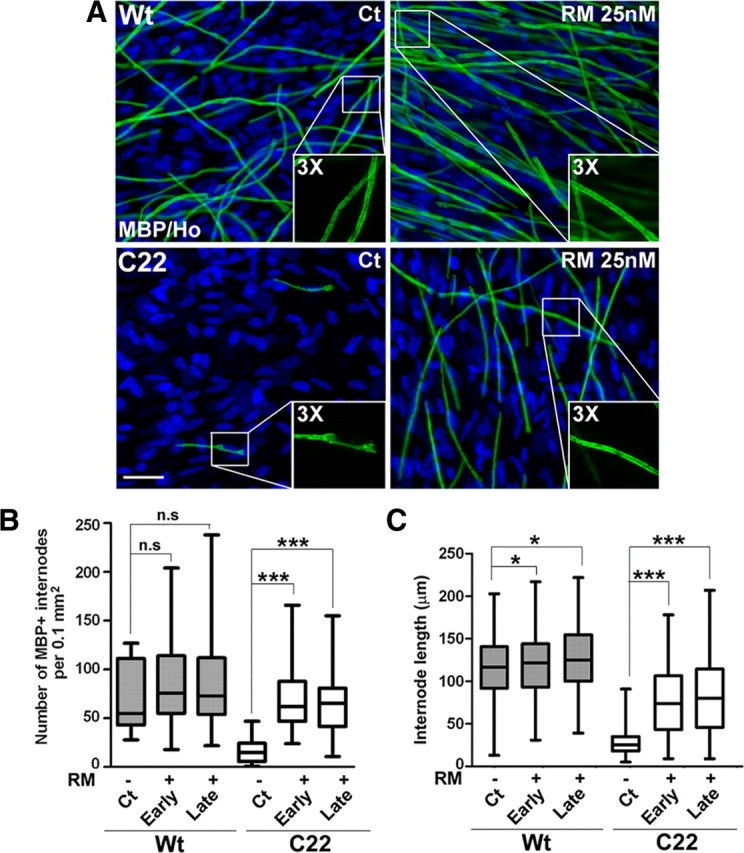
The abundance and length of myelin internodes are increased by rapamycin. A, Explant cultures from Wt and C22 mice were treated with Ct or RM at late stage, as described in Figure 3A, and stained for MBP (green) to visualize myelin. Scale bar, 40 μm. Hoechst dye stains the nuclei (blue). B, The abundance of myelin internodes was quantified in a fixed area [p > 0.2, not significant (n.s); ***p < 0.001, t test, mean ± SEM, n = 3). C, The lengths of the myelin internodes (n = 130–260) were measured and quantified in three independent experiments (*p < 0.05, **p < 0.01, ***p < 0.001, t test, mean ± SEM).
Autophagy is a critical pathway for the myelin-promoting effect of rapamycin
To assess the contribution of the autophagic pathway to the observed beneficial effects on myelination, we used lentiviral-mediated shRNA gene ablation of Atg12 (Dreux et al., 2009) in DRG explants (Bolis et al., 2009). First, we transduced DRG explants with lentiviral particles carrying a GFP reporter and visualized the long-term efficiency of transduction by direct GFP fluorescence (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). The transduction efficiency in DRG cultures on DIV11 (supplemental Fig. S4A, available at www.jneurosci.org as supplemental material) was nearly 65% (64.66 ± 9.04), and remained ∼62% (62.23 ± 11.88) on DIV26 (supplemental Fig. S4B, available at www.jneurosci.org as supplemental material). After establishing that lentivirus-mediated shRNA transduction is sustained throughout our myelinating culture paradigm, Neg. and Atg12 shRNA transduced Wt and C22 explants were treated with Ct or RM as described in Figure 3A. Similar to the nonmyelinating SCs (Fig. 2), the steady-state levels of the covalently linked Atg12-Atg5 conjugate are reduced by ∼50% in all samples transduced with Atg12 shRNA (*p < 0.05, n = 3), both in the absence and presence of RM (Fig. 5A). Within the same samples, the reduction in Atg12 is associated with a decrease in LC3 II/LC3 I ratios (Fig. 5A), as seen in the nonmyelinating samples (Fig. 2).
Figure 5.
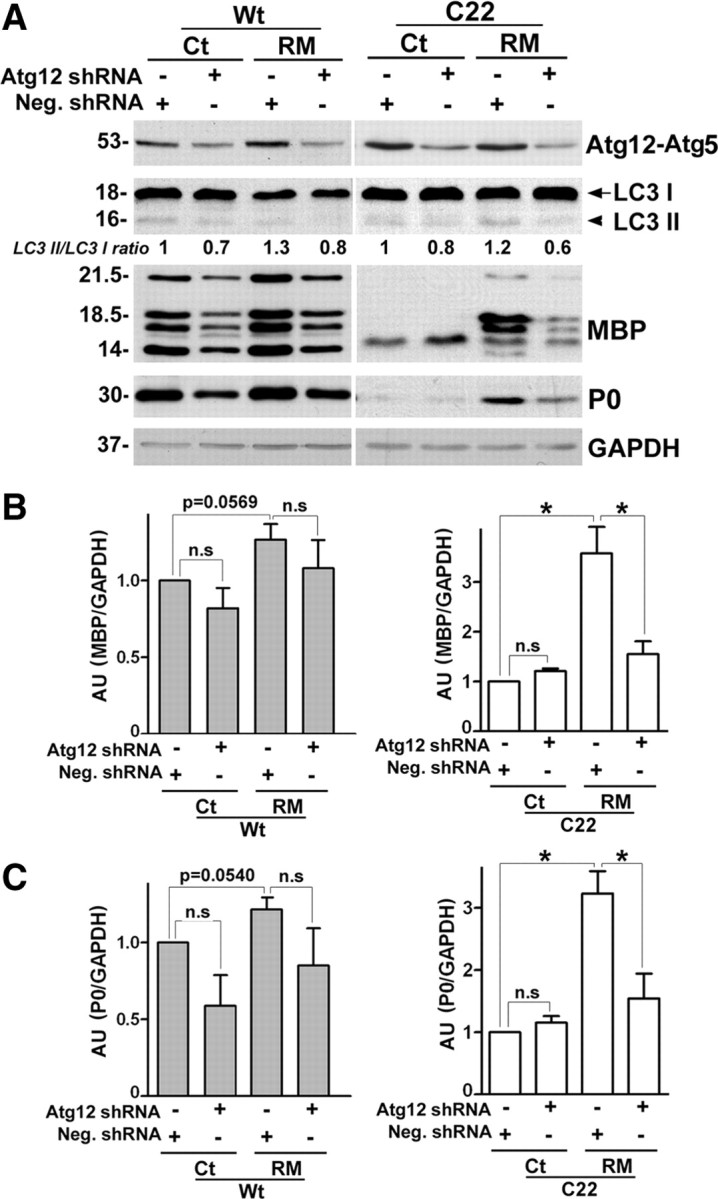
Atg12 is necessary for the myelin improvement by rapamycin. A, Wt and C22 DRG explants were transduced with scrambled Neg. shRNA or Atg12 shRNA. The shRNA transduced DRGs were given a late treatment (Fig. 3A) with Ct or RM, as indicated. Western blot analyses for Atg12-Atg5 conjugate, LC3 I and LC3 II and myelin proteins MBP, P0 are shown. Values of LC3 II/ LC3 I ratios at the end of the treatment paradigm is indicated below the representative blot. The value of LC3 II/LC3 I in Ct samples treated with Neg. shRNA was set as 1. GAPDH serves as protein loading control. Molecular mass in kDa. B, Semiquantitative analysis of MBP protein levels in Wt (left) and C22 (right) explants normalized to GAPDH. The value of MBP/GAPDH in Ct samples transduced with Neg. shRNA was set as 1 (n.s, not significant, p = 0.0569, *p < 0.05, t test, mean ± SEM, n = 3). C, Quantification of P0 protein levels in Wt (left panel) and C22 (right panel) samples normalized to GAPDH. The value of P0/GAPDH in Ct samples transduced with Neg. shRNA was set as 1 (n.s, not significant, p = 0.0540, *p < 0.05, t test, mean ± SEM, n = 3). AU, arbitrary units.
To verify the requirement for intact autophagy in RM-mediated myelin improvement in neuropathic samples, we analyzed the levels of MBP and P0 within cultures from independent isolations (Fig. 5A). Analyses of MBP levels normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH) reveal that downregulation of Atg12-Atg5 conjugate protein did not have a statistically significant effect on base-line MBP expression in Wt and C22 samples (Fig. 5B, n = 3). However, the effect of RM to slightly improve MBP levels in Wt DRGs (see supplemental Fig. S3, available at www.jneurosci.org as supplemental material), in this case transduced with Neg. shRNA, is reproducible (Fig. 5B, left, p = 0.0569). As seen previously (Fig. 6B), we detected a pronounced increase in MBP levels with RM treatment in Neg. shRNA transduced C22 samples, which is largely abolished by the reduction of Atg12 (Fig. 5B, right,*p < 0.05, n = 3). Similar to MBP, the RM-mediated improvements in P0 expression are attenuated by Atg12 deficiency (Fig. 5A,C, *p < 0.05, n = 3). Together, these results indicate that an intact autophagic pathway is necessary for the myelination-promoting activity of RM.
Figure 6.

The processing of PMP22 in myelinating SCs. A, Total protein lysates of DRG explants from Wt and C22 mice after treatment with Ct or RM were incubated with endo-H (H) or PNGase F (N) and PMP22 was detected by Western blots. Endo-H-resistant (R) (square bracket, ∼22 kDa) and endo-H-sensitive (S) (∼18 kDa) fractions of PMP22 are marked. Control (C) protein lysates for each sample without enzymatic treatment are also shown. B, Semiquantitative analyses of data from three independent experiments for endo-H-resistant (p = 0.0576, p = 0.0855, **p < 0.01, ***p < 0.001, t test, mean ± SD) and endo-H-sensitive fractions (##p < 0.01, ##p < 0.001, t test, mean ± SD) are shown.
As the myelinating explants contain DRG neurons, we tested the response of SC-depleted Wt and C22 neuron cultures (Einheber et al., 1993) to RM (supplemental Fig. S5, available at www.jneurosci.org as supplemental material). While RM is active in neurons, as judged from the pronounced dephosphorylation of pS6, the induction of autophagy in Neg. shRNA cultures is less apparent as deduced from p62 and LC3 levels (supplemental Fig. S5A, available at www.jneurosci.org as supplemental material). Quantification of the LC3 II/LC3 I ratios indicate that peripheral neurons are less responsive to RM (supplemental Fig. S5B, available at www.jneurosci.org as supplemental material) compared with SCs (Fig. 2B). The inhibition of Atg12 by shRNA (*p < 0.05, n = 3) results in a pronounced upregulation of p62 and reduction in LC3 II, indicating that basal neuronal autophagy is attenuated by ∼40–50% (supplemental Fig. S5A, available at www.jneurosci.org as supplemental material). Together, these studies indicate that basal autophagy, as well as RM-induced activation of this pathway is less robust within DRG neurons compared with SCs.
Activation of autophagy improves PMP22 trafficking
The accumulation of endo-H-resistant PMP22 at the SC membrane correlates with myelin synthesis (Pareek et al., 1997; Notterpek et al., 1999). Therefore, in addition to measuring the overall levels of PMP22 in our samples, we determined the subcellular trafficking of the protein (Fig. 5A). Consistent with the improvements in myelination (Figs. 3, 4), the overall levels of PMP22 are increased in neuropathic samples after early and late RM exposure (Fig. 6A). Significantly, we detected an increase in the fraction of endo-H-resistant PMP22 in RM-treated cultures, compared with vehicle. Since PMP22 exists in various glycosylated forms, we used N-glycosidase (PNGase F) to confirm that the identified protein bands are indeed PMP22 (Pareek et al., 1997). The C22 samples contain high levels of endo-H-sensitive PMP22 as detected by 35S-pulse labeling studies (Fortun et al., 2006) and on Western blots using an antibody against PMP22 (Pareek et al., 1997). As a result, the C22 samples have a low ratio of endo-H resistant to endo-H-sensitive PMP22 in the nerves and in the DRG explants, compared with Wt (Fortun et al., 2006; Rangaraju et al., 2008). There is a slight increase in the endo-H-resistant fraction of PMP22 in the Wt samples after early and late RM treatment, however, it is not significant (p = 0.0576, p = 0.0855, n = 4). In C22 neuropathic samples, semiquantitative analyses of three independent experiments confirmed the reduced levels of endo-H-resistant PMP22, compared with Wt (Fig. 6B, ***p < 0.001, n = 3–4) (Fortun et al., 2006), and its improvement after RM exposure at both early and late RM treatments (Fig. 6B, **p < 0.01, n = 3). Concurrently, the endo-H-sensitive PMP22 fraction in C22 samples decreases significantly (Fig. 6B, ##p < 0.01, n = 3) with RM treatment, suggesting the removal of immature PMP22 through the autophagy pathway. These results indicate that enhancement of autophagy within C22 neuropathic SCs is associated with an improvement in myelin formation and the subcellular processing of PMP22 (Figs. 3, 4).
The positive effect of RM is not genotype specific
A suitable therapeutic agent for hereditary neuropathies would not be genotype-specific, but rather show efficacy in multiple models. To assess whether RM could be beneficial for PMP22 point mutant neuropathies, we performed studies with SCs and myelinating explant cultures from TrJ mice (Fig. 7). Similar to the C22 model, DRGs from TrJ mice were incubated with RM at early or late stages of myelination and analyzed for MBP and P0 (Fig. 7A). Myelinating explants from TrJ mice respond positively to RM and show an increase in the steady-state levels of MBP and P0 (Fig. 7A,B). In agreement, DRG cultures immunolabeled with an MBP antibody reveal a pronounced increase in MBP-positive myelin segments in response to treatment (Fig. 7C). Quantification from three independent experiments reveals a significant improvement in the number (Fig. 7D, ***p < 0.001, n = 3) and length (Fig. 7E, **p < 0.01, n = 3) of myelin internodes, compared with vehicle. We confirmed the autophagic response in TrJ SCs, by analyzing protein lysates for pUb and LC3 after 48 h treatment with RM (supplemental Fig. S6A, available at www.jneurosci.org as supplemental material). As seen with the C22 sample (Fig. 2C), treatment with RM promotes a significant reduction in slow-migrating pUb substrates (supplemental Fig. S6B, available at www.jneurosci.org as supplemental material) and an approximately twofold increase in LC3 II/LC3 I ratios (supplemental Fig. S6C, available at www.jneurosci.org as supplemental material). Therefore, RM is an effective enhancer of autophagy in SCs from TrJ mice and improves their myelination capacity when in neuronal contact.
Figure 7.
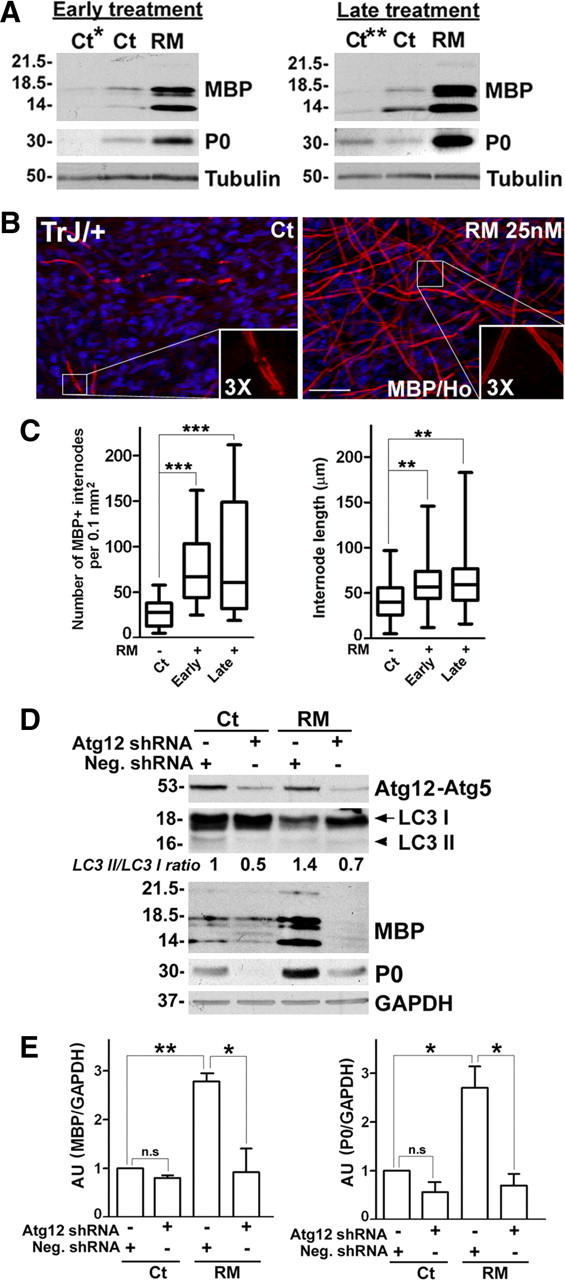
Rapamycin enhances myelination in cultures from TrJ mice. A, DRG explants from TrJ mice were treated as described in Figure 3A and whole protein lysates analyzed for the levels of myelin proteins, MBP and P0. Tubulin is shown as loading control. B, Cultures from TrJ mice were treated with vehicle Ct or RM at late stage as described in Figure 3A, and stained with an anti-MBP (red) antibody. Scale bar, 40 μm. Hoechst dye stains the nuclei (blue). C, The abundance (***p < 0.001, t test, mean ± SEM) and the lengths of the myelin internodes (n = 150–200) were quantified in explant cultures from TrJ mice treated without (−) or with (+) early or late RM treatment (**p < 0.01, t test, mean ± SEM, n = 3). D, TrJ DRG explants were transduced with scrambled Neg. shRNA or Atg12 shRNA and given a late treatment (Fig. 3A) with Ct or RM. Western blot analyses for Atg12-Atg5 conjugate, LC3 I and LC3 II and myelin proteins MBP, P0 at the end of the treatment paradigm are shown. Values of LC3 II/ LC3 I ratios is indicated below the representative blot. The value of LC3 II/LC3 I in Ct samples treated with Neg. shRNA was set as 1. GAPDH serves as protein loading control. Molecular mass, in kDa. E, Quantification of MBP (left) and P0 (right) protein levels in TrJ explants normalized to GAPDH. The value of MBP/GAPDH or P0/GAPDH in Ct samples transduced with Neg. shRNA was set as 1 (n.s, not significant, *p < 0.05, **p < 0.01, t test, mean ± SEM, n = 3). AU, arbitrary units.
To test whether the TrJ samples improve myelination as a result of the activated autophagy by RM treatment, we again performed shRNA-mediated inhibition of Atg12, as described in Figure 5. Analyses of the protein lysates at the end of the paradigm reveal a decline (*p < 0.05, n = 3) in the steady-state levels of Atg12-Atg5 conjugate, both in the absence and presence of RM (Fig. 7D). The reduction in Atg12 did not alter the level of MBP significantly in vehicle-treated TrJ samples (Fig. 7C, n = 3) and as shown (Fig. 7A), RM treatment enhanced MBP expression in Neg. shRNA transduced explants (Fig. 7E, **p < 0.01, n = 3). Significantly, this benefit is attenuated by the knockdown of Atg12 (Fig. 7E, *p < 0.05, n = 3). P0 follows a similar pattern of expression as MBP in response to the aforementioned RM treatment (Fig. 7D), which underscores the requirement for autophagy in myelin improvement.
Discussion
Hereditary neuropathies are common among the population, yet treatment options for affected individuals are limited. Currently, there is no effective drug therapy for CMT1A neuropathies and supportive care is limited to rehabilitation and surgical treatments (Pareyson and Marchesi, 2009). A recent 12-month clinical trial with ascorbic acid did not produce positive results, as efficacy of this antioxidant was not shown (Micallef et al., 2009). Here we tested RM, a widely used therapeutic compound for its ability to enhance autophagic response within neuropathic SCs, and improve their capacity to myelinate. We show that RM effectively activated autophagy in mouse SCs and lead to a significant reduction in pUb substrates in an autophagy-dependent manner. SCs from neuropathic mice responded to low concentrations (25 nm) of RM and produced more myelin, compared with vehicle controls. Significantly, RM improved myelination by SCs from two genetically distinct neuropathic models, including the gene duplication (C22) and the point mutation (TrJ) genotypes. Inhibition of a key autophagy gene abolished the improvements in myelination by RM in both C22 and TrJ models, suggesting a crucial role for autophagy in this beneficial effect.
CMT1A neuropathies associated with PMP22 gene duplication or mutations share characteristics with protein misfolding disorders, including the accumulation of ubiquitin-reactive protein aggregates and alterations in chaperone levels (Winklhofer et al., 2008). Once protein aggregates form they can interfere with essential cellular functions, such as myelination in the case of SCs. In this study, the reduction in pUb substrates with RM treatment is evident in SCs from both neuropathic models. The decline in the accumulation of pUb substrates is contingent upon the autophagy pathway (Fig. 2C) and may help in alleviating the burden on the proteasome (Fortun et al., 2005), which is the main degradative mechanism for the removal of newly synthesized PMP22 (Pareek et al., 1997). The concomitant decrease in p62 (Fig. 2B) along with pUb (Fig. 2C) in response to RM treatment in an autophagy-dependent fashion supports the idea that pUb substrates were indeed degraded by autophagy. Nonetheless, as RM reduces de novo protein synthesis in neuropathic SCs by ∼30% (supplemental Fig. S2, available at www.jneurosci.org as supplemental material), the observed 50% decrease in the levels of pUb substrates may have been assisted by this global affect. Restoration of protein homeostasis, including the improvement in the processing of PMP22 (Fig. 6) would likely provide a more permissive environment for myelination by SCs from neuropathic mice. Since P0 and MBP are coexpressed with PMP22 in myelinating SCs (Kuhn et al., 1993), and PMP22 and P0 are known to interact (Hasse et al., 2004), the improvement in PMP22 processing by RM most likely contributed to the concomitant increase in the expression of P0 and MBP (Figs. 3, 5, 7).
Compounds that enhance the activity of intracellular quality control mechanisms are possible treatment approaches for protein aggregation disorders. The roles of the chaperone and autophagy pathways in peripheral nerve biology have been less studied compared with neurons and glia of the CNS, nonetheless a decline in the activity and/or efficiency of protein-folding and/or degradation mechanisms likely contribute to age-associated degenerative changes in myelinated peripheral nerves (Verdú et al., 2000; Rangaraju et al., 2009). In a previous study, we chose the intermittent fasting regimen as proof-of-principle intervention to activate chaperones and autophagy in neuropathic mice (Madorsky et al., 2009). After a 5-month-long intervention, diet restricted mice showed an improvement in locomotor performance and myelination, compared with ad libitum fed littermates. Since such drastic dietary restriction is not suitable for humans, compounds that enhance chaperone production and/or autophagy will be more attractive for future therapies.
Autophagy is a particularly appealing target for therapeutic development for neurodegenerative diseases which are progressive with age, as activation of autophagy in normal aged mice appears to extend lifespan without negative side effects (Harrison et al., 2009). Upregulating autophagy by RM and other small molecules has been shown to suppress aggregation of disease-linked proteins, including huntingtin, α-synuclein, and to reduce cellular toxicity (Jaeger and Wyss-Coray, 2009). More recently, RM and its analog, have been shown to confer neuroprotection and ameliorate disease characteristics in a cellular model of spinocerebellar ataxia type 14 (Yamamoto et al., 2010) and in mouse models of Parkinson's disease (Malagelada et al., 2010), spinocerebellar ataxia type 3 (Menzies et al., 2010) and Alzheimer's disease (Caccamo et al., 2010; Spilman et al., 2010), which are a host of protein misfolding disorders. Independent studies using the transgenic approach confirm the involvement of autophagy in the removal of misfolded proteins, as mice deficient in autophagy genes within the CNS form intracellular inclusions and develop neurodegenerative symptoms (Komatsu et al., 2006; Mizushima and Hara, 2006). Consistent with these findings, the presence of misfolded aggregated PMP22 within peripheral nerves is associated with the neuropathic phenotype and demyelination (Fortun et al., 2003, 2006). In comparison, aggregates are reduced and myelin is improved in response to activation of autophagy and chaperones by long-term dietary restriction (Madorsky et al., 2009; Rangaraju et al., 2009), or by RM treatment as shown here. The results presented in this study are comparable to the increase in myelination in neuropathic samples by pharmacological activation of the heat shock pathway (Rangaraju et al., 2008), and suggest that combined pharmacological activation of the two protein quality control mechanisms, heat shock and autophagy pathways, might provide additive benefits.
RM is a widely used therapeutic compound that can elicit unique responses in different model systems and at given doses (Foster and Toschi, 2009). With regards to neural biology, in addition to its influence on autophagy in protein aggregation-linked neurodegenerative disorders (Jaeger and Wyss-Coray, 2009), RM has been tested in models of brain tumors and neuronal cell death. For example, in a mouse model of tuberous sclerosis the exposure of young mice to RM was associated with improved phenotype, including enhanced myelination (Meikle et al., 2008). In our PNS models, RM has a pronounced positive effect on activation of autophagy in SCs compared with DRG neurons, leading to an improvement in SC function, specifically myelination. We detected an increase in both the abundance and the lengths of myelin internodes indicating that RM supported the differentiation of myelinating SCs and their capacity to expand myelin sheaths (Figs. 4, 6). The Atg12 knockdown experiments indicate that an intact autophagy pathway is necessary for the myelin improvements in RM-treated C22 and TrJ cultures (Fig. 5, 7). This result reinstates our hypothesis that restoration of protein homeostasis within SCs improves the cells ability to myelinate axons. Nonetheless, contribution from other mechanisms such as the Akt pathway cannot be ruled out at this time. Akt is involved in neuregulin-mediated SC survival (Li et al., 2001) and in regulating myelination by oligodendrocytes (Narayanan et al., 2009). RM may have also influenced intracellular signaling pathways within neurons, and directly or indirectly through neuron-glial signaling mechanisms, facilitate myelination. In addition, RM is known to affect the endocytic pathway which appears to be important for myelination and membrane wrapping (Saito et al., 2005; Winterstein et al., 2008). Therefore, while the rationale for our studies with RM as a therapeutic agent for PMP22-associated neuropathies stem from our investigations of PMP22 trafficking and protein aggregation, it is possible that in addition to autophagy, RM will benefit neuropathic nerves through other mechanisms. The observed improvement in samples from Wt mice supports such hypothesis. In addition, in mice, RM may suppress the immune component of the neuropathy in vivo, which could offer an additional benefit to affected nerves (Hartung et al., 1992).
Based on the positive results presented here, we propose that RM is a suitable compound for further testing in neuropathic mice. As shown by our cellular studies, this drug is well tolerated by myelinating SCs as well as peripheral neurons and is effective at the low nm range. A recent study in aged mice showed that RM fed late in life extends lifespan without toxic side effects (Harrison et al., 2009). RM is an FDA-approved drug that is widely used in immunosuppression following organ transplantation; however, chronic use of RM can lead to adverse side-effects (Kahan, 2008). As SCs respond robustly to RM and the PNS is outside of the blood brain barrier, it is possible that peripheral nerves will respond to this compound more robustly than the CNS. In addition, small molecule enhancers of autophagy are being developed and identified and could offer alternatives for RM in the future (Sarkar and Rubinsztein, 2008). In summary, our results provide further support for the involvement of autophagy as a protective pathway in PMP22-associated neuropathies and provide a positive step toward identifying pharmacologic agents for these disorders.
Footnotes
These studies in part were supported by the National Institutes of Health–National Institute of Neurological Disorders and Stroke, the National Muscular Dystrophy Association, and a Pilot Project Award by the National Multiple Sclerosis Society. We thank Dr. Susie Zoltewicz for critical reading of the manuscript and Joseph Papin for technical assistance with the lentiviral transduction studies.
References
- Amici SA, Dunn WA, Jr, Notterpek L. Developmental abnormalities in the nerves of peripheral myelin protein 22-deficient mice. J Neurosci Res. 2007;85:238–249. doi: 10.1002/jnr.21118. [DOI] [PubMed] [Google Scholar]
- Archelos JJ, Roggenbuck K, Schneider-Schaulies J, Linington C, Toyka KV, Hartung HP. Production and characterization of monoclonal antibodies to the extracellular domain of P0. J Neurosci Res. 1993;35:46–53. doi: 10.1002/jnr.490350107. [DOI] [PubMed] [Google Scholar]
- Bolis A, Coviello S, Visigalli I, Taveggia C, Bachi A, Chishti AH, Hanada T, Quattrini A, Previtali SC, Biffi A, Bolino A. Dlg1, Sec8, and Mtmr2 regulate membrane homeostasis in Schwann cell myelination. J Neurosci. 2009;29:8858–8870. doi: 10.1523/JNEUROSCI.1423-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285:13107–13120. doi: 10.1074/jbc.M110.100420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreux M, Gastaminza P, Wieland SF, Chisari FV. The autophagy machinery is required to initiate hepatitis C virus replication. Proc Natl Acad Sci U S A. 2009;106:14046–14051. doi: 10.1073/pnas.0907344106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einheber S, Milner TA, Giancotti F, Salzer JL. Axonal regulation of Schwann cell integrin expression suggests a role for alpha 6 beta 4 in myelination. J Cell Biol. 1993;123:1223–1236. doi: 10.1083/jcb.123.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldridge CF, Bunge MB, Bunge RP, Wood PM. Differentiation of axon-related Schwann cells in vitro. I. Ascorbic acid regulates basal lamina assembly and myelin formation. J Cell Biol. 1987;105:1023–1034. doi: 10.1083/jcb.105.2.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortun J, Dunn WA, Jr, Joy S, Li J, Notterpek L. Emerging role for autophagy in the removal of aggresomes in Schwann cells. J Neurosci. 2003;23:10672–10680. doi: 10.1523/JNEUROSCI.23-33-10672.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortun J, Li J, Go J, Fenstermaker A, Fletcher BS, Notterpek L. Impaired proteasome activity and accumulation of ubiquitinated substrates in a hereditary neuropathy model. J Neurochem. 2005;92:1531–1541. doi: 10.1111/j.1471-4159.2004.02987.x. [DOI] [PubMed] [Google Scholar]
- Fortun J, Go JC, Li J, Amici SA, Dunn WA, Jr, Notterpek L. Alterations in degradative pathways and protein aggregation in a neuropathy model based on PMP22 overexpression. Neurobiol Dis. 2006;22:153–164. doi: 10.1016/j.nbd.2005.10.010. [DOI] [PubMed] [Google Scholar]
- Fortun J, Verrier JD, Go JC, Madorsky I, Dunn WA, Notterpek L. The formation of peripheral myelin protein 22 aggregates is hindered by the enhancement of autophagy and expression of cytoplasmic chaperones. Neurobiol Dis. 2007;25:252–265. doi: 10.1016/j.nbd.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DA, Toschi A. Targeting mTOR with rapamycin: one dose does not fit all. Cell Cycle. 2009;8:1026–1029. doi: 10.4161/cc.8.7.8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartung HP, Jung S, Stoll G, Zielasek J, Schmidt B, Archelos JJ, Toyka KV. Inflammatory mediators in demyelinating disorders of the CNS and PNS. J Neuroimmunol. 1992;40:197–210. doi: 10.1016/0165-5728(92)90134-7. [DOI] [PubMed] [Google Scholar]
- Hasse B, Bosse F, Hanenberg H, Müller HW. Peripheral myelin protein 22 kDa and protein zero: domain specific trans-interactions. Mol Cell Neurosci. 2004;27:370–378. doi: 10.1016/j.mcn.2004.06.009. [DOI] [PubMed] [Google Scholar]
- Huxley C, Passage E, Manson A, Putzu G, Figarella-Branger D, Pellissier JF, Fontés M. Construction of a mouse model of Charcot-Marie-Tooth disease type 1A by pronuclear injection of human YAC DNA. Hum Mol Genet. 1996;5:563–569. doi: 10.1093/hmg/5.5.563. [DOI] [PubMed] [Google Scholar]
- Huxley C, Passage E, Robertson AM, Youl B, Huston S, Manson A, Sabéran-Djoniedi D, Figarella-Branger D, Pellissier JF, Thomas PK, Fontés M. Correlation between varying levels of PMP22 expression and the degree of demyelination and reduction in nerve conduction velocity in transgenic mice. Hum Mol Genet. 1998;7:449–458. doi: 10.1093/hmg/7.3.449. [DOI] [PubMed] [Google Scholar]
- Jaeger PA, Wyss-Coray T. All-you-can-eat: autophagy in neurodegeneration and neuroprotection. Mol Neurodegener. 2009;4:16. doi: 10.1186/1750-1326-4-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahan BD. Fifteen years of clinical studies and clinical practice in renal transplantation: reviewing outcomes with de novo use of sirolimus in combination with cyclosporine. Transplant Proc. 2008;40:S17–20. doi: 10.1016/j.transproceed.2008.10.019. [DOI] [PubMed] [Google Scholar]
- King MA, Hands S, Hafiz F, Mizushima N, Tolkovsky AM, Wyttenbach A. Rapamycin inhibits polyglutamine aggregation independently of autophagy by reducing protein synthesis. Mol Pharmacol. 2008;73:1052–1063. doi: 10.1124/mol.107.043398. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Ichimura Y. Physiological significance of selective degradation of p62 by autophagy. FEBS Lett. 2010;584:1374–1378. doi: 10.1016/j.febslet.2010.02.017. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–884. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- Kuhn G, Lie A, Wilms S, Müller HW. Coexpression of PMP22 gene with MBP and P0 during de novo myelination and nerve repair. Glia. 1993;8:256–264. doi: 10.1002/glia.440080406. [DOI] [PubMed] [Google Scholar]
- Li Y, Tennekoon GI, Birnbaum M, Marchionni MA, Rutkowski JL. Neuregulin signaling through a PI3K/Akt/Bad pathway in Schwann cell survival. Mol Cell Neurosci. 2001;17:761–767. doi: 10.1006/mcne.2000.0967. [DOI] [PubMed] [Google Scholar]
- Madorsky I, Opalach K, Waber A, Verrier JD, Solmo C, Foster T, Dunn WA, Jr, Notterpek L. Intermittent fasting alleviates the neuropathic phenotype in a mouse model of Charcot-Marie-Tooth disease. Neurobiol Dis. 2009;34:146–154. doi: 10.1016/j.nbd.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. 2007;8:741–752. doi: 10.1038/nrm2239. [DOI] [PubMed] [Google Scholar]
- Malagelada C, Jin ZH, Jackson-Lewis V, Przedborski S, Greene LA. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson's disease. J Neurosci. 2010;30:1166–1175. doi: 10.1523/JNEUROSCI.3944-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meikle L, Pollizzi K, Egnor A, Kramvis I, Lane H, Sahin M, Kwiatkowski DJ. Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci. 2008;28:5422–5432. doi: 10.1523/JNEUROSCI.0955-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menzies FM, Huebener J, Renna M, Bonin M, Riess O, Rubinsztein DC. Autophagy induction reduces mutant ataxin-3 levels and toxicity in a mouse model of spinocerebellar ataxia type 3. Brain. 2010;133:93–104. doi: 10.1093/brain/awp292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Micallef J, Attarian S, Dubourg O, Gonnaud PM, Hogrel JY, Stojkovic T, Bernard R, Jouve E, Pitel S, Vacherot F, Remec JF, Jomir L, Azabou E, Al-Moussawi M, Lefebvre MN, Attolini L, Yaici S, Tanesse D, Fontes M, Pouget J, Blin O. Effect of ascorbic acid in patients with Charcot-Marie-Tooth disease type 1A: a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2009;8:1103–1110. doi: 10.1016/S1474-4422(09)70260-1. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Hara T. Intracellular quality control by autophagy: how does autophagy prevent neurodegeneration? Autophagy. 2006;2:302–304. doi: 10.4161/auto.2945. [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yoshimori T. How to interpret LC3 immunoblotting. Autophagy. 2007;3:542–545. doi: 10.4161/auto.4600. [DOI] [PubMed] [Google Scholar]
- Muchowski PJ, Wacker JL. Modulation of neurodegeneration by molecular chaperones. Nat Rev Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- Naef R, Suter U. Many facets of the peripheral myelin protein PMP22 in myelination and disease. Microsc Res Tech. 1998;41:359–371. doi: 10.1002/(SICI)1097-0029(19980601)41:5<359::AID-JEMT3>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Narayanan SP, Flores AI, Wang F, Macklin WB. Akt signals through the mammalian target of rapamycin pathway to regulate CNS myelination. J Neurosci. 2009;29:6860–6870. doi: 10.1523/JNEUROSCI.0232-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson SM, Gomès D, de Néchaud B, Bruzzone R. Altered gene expression in Schwann cells of connexin32 knockout animals. J Neurosci Res. 2001;66:23–36. doi: 10.1002/jnr.1194. [DOI] [PubMed] [Google Scholar]
- Notterpek L, Shooter EM, Snipes GJ. Upregulation of the endosomal-lysosomal pathway in the trembler-J neuropathy. J Neurosci. 1997;17:4190–4200. doi: 10.1523/JNEUROSCI.17-11-04190.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notterpek L, Snipes GJ, Shooter EM. Temporal expression pattern of peripheral myelin protein 22 during in vivo and in vitro myelination. Glia. 1999;25:358–369. [PubMed] [Google Scholar]
- Pareek S, Notterpek L, Snipes GJ, Naef R, Sossin W, Laliberté J, Iacampo S, Suter U, Shooter EM, Murphy RA. Neurons promote the translocation of peripheral myelin protein 22 into myelin. J Neurosci. 1997;17:7754–7762. doi: 10.1523/JNEUROSCI.17-20-07754.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pareyson D, Marchesi C. Diagnosis, natural history, and management of Charcot-Marie-Tooth disease. Lancet Neurol. 2009;8:654–667. doi: 10.1016/S1474-4422(09)70110-3. [DOI] [PubMed] [Google Scholar]
- Rangaraju S, Madorsky I, Pileggi JG, Kamal A, Notterpek L. Pharmacological induction of the heat shock response improves myelination in a neuropathic model. Neurobiol Dis. 2008;32:105–115. doi: 10.1016/j.nbd.2008.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangaraju S, Hankins D, Madorsky I, Madorsky E, Lee WH, Carter CS, Leeuwenburgh C, Notterpek L. Molecular architecture of myelinated peripheral nerves is supported by calorie restriction with aging. Aging Cell. 2009;8:178–191. doi: 10.1111/j.1474-9726.2009.00460.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabers CJ, Martin MM, Brunn GJ, Williams JM, Dumont FJ, Wiederrecht G, Abraham RT. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J Biol Chem. 1995;270:815–822. doi: 10.1074/jbc.270.2.815. [DOI] [PubMed] [Google Scholar]
- Saito K, Araki Y, Kontani K, Nishina H, Katada T. Novel role of the small GTPase Rheb: its implication in endocytic pathway independent of the activation of mammalian target of rapamycin. J Biochem. 2005;137:423–430. doi: 10.1093/jb/mvi046. [DOI] [PubMed] [Google Scholar]
- Sanders CR, Ismail-Beigi F, McEnery MW. Mutations of peripheral myelin protein 22 result in defective trafficking through mechanisms which may be common to diseases involving tetraspan membrane proteins. Biochemistry. 2001;40:9453–9459. doi: 10.1021/bi010894f. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–168. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Rubinsztein DC. Small molecule enhancers of autophagy for neurodegenerative diseases. Mol Biosyst. 2008;4:895–901. doi: 10.1039/b804606a. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Ravikumar B, Floto RA, Rubinsztein DC. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2009;16:46–56. doi: 10.1038/cdd.2008.110. [DOI] [PubMed] [Google Scholar]
- Shy ME, Balsamo J, Lilien J, Kamholz J. A molecular basis for hereditary motor and sensory neuropathy disorders. Curr Neurol Neurosci Rep. 2001;1:77–88. doi: 10.1007/s11910-001-0079-6. [DOI] [PubMed] [Google Scholar]
- Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer's disease. PLoS One. 2010;5:e9979. doi: 10.1371/journal.pone.0009979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani M, Dobson CM. Protein aggregation and aggregate toxicity: new insights into protein folding, misfolding diseases and biological evolution. J Mol Med. 2003;81:678–699. doi: 10.1007/s00109-003-0464-5. [DOI] [PubMed] [Google Scholar]
- Suter U, Moskow JJ, Welcher AA, Snipes GJ, Kosaras B, Sidman RL, Buchberg AM, Shooter EM. A leucine-to-proline mutation in the putative first transmembrane domain of the 22-kDa peripheral myelin protein in the trembler-J mouse. Proc Natl Acad Sci U S A. 1992;89:4382–4386. doi: 10.1073/pnas.89.10.4382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdú E, Ceballos D, Vilches JJ, Navarro X. Influence of aging on peripheral nerve function and regeneration. J Peripher Nerv Syst. 2000;5:191–208. doi: 10.1046/j.1529-8027.2000.00026.x. [DOI] [PubMed] [Google Scholar]
- Wakatsuki S, Yumoto N, Komatsu K, Araki T, Sehara-Fujisawa A. Roles of meltrin-beta/ADAM19 in progression of Schwann cell differentiation and myelination during sciatic nerve regeneration. J Biol Chem. 2009;284:2957–2966. doi: 10.1074/jbc.M803191200. [DOI] [PubMed] [Google Scholar]
- Winklhofer KF, Tatzelt J, Haass C. The two faces of protein misfolding: gain- and loss-of-function in neurodegenerative diseases. EMBO J. 2008;27:336–349. doi: 10.1038/sj.emboj.7601930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterstein C, Trotter J, Krämer-Albers EM. Distinct endocytic recycling of myelin proteins promotes oligodendroglial membrane remodeling. J Cell Sci. 2008;121:834–842. doi: 10.1242/jcs.022731. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Seki T, Adachi N, Takahashi T, Tanaka S, Hide I, Saito N, Sakai N. Mutant protein kinase C gamma that causes spinocerebellar ataxia type 14 (SCA14) is selectively degraded by autophagy. Genes Cells. 2010 doi: 10.1111/j.1365-2443.2010.01395.x. [DOI] [PubMed] [Google Scholar]
- Yang Z, Klionsky DJ. An overview of the molecular mechanism of autophagy. Curr Top Microbiol Immunol. 2009;335:1–32. doi: 10.1007/978-3-642-00302-8_1. [DOI] [PMC free article] [PubMed] [Google Scholar]