

Abstract
Purpose
Evaluation and comparison of 3’-[18F]-fluoro-3’-deoxy-L-thymidine (FLT) and 2-[18F]-fluoro-2-deoxyglucose (FDG)-PET to monitor early response following both cyclophosphamide and temsirolimus treatment in a mouse model of Burkitt lymphoma.
Methods
Daudi xenograft mice were treated with either cyclophosphamide or temsirolimus and imaged with FLT-PET and FDG-PET on appropriate days post therapy inititiation. Immunohistochemical (IHC) studies (H&E, TUNEL, CD20, PCNA and ki-67) and DNA flow cytometry studies were performed.
Results
FDG tumor uptake decreased immediately after cyclophosphamide treatment while FLT-PET showed only a late and less pronounced decrease. A fast induction of apoptosis was observed together with an early accumulation of cells in the S-phase of the cell cycle, suggesting DNA repair. Temsirolimus treatment reduced both FDG and FLT tumor uptake immediately after therapy and resulted in a fast induction of apoptosis and G0-G1 phase accumulation.
Conclusion
FLT response was less distinct than FDG response and may be controlled by DNA repair early after cyclophosphamide. Nevertheless, FLT-PET was able to reflect decreased proliferation following temsirolimus.
Keywords: FDG-PET, FLT-PET, Burkitt lymphoma, cyclophosphamide, mTOR inhibition, therapy response
Introduction
Positron emission tomography (PET) with 2-[18F]-fluoro-2-deoxyglucose (FDG) and 3’-[18F]-fluoro-3’-deoxy-L-thymidine (FLT) allows to report on tumor glucose metabolism and proliferation, respectively. These imaging modalities are clinically applied to diagnose cancer patients and characterize tumor properties [1-3]. Moreover these biomarkers can evaluate changes in glucose metabolism or proliferation in response to therapy [4-6]. This may improve patient therapy management since present methods to evaluate therapy response are still primarily based on morphological tumor shrinkage [7,8]. The disadvantage of tumor-size based therapy evaluation is not only the late sign of effective therapy but also the inability to distinguish fibrotic scarring from persisting viable tumor tissue [9]. Additionally new tumor-specific targeted agents mainly induce cell cycle arrest without pronounced tumor regression [10].
Previous studies have already demonstrated that drug-induced changes in FDG-PET occur before morphological changes are measured and allow to predict therapeutic outcome [11-14]. Nevertheless, the uptake of FDG in inflammatory cells may lead to an overestimation of the viable tumor cell fraction as inflammatory cells frequently infiltrate the tumor [15-17]. Additionally FDG measures cell viability which might be less relevant when cell cycle arrest is induced instead of tumor cell death. On the opposite, certain targeted agents might decrease glycolysis without altering cell proliferation. For example, a recently published study demonstrated that inhibition of mammalian target of rapamycin (mTOR), a regulator of cellular proliferation, induced a decreased FDG uptake in certain tumor subtypes independently of its effect on proliferation [18]. For these reasons, FLT is intensively studied as a valid alternative for FDG to predict therapy response [19]. Not only can FLT-PET measure direct effects on proliferation, FLT uptake might also be less disturbed by the inflammatory response because once inflammatory cells enter a tumor they have only minor tendency to proliferate [20,21]. Nevertheless, FLT uptake may be influenced by drug- and tumor-specific effects such as activation of the salvage pathway by pyrimidine metabolism [22]. Moreover, mTOR inhibition with temsirolimus in mantle cell lymphoma (MCL) induced a temporary rise in FLT uptake 7 days after therapy, which may be due to cyclin D1 driving cell-cycle progression of the tumor after release from mTOR inhibition [23].
In the current study we aimed to evaluate the ability of FLT-PET to measure early response following cyclophosphamide treatment in a mouse model of Burkitt lymphoma [17]. The second aim was to investigate whether FLT-PET was able to monitor a decreased proliferation following mTOR inhibition in comparison to FDG-PET.
Materials and methods
Cell line
The human B-lymphoblast cell line Daudi was derived from a Burkitt lymphoma [17]. Cells were cultured in DMEM medium without pyruvate and supplemented with 10 % fetal bovine serum (FBS), 1 % penicillin/streptomycin (P/S), 1 % L-glutamine, 1 % HEPES and 1 % sodium pyruvate. The cells were cultured in flasks in a humidified 5 % CO2 atmosphere at 37°C.
Animal model
Severe combined immune deficient (SCID)-mice (C.B-17/Icr scid/scid) were bred under germ-free conditions. Mice (6 to 8 weeks old) were inoculated with 5x106 Daudi cells in both shoulders. As soon as the tumor was visually perceptible caliper measurements were performed to follow up tumor growth. When the developing tumors had reached a diameter between 10 and 15 mm mice were subjected to treatment.
Experimental design
Table 1 schematically draws the experimental design of the study. The first treatment group was treated with cyclophosphamide (n=17); 125 mg/kg intraperitoneally (i.p.) (Endoxan®, Baxter). The second treatment group was treated with temsirolimus (n=17); 50 mg/kg i.p., which was provided by Wyeth (CCI-779, Torisel, Wyeth). A number of treated animals (n ≥ 4) were monitored with FDG-PET and FLT-PET on d0 (before treatment), d2, d4, d7, d9 and d14. To follow-up tumor growth, tumor dimensions were measured at each time point using a caliper. Tumor volumes (Volcaliper) were calculated using the equation: Volcaliper = (π/6) × a × b × c × 10-3 where a, b and c represent the three orthogonal axes of the tumors in millimeters. On corresponding time points following treatment, mice (n=2) were sacrificed to allow immunohistochemical (IHC) analysis of the tumors (2 tumors per mouse). The IHC analysis from these treated animals were compared to control tumors dissected from non-treated animals.
Table 1.
Study design of treatment groups, cyclophosphamide and temsirolimus, monitored with FDG-PET and FLT-PET on d0 (before treatment), d2, d4, d7, d9 and d14. Tumor volumes (Volcalip) were measured with a caliper. Immunohistochemical (IHC) and DNA FACS studies were performed on corresponding days following therapy.
| Cyclophosphamide (n=17) | d0 | d2 | d4 | d7 | d9 | d14 |
|
| ||||||
| FDG-PET + caliper (n=4) | X | X | X | X | X | X |
| FLT-PET + caliper (n=5) | X | X | X | X | X | X |
| IHC and DNA FACS (n=8) † | n=2 | n=2° | n=2 | n=2 | * | |
|
| ||||||
| Temsirolimus (n=17) | d0 | d2 | d4 | d7 | d9 | d14 |
|
| ||||||
| FDG-PET + caliper (n=5) | X | X | X | X | X | X |
| FLT-PET + caliper (n=4) | X | X | X | X | X | X |
| IHC and DNA FACS (n=8) † | n=2 | n=2° | n=2 | n=2 | * | |
|
| ||||||
| Control (n=2) | d0 | |||||
|
| ||||||
| IHC and DNA FACS (n=2) | n=2 | |||||
In total 8 mice were sacrificed for analysis on d2,4,7,9 (2 mice per time point);
tumors were dissected from mice following their final scan on d14;
Only IHC no DNA FACS
Small animal PET scanning
Small animal PET scanning was performed using a dedicated small animal PET (FOCUS 220 microPET; Concorde-CTI Siemens, Knoxville Tenessee, USA). For FDG the mice were fasted overnight. All animals were sedated by gas anesthesia with isoflurane, and body weight, tumor dimensions (caliper) and glycemia (in case of FDG) were determined. Then 8-11 MBq FDG or FLT was injected via a tail vein. Sixty minutes post-injection, PET imaging was performed (10 minutes/frame), on a single bed position with the tumor in the center of the view.
Parameters and statistical analysis
On PET images tumor tracer uptake was measured by delineating the tumors with in-house software (IDL viewer) using the isocontour tool which automatically contours regions from the data set such that the edge values of the regions of interest (ROIs) are always the same value [24]. When clicking inside the tumor the program generates a 2D isocontour surrounding the tumor. This is performed in all transversal sections containing tumor tissue. From the different 2D isocontours the program can generate a 3D delineation of the tumor. When no remaining tumor was present on PET, we used a standard region of interest at the previous localization of the tumor.
Tracer uptake was defined as standardized uptake value (SUV) which is calculated by the equation: SUV = measured activity concentration in the tumor (Bq/g) × body weight (g) / injected activity (Bq). The mean SUV (SUVmean), maximal SUV (SUVmax) and metabolic volume (Volmetab) were measured. Additionally, the total lesion glycolisis (TLG) was calculated as SUVmean × Volmetab. To correct for small variations in administered dose (paravenous injections), all SUVs were normalized by dividing SUV of the tumor (SUVmean and SUVmax) by the corresponding SUVmean of a standard region in the liver.
Within each animal the changes of the normalized parameters (SUVmean, SUVmax and TLG) and the changes in volumes (Volmetab and Volcalip) were expressed as percentage change (% change) compared to baseline values on d0. For example, changes in SUV were calculated by the following formula: % SUV change = (SUVx-SUV0)/SUV0 × 100 % with SUVx being the FDG-uptake at day x.
The statistical analysis was performed using GraphPad PRISM 5.04. In each treatment group mean % changes and standard error of the mean (SEM) were calculated and graphically expressed. For multiple comparisons, such as evaluation of mean changes over time, we performed one-way analysis of variances (ANOVA) and Bonferroni’s multiple comparisons. In order to detect significant differences between parameters on different time points we performed two-way ANOVA and Bonferroni’s multiple comparisons. A p-value <0.05 was defined as statistically significant.
Immunohistochemistry
Paraffin embedded sections were stained with haematoxylin and eosin (H&E). From these sections we assessed the amount of macrophages by semi-quantitative measurements performed in 10 high power fields (HPF) [<5 %, 5-10 %, 10-15 %, 15-20 % and 20-25 %]. The specific characteristics of macrophages (i.e. large cells, with vacuolated cytoplasm and an irregular nucleus) can easily be delineated by means of H&E stainings (see Fig. 5 for macrophage differentiation). IHC studies were performed with antiCD20 mAb, a pan B-cell marker, and with Mib-1, which recognizes an epitope of the ki-67 nuclear antigen that is present during DNA synthesis (all cells currently in the cell cycle, not in G0). Also IHC was carried out with proliferating cell nuclear antigen (PCNA) which is associated with late G1 and S phase in normal cells (S-phase). Tumor tissue was semi-quantitatively scored for the number of PCNA and ki-67 positive cells [50-60 %, 60-70 %, 70-80 %, 80-90 %, >90 %].
Also terminal deoxynucleotidyl transferase biotin-dUTP nick end labeling (TUNEL) was performed to assess the number of dying cells. TUNEL positive cells were counted in 10 HPF and expressed as positive cells/HPF.
DNA flow cytometry
To assess the cell cycle distribution of tumor cells following treatment with cyclophosphamide and temsirolimus, we performed DNA fluorescence-activating cell sorting (FACS) with propidium iodide (PI) [25]. After dissecting the tumor, a small piece was used for FACS analysis while the major part was fixed in 6% formol for IHC studies. To obtain a single cell suspension the tumor was first cut into pieces and cells were physically disaggregated with a BD Medimachine using 50 μm Medicon filters (BD Biosciences, Erembodegem, Belgium). Next the cell suspension was filtered over a 70 μm nylon cell strainer (BD Biosciences, Erembodegem, Belgium). Microscopy was performed to check the single-cell properties of the cell suspension. For analyses 1 million cells were fixed in 70 % ethanol for at least 1 day. Then cells were stained with PI (Sigma Aldrich, Bornem, Belgium) (40μg/mL) in PBS, containing RNase (DNase-free) (100 μg/ml) (Fermentas, St. Leon-Rot, Germany) and triton X-100 (0.1%) (Sigma Aldrich, Bornem, Belgium) and analyzed with FACSCanto (BD Biosciences, Erembodegem, Belgium). For each tumor the distribution in G0-G1, S and G2 phase was measured and expressed as percentage of the total viable tumor cell fraction (Cellquest software; Becton Dickinson Bioscience).
Ethical committee
All animals were treated in concordance with institutional guidelines and experiments were approved by the local ethical committee for animal experiments.
Results
Animal model
Between week 6 and week 7 following subcutaneous injection of Daudi tumor cells, 90 % of the animals developed a tumor with a diameter between 10 and 15 mm. All tumors showed a visible FLT and FDG-uptake. The baseline (non-normalized) mean SUVmean of FDG was 2.7 ± 0.08 while FLT uptake was significantly lower with a mean SUVmean value of 1.7 ± 0.04 at baseline.
Tumor volume measurement by caliper
Early after cyclophosphamide treatment (d2 and d4) the tumor volume increased moderately compared to baseline although not significantly (Figure 1A and Table 2). Between d4 and d7 a significant reduction in tumor size was measured (Figure 1A) while a significant reduction in tumor size compared to baseline was measured on d9 (-64±7%) (Table 2). The majority of mice demonstrated a complete disappearance of the tumors on d14 after cyclophosphamide (Figure 1A and Table 2).
Figure 1.
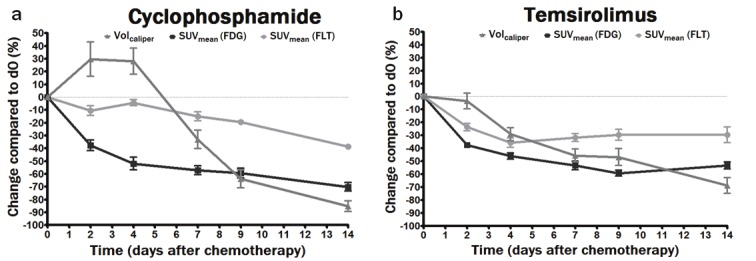
Graphical representation of mean % changes of Volcaliper (■) and SUVmean measured with FDG (▲) and FLT (▼) in cyclophosphamide treated animals (a) and temsirolimus treated animals (b) on d2, d4, d7, d9 and d14 as compared to do.
Table 2.
Mean % change in FDG and FLT uptake compared to baseline measurements following treatment with cyclophosphamide and temsirolimus as measured with the different parameters (SUVmean, SUVmax and TLG). Additionally tumor size mean % changes were measured with caliper measurements and expressed as Volcalip.
| Cyclophosphamide | FDG | FLT | Caliper | |||||
|
|
||||||||
| SUVmean | SUVmax | TLG | SUVmean | SUVmax | TLG | Volcalip | ||
|
|
||||||||
| d0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| d2 | -37,6 ± 4,2* | -33,8 ± 4,6* | -29,8 ± 2,8* | -8,6 ± 3,4 | -13,4 ± 5,1 | 2,1 ± 5,9 | 29,6 ± 13,4 | |
| d4 | -51,8 ± 5* | -50,7 ± 4,7* | -54,5 ± 6* | -5 ± 2 | -9,9 ± 2,5 | 0,7 ± 7 | 28,1 ± 10,1 | |
| d7 | -57,1 ± 3,5* | -57,7 ± 3,5* | -79,8 ± 2,9* | -13,5 ± 3* | -19,6 ± 3,6* | -46,7 ± 5,2* | -33,1 ± 7,1 | |
| d9 | -62,6 ± 2,3* | -58,2 ± 3,1* | -87,2 ± 1,6* | -15,49 ± 3* | -23,8 ± 4* | -68,6 ± 4,1* | -63,8 ± 7* | |
| d14 | -70,1 ± 3,4* | -70,8 ± 3,9* | -97,6 ± 0,8* | -38,7 ± 1,9* | -49,3 ± 2,2* | -94,3 ± 1,6* | -85,3 ± 4,2* | |
|
| ||||||||
|
| ||||||||
| Temsirolimus | FDG | FLT | Caliper | |||||
|
|
||||||||
| SUVmean | SUVmax | TLG | SUVmean | SUVmax | TLG | Volcalip | ||
|
|
||||||||
| d0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | |
| d2 | -37,6 ± 2* | -37,4 ± 1,6* | -59,3 ± 3,8* | -23,6 ± 2,9* | -28,4 ± 3,2* | -32,3 ± 7,1* | -1,9 ± 8,7 | |
| d4 | -46,1 ± 2,4* | -43,7 ± 2,2* | -68,2 ± 4* | -35,7 ± 3,6* | -44,2 ± 3,6* | -53,1 ± 7,2* | -23,8 ± 6,2 | |
| d7 | -53,3 ± 3,3* | -51,5 ± 4* | -84,3 ± 2,1* | -31,7 ± 3,4* | -42,9 ± 5* | -68 ± 6,2* | -48,5 ± 7,3* | |
| d9 | -59,3 ± 2,3* | -57,8 ± 3,1* | -78,7 ± 4* | -29,6 ± 4,1* | -41 ± 5* | -64,6 ± 7* | -50,2 ± 8,8* | |
| d14 | -53,3 ± 2,7* | -50,2 ± 3,9* | -79,9 ± 3,7* | -29,6 ± 6,2* | -37,8 ± 6* | -69,8 ± 5,9* | -71,7 ± 6,1* | |
significantly different from d0
Bold = most pronounced decrease measured for this parameter
Caliper measurements in temsirolimus treated mice revealed a significant reduction in Volcalip on d7 (-49±7%) with a further decrease until d14 (-72±6%). Unlike cyclophosphamide treated animals, none of the temsirolimus treated animals demonstrate a complete remission of the tumors on d14 following treatment (Figure 1B, Table 2).
Changes in FDG and FLT uptake following cyclophosphamide
From Figure 1A it is clear that FDG uptake decreased immediately after cyclophosphamide administration (SUVmean change of -38±4 % on d2 and -52±5 % on d4 compared to baseline, Table 2). Unlike FDG uptake, FLT uptake was not significantly decreased at these early time points. The FLT uptake was significantly reduced on d7 (SUVmean change of -14±3 %) and was lowest on d14 following therapy (SUVmean change of -39±2 %) (Table 2).
Figure 2 gives an example of FDG and FLT images following cyclophosphamide treatment. A fast decrease in FDG uptake in the tumors was visualized (Figure 2A) while FLT uptake remained unchanged until d7 (Figure 2B). Concordant with the decrease in tracer uptake cyclophosphamide therapy led to tumor size reduction (Volcalip) starting on d7 while on d14 almost no tumor tissue remained visible.
Figure 2.

Serial FDG-PET (a) and FLT-PET (b) images acquired on d0, d2, d4, d7, d9 and d14 following cyclophosphamide treatment. Images are scaled to the same SUV scale ranging from 0-5 for FDG-PET and from 0-3 for FLT-PET. Matching coronal views are depicted to demonstrate the FDG uptake (a) and FLT uptake (b) in the tumors on both shoulders (indicated with arrow).
Changes in FDG and FLT uptake following temsirolimus
Both FDG and FLT uptake decreased immediately after temsirolimus administration (SUVmean change of -38±2 % and -24±3 % on d2 respectively) (Figure 1B, Table 2). FDG uptake decreased further on d4 and d7 and reached a minimum on d9 (SUVmean change of -59±2 %) after which it stabilized (SUVmean change of - 53±3 % on d14). FLT uptake on the other hand was minimal on d4 (SUVmean change of -36±4 %) and remained unchanged until d14 (SUVmean change of -30±6 %) (Table 2).
In Figure 3 an example is given for FDG and FLT images following temsirolimus treatment. A fast reduction of both FDG and FLT uptake was observed after temsirolimus (Figure 3A and B respectively). Tumor volumes (Volcalip) decreased on d4 after which the volumes further remained approximately the same.
Figure 3.

Serial FDG-PET (a) and FLT-PET (b) images acquired on d0, d2, d4, d7, d9 and d14 following temsirolimus treatment. Images are scaled to the same SUV scale ranging from 0-5 for FDG-PET and from 0-3 for FLT-PET. Matching coronal views are depicted to demonstrate the FDG uptake (a) and FLT uptake (b) in the tumors on both shoulders (indicated with arrow).
SUVmean, SUVmax and TLG
In this study we compared 3 different parameters to evaluate therapy response (SUVmean, SUVmax and TLG). No significant differences were observed between decreases in SUVmean and decreases in SUVmax in both treatment groups on the different follow-up time points. When comparing decreases in TLG with both decreases in SUVmean and SUVmax we observed significant differences mainly late after therapy. Following cyclophosphamide we measured significantly more pronounced TLG decreases compared to decreases in both SUVmean and SUVmax on d7, d9 and d14 measured for both FDG and FLT uptake. Temsirolimus treated mice demonstrated significantly more pronounced TLG decreases compared to both SUVmean and SUVmax decreases for FDG on d4, d7, d9 and d14 while for FLT this was observed on d7, d9 and d14.
Immunohistochemistry
Untreated tumors revealed a massive proliferation of CD20 positive cells with a high proliferation rate (ki-67 highly positive) (Figure 4). Only few (4/HPF) TUNEL positive cells were observed indicating a low level of spontaneous apoptosis (Table 3).
Figure 4.

Overview of IHC studies with ki-67, H&E and TUNEL for non-treated tumors (d0) and cyclophosphamide and temsirolimus treated tumors on d2, d4, d7, d9 and d14 following therapy.
Table 3.
Histological analysis in both treatment groups at indicated time points following therapy. Tumor tissue was semi-quantitatively scored for the number of macrophages [<5%, 5-10%, 10-15%, 15-20% and 20-25%] and ki-67 positive cells [50-60%, 60-70%, 70-80%, 80-90%, >90%] while TUNEL positive cells were counted in 10 HPF and expressed as positive cells/HPF (bold= most pronounced level of apoptosis).
| Cyclophosphamide | Temsirolimus | |||||
|---|---|---|---|---|---|---|
|
| ||||||
| Macrophages | Ki-67 | TUNEL | Macrophages | Ki-67 | TUNEL | |
| d0 | 5-10% | >90% | 3.9±0.3 | 5-10% | >90% | 3.9±0.3 |
| d2 | 10-15% | 80-90% | 8.7±1.7 | <5% | 80-90% | 9.4±4.6 |
| d4 | 15-20% | 80-90% | 9.2±2.8 | <5% | 50-60% | 11.5±4.2 |
| d7 | 20-25% | 60-70% | 15.5±0.3 | <5% | 70-80% | 11.6±0.8 |
| d9 | 20-25% | 70-80% | 7.4±5.2 | 5-10% | 80-90% | 6.4±2.6 |
| d14 | 15-20% | >90% | 4.4±1.3 | 20-25% | 80-90% | 3.3±0.4 |
Upon cyclophosphamide treatment we observed a direct induction of cell death with TUNEL. However only from d7 on the amount of TUNEL positive cells became significant (Figure 4). Following cyclophosphamide treatment an infiltration of macrophages (see Figure 5 for macrophage differentiation) was observed which was most pronounced on d9 (Table 3). The ki-67 staining revealed a modest decrease in the number of positive cells which was most pronounced on d7 (Figure 4).
Figure 5.

Example of macrophage detection on H&E stained tumor tissue. Macrophages were distinguished from tumor tissue by their size, vacuolated cytoplasm, irregular nucleus and “foamy” appearance (indicated with arrow).
Following temsirolimus therapy a raising number of TUNEL positive cells was measured and became most pronounced between d4 and d7 (Table 3). Contrary to the situation after cyclophosphamide treatment, no clear infiltration of inflammatory cells was observed. Only on d14 macrophages were observed on H&E. The ki-67 showed a clear decrease in positive cells mainly on d4 following therapy (Figure 4).
No clear responses were seen in the number of PCNA positive cells for both treatment groups.
DNA flow cytometry
The FACS data in Figure 6 demonstrate a significant accumulation of cells in the S-phase early after cyclophosphamide therapy (45.1 ± 2.3% cells in the S-phase on d2). ANOVA and bonferroni’s multiple comparison demonstrate significantly higher amounts of cells in the S-phase on d2 compared to d0, d7, d9 and d14. The analysis also revealed significantly increased levels of cells in the G2 phase on d7 following therapy when compared to d0 and d2.
Figure 6.
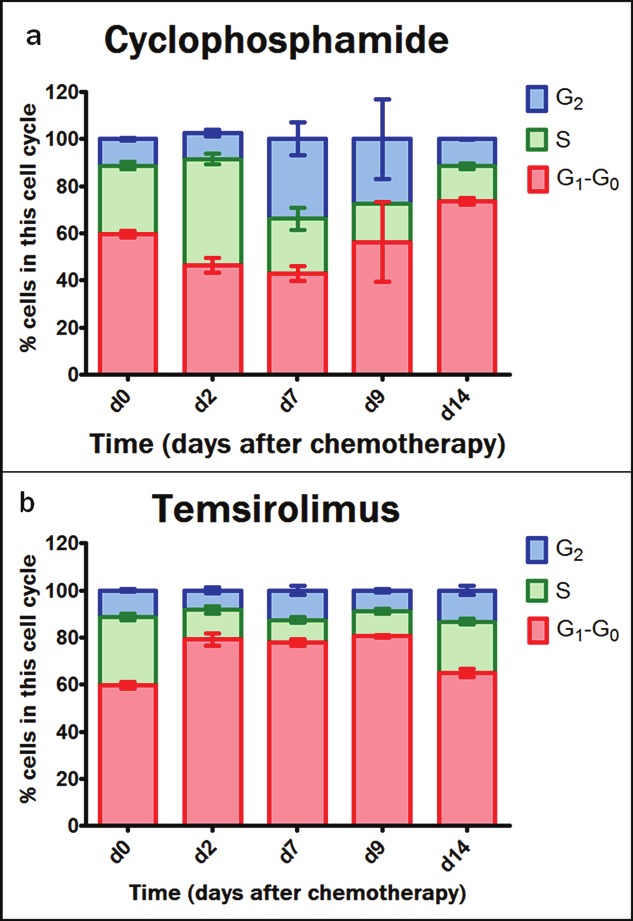
Cell cycle distribution in the G0-G1 phase, S phase and G2 phase as measured with DNA flow cytometry and expressed as the percentage of total viable tumor cell fraction. This was performed for cyclophosphamide treated tumors (a) and temsirolimus treated tumors (b) over time (no data for d4 following therapy).
Following temsirolimus treatment an immediate increase was measured in the amount of cells in the G0-G1 phase (79.2 ± 2.6% cells in the G0-G1 phase on d2) compared to d0 (59.8 ± 1.4% cells in the G0-G1 phase on d0). This significant increase in G0-G1 phase was also measured on d7 and d9 following temsirolimus (significant compared to d0) while this was not measured on d14.
Discussion
Nowadays clinical tumor response is still primarily evaluated by conventional imaging techniques (magnetic resonance imaging (MRI), computed tomography (CT)) measuring anatomical tumor size reduction. Unlike the late effects of anti-cancer therapy on tumor size, molecular and metabolic changes are induced much earlier after the start of therapy. This has already been demonstrated in several studies for FDG-PET reflecting decreased metabolism before morphological changes have occurred [7,16]. However, due to the aspecific accumulation of FDG in inflammatory cells, other potentially more specific PET probes are currently explored [15]. Although FLT was believed to be more closely related to the number of viable tumor cells, tumor-and drug-specific effects may be critical [20,22]. In the present study, we aimed to evaluate and compare FLT-PET with FDG-PET in a mouse model of Burkitt lymphoma treated with cyclophosphamide and temsirolimus. Therapy response was confirmed with caliper measurements of decreased tumor size together with a series of histological studies to assess apoptosis (TUNEL), proliferation (ki67) and macrophage infiltration (H&E). Additionally DNA flow cytometry was performed to measure cell cycle distribution following therapy.
Our study showed that early after cyclophosphamide treatment FDG-PET revealed an immediate and marked decrease in the glucose tumor metabolism several days before the tumor had reduced in size. Even though a stunning effect, resulting in decreased FDG uptake, cannot be completely ruled out the induction of apoptosis suggests a fast killing effect of cyclophosphamide.
Unlike FDG, FLT tumor uptake was not decreased on early time points after cyclophosphamide therapy and decreased only late after therapy. Though FLT-PET measured a decreased proliferation late in the course of cyclophosphamide therapy (from d7), the decrease was less pronounced when compared to the FDG-PET. A possible explanation may be increased uptake of FLT due to DNA repairing processes that can offset decreased FLT uptake from cyclophosphamide-induced cell death. Indeed, DNA flow cytometry data showed an accumulation of cells in the S-phase 2 days after cyclophosphamide treatment which suggests repair of cyclophosphamide induced DNA crosslinks [26,27]. The results obtained in cycloposphamide treated animals indicate that, compared to FLT, FDG-PET is more suitable to evaluate therapy response following this type of treatment.
However, the contribution of inflammatory cells to the FDG signal remains the major limitation. In the studied Burkitt lymphoma xenograft model an influx of inflammatory cells was described from d6 until d13 with a peak of 24 % of the total cell fraction on d9 following cyclophosphamide [23]. In the current study the infiltration of macrophages was semi-quantitatively confirmed with H&E and showed to be most pronounced between d7 and d9. On these time points no temporary rise was observed for both FDG and FLT, although for FDG we did observe a transient plateau between d7 and d9 after which the FDG decreased further. Although FLT uptake seemed to be less influenced by the inflammatory response in this and other studies [23], we must take into account the limited immune system of scid mice used in our study. Since the mice lack a functional B or T cell compartment and only have a functional macrophage, granulocyte and natural killer (NK) function the potential effect of proliferating B and T cells on FLT uptake was not taken into account. However, since inflammatory cells once inside the tumor have only minor tendency to proliferate, the effect on tumor uptake is expected to be minimal [21].
Although cyclophosphamide is routinely used together with other chemotherapeutic agent to treat lymphoma, leukemia and some solid tumors, certain tumor types may be resistant to chemotherapy or may acquire resistance during the course of treatment. Therefore other disease-specific molecularly targeted agents are increasingly recognized. One of these targets is mTOR, a protein kinase that exerts its regulatory effects on cell proliferation by controlling the production of cyclinD1, which induces cell cycle G1-S transition. Consequently, inhibition of mTOR increases the number of cells in the G0-G1 phase and leads to growth inhibition which may eventually lead to tumor cell death and in some tumor types results in tumor shrinkage. Although tumor shrinkage is rarely seen following mTOR inhibition [10,28], a single dose of everolimus has shown to induce remarkable tumor regression in squamous cell carcinoma of the head and the neck (HNSCC) [29]. In our animal model, temsirolimus resulted in apoptosis and relatively fast tumor shrinkage, suggesting that Burkitt lymphoma are sensitive to mTOR inhibitors. Although beyond the scope of this research, this opens new perspectives for novel combination therapies, potentially including combinations of temsirolimus with conventional chemotherapy agents [30].
Since FDG-PET was not predictive in patients with various types of cancer treated with mTOR inhibitors [18], new imaging approaches are required to follow up response to mTOR inhibitors. Therefore this study aimed to evaluate the ability of FLT-PET to predict response to temsirolimus in a mouse model of Burkitt lymphoma. We demonstrated that FLT was able to reflect a reduced proliferation early after mTOR inhibition. Ki67 stainings showed a decreased proliferation but also DNA FACS data demonstrate that cells accumulate in the G0-G1 phase following temsirolimus administration. Ki67 stainings were immediately reduced and the reduction was most pronounced on d4. Unfortunately no DNA FACS data exist for d4 following therapy.
In a similar study performed in MCL, a temporary rise in FLT uptake was observed on d7 following mTOR inhibition probably because MCL have cyclinD1 overexpression and still-viable tumor cells will reenter the S phase after removal of the drug (biological half-life 9-17h) [23]. In our study no temporary rise was observed but FLT uptake decrease stabilized from d7 which most likely results from the release of mTOR inhibition. Unlike FLT uptake, FDG uptake was minimal on d9 after which it stabilized. This suggests that the effect of mTOR inhibition on glucose metabolism was slightly more extended (until d9) than the effect on proliferation (until d4). As already observed by other groups the effects of mTOR inhibition on glycolysis may be independent on the effects of proliferation in certain cell lines [18]. This is most likely because mTOR forms two complexes of which the mTOR complex 1 (mTORC1) regulates growth and cell cycle progression. The mTORC2 is part of the upstream pathway PI3k/Akt pathway and regulates Akt activity [18,31]. Taking together our results obtained in temsirolimus mice and the data reported from patients [18], we suggest that FLT-PET can be a valid alternative for FDG-PET to follow-up target specific drugs such as mTOR inhibitors.
Overall our study demonstrates that drug and tumor-specific effects need to be considered when selecting the most suitable molecular imaging probe for therapy response evaluation. Depending on the clinical question the optimal parameter should also be determined. SUVmax accounts for resistant foci while SUVmean averages the uptake in the tumor and may fail to notice chemoresistant tumor tissue foci. In this study we compared three semiquantitative PET parameters in the different treatment groups and revealed that SUVmean and SUVmax show a similar pattern following therapy indicating that there are no major resistant foci present in our tumor model. Besides these commonly used response parameters, TLG may provide complementary information on the global response of the tumor as it accounts for both the FDG uptake and tumor volume. In our study changes in TLG became more pronounced in the course of therapy than SUV. Although TLG may be more susceptible to contouring variations which can complicate standardization of quantification methods, we and other research groups believe that TLG can more clearly demonstrate relevant changes following effective therapy [11,32].
This study, in Burkitt lymhoma xenografts, demonstrated a delayed and less pronounced reduction in FLT uptake compared to FDG uptake following cyclophosphamide treatment. This is most likely because the potential decrease in FLT uptake was offset by DNA repair processes. Although the influx of inflammatory cells should be considered when using FDG-PET to evauate response, we suggest that FDG-PET is more suitable to monitor cyclophosphamide.
Secondly, mTOR inhibition immediatelly induces both FDG and FLT reduction in our animal model while the effect on FDG was more extended (until d9) than on FLT (until d4). The study demonstrated that FLT-PET was able to reflect decreased proliferation following mTOR induced cell cycle arrest and may therefore be a valid alternative to FDG-PET to follow-up target specific drugs such as mTOR inhibitors.
Acknowledgements
We would like to thank Ann Van Santvoort for her contribution in this study. This research was supported by the Euregional PACT II project (IVAVLANED-1.20) and EC-FP6-Project DIMI. A further grant was received from the center of excellence ‘MoSAIC’ project number EF/05/008.
References
- 1.Juweid ME, Cheson BD. Role of positron emission tomography in lymphoma. J. Clin. Oncol. 2005;23:4577–4580. doi: 10.1200/JCO.2005.01.904. [DOI] [PubMed] [Google Scholar]
- 2.Juweid ME, Cheson BD. Positron-emission tomography and assessment of cancer therapy. N Engl J Med. 2006;354:496–507. doi: 10.1056/NEJMra050276. [DOI] [PubMed] [Google Scholar]
- 3.Herrmann K, Buck AK, Schuster T, Junger A, Wieder HA, Graf N, Ringshausen I, Rudelius M, Wester HJ, Schwaiger M, Keller U, Dechow T. Predictive value of initial 18F-FLT uptake in patients with aggressive non-Hodgkin lymphoma receiving R-CHOP treatment. J Nucl Med. 2011;52:690–696. doi: 10.2967/jnumed.110.084566. [DOI] [PubMed] [Google Scholar]
- 4.Juweid ME, Stroobants S, Hoekstra OS, Mottaghy FM, Dietlein M, Guermazi A, Wiseman GA, Kostakoglu L, Scheidhauer K, Buck A, Naumann R, Spaepen K, Hicks RJ, Weber WA, Reske SN, Schwaiger M, Schwartz LH, Zijlstra JM, Siegel BA, Cheson BD. Use of positron emission tomography for response assessment of lymphoma: consensus of the Imaging Subcommittee of International Harmonization Project in Lymphoma. J. Clin. Oncol. 2007;25:571–578. doi: 10.1200/JCO.2006.08.2305. [DOI] [PubMed] [Google Scholar]
- 5.Boellaard R, O'Doherty MJ, Weber WA, Mottaghy FM, Lonsdale MN, Stroobants SG, Oyen WJ, Kotzerke J, Hoekstra OS, Pruim J, Marsden PK, Tatsch K, Hoekstra CJ, Visser EP, Arends B, Verzijlbergen FJ, Zijlstra JM, Comans EF, Lammertsma AA, Paans AM, Willemsen AT, Beyer T, Bockisch A, Schaefer-Prokop C, Delbeke D, Baum RP, Chiti A, Krause BJ. FDG PET and PET/CT: EANM procedure guidelines for tumour PET imaging: version 1.0. Eur J Nucl Med Mol Imaging. 2010;37:181–200. doi: 10.1007/s00259-009-1297-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herrmann K, Wieder HA, Buck AK, Schoffel M, Krause BJ, Fend F, Schuster T, Meyer zum Buschenfelde C, Wester HJ, Duyster J, Peschel C, Schwaiger M, Dechow T. Early response assessment using 3'-deoxy-3'-[18F] fluorothymidine-positron emission tomography in high-grade non-Hodgkin's lymphoma. Clin Cancer Res. 2007;13:3552–3558. doi: 10.1158/1078-0432.CCR-06-3025. [DOI] [PubMed] [Google Scholar]
- 7.Wahl RL, Jacene H, Kasamon Y, Lodge MA. From RECIST to PERCIST: Evolving Considerations for PET response criteria in solid tumors. J Nucl Med. 2009;50(Suppl 1):122S–150S. doi: 10.2967/jnumed.108.057307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, Rubinstein L, Shankar L, Dodd L, Kaplan R, Lacombe D, Verweij J. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1) Eur J Cancer. 2009;45:228–247. doi: 10.1016/j.ejca.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 9.Weber WA. Use of PET for monitoring cancer therapy and for predicting outcome. J Nucl Med. 2005;46:983–995. [PubMed] [Google Scholar]
- 10.Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grunwald V, Thompson JA, Figlin RA, Hollaender N, Urbanowitz G, Berg WJ, Kay A, Lebwohl D, Ravaud A. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet. 2008;372:449–456. doi: 10.1016/S0140-6736(08)61039-9. [DOI] [PubMed] [Google Scholar]
- 11.Brepoels L, De Saint-Hubert M, Stroobants S, Verhoef G, Balzarini J, Mortelmans L, Mottaghy FM. Dose-response relationship in cyclophosphamide-treated B-cell lymphoma xenografts monitored with [18F] FDG PET. Eur J Nucl Med Mol Imaging. 2010;37:1688–1695. doi: 10.1007/s00259-010-1479-0. [DOI] [PubMed] [Google Scholar]
- 12.Brepoels L, Stroobants S, De Wever W, Spaepen K, Vandenberghe P, Thomas J, Uyttebroeck A, Mortelmans L, De Wolf-Peeters C, Verhoef G. Aggressive and indolent non-Hodgkin's lymphoma: response assessment by integrated international workshop criteria. Leuk Lymphoma. 2007;48:1522–1530. doi: 10.1080/10428190701474365. [DOI] [PubMed] [Google Scholar]
- 13.Brepoels L, Stroobants S, De Wever W, Spaepen K, Vandenberghe P, Thomas J, Uyttebroeck A, Mortelmans L, De Wolf-Peeters C, Verhoef G. Hodgkin lymphoma: Response assessment by revised International Workshop Criteria. Leuk Lymphoma. 2007;48:1539–1547. doi: 10.1080/10428190701422414. [DOI] [PubMed] [Google Scholar]
- 14.Zinzani PL, Stefoni V, Tani M, Fanti S, Musuraca G, Castellucci P, Marchi E, Fina M, Ambrosini V, Pellegrini C, Alinari L, Derenzini E, Montini G, Broccoli A, Bacci F, Pileri S, Baccarani M. Role of [18F] fluorodeoxyglucose positron emission tomography scan in the follow-up of lymphoma. J. Clin. Oncol. 2009;27:1781–1787. doi: 10.1200/JCO.2008.16.1513. [DOI] [PubMed] [Google Scholar]
- 15.Kubota R, Yamada S, Kubota K, Ishiwata K, Tamahashi N, Ido T. Intratumoral distribution of fluorine-18-fluorodeoxyglucose in vivo: high accumulation in macrophages and granulation tissues studied by microautoradiography. J Nucl Med. 1992;33:1972–1980. [PubMed] [Google Scholar]
- 16.Spaepen K, Stroobants S, Dupont P, Bormans G, Balzarini J, Verhoef G, Mortelmans L, Vandenberghe P, De Wolf-Peeters C. [(18)F] FDG PET monitoring of tumour response to chemotherapy: does [(18)F] FDG uptake correlate with the viable tumour cell fraction? Eur J Nucl Med Mol Imaging. 2003;30:682–688. doi: 10.1007/s00259-003-1120-6. [DOI] [PubMed] [Google Scholar]
- 17.Brepoels L, Stroobants S, Vandenberghe P, Spaepen K, Dupont P, Nuyts J, Bormans G, Mortelmans L, Verhoef G, De Wolf-Peeters C. Effect of corticosteroids on 18F-FDG uptake in tumor lesions after chemotherapy. J Nucl Med. 2007;48:390–397. [PubMed] [Google Scholar]
- 18.Ma WW, Jacene H, Song D, Vilardell F, Messersmith WA, Laheru D, Wahl R, Endres C, Jimeno A, Pomper MG, Hidalgo M. [18F] fluorodeoxyglucose positron emission tomography correlates with Akt pathway activity but is not predictive of clinical outcome during mTOR inhibitor therapy. J. Clin. Oncol. 2009;27:2697–2704. doi: 10.1200/JCO.2008.18.8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mier W, Haberkorn U, Eisenhut M. [18F] FLT; portrait of a proliferation marker. Eur J Nucl Med Mol Imaging. 2002;29:165–169. doi: 10.1007/s00259-001-0675-3. [DOI] [PubMed] [Google Scholar]
- 20.Been LB, Suurmeijer AJ, Cobben DC, Jager PL, Hoekstra HJ, Elsinga PH. [18F] FLT-PET in oncology: current status and opportunities. Eur J Nucl Med Mol Imaging. 2004;31:1659–1672. doi: 10.1007/s00259-004-1687-6. [DOI] [PubMed] [Google Scholar]
- 21.van Waarde A, Cobben DC, Suurmeijer AJ, Maas B, Vaalburg W, de Vries EF, Jager PL, Hoekstra HJ, Elsinga PH. Selectivity of 18F-FLT and 18F-FDG for differentiating tumor from inflammation in a rodent model. J Nucl Med. 2004;45:695–700. [PubMed] [Google Scholar]
- 22.Dittmann H, Dohmen BM, Kehlbach R, Bartusek G, Pritzkow M, Sarbia M, Bares R. Early changes in [18F] FLT uptake after chemotherapy: an experimental study. Eur J Nucl Med Mol Imaging. 2002;29:1462–1469. doi: 10.1007/s00259-002-0925-z. [DOI] [PubMed] [Google Scholar]
- 23.Brepoels L, Stroobants S, Verhoef G, De Groot T, Mortelmans L, De Wolf-Peeters C. (18)F-FDG and (18)F-FLT uptake early after cyclophosphamide and mTOR inhibition in an experimental lymphoma model. J Nucl Med. 2009;50:1102–1109. doi: 10.2967/jnumed.109.062208. [DOI] [PubMed] [Google Scholar]
- 24.Krak NC, Boellaard R, Hoekstra OS, Twisk JW, Hoekstra CJ, Lammertsma AA. Effects of ROI definition and reconstruction method on quantitative outcome and applicability in a response monitoring trial. Eur J Nucl Med Mol Imaging. 2005;32:294–301. doi: 10.1007/s00259-004-1566-1. [DOI] [PubMed] [Google Scholar]
- 25.Ormerod MG, editor. Flow cytometry: a practical approach. Vol Second edition. NY: Oxford University Press; 1992. [Google Scholar]
- 26.Dronkert ML, Kanaar R. Repair of DNA interstrand cross-links. Mutat Res. 2001;486:217–247. doi: 10.1016/s0921-8777(01)00092-1. [DOI] [PubMed] [Google Scholar]
- 27.Aide N, Poulain L, Briand M, Dutoit S, Allouche S, Labiche A, Ngo-Van Do A, Nataf V, Batalla A, Gauduchon P, Talbot JN, Montravers F. Early evaluation of the effects of chemotherapy with longitudinal FDG small-animal PET in human testicular cancer xenografts: early flare response does not reflect refractory disease. Eur J Nucl Med Mol Imaging. 2009;36:396–405. doi: 10.1007/s00259-008-0984-x. [DOI] [PubMed] [Google Scholar]
- 28.O'Reilly T, McSheehy PM. Biomarker Development for the Clinical Activity of the mTOR Inhibitor Everolimus (RAD001): Processes, Limitations, and Further Proposals. Transl Oncol. 2010;3:65–79. doi: 10.1593/tlo.09277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amornphimoltham P, Leelahavanichkul K, Molinolo A, Patel V, Gutkind JS. Inhibition of Mammalian target of rapamycin by rapamycin causes the regression of carcinogen-induced skin tumor lesions. Clin Cancer Res. 2008;14:8094–8101. doi: 10.1158/1078-0432.CCR-08-0703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.A phase I trial of Doxil, Bevacizumab and Temsirolimus. http://clinicaltrials.gov/ct2/show/NCT00761644, 2008(start date) [Google Scholar]
- 31.Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–734. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 32.Larson SM, Erdi Y, Akhurst T, Mazumdar M, Macapinlac HA, Finn RD, Casilla C, Fazzari M, Srivastava N, Yeung HW, Humm JL, Guillem J, Downey R, Karpeh M, Cohen AE, Ginsberg R. Tumor Treatment Response Based on Visual and Quantitative Changes in Global Tumor Glycolysis Using PET-FDG Imaging. The Visual Response Score and the Change in Total Lesion Glycolysis. Clin Positron Imaging. 1999;2:159–171. doi: 10.1016/s1095-0397(99)00016-3. [DOI] [PubMed] [Google Scholar]