

Abstract
During the past five years many patients suffering from congenital myasthenic syndromes (CMS) have been identified worldwide and novel causative genes and mutations have been discovered. The disease genes now include those encoding each subunit of the acetylcholine receptor (AChR), the ColQ part of acetylcholinesterase (AChE), choline acetyltransferase, Nav 1.4, MuSK, and Dok-7. Moreover, emerging genotype-phenotype correlations are providing clues for targeted mutation analysis. This review focuses on the recent observations in selected CMS.
Keywords: congenital myasthenic syndromes, acetylcholinesterase, choline acetyltransferase, acetylcholine receptor, Dok-7
Introduction
The congenital myasthenic syndromes (CMS) have now been traced to an array of molecular targets at the neuromuscular junction encoded by no fewer than 10 disease genes. The disease genes were identified by the candidate gene approach, using clues derived from clinical, electrophysiologic, cytochemical, and ultrastructural features. For example, electrophysiologic studies in patients suffering from sudden episodes of apnea pointed to a defect in acetylcholine (ACh) resynthesis and CHAT as the candidate gene1; refractoriness to anticholinesterase medications and partial or complete absence of acetylcholinesterase (AChE) from the endplates (EPs) has pointed to one of the two genes (COLQ and ACHET) encoding AChE, though mutations were observed only in COLQ. After a series of patients carrying mutations in a disease gene have been identified, the emerging genotype-phenotype correlations provided clues for targeted mutation analysis in other patients. Mutations in EP-specific proteins also prompted expression studies that proved pathogenicity, highlighted important functional domains of the abnormal proteins, and pointed to rational therapy.
Table 1 shows a classification of the 276 CMS kinships investigated at the Mayo Clinic to date. The table indicates that the postsynaptic CMS are far more frequent than the presynaptic or synaptic-space-associated CMS, and that most postsynaptic CMS stem from defects in the acetylcholine receptor (AChR). This review focuses on selected CMS investigated in the Mayo patient cohort since 2002 when the last International Conference on Myasthenia Gravis was held.
TABLE 1. Site-of-defect classification of the CMSa.
| Index cases |
|
|---|---|
| Presynaptic defects (7%) | |
| Choline acetyltransferase deficiencyb | 15 |
| Paucity of synaptic vesicles and reduced quantal release | 1 |
| Lambert–Eaton syndrome like | 2 |
| Other presynaptic defects | 2 |
| Synaptic basal lamina-associated defects (13%) | |
| Endplate AChE deficiencyb | 37 |
| Postsynaptic defects (80%) | |
| Kinetic abnormality of AChR with/without AChR deficiencyb (slow- and fast-channel syndromes) |
52 |
| AChR deficiency with/without minor kinetic abnormalityb |
107 |
| Rapsyn deficiencyb | 40 |
| Dok-7 myastheniab | 18 |
| Na-channel myastheniab | 1 |
| Plectin deficiency | 1 |
| Total (100%) | 276 |
Classification based on cohort of CMS patients investigated at the Mayo Clinic between 1988 and 2007. (One hundred seventeen of the 276 patients also underwent intercostal muscle biopsies)
Gene defects identified.
Endplate AChE Deficiency Due to Mutations in COLQ
Since 2002, we identified 10 additional patients with EP AChE deficiency caused by 10 mutations in COLQ, of which seven are novel. The main function of ColQ is to anchor the enzyme in the synaptic basal lamina. In Figure 1A the left image is a schematic diagram of ColQ; the right image shows the asymmetric enzyme composed of three ColQ strands, each of which can bind four catalytic subunits (AChET) to produce A4, A8, and A12 species of the asymmetric enzyme, as well as the positions of the ColQ mutations identified in our patients. Figure 1B shows the consequences of the mutations in different ColQ domains revealed by density gradient analysis of extracts of COS cells transfected with mutant ColQ cDNAs along with wild-type AChET cDNA. The proline-rich attachment domain (PRAD) of ColQ binds the catalytic subunits; mutations in this domain prevent assembly of a functional asymmetric enzyme. Nonsense mutations in the collagen domain result in formation of a truncated single-stranded ColQ associated with a single catalytic subunit tetramer which is insertion incompetent. Interestingly, seven previously recognized mutations in the globular C-terminal region comprising two nonsense mutations (R315X and Q371X) and five missense mutations (S312G, D342E, R410Q, R410P, and C444Y) did not prevent the assembly of the asymmetric enzyme (see Fig. 1B, rightmost panels). This suggested that some C-terminal mutations act by hindering insertion of ColQ into the synaptic basal lamina. We tested this notion in collaboration with Richard Rotundo by transplanting genetically engineered C-terminal mutants of ColQ together with wild-type AChET on frog EPs. Attachment of the human enzyme to the frog EP was monitored with a mouse monoclonal antibody that recognizes human but not frog AChE. Five of the seven C-terminal mutants failed to insert into the frog EP, but two mutants (S312G and C444Y) did. S312G was on the same allele as R410Q and proved to be a benign mutation. The insertion competence of the C444Y mutant remains unexplained.2 In parallel studies, we also examined the effects of mutating residues in the heparin sulfate proteoglycan binding domain (HSPD) of ColQ, which had previously been implicated in anchoring ColQ at the EP.3 These mutants were also insertion incompetent, indicating that both the C-terminal and HSPD domains of ColQ are required for anchoring the enzyme in the synaptic basal lamina.
FIGURE 1.

(A) Schematic diagram of a ColQ strand and of asymmetric AChE (A12) indicating mutations identified in the Mayo patient cohort. Asterisks indicate seven recently identified mutations. HSPBD, heparin sulfate proteoglycan binding domain. (B) Consequences of mutations in different ColQ domains revealed by density gradient analysis. Leftmost panel: wild-type gradient profile with A12, A8, and A4 peaks corresponding to triple helical ColQ binding 3, 2, and 1 AChET homotetramer, and G peaks corresponding to monomers and dimers of free AChET. Second panel from left: PRAD mutations prevent assembly of the asymmetric enzyme; the gradient profile only shows G peaks. Second panel from right: Nonsense mutation in the ColQ collagen domain produces a single truncated ColQ strand that binds one AChET tetramer reflected by a mutant M peak in the gradient profile. Rightmost panel: Most C-terminal mutations do not prevent assembly of A12, and may or may not produce an M peak in the gradient profile. (In color in Annals online.) (Reproduced by permission from Ref. 26.)
Ephedrine Is Useful in the Treatment of EP AChE Deficiency
In collaboration with Bestue-Cardiel in Spain and Menachem Sadeh in Israel, we found that seven patients (two North American, one South American, three Spanish, and one Israeli) suffering from EP AChE deficiency due to mutations in different parts of ColQ responded favorably to therapy with ephedrine at doses of 150-200 mg per day.4,5 Patients 1–4 responded dramatically as judged by muscle testing, endurance, and electromyographic (EMG) criteria. Patients 5 and 6 were also improved in endurance but could not be reexamined. Patient 7 improved for several years; ephedrine was then discontinued and she gradually became much weaker. Several years later, ephedrine was restarted but she no longer responded to it. The findings point to a need for a prospective cooperative clinical trial to define the long-term safety and efficacy of ephedrine in EP AChE deficiency.
Kinetic Mutations in AChR
Novel Fast-channel Mutations
Fast-channel CMS are caused by recessive loss-of-function mutations usually accompanied by a low-expressor or null mutation in the second allele, so that the fast-channel mutation determines the phenotype. The name of the syndrome stems from abnormally rapid decay of the synaptic response caused by abnormally brief opening burst of the AChR channel. The underlying mechanisms include a decreased opening rate (β) or an increased closing rate (α) of the AChR channel, decreased agonist affinity, or a combination of these factors. A decrease in β or an increase in α decreases the gating efficiency (θ) which is determined by β/α.
The diagnosis of a fast-channel syndrome depends on in vitro microelectrode recordings because there are no clinical, EMG, or anatomic clues to the diagnosis. Accurate diagnosis, however, is important because this CMS responds well to combined therapy with 3,4-diaminopyridine, which increases the number of quanta released by nerve impulse, and pyridostigmine, which increases the number of receptors activated by each quantum, provided that the kinetic mutation does not significantly curtail AChR expression at the EP.
Fast-channel Mutation in the AChR α Subunit
An αV132L mutation was detected in the signature cys-loop of the AChR α subunit in a 4-year-old girl who could not speak, swallow, sit, or stand, and required respiratory support (Fig. 2A).6 EP fine structure and AChR expression were normal (Fig. 2B), but the amplitude of the synaptic response to ACh as well as the length of the channel opening events were decreased to 10% of normal. Single-channel kinetic analysis revealed that αV132L decreases affinity of the resting closed state of the receptor 30-fold and the gating efficiency ~two-fold (see right upper panel in Fig. 3). Mutation of the equivalent Val in the δ subunit has little effect on affinity but impairs gating efficiency ~four-fold. Mutation of the corresponding Val in the β and ε subunits is without effect, indicating that the effects are subunit specific. We attribute the deleterious consequences of αV132L to an allosteric effect transmitted via β-strand 7 that links the cys-loop to Trp149 at the center of the ligand binding site (Fig. 2C).
FIGURE 2.

(A) Patient at age 4. Note eyelid ptosis, facial diplegia, open mouth, gothic palate, and gastrostomy. (B) Ultrastructural localization of EP AChR with peroxidase-labeled α-bungarotoxin. The density and distribution of AChR on the junctional folds is normal. x9500. (C) Structural model of the AChR α subunit (Protein Data Bank code 2BG9) shown in magenta with the Cys-loop and contiguous β-strand 7 highlighted in green. Ball and stick representations indicate V132 within the Cys-loop, Cys128 and 142 that form the loop, and W149 at the center of the α subunit portion of the binding pocket. (In color in Annals online.) (Reproduced by permission from Ref. 6.)
FIGURE 3.
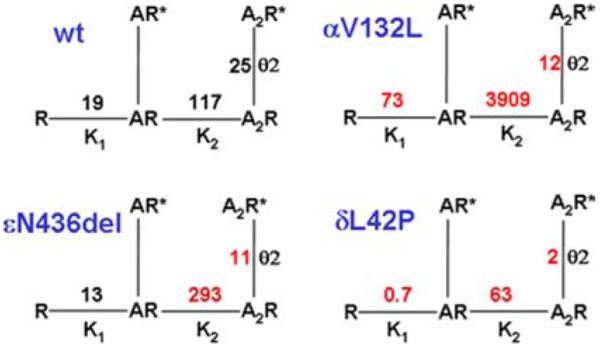
Schemes of activation of wild-type AChR and mutant AChRs expressed in HEK cells based on single-channel closed and open dwell times elicited over a range of ACh concentrations. Lines indicate reversible transitions between allosteric states. R and R* represent closed and open states of the receptor; A indicates agonist. K1 and K2 are equilibrium constants in μM for the first and second agonist binding steps; Kn is defined as agonist dissociation rate k-n/agonist association rate k+n. θ2 is the gating equilibrium constant of the diliganded receptor and a measure of its gating efficiency; θ1 is not shown. θn, channel opening rate/channel closing rate. Transition to the nonconducting blocked state occurring at high ACh concentration is not indicated. (In color in Annals online.)
Fast-channel Mutation in the AChR ε Subunit
A 25-year-old woman and an unrelated 12-year-old girl had severe myasthenic symptoms since birth. Each carried an εN346del fast-channel mutation on one ε allele, and a splice site or a frameshift mutation on the second ε allele.7 The deleted Asn residue is at the C terminus of the long cytoplasmic link between the third (M3) and fourth (M4) transmembrane domains of the ε subunit (Fig. 4). The deletion shortens the M3-M4 link and shifts a negatively charged Asp residue against M4. EP studies showed reduced expression of εN346del-AChR and compensatory accumulation of fetal γ-AChR. Single-channel kinetic analysis of εN346del-AChR expressed in HEK cells revealed that εN346del-AChR decreases the duration of channel opening bursts 2.7-fold compared to wild-type AChR; this can be attributed to a 2.3-fold decrease in gating efficiency and a 2.5-fold decrease in agonist affinity of the diliganded closed state (see left lower panel in Fig. 3). Mutagenesis studies established that the effects of εN346del are not due to juxtaposition of a negative charge against M4 but to shortening of the M3–M4 linker. Deletion of the C-terminal residue of the M3–M4 link in the β and δ subunits also results in fast-channel kinetics, but in the α subunit it dictates slow-channel kinetics. The overall findings indicate that the M3–M4 linkers contribute in an asymmetric manner to optimizing the activation of AChR through allosteric links to the channel and the agonist binding site.7
FIGURE 4.

(A) Scheme of the ε subunit showing positions of mutations. ε911delT and εIVS9-1G→C are null mutations in patients 1 and 2, respectively. Accordingly, εN436del determines each patient’s phenotype. (B) and (C), Family analysis in patients 1 and 2. (Reproduced by permission for Ref. 7.)
Fast-channel Mutation in the AChR δ Subunit
A δL42P mutation was identified in a 20-year-old woman with moderately severe to severe myasthenic symptoms since birth.8 The mutated Leu-42 is in a β1 strand at a ligand binding interface with an α subunit, and positioned in the transitional zone where β strands of the extracellular domain merge with the α-helices of the transmembrane domain (see Fig. 5). Expression studies in HEK cells revealed that δL42P decreases gating efficiency 12-fold, has a modest effect on agonist affinity, and shortens the channel burst length to 20% of normal (see right lower panel in Fig. 3). Replacing δ Leu-42 with a small flexible Gly, or charged Lys or Asp, or substitutions of Pro along the β1 strand around δ Leu-42 have similar kinetic consequences. Pro substitution of the corresponding Leu in the ε subunit also has similar effects, in the β subunit it has no kinetic effect, but in the α subunits it markedly prolongs the channel burst length and results in slow-channel kinetics.
FIGURE 5.
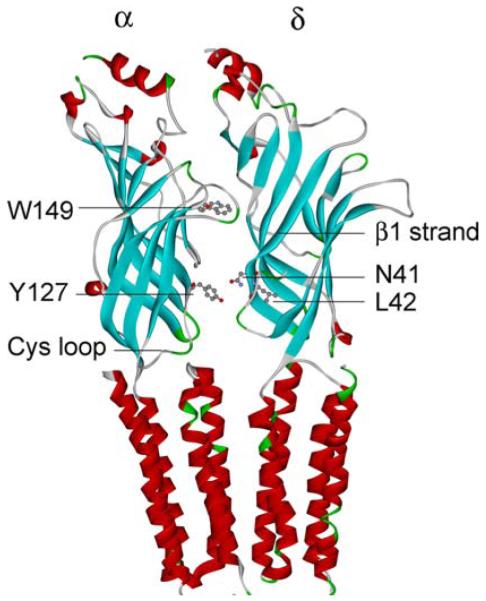
Structural model of the AChR α and δ subunits based on Protein Data Bank code 2BG9 and Ref. 27. The scheme indicates αW149 at the center of the ligand binding site, and αY127, δN41, and δL42 on β strands in the transition zone between the ligand-binding and transmembrane domains. (In color in Annals online.)
Because δ Asn-41 and ε Asn-39 were previously shown to be energetically coupled to α Tyr-127 in effecting rapid and efficient opening of the AChR channel,9 we postulated that δL42P and the corresponding substitution in the ε subunit, εL40P, could also hinder interaction with αY127. To examine how mutation of one residue in an AChR subunit affects the functional property of another residue in the same or another subunit, we performed mutant cycle experiments.10 Using −RTlnθ2 to compute the free energy of channel gating of each moiety of AChR, we generated a two-dimensional mutant cycle composed of gating free energies for wild-type, δL42P/εL40P, αY127T, and αY127T/δL42P/εL40P AChRs (see Fig. 6). These experiments revealed that δL42 and εL40 are energetically coupled to αY127 with a free energy of 3.9 kcal/mol. This value approaches the 5.8 kcal/mol coupling energy of δN41A and εN39A with αY127T revealed by previous mutant cycle experiments.9
FIGURE 6.

Mutant cycle experiments to examine interactions of δL42 and εL40 with αY127, and representative single-channel currents for the indicated AChRs elicited by 100 μM ACh. Changes in gating free energy are shown along each limb of the cycle. The overall coupling free energy (ΔΔGint) of δ Leu-42 and εLeu-40 with α Tyr-127 is expressed in kcal/mol. This value was computed from −RTln[(θwwθmm)/(θwmθmw)], where the θ (β/α) are gating equilibrium constants for diliganded wild-type (ww), single-mutant (wm, mw), or double-mutant (mm) AChRs. Horizontal line, 20 ms for wild-type and 100 ms for mutant AChRs; vertical line, 5 pA.
The overall findings indicate that (1) δL42 together with εL40 are functionally coupled to the proximal αY127 and thus essential for efficient gating and (2) the equivalent α L40 residues positioned at nonreceptor binding interfaces of the α-subunits also contribute to channel gating but in a novel manner.
Silencing a Slow-channel Mutation
The slow-channel syndromes stem from dominant gain-of-function mutations of AChR that increase agonist affinity, gating efficiency, or both. They can be effectively treated with quinidine11, 12 or fluoxetine,13 both long-lived open-channel blockers of AChR. Recently, advances in the use of small interfering RNAs (siRNAs)14 and short hairpin RNAs (shRNA)15-17 established the feasibility of silencing dominant mutations in model systems. However, there are a number of problems inherent in the use of these constructs even in model systems: first, each mutation requires a specific silencer; second, the mutant nucleotide as well as its neighboring nucleotides affect silencing efficiency; and third, the silencing construct must selectively inhibit the mutant but not the wild-type allele, but only about 20% of designed siRNAs or shRNAs silence the mutant allele selectively.18, 19 Fourth, an efficient method is needed to screen for suitable silencing constructs.
In vitro silencing of the αS226F slow-channel mutation has already been evaluated,17 but the mutant allele was suppressed to only 38% of the control level. We explored the feasibility of silencing another slow-channel allele at a high efficiency. The mutant allele harbored a Cys to Trp substitution at codon 418 in the M4 domain of the α subunit (αC418W). This mutation was observed in a 24-year-old man with eyelid ptosis, oculoparesis, selectively severe distal limb weakness, a decremental EMG response, and a repetitive compound muscle action potential. The mutation increases gating efficiency ~26-fold and prolongs the duration of the channel opening bursts ~seven-fold.20
Quantitative screening for suitable siRNAs was facilitated by the use of check-plasmids harboring siRNAs flanked by a reporter sequence.21 We placed two rather than one mismatch in our siRNA at varied positions, and found that one of the eight designed siRNAs, D6, suppressed transcription of the αC418W-allele to ~5% and of the wild-type allele to 84% of the control level (Fig. 7). The same siRNA had a comparable effect on expression of the mutant protein in HEK cells (Fig. 7B) and abolished the activity of nearly all high-probability channel openings typical of αC418W-receptor (Fig. 8). Thus, the selected siRNA was effective at the mRNA, protein, and functional level.20 Despite this theoretical advance, numerous obstacles need to be overcome before gene therapy with siRNA constructs can become feasible in clinical practice. These include design and large-scale production of highly efficient mutation-specific siRNAs or shRNAs and safe repeated delivery of the specific constructs to the patient at an affordable cost.
FIGURE 7.
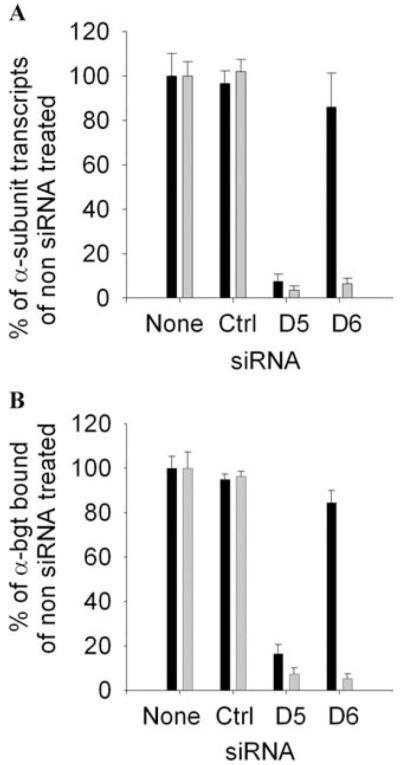
(A) Real-time PCR analysis of the effects of siRNA on the transcription of wild-type and mutant α subunits by HEK cells relative to transcription of the wild-type ε subunit by HEK cells. The siRNA D6 knocks down transcription of the mutant α subunit allele to 6% and of the wild-type allele to 86% observed in HEK cells not transfected with siRNA. Bars and lines indicate mean and SD of five experiments. Black bars, wild-type; gray bars, mutant. (B) Quantitation of the surface expressions of wild-type AChR and αC418W-AChR with [125I]α-bgt. HEK cells were cotransfected with indicated siRNA plus wild-type or mutant α subunit cDNA and complementary wild-type β, δ, and ε subunit cDNAs. The results are normalized for [125I]α-bgt binding by HEK cells transfected with AChR subunit cDNAs without siRNA. The Renilla luciferase signal was used as an internal control to normalize for transfection efficiency. siRNA D6 reduces surface expression of mutant AChR to 5% and of the wild-type AChR to 84% of the surface expression by HEK cells not exposed to siRNA. Bars represent mean and SD of three experiments. Black bars, wild-type; gray bars, mutant. (Reproduced by permission from Ref. 20.)
FIGURE 8.

siRNA D6 silences the functional activity of αC418W-AChR. Histograms show distribution of the open probability (Popen) of clusters of AChR channel openings in the presence of 10 μM ACh. Each cluster represents the activity of one channel. (A) Wild-type AChR. (B) αC418W-AChR. (C) Wild-type AChR coexpressed with αC418W-AChR. (D) Wild-type AChR coexpressed with αC418W-AChR in presence of siRNA D6. Note disappearance of nearly all high-probability channel openings typical of αC418W-AChR. (Reproduced by permission from Ref. 20.)
DOK-7 Myasthenia
In 2006, Okada and co-workers identified Dok-7 as a muscle-intrinsic activator of MuSK required for synaptogenesis.22 Dok-7 harbors N-terminal pleck-strin homology and phosphotyrosine-binding domains and is strongly expressed at the postsynaptic region of skeletal muscle and in heart. Subsequently, Beeson and co-workers detected recessive DOK7 mutations in CMS patients whose weakness was mostly in a limbgirdle distribution.23 The majority of patients were heterozygous or homozygous for a common 1124-1127dupTGCC mutation, but in two patients only a single mutation was detected, and only the common mutation was functionally characterized. In six patients eventually shown to carry DOK7 mutations by Beeson, Slater and co-workers described nonfatigable weakness largely confined to the proximal limb muscles. These patients had small EPs relative to muscle fiber size with few and simple junctional folds, which the authors considered a constitutive feature of the disease; AChR counts per EP were also reduced, but were deemed appropriate for small EPs.24 The amplitude of the miniature EP potentials (MEPPs) was decreased; this was largely attributed to the decreased input resistance of the simplified postsynaptic architecture. The quantal content (m) and the amplitude of the EP potential (EPP) were decreased but the amplitude of the miniature EP currents (MEPCs) was not.
After Okada’s discovery, we considered DOK7 a good candidate gene in patients who had small EPs, or a reduced number of AChRs per EP, but no mutations in AChR or rapsyn. Using these criteria, we first identified 1325 and then three additional patients who carry DOK7 mutations. Fourteen of these had had intercostal muscle biopsies with detailed in vitro electrophysiology and electron microscopy studies of the neuromuscular junction.25
All 16 patients had a significant EMG decrement, limb-girdle weakness, and short-term fatigability. In eight patients the disease presented in the neonatal period. Eyelid ptosis was present in 14 patients; it was frequently asymmetric but was severe only in one patient. Bulbar symptoms occurred in 11, oculoparesis in six, respiratory muscle involvement in 12 with evidence of ventilatory failure in six. Eleven patients experienced intermittent worsenings, and 12 had a progressive course with disability ranging from mild to severe at the time of the last evaluation.
Type-1 fiber preponderance was noted in 13 patients, type-2 fiber atrophy in eight, isolated necrotic or regenerating fibers in four, and pleomorphic oxidative-enzyme decreases or target formations in 12. All EPs consisted of one to multiple small synaptic contacts.
The mean MEPP, the MEPC amplitude, and the quantal content of the EPP were reduced by ~25%, but the distribution of patient and control values over-lapped. Electron microscopy of 613 EP regions of 409 EPs revealed disintegrating junctional folds in 33% (Fig. 9B), denuded postsynaptic regions with nearby nerve sprouts in 18%, and degenerating subsynaptic organelles in 10% (Fig. 9B). Importantly, however, some EPs in each patient had a normal postsynaptic architecture (Fig. 10), and the density and distribution of AChR on the crests of the preserved junctional folds was normal (Fig. 9A); therefore, a simplified postsynaptic architecture is not a constitutive feature of Dok-7 myasthenia, and the defects in Dok-7 do not cause a decreased concentration of AChR on the preserved junctional folds of the innervated human neuromuscular junction.
FIGURE 9.

Dok-7 myasthenia. (A) AChR localization at a structurally normal EP region with peroxidase-labeled α-bungarotoxin. The density and distribution of AChR on the junctional folds are normal. (B) A structurally abnormal EP region. Several junctional folds are replaced by globular debris (asterisk) causing widening of the synaptic space and simplification of the folds. These findings predict a decreased synaptic response to ACh due to loss of AChR from tips of the destroyed folds, loss of ACh by diffusion from the widened synaptic space, and decreased input resistance of the remaining simple folds. A large myeloid structure in the junctional sarcoplasm indicates concomitant destruction of junctional organelles. Bars, 1 μm.
FIGURE 10.

Dok-7 myasthenia. A normal EP region displays elongated, narrow, and complex junctional folds predicting a high input resistance. A number of EP regions with normal postsynaptic architecture were present in each patient. Bar, 1 μm.
Each patient harbored hetero-allelic pathogenic mutations: 11 of these were detected in genomic DNA and six in cDNA isolated from EP-enriched muscle specimens. Four mutations in genomic DNA were previously reported.23 No consistent phenotype—genotype correlations emerged.
We conclude that (1) the clinical features of Dok-7 myasthenia are highly variable, (2) the morphologicfeatures bespeak of ongoing destruction and remodeling of the EPs which likely contribute importantly to defective neuromuscular transmission, and (3) some mutations are complex and can only be identified and analyzed in cDNA or cloned cDNA.25
Footnotes
Conflicts of Interest
The authors declare no conflicts of interest.
References
- 1.Ohno K, Tsujino A, Brengman JM, et al. Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proc. Natl. Acad. Sci. USA. 2001;98:2017–2022. doi: 10.1073/pnas.98.4.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kimbell LM, Ohno K, Engel AG, et al. C-terminal and heparin-binding domains of collagenic tail subunit are both essential for anchoring acetyl-cholinesterase at the synapse. J. Biol. Chem. 2004;279:10997–11005. doi: 10.1074/jbc.M305462200. [DOI] [PubMed] [Google Scholar]
- 3.Deprez PN, Inestrosa NC. Two heparin-binding domains are present on the collagenic tail of asymmetric acetylcholinesterase. J. Biol. Chem. 1995;270:11043–11046. doi: 10.1074/jbc.270.19.11043. [DOI] [PubMed] [Google Scholar]
- 4.Bestue-Cardiel M, de-Cabazon-Alvarez AS, Capablo-Liesa JL, et al. Congenital endplate acetylcholinesterase deficiency responsive to ephedrine. Neurology. 2005;65:144–146. doi: 10.1212/01.wnl.0000167132.35865.31. [DOI] [PubMed] [Google Scholar]
- 5.Brengman JM, Capablo-Liesa JL, Lopez-Pison J, et al. Ephedrine treatment of seven patients with congenital endplate acetylcholinesterase deficiency. Neuromuscul. Disord. 2006;16(Suppl 1):S129. [Google Scholar]
- 6.Shen X-M, Ohno K, Tsujino A, et al. Mutation causing severe myasthenia reveals functional asymmetry of AChR signature Cys-loops in agonist binding and gating. J. Clin. Invest. 2003;111:497–505. doi: 10.1172/JCI16997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shen X-M, Ohno K, Sine SM, et al. Subunit-specific contribution to agonist binding and channel gating revealed by inherited mutation in muscle AChR M3-M4 linker. Brain. 2005;128:345–355. doi: 10.1093/brain/awh364. [DOI] [PubMed] [Google Scholar]
- 8.Shen X-M, Fukuda T, Ohno K, Sine SM, Engel AG. Novel AChR δ subunit mutation causes myasthenia by interfering with inter-subunit communication essential for channel gating. J. Clin. Invest. 2008;118:1867–1876. doi: 10.1172/JCI34527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mukhtasimova N, Sine SM. An inter-subunit trigger of channel gating in the muscle nicotinic receptor. J. Neurol. Sci. 2007;27:4110–4119. doi: 10.1523/JNEUROSCI.0025-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horovitz A, Fersht A. Strategy for analyzing the co-operativity of intramolecular interactions in peptides and proteins. J. Mol. Biol. 1990;214:613–617. doi: 10.1016/0022-2836(90)90275-Q. [DOI] [PubMed] [Google Scholar]
- 11.Fukudome T, Ohno K, Brengman JM, et al. Quinidine normalizes the open duration of slow-channel mutants of the acetylcholine receptor. Neuroreport. 1998;9:1907–1911. doi: 10.1097/00001756-199806010-00044. [DOI] [PubMed] [Google Scholar]
- 12.Harper CM, Engel AG. Safety and efficacy of quinidine sulfate in slow-channel congenital myasthenic syndrome. Ann. N.Y. Acad. Sci. 1998;841:203–206. doi: 10.1111/j.1749-6632.1998.tb10929.x. [DOI] [PubMed] [Google Scholar]
- 13.Harper CM, Fukudome T, Engel AG. Treatment of slow channel congenital myasthenic syndrome with fluoxetine. Neurology. 2003;60:170–173. doi: 10.1212/01.wnl.0000061483.11417.1b. [DOI] [PubMed] [Google Scholar]
- 14.Elbashir SM, Harborth J, Lendeckel W, et al. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001;411:494–498. doi: 10.1038/35078107. [DOI] [PubMed] [Google Scholar]
- 15.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. doi: 10.1126/science.1068999. [DOI] [PubMed] [Google Scholar]
- 16.Paddison PJ, Caudy AA, Hannon GJ. Stable suppression of gene expression by RNAi in mammalian cells. Proc. Natl. Acad. Sci. USA. 2002;99:1443–1448. doi: 10.1073/pnas.032652399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Abdelgany A, Wood M, Beeson D. Allele-specific silencing of a pathogenic mutant acetylcholine receptor subunit by RNA interference. Hum. Mol. Genet. 2003;12:2637–2644. doi: 10.1093/hmg/ddg280. [DOI] [PubMed] [Google Scholar]
- 18.Kapadia SB, Brideau-Andersen A, Chisari FV. Interference of hepatitis C virus RNA replication by short interfering RNAs. Proc. Natl. Acad. Sci. USA. 2003;100:2014–2018. doi: 10.1073/pnas.252783999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McManus MT, Sharp PA. Gene silencing in mammals by small interfering RNAs. Nat. Rev. Genet. 2002;3:737–747. doi: 10.1038/nrg908. [DOI] [PubMed] [Google Scholar]
- 20.Shen X-M, Deymeer F, Sine SM, et al. Slow-channel mutation in AChR αM4 domain and its efficient knockdown. Ann. Neurol. 2006;60:128–136. doi: 10.1002/ana.20861. [DOI] [PubMed] [Google Scholar]
- 21.Kumar R, Conklin DS, Mittal V. High-throughput selection of effective RNAi probes for gene silencing. Genome Res. 2003;13:2333–2340. doi: 10.1101/gr.1575003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Okada K, Inoue A, Okada M, et al. The muscle protein Dok-7 is essential for neuromuscular synaptogenesis. Science. 2006;312:1802–1805. doi: 10.1126/science.1127142. [DOI] [PubMed] [Google Scholar]
- 23.Beeson D, Higuchi O, Palace J, et al. Dok-7 mutations underlie a neuromuscular junction synaptopathy. Science. 2006;313:1975–1978. doi: 10.1126/science.1130837. [DOI] [PubMed] [Google Scholar]
- 24.Slater CR, Fawcett PRW, Walls TJ, et al. Pre- and postsynaptic abnormalities associated with impaired neuromuscular transmission in a group of patients with ‘limb-girdle myasthenia’. Brain. 2006;127:2061–2076. doi: 10.1093/brain/awl200. [DOI] [PubMed] [Google Scholar]
- 25.Selcen D, Milone M, Shen X-M, et al. Dok-7 myasthenia: clinical spectrum, endplate electrophysiology and morphology, 12 novel DNA rearrangements, and genotype-phenotype relations in a Mayo cohort of 13 patients [abstract] Neurology. 2007;68(Suppl 1):A106–A107. [Google Scholar]
- 26.Engel AG, Ohno K, Sine SM. Congenital myasthenic syndromes. In: Engel AG, Franzini-Armstrong C, editors. Myology. 3rd ed McGraw-Hill; New York: 2004. pp. 1755–1790. [Google Scholar]
- 27.Unwin N. Refined structure of the nicotinic acetylcholine receptor at 4 Å resolution. J. Mol. Biol. 2005;346:967–989. doi: 10.1016/j.jmb.2004.12.031. [DOI] [PubMed] [Google Scholar]