

Abstract
Several genes and proteins of the complement cascade are present at elevated levels in brains of patients with Huntington's disease (HD). The complement cascade is well characterized as an effector arm of the immune system, and in the brain it is important for developmental synapse elimination. We hypothesized that increased levels of complement in HD brains contributes to disease progression, perhaps by contributing to synapse elimination or inflammatory signaling. We tested this hypothesis in the R6/2 mouse model of HD by crossing mice deficient in complement component 3 (C3), a crucial complement protein found at increased levels in HD brains, to R6/2 mice and monitoring behavioral and neuropathological disease progression. We found no alterations in multiple behavioral assays, weight or survival in R6/2 mice lacking C3. We also quantified the expression of several complement cascade genes in R6/2 brains and found that the large scale upregulation of complement genes observed in HD brains is not mirrored in R6/2 brains. These data show that C3 deficiency does not alter disease progression in the R6/2 mouse model of HD.
Keywords: Complement C3, Huntington Disease, Neurodegenerative Diseases, Immunity, Innate
Introduction
Huntington's Disease (HD) is an adult-onset neurodegenerative disease characterized by impaired motor control and cognitive and emotional changes [1]. The disease is inherited in an autosomal dominant fashion and is caused by expansion of a polyglutamine tract in the ubiquitously expressed protein huntingtin. Neurodegeneration is most severe in striatum and cortex, but is ultimately observed throughout the brain.
In addition to these pathological hallmarks of HD, several studies suggest that the innate immune system is activated in HD. In blood from HD patients, several cytokines and other proteins associated with innate immune activation are elevated before—in some cases several years before—disease onset, and their presence may predict disease progression [2—4]. Inflammation in the brain also predicts and correlates with disease progression. For example, microglial activation occurs before symptoms appear and increases as disease progresses [5–7]. One aspect of the innate immune response that is elevated both in the brain and in the periphery of HD patients is the complement cascade. Several proteins of this family, including C3, C4, C7 and C9 are elevated in blood from HD patients [2,3]. Microarray experiments reveal elevated transcription of multiple complement proteins in brains from early stages HD patients (Grades 0–2) [8] and immunostaining and in situ hybridization experiments show that complement proteins are produced by microglia and deposited on neurons in late stages of disease (Grades 3–4) [9,10]. Despite this accumulating evidence of elevated levels of complement proteins in HD, cause and effect relationships between complement and disease progression in vivo have not yet been investigated.
The complement cascade has been well studied outside of the brain. It consists of soluble proteases that can bind to pathogens and signal for their destruction, thus contributing to elimination of a variety of infections. Upon binding, the initiating protease of the complement cascade, generally either C3 or complement component 1q (C1q), is activated and cleaves the next protein in the cascade, thereby activating that second protease. Sequential activation of further complement proteins leads to the formation of a “C3 convertase” that drives a common series of downstream events. These events include proinflammatory signaling, receptor-mediated phagocytosis, and destruction of cell membranes by the end product of the complement cascade: the membrane attack complex. Several complement proteins can initiate the cascade, but regardless of which one does so, activation proceeds through a C3 convertase complex that contains C3 protein. Mice lacking a functional C3 gene (C3−/− mice) are unable to undergo normal activation of the downstream effector functions of the complement cascade and are less adept at eliminating pathogens [11]. These mice do show some downstream complement activation because other proteases can compensate for lack of C3 [12], but this compensation is incomplete.
C3−/− mice show several interesting brain phenotypes that may bear on the role of C3 in HD. Notably, normal developmental synapse elimination is impaired in C3−/− mice and in C1q−/− mice [13]. Wild-type (WT) mice normally have low levels of C3 and C1q in the brain, but they can increase expression during inflammation, likely to control infection [14], and during development [13]. Increased levels of C3 and C1q in the brain during development may contribute to synapse elimination via mechanisms similar to those by which complement in the immune system mediates phagocytosis of pathogens. Recent in vitro data shows that C1q and C3 can bind to neurites and trigger their phagoycytic removal by microglia [15]. Even when initiated by C1q binding, this process likely depends on fragments of C3 protein that are deposited on the neurite and recognized by microglial complement receptor 3 (CR3) [15].
In addition to complement upregulation during developmental synapse elimination, complement synthesis in the brain is also upregulated in other neurodegenerative diseases, including Alzheimer's disease (AD) [16], experimental autoimmune encephalomyelitis and multiple sclerosis [17], nerve injury models [18], AIDS-associated dementia [19] and glaucoma [13]. Studies in C3−/− mice show that complement can have a positive or a negative role in progression of brain diseases. For example, when C3−/− mice were crossed to a mouse model of AD, there was increased plaque deposition and exacerbated neurodegeneration, quantified by loss of neuronal-specific nuclear protein positive neurons in the hippocampus, implying that complement C3 is beneficial in AD [20]. On the other hand, retinal ganglion cells in C3−/− mice are protected from retinal ischemia-reperfusion induced cell death [21], illustrating that C3 is detrimental in this context. Further, C3 outside of the brain can also impact brain health. For example, in a mouse model of multiple sclerosis, genetic ablation of the complement C3a receptor, which mediates some of the effects of C3, attenuates disease, likely by impairing signaling in macrophages and/or T cells outside of the brain [17]. Given these mixed results, it is difficult to predict a priori what role the complement cascade might be playing in the brains of HD patients.
The time course of complement upregulation could offer insight into the role of the complement cascade in HD, but again the evidence is mixed, as different complement proteins are upregulated with different time courses in HD patients. Proteomic analysis of the blood of HD patients revealed that premanifest HD patients have elevated levels of complement components C4 and C7, but levels of C9 and clusterin are not altered until advanced stages of disease [3]. Similarly, microarray studies comparing brains from early stage (Grades 0–2) HD patients to controls show that patients have increased expression of several complement mRNAs, including C3 [10,22]. However, late-grade HD brains (Grades 3–4) show upregulation of a wider variety of complement related proteins, including C3 and several other components of the complement cascade, complement regulatory factors, and complement receptors [9].
The complement cascade is one of many aspects of the innate immune system that is altered in HD [23]. Any of these interconnected elements could have beneficial or detrimental effects on brain health. We chose to focus our experiments on the complement cascade because there is strong evidence that complement in the brain can play a role in neurodegeneration and because the intriguing functional role of complement in synaptic maintenance is beginning to be elucidated. Further, complement is an attractive target because it is upregulated in HD and because efforts to develop complement inhibitors for other inflammatory diseases are already under way [24,25]. Within the complement cascade, we chose to focus on C3 because it is central to many pathways of complement activation and signaling and because several studies have shown that C3−/− mice are a powerful tool for manipulating brain complement activity.
To explore the role of C3 in HD, we crossed C3−/− mice to the R6/2 mouse model of HD and monitored disease progression. R6/2 mice transgenically express exon 1 of the mutant human gene that causes HD. These mice exhibit progressive behavioral and neuropathological deficits that culminate in premature death. While neuronal loss is not apparent in these mice, they do lose synapses over time [26,27] and this likely contributes to behavioral phenotypes. We hypothesized that R6/2;C3−/− mice would benefit from the absence of this powerful destructive force and would show slower disease progression than R6/2;C3+/+ mice. However, we found that motor deficits, weight, survival and disease associated gene expression changes in R6/2 mice were unaffected by genetic deficiency of C3.
Materials and Methods
Mice
All animals were housed and handled in accordance with the National Institutes of Health “Guide for the Care and Use of Laboratory Animals.” All studies were approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco. All animals were housed in a pathogen-free barrier facility on a 12 h light/dark cycle.
R6/2 and WT mice on a mixed CBA and C57BL/6 background (stock # 002810) and C3−/− mice on a C57BL/6 background (stock # 003641) were obtained from The Jackson Laboratory (Bar Harbor, ME). These mice were bred and progeny were used for behavioral studies and HD associated gene expression studies. R6/2 and WT mice used for characterization of complement related genes were generated from the sixth generation backcross of male R6/2 breeders to WT C57BL/6 females.
Genotyping
Mouse tail DNA was analyzed by PCR to determine genotype. The R6/2 transgene was amplified using the forward primer [CGC AGG CTA GGG CTG TCA ATC ATG CT] and the reverse primer [TCA TCA GCT TTT CCA GGG TCG CCA T]. The wild-type and mutant C3 alleles were amplified separately. The forward primer [GGT TGC AGC AGT CTA TGA AGG] and the reverse primer [ATC TTG AGT GCA CCA AGC C] were used to amplify the wild-type C3 allele while the forward primer [CTT GGG TGG AGA GGC TAT TC] and the reverse primer [AGG TGA GAT GAC AGG AGA TC] were used to amplify the Neo cassette in the mutant C3 allele.
Behavioral assays
Open field
Spontaneous locomotor activity was measured using an automated Photobeam Activity System (San Diego Instruments, San Diego, CA). Before testing, mice were acclimated in the testing room for 1 h. During testing, mice were placed into a clear plastic testing chamber (41 × 41 × 30 cm) for 15 min. Horizontal and vertical movements were detected by photobeam arrays traversing the testing chamber. Testing chambers were cleaned with 70% alcohol after each session.
Balance beam
The balance beam apparatus consisted of two elevated plastic platforms, approximately 53 cm high, joined by a 38 cm long plastic beam. The starting platform was open, while the finishing platform was enclosed by an opaque plastic box. To determine performance on this assay, mice were placed on the starting platform and the experimenter measured the time required for the mouse to walk across the beam to the finishing platform.
At each time point (4, 8, and 12 weeks of age), the balance beam assay began with two training trials, where the mouse was placed on the starting platform and guided across the beam by the experimenter. Testing trials were similar, except the mouse was left to navigate the beam unaided. Each mouse received three testing trials with a large cylindrical beam (approximately 1.59 cm diameter) on the first day of training/testing, three testing trials with a medium cylindrical beam (approximately 1.27 cm diameter) on the following day and three testing trials with a small square beam (approximately 0.6 cm on each face) on the final day of the assay. Time to cross the beam was recorded for each trial and the three trials on each beam were averaged to determine the average time to cross for a given mouse on a given beam. If the mouse fell off the beam, it was placed back on the beam at the point where it fell and the time it took to fall and be replaced was subtracted from the total time for that trial. Data with the three different sized beams gave similar results, so only results from the medium sized beam are shown.
Rotarod
The same rotarod (Med Associates ENV-577M) was used for all rotarod studies. At each time (4, 8, and 12 weeks of age), the rotarod assay began with three training trials where the rotarod was programmed to spin at a constant rate of 16 rpm. During each training trial, mice were allowed to remain on the rotarod until they fell off or until 5 min had elapsed. Testing occurred on the 4 days after training. On each day of testing, mice received three trials during which the rotarod was programmed to accelerate from 4 to 40 rpm over a period of 5 min. Mice were allowed to remain on the rotarod until they fell off or until 5 min had elapsed. Latency to fall was recorded automatically by a photobeam beneath the rotarod. Latency to fall was averaged across all four days of testing (twelve trials total for each mouse at a given age).
Survival
For survival studies mice were observed daily. Survival was evaluated as the time to which the mice either died spontaneously, or lost > 15% of their maximal weight.
Quantitative real-time PCR
RNA was harvested from frozen mouse forebrain (i.e. whole brain minus cerebellum) or striatum [microdissected as described [28]] with an RNeasy Mini kit (Qiagen, USA), according to the manufacturer's instructions. RNA was immediately reverse transcribed to cDNA with Multiscribe™ reverse transcriptase (Applied Biosystems, USA) and stored at −20 °C.
For HD associated gene expression studies and quantification of C3 mRNA, quantitative real-time PCR (qPCR) was performed using FAST SYBR Green Mastermix (Applied Biosystems, USA) as provided by the manufacturer and thermal cycling was performed using an ABI Prism 7900 HT Sequence Detector (Applied Biosystems, USA) with the following program: UNG activation (50 °C for 2 min), initial denaturation (95 °C for 10 min), and then 40 cycles of denaturation (95 °C for 15 seconds) then annealing and extension (60 °C for 1 min), followed by a disassociation stage for melting curve analysis. Analysis of fluorescence data from genes of interest and from the control genes ATP synthase subunit 5b (Atp5b), ubiquitin C (UbC) and eukaryotic initiation factor 4a2 (Aif4a2) [29] was performed using the online analysis tool QPCR [30].
For characterization of complement related genes other than C3, qPCR was performed using SYBR Green Mastermix (Applied Biosystems, USA) as provided by the manufacturer. Thermal cycling was performed using an ABI Prism 7700 Sequence Detector (Applied Biosystems, USA) with the same program described above. Amplification efficiency was calculated for each primer pair using serially diluted standards. This efficiency was used to convert threshold cycle (number of PCR cycles required to reach an arbitrary threshold fluorescence value) into relative gene expression using the efficiency corrected delta Ct method as described [31]. For each sample, expression of the gene of interest was normalized to the control gene Atp5b.
All data are expressed as a fold change compared to WT mice, and all primers are listed in Table 1. Primers were designed using the online Roche Universal Probe Library Assay Design Center (Roche Applied Science, USA) or the online tool Primer-BLAST (NCBI) except complement component C5a receptor 1 (C5aR) [32], C3 [33] and Factor B [33] primers whose sequences were taken from published literature. Primer specificity was evaluated using Primer-BLAST (NCBI) and melting curve analysis.
Table 1.
Primers used for qPCR.
| Gene | Accession # | Forward Primer | Reverse Primer |
|---|---|---|---|
| 1. Atp5b | NM_016774 | TGA GAG AGG TCC TAT CAA AAC CA | ACC AGA ATC TCC TGC TCA ACA |
| 2. C1 Inh | NM_009776 | GAA GCT GCC TAG TGA CCA AGA | CAG CAG GAG GGT CAG TGG |
| 3. C1q | NM_007572 | CAA GGA CTG AAG GGC GTG AA | CAA GCG TCA TTG GGT TCT GC |
| 4. C1qR | NM_010740 | TGA AAT AGA CGC CCT GAA AAC | AAT CAA AGC CTG GGT TTA GGA |
| 5. C4 | NM_009780 | TCT CAC AAA CCC CTC GAC AT | AGC ATC CTG GAA CAC CTG AA |
| 6. C5 | NM_010406 | GGA TTC AAG CGC ATA ATA GCA | ACC CGG ATG TTG ACT CCT C |
| 7. C6 | NM_016704 | TCC AGT ACT TGA GAT GTT TAC CAG A | TTG AGG CAC GAG GTC CTT |
| 8. C9 | NM_013485 | CCG AGT AGC GGA AGA ATC AG | GGG CTC CAT CCC TAA GAT GT |
| 9. Calreticulin | NM_007591 | TGA AGC TGT TTC CGA GTG GT | GAT GAC ATG AAC CTT CTT GGT G |
| 10. CD91 | NM_008512 | GGA CCA CCA TCG TGG AAA | TCC CAG CCA CGG TGA TAG |
| 11. Crry | NM_013499 | GCC TTC AGT CTC TGC TCA CA | AAT TCC GTG CTG GGC TAG T |
| 12. DAF | NM_010016 | ACT GTT GAT TGG GAC GAT GAG | TGG TGG CTC TGG ACA ATG TA |
| 13. Factor B | NM_001142706 | GCT ACA GTC CCC AAA GTG TT | CAT GCT ATA CAC AGC CTG GA |
| 14. Protectin | NM_007652 | GAT GCT GCC AGT TTA ACT TGT G | GAG GTC CCC AGC AAT GGT |
|
| |||
| 15. C3 | NM_009778 | CAC CGC CAA GAA TCG CTA C | GAT CAG GTG TTT CAG CCG C |
| 16. C5aR | NM_007577 | GGG ATG TTG CAG CCC TTA TCA | CGC CAG ATT CAG AAA CC AG ATG |
| 17. Caspase 3 | NM_009810 | TCA TTC AGG CCT GCC GGG GT | TGG ATG AAC CAC GAC CCG TCC |
| 18. D1 DAR | NM_010076 | TGT TTG AAA TGT TTA CAA GGT GTT C | CAG TCA GCC CTT CCT TCA GT |
| 19. Eif4a2 | NM_013506 | GAA GCC CCT CAC ATT GTT GT | GCT TCG TCC AAA ACG AAC AT |
| 20. Hsp90 | NM_010480 | AGG CTT TGC AGG CTG GTG CAG | TCC ACG ACC CAT TGG TTC ACC TGT |
| 21. Penk | NM_001002927 | CCC AGG CGA CAT CAA TTT | TCT CCC AGA TTT TGA AAG AAG G |
| 22. Thrombin | NM_010168 | TGG CCT GCG GCC TTT GTT CG | TCA CCT GCC AGG GGG CGA T |
| 23. UbC | NM_019639 | AGC CCA GTG TTA CCA CCA AG | CTA AGA CAC CTC CCC CAT CA |
Primers 1–14 were run with SYBR Green mastermix and an ABI 7700 thermal cycler. Primers 15–23 were run with Fast SYBR Green mastermix and an ABI 7900 HT thermal cycler. Atp5b and C3 primers were run in both sets of conditions.
C3 protein levels
To isolate protein, fresh frozen brain was homogenized in 1X TBS buffer with 1X protease inhibitor (Roche Applied Science, USA) and spun at 10,000 × g for 90 min at 4 °C. A Pierce BCA protein assay kit (Thermo Scientific, USA) was used to determine protein concentration in the supernatant. Blood was collected via cardiac puncture and spun in the presence of EDTA. C3 protein levels in the supernatant and in plasma were quantified using a C3 ELISA kit (ICL Inc, Portland, OR) per the manufacturer's instructions.
Statistics
All statistical analysis was performed using Prism (Graphpad, La Jolla, CA).
Results
We sought to determine the effect of genetic deficiency of complement protein C3 on disease progression in the R6/2 transgenic mouse model of HD. To accomplish this, we crossed R6/2 mice to C3−/− mice. Breeding the progeny of this cross (R6/2;C3+/− males crossed to WT;C3+/− females) gave us mice of the four different genotypes we studied: WT;C3+/+, WT;C3−/−, R6/2;C3+/+, and R6/2;C3−/−.
C3−/− mice do not have detectable levels of C3 protein in brain
In order to verify the efficacy of genetic deletion of C3, we used an ELISA to measure C3 protein levels in brains from mice of all four genotypes we studied (n=3/genotype). These brains were dissected and striatum was used for qPCR based detection of genes associated with disease progression, while the remaining brain material was used for C3 protein detection. As expected, we were unable to detect C3 protein in brains from C3−/− mice on either a WT or R6/2 background (Fig. 1A).
Fig. 1. Complement component 3 does not influence behavioral phenotypes in R6/2 mice.

Behavioral readouts of disease progression were measured at 4, 8, and 12 weeks of age using balance beam, rotarod and open field testing. Data at each time point were tested for statistically significant differences using two-way ANOVA followed by Bonferroni post-hoc tests. Survival was monitored throughout the study and survival curves were tested for statistically significant differences using a Log-rank (Mantel-Cox) test. At 4 weeks of age, group sizes were as follows: WT;C3+/+ (n=15), WT;C3−/− (n=11), R6/2;C3+/+ (n=7), R6/2;C3−/− (n=14). Error bars represent SEM. ** = p<0.01 or ****=p<0.0001 for main effect of the R6/2 transgene by two-way ANOVA.
A) C3 protein levels in brains from 12 week old mice. As expected, we were unable to detect C3 protein in brain from C3−/− mice.
B) Rotarod. R6/2 mice fall off an accelerating rotarod sooner than WT littermates (significant main effect of the R6/2 transgene at 4 weeks, F(1,43)=7.581, p=0.0086, 8 weeks F(1,42)=38.91, p<0.0001 and 12 weeks F(1,38)=87.62, p<0.0001 of age), but this is not affected by C3 genotype (no significant effect of C3 genotype at any age) and there is no significant interaction between the two factors.
C) Balance Beam. R6/2 mice are slower to cross an elevated balance beam than WT littermates at 8 and 12 weeks of age (significant main effect of the R6/2 transgene at 8 weeks F(1,43)=68.36, p<0.0001 and 12 weeks F(1,40)=47.41, p<0.0001 of age), but this is not affected by C3 genotype (no significant main effect of C3 genotype at any age) and there is no significant interaction between the two factors.
D) Open field. R6/2 mice are less active in an open field than WT littermates (significant main effect of the R6/2 transgene at 4 weeks F(1,43)=38.47, p<0.0001, 8 weeks F(1,43)=36.83, p<0.0001 and 12 weeks F(1,40)=45.84, p<0.0001 of age), but this is not affected by C3 genotype (no significant effect of C3 genotype at any age) and there is no significant interaction between the two factors.
E) Weight. Genotype does not significantly impact weight at early time points, but R6/2 mice weigh significantly less than WT mice starting at 8.3 weeks of age (significant main effect of the R6/2 transgene F(1,43)=6.07, p=0.0178) and continuing through the course of the study. There is not a significant effect of C3 genotype at any age, and there is no significant interaction between the two factors.
F) Survival. Survival curves for R6/2;C3+/+ and R6/2;C3−/− were not significantly different (p=0.69).
C3 genotype does not alter behavioral deficits in R6/2 mice
We chose to evaluate behaviors at 4, 8, and 12 weeks of age. These ages correspond to early-, mid- and late-stage disease, respectively [34]. The balance beam and rotarod assays are tests of motor coordination, while the open field test affords insight into general activity levels and anxiety related behavior. In each assay, the R6/2 transgene had a dramatic and age-dependent effect, as expected (Fig. 1). However, R6/2;C3−/− mice were not significantly different from R6/2;C3+/+ mice in any of the behavioral outcomes measured at any time (Fig. 1). In addition to these behavioral readouts, we saw no significant effects of C3 genotype on weight loss or survival in these mice (Fig. 1).
C3 genotype does not alter expression of genes associated with disease progression in R6/2 mice
We used qPCR to measure mRNA levels of genes associated with disease progression in the striatum of 12 week old mice. Transcriptional dysregulation is a widely recognized feature of HD and mouse models of HD. It is not clear whether or not these gene expression changes are causative to pathogenesis, but they are correlated with disease progression and therefore offer another surrogate marker of disease progression. We chose to evaluate two genes whose expression is increased and two genes whose expression is decreased by the R6/2 transgene.
Expression levels of D1 dopamine receptor (D1 DA R) and preproenkephalin (Penk) decline sharply over the course of disease in R6/2 mice and in human HD patients [35,36]. Both of these genes are expressed by medium spiny neurons that are lost in disease, and each gene could contribute to dysfunctional signaling in those neurons.
Heat shock protein 90 (Hsp90) is a member of the heat shock family of proteins and its activity may play a role in disease progression in R6/2 mice [37]. Microarray data suggests that it is upregulated in brains of R6/2 mice [22]. Similarly, caspase 3 expression is upregulated in brains of R6/2 mice and may be important for disease progression [38].
Brains from 12 week old R6/2 mice (late-stage disease) should have lower levels of D1 DA R and Penk but higher levels of Hsp90 and caspase 3 than brains from 12 week old WT mice. Our studies reproduced this expected effect of the R6/2 transgene, but also showed that, in R6/2 mice, C3 genotype did not modulate expression of these genes (Fig. 2). Unexpectedly, C3 genotype decreased expression of D1 DA R and Penk in WT mice.
Fig. 2. C3 genotype does not alter expression of genes associated with disease progression in R6/2 mice.
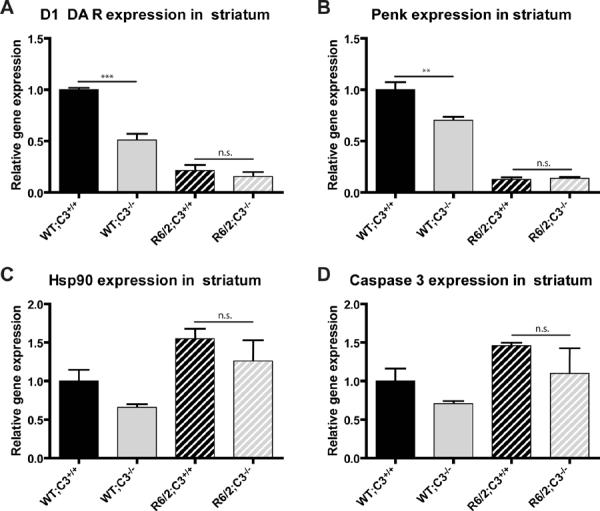
Littermates from each of the four genotypes tested (n=3/group) were sacrificed at 12 weeks of age (late stage of disease), and RNA was isolated from striatum. Expression of genes relevant to disease progression was measured by qPCR. Two-way ANOVA followed by Bonferroni post hoc tests shows that expression of these genes is not significantly different between R6/2;C3+/+ and R6/2;C3−/− mice at this age. Error bars represent SEM. **=p<0.01 or ***=p<0.001 by Bonferroni post hoc test. n.s. = not significant.
A and B) Expression levels of D1 dopamine receptor (D1 DAR) and preproenkephalin (Penk) mRNA were lower in R6/2 mice than in WT mice, as expected (significant main effect of the R6/2 transgene, D1 DAR F(1,8)=151.0, p<0.0001, Penk F(1,8)=278.9, p<0.0001). Unexpectedly, C3 deficiency also significantly decreased gene expression (significant main effect of C3 genotype, D1 DAR F(1,8)=34.6, p=0.0004, Penk F(1,8)=11.12, p=0.01) and there was a significant interaction between the R6/2 transgene and C3 genotype (D1 DAR F(1,8)=21.3, p=0.0017, Penk F(1,8)=12.83, p=0.0072). Using Bonferroni post hoc tests, we found that the effect of C3 genotype was confined to WT mice, as WT;C3−/− mice have lower levels of gene expression than WT;C3+/+ mice (D1 DA R p<0.001, Penk p<0.01) while R6/2;C3−/− mice are not significantly different from R6/2;C3+/+ mice (D1 DA R and Penk p>0.05).
C and D) Expression levels of Heat shock protein 90 (Hsp90) and Caspase 3 were higher in R6/2 mice than in WT mice (significant main effect of the R6/2 transgene Hsp90 F(1,8)=11.89, p=0.009, Caspase 3 F(1,8)=5.37, p=0.049) but there was no statistically significant effect of C3 genotype and no significant interaction between the R6/2 transgene and C3 genotype.
In addition to genes associated with HD, we attempted to measure prothrombin gene expression because previous studies have shown that thrombin can partially compensate for loss of C3 in C3−/− mice [12]. However, when using primers that reliably detected prothrombin in positive control tissue (WT liver) the brain levels of this gene were below the limit of reliable detection in mice of all four genotypes (data not shown).
Complement C3 mRNA and protein levels are not altered in R6/2 mice
We used qPCR to quantify C3 mRNA expression in striatum from 4, 8, and 12 week old R6/2 and WT mice. Low levels of C3 mRNA were found at each age, but the presence of the R6/2 transgene did not significantly alter C3 expression at any age (Fig. 3; data from 4 weeks not shown).
Fig. 3. Complement C3 mRNA and protein levels are not altered in R6/2 mice.

R6/2 and WT littermates were sacrificed at 8 and 12 weeks of age, and mRNA and protein levels were measured. Protein levels were determined in whole brain and in plasma, while mRNA levels were measured in striatum. No significant differences were detected. Data were tested for statistically significant differences using t tests. Error bars represent SEM.
A and B) R6/2 and WT mice have similar levels of C3 mRNA expression in striatum at 8 weeks (n=4/group) and 12 weeks (n=6/group) of age.
C and D) R6/2 and WT mice have similar levels of C3 protein in brain at 8 weeks (n=5/group) and 12 weeks (n=8/group) of age.
E and F) R6/2 and WT mice have similar levels of C3 protein in plasma at 8 weeks (n=7–9/group) and 12 weeks (n=6/group) of age.
We used a C3 ELISA to quantify C3 protein levels in brains and plasma from 8 and 12 week old R6/2 and WT mice. In both 8 and 12 week old mice, R6/2 genotype did not significantly alter C3 protein levels (Fig. 3).
Expression of many complement related genes is not altered in the brains of R6/2 mice
To gain a broader understanding of the status of the complement cascade, we used qPCR to quantify mRNA levels of complement genes in forebrains from 12 week old R6/2 and WT mice. We chose the genes to analyze from several types of genes: complement components that initiate and/or propagate activation of the complement cascade [complement components 1q, 3, 4 (C1q, C3, C4) and Factor B], downstream complement components that form the membrane attack complex [complement components 5, 6 and 9 (C5, C6, C9)], soluble and membrane-bound inhibitors of complement activation [C1 Inhibitor (C1 Inh), complement receptor-related protein y (Crry), decay accelerating factor (DAF) and Protectin], and receptors thought to be involved in complement signaling [C1q receptor 1 (C1qR aka CD93), C5aR, calreticulin, and low density lipoprotein receptor-related protein 1 (CD91)]. We observed few significant differences in gene expression (Table 2). Of note, C4, a complement gene that is highly upregulated in human HD brain samples [9,10,39], was not significantly upregulated in R6/2 mice. Generally, the large-scale upregulation of complement-related genes that is observed in human HD brains is not paralleled in late-stage R6/2 brains.
Table 2.
Expression of complement related genes in brain from 12 week old R6/2 and WT mice.
| Gene | Function in complement cascade | Fold change in R6/2 vs. WT | p value |
|---|---|---|---|
| C1q | initiation/propagation | 0.83 | 0.105 |
| C3 | initiation/propagation | 0.94 | 0.861 |
| C4 | propagation of activation | 1.24 | 0.282 |
| Factor B | propagation of activation | 0.87 | 0.460 |
| C5 | signaling, part of MAC | not detectable | |
| C6 | part of MAC | not detectable | |
| C9 | part of MAC | not detectable | |
| C1 Inh | soluble inhibitor | 1.24 | 0.097 |
| Crry | membrane bound inhibitor | 0.77 | 0.146 |
| DAF | membrane bound inhibitor | 0.85 | 0.031* |
| Protectin | membrane bound inhibitor | 0.74 | 0.083 |
| C1qR | phagocytosis, receptor for C1q, MBL | 0.92 | 0.329 |
| C5aR | inflammatory signaling, receptor for C5a | 0.72 | 0.005** |
| Calreticulin | phagocytosis, receptor for C1q, MBL | 1.23 | 0.221 |
| CD91 | phagocytosis, receptor for C1q, MBL | 1.05 | 0.814 |
Comparison of gene expression in late-stage R6/2 (n=5) and age-matched WT (n=4) forebrain. Listed p values are from t tests (*=p<0.05, **=p<0.01). For genes described as “not detectable” in these brain samples, primers were verified by their ability to detect gene expression in positive control tissue (liver) [47]. Abbreviations: MAC (Membrane Attack Complex), MBL (Mannose Binding Lectin).
Discussion
The complement cascade is a powerful effector arm of the immune system and exerts a profound influence on disease progression in several mouse models of neurodegenerative disease. Elements of the complement cascade are upregulated in the brains of human HD patients at early and late stages of disease progression. However, our behavioral and gene expression experiments conclusively show that genetic deletion of C3 does not alter these aspects of disease progression in the R6/2 mouse model of HD.
Our data stand in contrast to work showing that C3−/− mice modulate pathogenesis in other models of neurodegeneration. In one example, genetic deletion of C3 exacerbates neurodegeneration in a mouse model of AD [20]. This illustrates that ubiquitous, lifelong C3 deficiency can strongly influence brain health in a setting of chronic neurodegeneration. We hypothesized that this would also be true for chronic neurodegeneration induced by expression of a fragment of the mutant htt gene that causes HD. Further, we hypothesized that C3 deficiency would likely be protective, rather than detrimental, in this model of HD because blocking complement signaling downstream of C3 using a C5aR antagonist reduces lesion size in rats injected with 3-nitroproprionic acid (3-NP), a mitochondrial toxin used to model HD [40]. Interestingly, one of the few significant complement related changes we observed in R6/2 mice is a downregulation of the C5aR. Since antagonizing this receptor is protective in the 3-NP model of HD, perhaps this decrease in C5aR expression is a compensatory response. If so, administration of a C5aR antagonist may enhance this effect and offer protection in R6/2 mice as well.
It is possible that C3 deficiency alters disease progression in other studies but not in ours because C3 deficiency has multiple positive and negative effects that offset each other in R6/2 mice. We hypothesized that C3 could exacerbate pathogenesis by contributing to synapse loss or detrimental pro-inflammatory signaling, but C3 in the brain may also have protective effects. For example, C3−/− mice have reduced neurogenesis [41], while increased neurogenesis seems to be beneficial in mouse models of HD [42]. Also, C3−/− mice are unable to produce C3a and several studies have shown that C3a signaling can protect cells from inflammatory injury [43–45]. Alternatively, any effects of C3 deficiency, positive or negative, may be masked in R6/2 mice because immune cells, including microglia, are abnormal in HD [4]. The consequences of atypical immune cells in HD have not yet been fully explored, but they could include altered responses to complement. If such changes are present in HD but not in other neurodegenerative conditions, then this might help explain why C3 deficiency modulates pathogenesis in other degenerative conditions but not in the R6/2 model of HD.
The lack of effect of C3 deficiency in our study implies that there are important differences between our study and related studies where manipulations of complement do impact neurodegeneration. One feature that is seen in many such studies but absent in our experiments is significant complement upregulation. In AD and mouse models of AD, many complement components in the brain will bind to aggregated extracellular amyloid beta and indeed studies suggest that altering complement activity in AD brain alters removal of these plaques [46]. Though mutant huntingtin does aggregate, the intracellular location of these aggregates decreases their availability for binding by complement and thus removes one stimulus that could trigger complement activation. However, there is complement upregulation both in human HD and in the 3NP model of HD despite the presumed absence of extracellular protein aggregates. This complement cascade upregulation may be in response to unknown cues, perhaps similar to those that activate complement synthesis during development. A second possibility is that complement in HD brains and in the 3NP model of HD is upregulated in response to neuronal death. The complement cascade is involved in removing dead or damaged cells throughout the body, and may play that role in the brain as well. There is significant neuronal death in human HD brains and in brains of rats injected with 3-NP, but there is little neuronal death in the R6/2 mouse model of HD. A third possible explanation for the lack of complement upregulation in our study is that we housed our mice in sterile conditions. Immune cells from HD patients and models of HD respond abnormally to immune stimulation [4] and this initial abnormal response may be necessary for other immune system changes. If so, complement upregulation in HD patients may be the result of an interplay between the disease causing mutation and normal bouts of immune system activation that are presumably lacking in mice housed in pathogen-free colonies.
Regardless of the mechanisms governing complement expression, the discrepancy between large scale complement upregulation in HD patients and few alterations in the complement system in R6/2 mice highlights the need to use multiple animal models of disease in order to explore disease mechanisms. Specifically, any future studies of the complement system in HD should consider models other than the R6/2 mouse model in the hope that other models will more accurately reflect the status of the complement system in human HD patients. Our qPCR data, along with microarray data from other studies [22], illustrate that the complement upregulation seen in HD is not a phenomenon that is mirrored in the R6/2 mouse model of HD. Here, we have also shown that completely removing C3 does not alter disease associated behavioral and gene expression changes in R6/2 mice.
Acknowledgements
This work was supported by NS057715 and the Taube-Koret Center for Huntington's Disease Research. We are grateful to Dr. S. Pabinger (Innsbruck Medical University) for valuable assistance with the online analysis program QPCR and to G. Howard for editorial review. Behavioral data were obtained with the help of the Gladstone Institutes' Behavioral Core (supported by NIH grant P30NS065780).
References
- [1].Gil JM, Rego AC. Mechanisms of neurodegeneration in Huntington's disease. Eur J Neurosci. 2008;27(11):2803–20. doi: 10.1111/j.1460-9568.2008.06310.x. [DOI] [PubMed] [Google Scholar]
- [2].Leblhuber F, Walli J, Jellinger K, Tilz GP, Widner B, Laccone F, Fuchs D. Activated immune system in patients with Huntington's disease. Clin Chem Lab Med. 1998;36(10):747–50. doi: 10.1515/CCLM.1998.132. [DOI] [PubMed] [Google Scholar]
- [3].Dalrymple A, Wild EJ, Joubert R, Sathasivam K, Björkqvist M, Petersén A, Jackson GS, Isaacs JD, Kristiansen M, Bates GP, Leavitt BR, Keir G, Ward M, Tabrizi SJ. Proteomic profiling of plasma in Huntington's disease reveals neuroinflammatory activation and biomarker candidates. J Proteome Res. 2007;6(7):2833–40. doi: 10.1021/pr0700753. [DOI] [PubMed] [Google Scholar]
- [4].Björkqvist M, Wild EJ, Thiele J, Silvestroni A, Andre R, Lahiri N, Raibon E, Lee RV, Benn CL, Soulet D, Magnusson A, Woodman B, Landles C, Pouladi MA, Hayden MR, Khalili-Shirazi A, Lowdell MW, Brundin P, Bates GP, Leavitt BR, Möller T, Tabrizi SJ. A novel pathogenic pathway of immune activation detectable before clinical onset in Huntington's disease. J. Exp. Med. 2008;205(8):1869–77. doi: 10.1084/jem.20080178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sapp E, Kegel KB, Aronin N, Hashikawa T, Uchiyama Y, Tohyama K, Bhide PG, Vonsattel JP, DiFiglia M. Early and progressive accumulation of reactive microglia in the Huntington disease brain. 2001:161–72. doi: 10.1093/jnen/60.2.161. [DOI] [PubMed] [Google Scholar]
- [6].Tai YF, Pavese N, Gerhard A, Tabrizi SJ, Barker RA, Brooks DJ, Piccini P. Microglial activation in presymptomatic Huntington's disease gene carriers. Brain. 2007;130(Pt 7):1759–66. doi: 10.1093/brain/awm044. [DOI] [PubMed] [Google Scholar]
- [7].Politis M, Pavese N, Tai YF, Kiferle L, Mason SL, Brooks DJ, Tabrizi SJ, Barker RA, Piccini P. Microglial activation in regions related to cognitive function predicts disease onset in Huntington's disease: a multimodal imaging study. Hum Brain Mapp. 2011;32(2):258–70. doi: 10.1002/hbm.21008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Vonsattel JP, Myers RH, Stevens TJ, Ferrante RJ, Bird ED, Richardson EP., Jr Neuropathological classification of Huntington's disease. J. Neuropathol. Exp. Neurol. 1985;44(6):559–77. doi: 10.1097/00005072-198511000-00003. [DOI] [PubMed] [Google Scholar]
- [9].Singhrao SK, Neal JW, Morgan BP, Gasque P. Increased complement biosynthesis by microglia and complement activation on neurons in Huntington's disease. Exp Neurol. 1999:362–76. doi: 10.1006/exnr.1999.7170. [DOI] [PubMed] [Google Scholar]
- [10].Hodges A, Strand AD, Aragaki AK, Kuhn A, Sengstag T, Hughes G, Elliston LA, Hartog C, Goldstein DR, Thu D, Hollingsworth ZR, Collin F, Synek B, Holmans PA, Young AB, Wexler NS, Delorenzi M, Kooperberg C, Augood SJ, Faull RLM, Olson JM, Jones L, Luthi-Carter R. Regional and cellular gene expression changes in human Huntington's disease brain. Hum Mol Genet. 2006;15(6):965–77. doi: 10.1093/hmg/ddl013. [DOI] [PubMed] [Google Scholar]
- [11].Wessels MR, Butko P, Ma M, Warren HB, Lage AL, Carroll MC. Studies of group B streptococcal infection in mice deficient in complement component C3 or C4 demonstrate an essential role for complement in both innate and acquired immunity. Proc Natl Acad Sci U S A. 1995;92(25):11490–4. doi: 10.1073/pnas.92.25.11490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM, Gebhard F, Younger JG, Drouin SM, Wetsel RA, Ward PA. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. 2006;12(6):682–7. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- [13].Stevens B, Allen NJ, Vazquez LE, Howell GR, Christopherson KS, Nouri N, Micheva KD, Mehalow AK, Huberman AD, Stafford B, Sher A, Litke AM, Lambris JD, Smith SJ, John SWM, Barres BA. The classical complement cascade mediates CNS synapse elimination. Cell. 2007;131(6):1164–78. doi: 10.1016/j.cell.2007.10.036. [DOI] [PubMed] [Google Scholar]
- [14].Francis K, van Beek J, Canova C, Neal JW, Gasque P. Innate immunity and brain inflammation: the key role of complement. Expert Rev Mol Med. 2003;5(15):1–19. doi: 10.1017/S1462399403006252. [DOI] [PubMed] [Google Scholar]
- [15].Linnartz B, Kopatz J, Tenner AJ, Neumann H. Sialic acid on the neuronal glycocalyx prevents complement C1 binding and complement receptor-3-mediated removal by microglia. J. Neurosci. 2012;32(3):946–52. doi: 10.1523/JNEUROSCI.3830-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21(3):383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Boos L, Campbell IL, Ames R, Wetsel RA, Barnum SR. Deletion of the complement anaphylatoxin C3a receptor attenuates, whereas ectopic expression of C3a in the brain exacerbates, experimental autoimmune encephalomyelitis. J. Immunol. 2004;173(7):4708–14. doi: 10.4049/jimmunol.173.7.4708. [DOI] [PubMed] [Google Scholar]
- [18].Aldskogius H, Liu L, Svensson M. Glial responses to synaptic damage and plasticity. J Neurosci Res. 1999;58(1):33–41. [PubMed] [Google Scholar]
- [19].Bruder C, Hagleitner M, Darlington G, Mohsenipour I, Würzner R, Höllmüller I, Stoiber H, Lass-Flörl C, Dierich MP, Speth C. HIV-1 induces complement factor C3 synthesis in astrocytes and neurons by modulation of promoter activity. Mol Immunol. 2004;40(13):949–61. doi: 10.1016/j.molimm.2003.10.016. [DOI] [PubMed] [Google Scholar]
- [20].Maier M, Peng Y, Jiang L, Seabrook TJ, Carroll MC, Lemere CA. Complement C3 Deficiency Leads to Accelerated Amyloid {beta} Plaque Deposition and Neurodegeneration and Modulation of the Microglia/Macrophage Phenotype in Amyloid Precursor Protein Transgenic Mice. J. Neurosci. 2008;28(25):6333–41. doi: 10.1523/JNEUROSCI.0829-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Kuehn MH, Kim CY, Jiang B, Dumitrescu AV, Kwon YH. Disruption of the complement cascade delays retinal ganglion cell death following retinal ischemia-reperfusion. Exp Eye Res. 2008;87(2):89–95. doi: 10.1016/j.exer.2008.04.012. [DOI] [PubMed] [Google Scholar]
- [22].Kuhn A, Goldstein DR, Hodges A, Strand AD, Sengstag T, Kooperberg C, Becanovic K, Pouladi MA, Sathasivam K, Cha J-HJ, Hannan AJ, Hayden MR, Leavitt BR, Dunnett SB, Ferrante RJ, Albin R, Shelbourne P, Delorenzi M, Augood SJ, Faull RLM, Olson JM, Bates GP, Jones L, Luthi-Carter R. Mutant huntingtin's effects on striatal gene expression in mice recapitulate changes observed in human Huntington's disease brain and do not differ with mutant huntingtin length or wild-type huntingtin dosage. Hum Mol Genet. 2007;16(15):1845–61. doi: 10.1093/hmg/ddm133. [DOI] [PubMed] [Google Scholar]
- [23].Soulet D, Cicchetti F. The role of immunity in Huntington's disease. Mol. Psychiatry. 2011;16(9):889–902. doi: 10.1038/mp.2011.28. [DOI] [PubMed] [Google Scholar]
- [24].Ricklin D, Lambris JD. Complement-targeted therapeutics. Nat Biotechnol. 2007;25(11):1265–75. doi: 10.1038/nbt1342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Qu H, Ricklin D, Lambris JD. Recent developments in low molecular weight complement inhibitors. Mol. Immunol. 2009;47(2–3):185–95. doi: 10.1016/j.molimm.2009.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Klapstein GJ, Fisher RS, Zanjani H, Cepeda C, Jokel ES, Chesselet MF, Levine MS. Electrophysiological and morphological changes in striatal spiny neurons in R6/2 Huntington's disease transgenic mice. J Neurophysiol. 2001;86(6):2667–77. doi: 10.1152/jn.2001.86.6.2667. [DOI] [PubMed] [Google Scholar]
- [27].Cepeda C, Hurst RS, Calvert CR, Hernández-Echeagaray E, Nguyen OK, Jocoy E, Christian LJ, Ariano MA, Levine MS. Transient and progressive electrophysiological alterations in the corticostriatal pathway in a mouse model of Huntington's disease. J Neurosci. 2003;23(3):961–9. doi: 10.1523/JNEUROSCI.23-03-00961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Chiu K, Lau WM, Lau HT, So K-F, Chang RC-C. Micro-dissection of rat brain for RNA or protein extraction from specific brain region. J Vis Exp. 2007;(7):269. doi: 10.3791/269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Benn CL, Fox H, Bates GP. Optimisation of region-specific reference gene selection and relative gene expression analysis methods for pre-clinical trials of Huntington's disease. Mol Neurodegener. 2008;3:17. doi: 10.1186/1750-1326-3-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Pabinger S, Thallinger GG, Snajder R, Eichhorn H, Rader R, Trajanoski Z. QPCR: Application for real-time PCR data management and analysis. BMC Bioinformatics. 2009;10:268–268. doi: 10.1186/1471-2105-10-268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Bookout AL, Cummins CL, Mangelsdorf DJ, Pesola JM, Kramer MF. High-Throughput Real-Time Quantitative Reverse Transcription PCR. Current Protocols in Molecular Biology. 2006:15.8.1–15.8.28. doi: 10.1002/0471142727.mb1508s73. [DOI] [PubMed] [Google Scholar]
- [32].Manthey HD, Thomas AC, Shiels IA, Zernecke A, Woodruff TM, Rolfe B, Taylor SM. Complement C5a inhibition reduces atherosclerosis in ApoE−/− mice. FASEB J. 2011;25(7):2447–55. doi: 10.1096/fj.10-174284. [DOI] [PubMed] [Google Scholar]
- [33].Bolger MS, Ross DS, Jiang H, Frank MM, Ghio AJ, Schwartz DA, Wright JR. Complement levels and activity in the normal and LPS-injured lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2007;292(3):L748–59. doi: 10.1152/ajplung.00127.2006. [DOI] [PubMed] [Google Scholar]
- [34].Menalled L, El-Khodor BF, Patry M, Suárez-Fariñas M, Orenstein SJ, Zahasky B, Leahy C, Wheeler V, Yang XW, MacDonald M, Morton AJ, Bates G, Leeds J, Park L, Howland D, Signer E, Tobin A, Brunner D. Systematic behavioral evaluation of Huntington's disease transgenic and knock-in mouse models. Neurobiol. Dis. 2009;35(3):319–36. doi: 10.1016/j.nbd.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Cha JH, Kosinski CM, Kerner JA, Alsdorf SA, Mangiarini L, Davies SW, Penney JB, Bates GP, Young AB. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human huntington disease gene. Proc. Natl. Acad. Sci. U.S.A. 1998;95(11):6480–5. doi: 10.1073/pnas.95.11.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Benn CL, Butler R, Mariner L, Nixon J, Moffitt H, Mielcarek M, Woodman B, Bates GP. Genetic knock-down of HDAC7 does not ameliorate disease pathogenesis in the R6/2 mouse model of Huntington's disease. PLoS ONE. 2009;4(6):e5747. doi: 10.1371/journal.pone.0005747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Labbadia J, Cunliffe H, Weiss A, Katsyuba E, Sathasivam K, Seredenina T, Woodman B, Moussaoui S, Frentzel S, Luthi-Carter R, Paganetti P, Bates GP. Altered chromatin architecture underlies progressive impairment of the heat shock response in mouse models of Huntington disease. J. Clin. Invest. 2011;121(8):3306–19. doi: 10.1172/JCI57413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Chen M, Ona VO, Li M, Ferrante RJ, Fink KB, Zhu S, Bian J, Guo L, Farrell LA, Hersch SM, Hobbs W, Vonsattel JP, Cha JH, Friedlander RM. Minocycline inhibits caspase-1 and caspase-3 expression and delays mortality in a transgenic mouse model of Huntington disease. Nat. Med. 2000;6(7):797–801. doi: 10.1038/77528. [DOI] [PubMed] [Google Scholar]
- [39].Silvestroni A, Faull RLM, Strand AD, Möller T. Distinct neuroinflammatory profile in post mortem human Huntington's disease. Neuroreport. 2009;20(12):1098–103. doi: 10.1097/WNR.0b013e32832e34ee. [DOI] [PubMed] [Google Scholar]
- [40].Woodruff TM, Crane JW, Proctor LM, Buller KM, Shek AB, de Vos K, Pollitt S, Williams HM, Shiels IA, Monk PN, Taylor SM. Therapeutic activity of C5a receptor antagonists in a rat model of neurodegeneration. FASEB J. 2006;20(9):1407–17. doi: 10.1096/fj.05-5814com. [DOI] [PubMed] [Google Scholar]
- [41].Rahpeymai Y, Hietala MA, Wilhelmsson U, Fotheringham A, Davies I, Nilsson A-K, Zwirner J, Wetsel RA, Gerard C, Pekny M, Pekna M. Complement: a novel factor in basal and ischemia-induced neurogenesis. EMBO J. 2006;25(6):1364–74. doi: 10.1038/sj.emboj.7601004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Winner B, Kohl Z, Gage FH. Neurodegenerative disease and adult neurogenesis. Eur. J. Neurosci. 2011;33(6):1139–51. doi: 10.1111/j.1460-9568.2011.07613.x. [DOI] [PubMed] [Google Scholar]
- [43].Boos L, Szalai AJ, Barnum SR. C3a expressed in the central nervous system protects against LPS-induced shock. Neurosci. Lett. 2005;387(2):68–71. doi: 10.1016/j.neulet.2005.07.015. [DOI] [PubMed] [Google Scholar]
- [44].Kildsgaard J, Hollmann TJ, Matthews KW, Bian K, Murad F, Wetsel RA. Cutting edge: targeted disruption of the C3a receptor gene demonstrates a novel protective anti-inflammatory role for C3a in endotoxin-shock. J. Immunol. 2000;165(10):5406–9. doi: 10.4049/jimmunol.165.10.5406. [DOI] [PubMed] [Google Scholar]
- [45].Wenderfer SE, Wang H, Ke B, Wetsel RA, Braun MC. C3a receptor deficiency accelerates the onset of renal injury in the MRL/lpr mouse. Mol. Immunol. 2009;46(7):1397–404. doi: 10.1016/j.molimm.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Wyss-Coray T, Yan F, Lin AH-T, Lambris JD, Alexander JJ, Quigg RJ, Masliah E. Prominent neurodegeneration and increased plaque formation in complement-inhibited Alzheimer's mice. Proc Natl Acad Sci U S A. 2002;99(16):10837–42. doi: 10.1073/pnas.162350199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Qin X, Gao B. The complement system in liver diseases. Cell. Mol. Immunol. 2006;3(5):333–40. [PubMed] [Google Scholar]