

Abstract
The Foxp3 transcription factor is the master regulator of regulatory T cell (Treg) differentiation and function. Its activity is regulated by reversible acetylation. Using mass spectrometry of immunoprecipitated proteins, we identify three novel acetylation sites in murine Foxp3 (K31, K262, and K267) and the corresponding sites in human FoxP3 proteins. Newly raised modification-specific antibodies against acetylated K31 and K267 confirm acetylation of these residues in murine Tregs. Mutant Foxp3 proteins carrying arginine substitutions at the three acetylation sites (3KR) accumulate in T cells to higher levels than wildtype Foxp3 and exert better suppressive activity in co-culture experiments. Acetylation and stability of wildtype, but not mutant, Foxp3 is enhanced when cells are treated with Ex-527, an inhibitor of the NAD+-dependent deacetylase SIRT1. Treatment with Ex-527 promotes Foxp3 expression during induced Treg differentiation, enhances Foxp3 levels in natural Tregs, and prevents loss of Foxp3 expression in adoptively transferred Tregs in mice. Our data identify SIRT1 as a negative regulator of Treg function via deacetylation of three novel target sites in Foxp3. SIRT1 inhibitors strengthen the suppressive activity of Tregs and may be useful in enhancing Treg-based therapeutic approaches to autoimmune diseases or graft rejections.
Introduction
Regulatory T cells (Tregs) are CD4+ suppressor T cells that prevent pathological self-reactivity in the immune system (1,2). Expression of the forkhead transcription factor Foxp3 is both essential and sufficient for the development and immunosuppressive function of Tregs (1,2). Foxp3 positively and negatively regulates gene expression. It suppresses the production of effector cytokines, such as IL-2, IFN-γ, and IL-4, but activates the expression of IL-10 as well as cell-surface-associated markers, including the high-affinity IL-2 receptor CD25, the cytotoxic T cell-associated antigen-4 (CTLA-4), and the glucocorticoid-induced TNF receptor family-related gene/protein (GITR) (1,3). Although Foxp3 represses production of IL-2 in Tregs themselves, exogenous IL-2 is critical for sustained expression of Foxp3 and CD25 and for the suppressive function of Tregs (1).
Two major populations of Foxp3+ Treg cells exist (4). Naturally occurring CD4+CD25+Foxp3+ Treg cells (nTregs) originate in the thymus where they develop after T-cell receptor (TCR) stimulation through MHC/self-peptide complex engagement, in combination with signaling through CD28 and CD25 (5). Induced Treg cells (iTregs) develop in the periphery from naive CD4+ T cells, acquiring Foxp3 expression and associated suppressive functions. Generation of iTregs has been described in the gut-associated lymphoid tissue, spleen, lymph node, chronically inflamed tissues and transplanted tissues (4). Suboptimal costimulation of T-cell receptor-mediated signals in combination with exposure to transforming growth factor-beta (TGF-β) is critical for the differentiation of iTregs in peripheral tissues (4). These conditions can be recapitulated in cell culture, and iTregs can be differentiated from naive CD4+ T cells in vitro.
The suppressive functions of nTregs and iTregs are dependent on continuous high-level expression of Foxp3. Expression levels of Foxp3 are controlled by transcriptional and posttranscriptional events. Epigenetic modifications at the Foxp3 gene locus prevent expression of Foxp3 in nonTreg cells (6,7). DNA methylation patterns at the Foxp3 locus differ in the different subtypes of Tregs: while nTregs maintain relatively demethylated CpG islands at the Foxp3 locus to ensure stable Foxp3 expression, the degree of CpG demethylation and levels of Foxp3 expression are more variable in iTregs (8). So called ‘Ex-Treg cells’ have been detected in inflamed tissues during autoimmune conditions (9). These cells have lost Foxp3 expression and contain highly methylated CpG islands at the Foxp3 locus (9).
Expression levels of Foxp3 are also controlled by reversible acetylation of the Foxp3 protein. Various cellular acetyltransferases and deacetylases have been implicated in Foxp3 acetylation/deactylation in the past but precise acetylation sites have not been conclusively determined. Intraperitoneal injection of trichostatin A (TSA), an inhibitor specific for class I and II histone deacetylases (HDACs), or knockout of the HDAC9 gene in mice was shown to cause an increase in number and suppressive activity of Tregs (10). In addition, acetylation levels of Foxp3 were found to be increased in CD4+CD25+ T cells isolated from TSA-treated mice (10). Foxp3 was shown to interact with the acetyltransferase Tip60 and with HDAC7, and this interaction was found necessary for the suppression of IL-2 production in Jurkat T cells (11). TCR ligation and TGF-β signaling enhanced binding of acetylated FoxP3 to the IL-2 promoter in human Tregs (12). Recent studies have also implicated the p300 acetyltransferase and the class III HDAC SIRT1 in the regulation of Foxp3 acetylation (13-15).
SIRT1 is one of seven mammalian homologs of the yeast transcriptional repressor silent information regulator 2 (sir2), an important regulator of aging in yeast (16). Like sir2, SIRT1 is a NAD+-dependent histone deacetylase with many nonhistone targets, including transcription factors NF-κB, p53, and Foxo proteins (16). Deacetylation by SIRT1 modulates the catalytic activity of substrate proteins as well as protein stability and protein:protein interactions, thereby participating in many important cellular responses such as cell cycle, senescence, apoptosis, inflammation, and stress responses (16-18). In the immune system, SIRT1 functions as a negative regulator of CD4+ T-cell activation by repressing the pro-inflammatory activity of the transcription factor NF-κB, and this negative regulatory function was antagonized by the viral Tat protein during HIV infection (19). In mice, SIRT1 suppresses activation and proliferation of CD4+ effector T cells through deacetylation and inactivation of AP-1 transcription factors (20).
To gain insight into the physiological role of Foxp3 acetylation, we used mass spectrometry to determine acetylation sites in the cellular Foxp3 protein. We identify three novel lysine residues that are targets of the deacetylase activity of SIRT1. We performed detailed in vitro and in vivo studies and show that these sites regulate the stability of the Foxp3 protein, thus directly influencing the suppressive function of Tregs.
Materials and Methods
Cells and plasmids
293T, Jurkat cells were cultured in DMEM and RPMI media containing 10 % FBS and 1% penicillin/streptomycine (v/v), respectively. Primary mouse T cells were cultured in RPMI-1640 media containing 50 μM β-mercaptoethanol, 1% penicillin/streptomycine (v/v), 2mM HEPES, and 10% FBS (v/v), all at 37°C in a 5% CO2-humidified incubator. Mutant constructs for Foxp3 acetylation sites were generated by site-directed mutagenesis with FLAG-tagged Foxp3 as a template. The FLAG-tagged Foxp3 deletion constructs and AML1/Runx1 were kindly provided by S. Sakaguchi (Kyoto University), the pSico-GFP plasmids by T. Jacks (MIT), and the MSCV-IRES-Thy1.1 construct by J. Bluestone (UCSF).
Mass spectrometric analysis of Foxp3
To determine target acetylation sites of Foxp3, cellular Foxp3 was prepared in 293T cells. Mouse and human FLAG-Foxp3 were expressed in 293T cells by transient transfection and immunoprecipitated by α-FLAG agarose. Foxp3 proteins were released from beads by incubating with excessive FLAG peptides. Eluted Foxp3 protein was resolved in SDS-PAGE gels, and Foxp3 bands were excised from the gel for analysis. Tryptic digestions and extractions were performed as described with adaptation to the volume of the gel plugs (21). After extraction, the samples were vacuum dried and dissolved in 0.1% TFA by sonication for 15 min. Peptide separation was achieved using a nanoAcquity UPLC system (Waters GmbH, Eschborn, Germany). Briefly, peptides were trapped on a nanoAcquity C18 column, 180 mm × 20 mm, particle size 5 μm (Waters GmbH). The liquid chromatography separation was performed at a flow rate of 400 nl/min on a BEH 130 C18 column, 100 mm × 100 μm, particle size 1.7 μm (Waters GmbH). The following linear gradient was applied: from 0 to 4% B in 1 min, from 4 to 30% B in 80 min, from 30 to 45% B in 10 min, from 45 to 90% B in 10 min, 10 min at 90% B and from 90 to 0% B in 0.1 min. Solvent A contained 98.9% water, 1% acetonitrile, 0.1 % formic acid; solvent B contained 99.9% acetonitrile and 0.1% formic acid. The nanoAcquity UPLC system was coupled online to a LTQ Orbitrap XL mass spectrometer (Thermo Fisher Scientific, Waltham, MA, USA). Electrospray ionization was achieved using a PicoTip Emitter Silica Tip (New Objective, Woburn, USA) and a spray voltage of 1.7 kV. Data dependent acquisition using Xcalibur 2.0.6 (Thermo Fisher Scientific) was performed by one FTMS scan with a resolution of 60,000 and a range from 370 to 2000 m/z in parallel with six MS/MS scans of the most intense precursor ions in the ion trap. The mgf files were used for database searches with the MASCOT search engine (Matrix Science, London, UK) against the NCBInr database. The peptide mass tolerance for database searches was set to 5 ppm and fragment mass tolerance to 0.6 Da. Carbamidomethylation of C was set as a fixed modification. Variable modifications included oxidation of M and acetylation at K. Two missed cleavage sites were allowed in case of incomplete trypsin hydrolysis.
Generation of modification-specific Foxp3 antibodies
The α-AcK31 and α-AcK262 Foxp3 antibodies were generated in rabbits immunized with chemically synthesized K31 and K262 acetylated peptides (AcK31 peptide, KTAPKAcGSELLC; AcK262 peptide, AHLAGKAcMALAC) in a 77-day rabbit peptide protocol (Covance, Princeton NJ). Antibodies were purified on affinity columns (Affi-Gel 10 and 15, BioRad)(Hercules, CA, USA) loaded with the respective immunogen as described (22).
Infection with lentiviral vectors expressing wildtype and mutant Foxp3
To express wildtype (WT) or 3KR mutant Foxp3 in Jurkat T cells, lentiviral vectors were constructed using pSico-GFP as a backbone (23). The EF-1α promoter was inserted upstream of U6 promoter and the U6-CMV promoter-EGFP fragment was replaced with either IRES-Thy1.1 or wildtype or 3KR mutant Foxp3-IRES-Thy1.1 fragments. All clones were fully sequenced. Pseudotyped Foxp3-expressing lentiviral vectors as well as empty control vectors were prepared in 293T cells and were used in infections of Jurkat T cells as described (19). Four days after infection, Jurkat T cells (102) were treated with cycloheximide (CHX, 12.5 μM) and incubated for 2, 4, or 6 hrs. Foxp3 expression was measured from infected cells by intracellular staining using α-Foxp3-APC antibodies (eBioscience, San Diego, CA) after surface staining with α-Thy1.1-PE antibodies. Thy1.1+ cells were gated and MFI values of Foxp3 were measured using FlowJo program (Tristar Inc, Phoenix, AZ).
Retroviral transduction with wildtype or 3KR mutant Foxp3, cytokine staining, and suppression assays
The open reading frames corresponding to wildtype and 3KR mutant Foxp3 were inserted upstream of the IRES into the MSCV-IRES-Thy1.1 vector, which is a murine stem cell virus (MSCV)-based retroviral vector that expresses Thy1.1 as a surface marker. To obtain recombinant virus, 10 μg of each construct or empty control vector were transfected into phoenix-eco cells, a retroviral packaging cell line derived from 293T cells. The supernatants were collected 48 hrs after transfection and filtered through a 0.45 μm membrane. Naïve CD4+ T cells (>97% purity) isolated from spleen and lymph nodes (SP/LN) of C57BL/6 mice were activated with α-CD3/CD28 antibodies for 24 hrs. Activated T cells were infected with retroviral supernatants in the presence of polybrene (6 μg/ml) by spinoculation and further activated with α-CD3/CD28 antibodies and recombinant human IL-2 (rhIL-2, 100 U/ml) for 5 days. Cells were stained with α-CD4, α-CD25, α-CTLA-4 and α-Thy1.1 antibodies for surface staining as well α-Foxp3-PE antibodies for intracellular staining (eBioscience). For staining of cytokine-secreting cells, infected cells were further activated with phorbol myristate acetate (PMA, 50 ng/ml; Sigma-Aldrich)(St. Louis, MO, USA) and ionomycin (1 μM; Sigma-Aldrich) for 4 hrs in the presence of brefeldin A (Sigma-Aldrich). Cells were fixed, permeabilized, and stained with PE-conjugated antibodies against IL-2, IL-10, and IL-13 (all from eBioscience). For suppression assays, Thy1.1+ T cells were positively isolated using α-Thy1.1-PE and α-PE Microbeads (Miltenyi Biotech, Bergisch Gladbach, Germany) and further sorted by FACSAria III (BD Biosciences) with 98% purity. Naive CD4+ T cells (>97% purity) were isolated from SP/LN of Boy/J mice as responder T cells and labeled with CFSE (5-(and-6)-carboxyfluorescein diacetate succinimidyl ester, (Invitrogen, Grand Island, NY). 50,000 sorted Thy1.1+ cells were co-cultured with 50,000 responder T cells (1:1 ratio) in a 96-well round bottom plate in the presence of α-CD3/CD28 antibodies and rhIL-2 (100 U/ml) for 3 days. Cells were then stained with α-CD4 and α-CD45.1 and analyzed by FACS.
Differentiation of mouse CD4+ T cells into iTregs in vitro
All animal studies have been reviewed and approved by the UCSF IACUC. Naive T cells (CD4+CD25−CD69−CD44−) were isolated from SP/LN of mice by depletion of non-CD4+ T cells with the CD4+ T Cell Isolation Kit (Miltenyi Biotech), and subsequent depletion of CD4+CD25+CD69+CD44+ T cells by α-CD25-PE, α-CD69-PE, α-CD44-PE, and α-PE Microbeads (Miltenyi Biotech). Naive CD4+ T cells were differentiated into iTregs with plate-bound α-CD3 (5 μg/ml), soluble α-CD28 (2 μg/ml), rhIL-2 (20 U/ml), and TGF-β (concentration?) for 3 or 5 days. In some experiments, CFSE-labeled naive CD4+ T cells were differentiated into iTregs in the presence of different concentrations of TGF-β (0.1–10 ng/ml) to achieve suboptimal conditions for iTreg differentiation for 3 days. To evaluate the effects of SIRT1 inhibition on Treg differentiation, Ex-527 and DMSO was added during iTreg differentiation as indicated.
Measurement of Foxp3 mRNA
RNA purification, cDNA synthesis, and real-time RT-PCR was performed as described (19). Mouse Foxp3 mRNA was quantified by QuantiTect gene expression assays (Qiagen) with SYBR Green I master mix (MCLab). Relative Foxp3 gene expression ratios between Ex-527 treated and non-treated samples was normalized to GAPDH expression and then calculated by the equation, ratio = 2-ΔΔCt, comparative Ct method (Applied Biosystems). Real-time RT-PCR analysis was performed in duplicate on three independent samples.
Transfections and coimmunoprecipitations
293T cells grown in 6-well plates at 70% confluency were cotransfected with expression vectors indicated in the figure legends with Lipofectamine (Invitrogen) or FuGene 6 (Roche). 24 hrs after transfection, cells were lysed and processed for coimmunoprecipitation and western blotting as described (24). Statistical analysis was carried out using paired T-tests (significance = P<0.05).
Adoptive transfer of iTregs into mice
Naive CD4+ T cells isolated from SP/LN of GFP-Foxp3 knock in mice in a (C57BL/6 background) were differentiated into iTregs as described above. CD4+GFP+ T cells were sorted by FACSAria III with >98% purity. 105 sorted cells were transferred into Boy/J recipient mice as a single retro-orbital injection. DMSO or Ex-527 (40 mg/kg) were injected intraperitoneally into adoptively transferred mice every day for 2 weeks. CD4+ T cells were isolated from the draining lymph nodes (pancreatic and mesenteric lymph nodes) of these mice. To enrich adoptively transferred iTregs (CD4+CD45.2+ T cells), cells from recipient mice (CD4+CD45.1+ T cells) were depleted by positive selection after staining cells with α-CD45.1 antibodies. The cells were then stained with α-CD4, α-CD45.1, α-CD45.2, and α-Foxp3 antibodies. Foxp3 expression was analyzed by FACS and FlowJo program.
Isolation and ex vivo expansion of mouse nTregs
GFP-Foxp3 transgenic mice were used to isolate nTregs (25). CD4+GFP+ cells were sorted from total CD4+ T cells isolated from SP/LN of GFP-Foxp3 knock in mice by FACSAria III high-speed cell sorter (BD Biosciences) with 95% purity. FACS isolated GFP+ nTregs were plated at 4 × 105 cells per well in a 24-well plate and activated with RPMI complete medium containing rhIL-2 (2000 U/ml) and α-CD3/CD28-coated microbeads (Invitrogen) at a 2:1 bead-to-cell ratio. Five days after incubation, cells were treated with either Ex-527 (50 μM) or DMSO and continuously cultured for an additional 2 or 5 days. Foxp3 levels were measured by intracellular staining as described above from GFP+ cells.
Results
K31, K262, and K267 are acetylated in Foxp3
To map the acetylation sites in Foxp3, we performed mass spectrometry on cellular Foxp3 proteins isolated from 293T cells. Both, mouse and human, Foxp3 proteins were transiently overexpressed as FLAG-tagged fusion proteins. To enhance acetylation levels, the p300 acetyltransferase was coexpressed, and cells were treated with a combination of TSA and nicotinamide to block class I–III HDACs. Foxp3 was immunoprecipitated using α-FLAG agarose, eluted with FLAG peptides and analyzed by mass spectrometry. Peptides recovered in the analysis corresponded to 78 or 82 % of human or mouse Foxp3, respectively, and corresponding acetylation sites were identified in both samples. Three acetylated lysines were identified: K31, K262, K267 in mouse Foxp3 and the corresponding sites K31, K263 and K268 in human Foxp3 (Figure 1).
Figure 1. Identification of acetylation sites in Foxp3 by mass spectrometry.

Sequence of murine and human Foxp3. The letters in bold black indicate the peptides recovered by mass spectrometry. Boxed residues mark acetylated residues identified in the analysis. The underlined regions represent the subdomains of Foxp3 as indicated. * : lysine residues not covered by our analysis. Numbers for acetylated lysine residues correspond to mouse Foxp3 protein.
Next, we generated modification-specific antibodies against acetylated K31 in mouse Foxp3. Antigen-purified antibodies were tested in a dot blot analysis; they recognized only the acetylated antigen, and did not cross-react with unmodified peptide or acetylated K262 or K267 peptides (Figure 2A). We then expressed FLAG-Foxp3 or a K31R mutant protein in 293T cells and treated cells with TSA, nicotinamide or Ex-527, the latter a specific inhibitor of SIRT1(26). Foxp3 proteins were immunoprecipitated with α-FLAG agarose, followed by western blotting with α-AcK31 antibodies. Acetylation at K31 was detected in cells treated with Ex-527 or nicotinamide, but not with TSA, indicating that K31 is a target site of SIRT1 (Figure 2A, lanes 1–4). No signal was detected in lysates expressing mutant K31R Foxp3, demonstrating the specificity of the antiserum for acetylated Foxp3 in cells (Figure 2B, lanes 5–8). Similar results were obtained with an antiserum raised against acetylated K262 in Foxp3 (Supplemental Figure 1A and B). Attempts to generate a specific antiserum for acetylated K267 were unsuccessful (not shown).
Figure 2. Generation of a modification-specific antiserum against acetylated Foxp3.
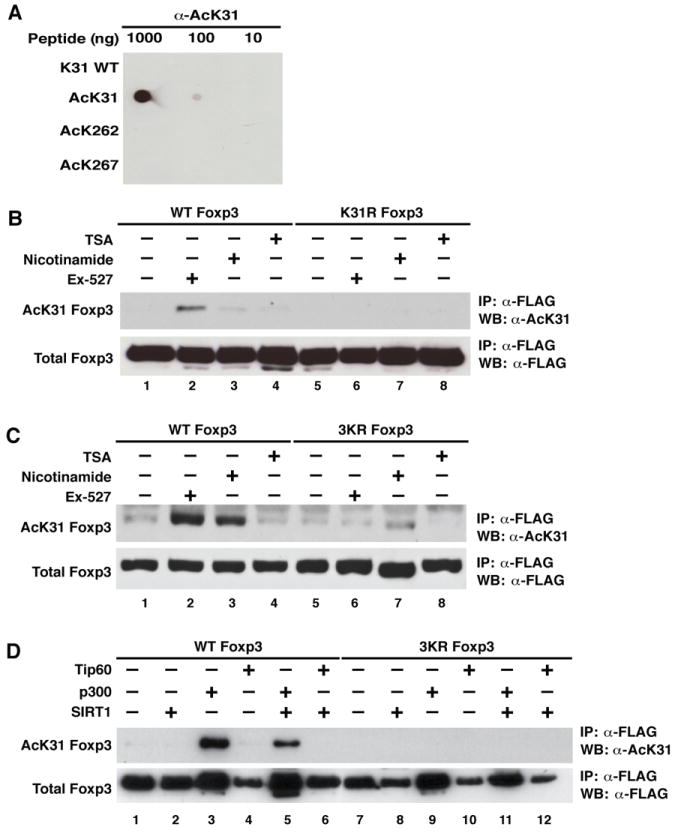
(A) Dot blotting of acetylated and unacetylated Foxp3 peptides using affinity-purified acetylated K31 Foxp3 antibodies (α-AcK31). Decreasing amounts (1000, 100, and 10 ng) of each peptide were spotted onto nitrocellulose membranes and processed by western blotting (WB) with α-AcK31 Foxp3 antibody. (B) Immunoprecipitation/western blot analysis of acetylated FLAG-Foxp3. Expression vectors for wildtype or K31R mutant FLAG-Foxp3 were transfected into 293T cells, which were subsequently treated overnight with Ex-527 (50 μM), an inhibitor of SIRT1; nicotinamide (5 mM), an inhibitor of sirtuins; and trichostatin A (TSA, 400 nM), an inhibitor of class I and II HDACs. Immunoprecipitations were performed with α-FLAG agarose followed by western blotting with α-AcK31 Foxp3 and α-FLAG antibodies. (C) Same procedure as in (B) performed in Jurkat T cells infected at a high multiplicity of infection (m.o.i.) with lentiviral vectors expressing FLAG-tagged wildtype or 3KR mutant Foxp3. Four days after infection, cells were treated as in (B). (D) Same procedure as in (B) performed in 293T cells after co-transfection of expression vectors for FLAG-tagged wildtype or 3KR mutant Foxp3 together with p300, SIRT1, and Tip60. One representative of three independent experiments is shown.
Similar results were obtained in Jurkat T cells infected with lentiviral vectors expressing wildtype Foxp3 or a mutant protein in which all three acetylation sites were replaced by arginines (3KR). Acetylation of K31 was robustly induced in response to treatment with Ex-527 or nicotinamide in wildtype Foxp3-expressing cells (Figure 2C, lanes 1–4), while only background signal was detected in lysates expressing the 3KR mutant (Figure 2C, lanes 5–8). No effect was observed after TSA treatment, indicating the K31 is deacetylated by SIRT1 in T cells.
To determine which acetyltransferase enzyme acetylates K31 in cells, we coexpressed FLAG-Foxp3 or the 3KR mutant Foxp3 in 293T cells together with p300 and Tip60, two acetyltransferases previously implicated in Foxp3 acetylation (11,13,14). Acetylation of K31 in Foxp3 was enhanced by coexpression of p300, but not by Tip60, pointing to p300 as the K31 acetyltransferase in cells (Figure 2D, lanes 3 and 4). Coexpresssion of SIRT1 decreased p300-induced acetylation of K31 (lane 5), while no acetylation was observed in 3KR-expressing cells, as expected (lanes 7–12). Similar results were obtained when K262 acetylation was analyzed, indicating that p300 and SIRT1 acetylate and deacetylate these two sites in Foxp3, respectively (Supplemental Figure 1C).
Our data identify three new acetylation sites in Foxp3: K31, K262 and K267. Acetylation of K31 and K262 was confirmed by modification-specific antibodies for Foxp3 and is regulated by p300 and SIRT1. Notably, two of the newly identified sites, K262 and K267, are located close to a C-terminal SIRT1-binding region in Foxp3 (amino acids (aa) 279–337) that we identified in coimmunoprecipitation experiments (Supplemental Figure 2).
Dynamic regulation of Foxp3 acetylation during iTreg differentiation
Next, we examined physiological acetylation levels of Foxp3 in iTregs cultures. iTregs were ex vivo differentiated from naive CD4+ T cells in the presence of α-CD3/CD28 antibodies and TGF-β as described (27). At different time points during the differentiation process, samples of the T cells were harvested, and endogenous Foxp3 protein was immunoprecipitated and subjected to western blotting with α-AcK31 and Foxp3 antibodies. Foxp3 expression was detected after 24 hrs of differentiation and peaked at 72–96 hrs (Figure 3A). Acetylation of K31 was detected at 72 and 96 h, coinciding with maximal cellular Foxp3 accumulation (Figure 3A). At 120 hrs, acetylation of K31 disappeared, but was rescued by treatment with Ex-527 (Figure 3B). These data demonstrate physiological acetylation of K31 in Treg cells and underscore the critical role of SIRT1 in Foxp3 deacetylation.
Figure 3. The acetylation level of Foxp3 is regulated in iTregs.
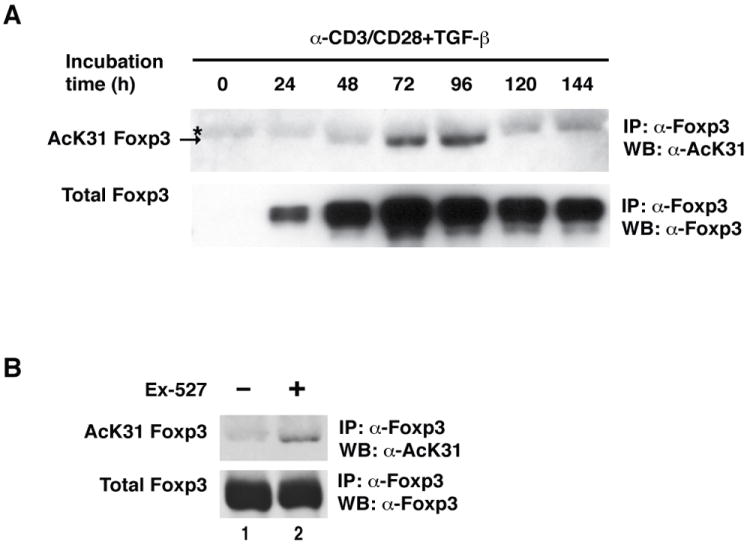
(A) Immunoprecipitation/western blot analysis of endogenous acetylated Foxp3 in iTregs. Mouse iTregs were differentiated from naive CD4+ T cells as described in Materials and Methods. Cells were harvested at the indicated time points and subjected to immunoprecipitation with α-mouse Foxp3 antibodies and analyzed by western blotting with α-Foxp3 and α-AcK31 Foxp3 antibodies. * Indicates non-specific bands. The arrow points to acetylated Foxp3. (B) iTreg cells were differentiated for 120 hrs and treated with Ex-527 (50 μM) overnight. Data are representative of two independent experiments.
Deacetylation by SIRT1 destabilizes Foxp3
We were intrigued by the finding that Foxp3 acetylation and Foxp3 levels correlated in differentiating iTregs. In addition, we noticed in the experiments depicted in Figure 2 that the 3KR mutant protein accumulated to higher levels than the wildtype protein. Protein levels of wildtype and mutant Foxp3 were equalized in these experiments to allow proper assessment of acetylation. We speculated that the newly identified acetylation sites may be subject to an “acetylation/ubiquitinylation switch” as described for PER2, BAML1, and Tau proteins (28-30) as well as recently for Foxp3 (13). These studies showed that acetylation and ubiquitinylation target the same lysine residue, whereby acetylation blocks ubiquitinylation and prevents protein degradation. Conversely, deacetylation of lysine residues enhances their susceptibility to ubiquitinylation, leading to accelerated protein degradation. Mutation of such lysine residue prevents both acetylation and ubiquitinylation and may result in a paradoxical increase in protein stability.
To test the hypothesis that the three lysines are involved in Foxp3 protein stabilization, we quantified protein expression of wildtype and 3KR mutant Foxp3 proteins in infected Jurkat T cells by intracellular staining and flow cytometry. Levels of Foxp3 protein per cell (measured as mean fluorescence intensity (MFI)) was ~twofold increased in cells expressing the 3KR mutant as compared to wildtype Foxp3 (Figure 4A). This corresponded to a ~fivefold increase in protein expression measured by western blotting (Figure 4B). Because cells were infected with a bicistronic vector simultaneously expressing Foxp3 and the Thy1.1 surface marker from the same mRNA, we also measured Thy1.1 surface levels by flow cytometry. MFI values of Thy1.1 were unchanged in cells expressing wildtype or mutant Foxp3 supporting the model that changes in Foxp3 expression occurred at a posttranscriptional level (Figure 4A).
Figure 4. SIRT1 destabilizes Foxp3 protein through deacetylation of three newly identified acetylation sites.
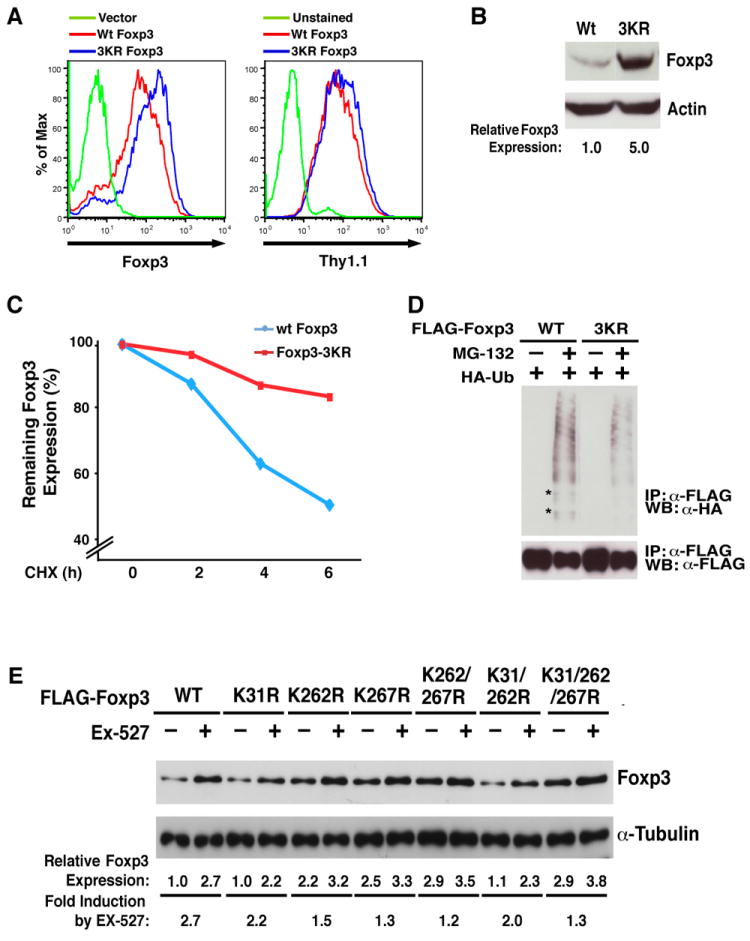
(A) Intracellular staining of wildtype and 3KR mutant Foxp3 proteins expressed in Jurkat T cells after lentiviral transduction. Four days after infection, Foxp3 was measured by intracellular staining using α-Foxp3-APC antibody after staining of cell surface Thy1.1 using α-Thy1.1-PE antibody. Data are representative of three independent experiments in which infection efficiencies ranged from 85–95% as measured by Thy1.1+ cells. (B) Western blot analysis of Foxp3 in Jurkat T cell lysates from (A) with α-Foxp3 and α-actin antibodies. (C) Pulse-chase experiment in Jurkat T cells infected with lentiviral vectors expressing wildtype or 3KR mutant Foxp3 along with Thy1.1. Four days after infection, infected cells were treated with cycloheximide (CHX, 12.5 μM) for 2, 4, or 6 hrs. Foxp3 protein expression was measured by intracellular staining using α-Foxp3-APC antibodies after staining of cell surface Thy1.1 using anti-Thy1.1-PE. The MFI values of Foxp3 were measured in Thy1.1+ populations. Data are presented relative to MFI values obtained in cells without CHX (100%). (D) 3KR mutant Foxp3 protein is less ubiquitinylated. Expression vectors for wildtype and 3KR Foxp3 were co-transfected with HA-Ubiquitin (HA-Ub) into 293T cells. 16 hrs after transfection, cells were treated with MG-132 (10 μM), a proteasome inhibitor for 4 hrs. Foxp3 proteins were immunoprecipitated with α-FLAG agarose followed by western blotting with α-HA (ubiquitin) and α-FLAG (Foxp3) antibodies. Some ubiquitinylated bands that were underrepresented in cells expressing the 3KR mutant are highlighted by *. (D) Western blot analysis of wildtype or mutant FLAG-Foxp3 proteins in 293T cell lysates treated with Ex-527 (50 μM) or DMSO as a control with α-FLAG and α-Tubulin antibodies. One representative experiment of 4 independent experiments is shown.
This model was further confirmed when we measured the half-life of wildtype and 3KR mutant proteins in Jurkat cells. While 50% of wildtype Foxp3 protein was degraded after 6 hrs (t1/2 = 6 hrs), 80% of 3KR mutant protein remained in cells at that time (Figure 4C). Consistent with the hypothesis that the three lysines are also subject to ubiquitinylation, the 3KR mutant was less ubiquitinylated in cells than wildtype Foxp3 (Figure 4D). These data demonstrate that the three newly identified lysines function as targets for the described acetylation/ubiquitinylation switch in Foxp3.
To study the role of SIRT1 in this process, we treated 293T cells transiently expressing a series of mutant Foxp3 proteins with Ex-527. Ex-527 increased expression of wildtype Foxp3 protein by 2.7-fold, supporting the model that hyperacetylation induced by SIRT1 inhibition stabilized Foxp3 (Figure 4E; WT). The 3KR mutant FoxP3 protein showed a ~threefold increase in basal expression in this experiment; expression only increased by 1.3-fold in response to Ex-527 treatment, consistent with the notion that these sites are major SIRT1 deacetylation sites in Foxp3 (Figure 4E, last two lanes). Expression of Foxp3 was also increased when single mutations were introduced at position 262 and 267, and the double mutation reached similar expression levels as the 3KR mutant, pointing to these two sites as the main residues involved in the stabilization of Foxp3 (Figure 4E). Of note, expression levels of the K31 single mutant protein varied; in half of the experiments, no changes were observed as depicted in the Figure, while in the other half, a twofold increase was observed (not shown).
Enhanced suppressive function of 3KR mutant Foxp3
To examine the influence of the three sites on Foxp3 suppressive activity, primary CD4+ T cells were purified from mice and transduced with lentiviral vectors expressing wildtype or 3KR mutant Foxp3. Thy1.1 was bicistronically coexpressed along with Foxp3, and control cells were transduced with vectors expressing Thy1.1 alone. The 3KR mutant accumulated to higher levels than wildtype protein within CD4+ T cells as shown by intracellular staining and flow cytometry of Thy1.1+ cell populations (Figure 5A). Similarly, the MFI of CD25 and CTLA-4, two characteristic Treg-associated cell-surface markers, was increased in cells expressing 3KR mutant Foxp3 (Figure 5A). Infected cells were also activated with a combination of phorbol esters and ionomycin, and intracellular staining for several cytokines associated with Treg function was performed. The secretion of IL-2 and IL-13 was more potently suppressed in cells expressing the 3KR mutant than in wildtype Foxp3-expressing cells, pointing to increased suppressive activity of the 3KR mutant (Figure 5B). In agreement with this model, more IL-10 producing cells were detected in cell populations transduced with the 3KR mutant (Figure 5B).
Figure 5. The suppressive function of Foxp3 is enhanced by the 3KR mutation.
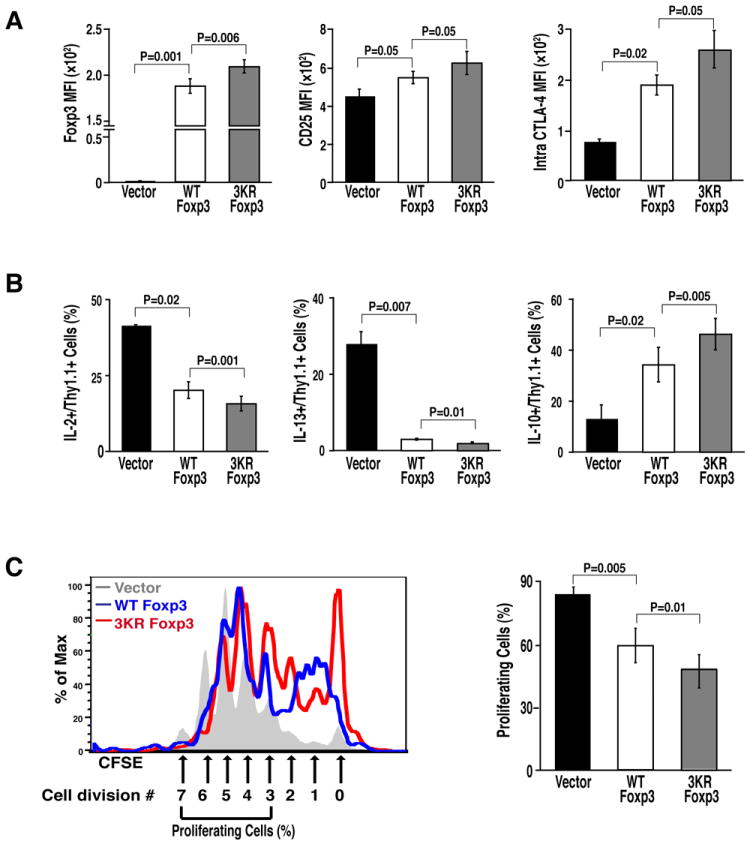
(A) Flow cytometric analysis (MFI) of Foxp3, CD25, and intracellular CTLA-4 in murine CD4+ T cells transduced with retroviral vectors expressing wildtype or 3KR mutant Foxp3. (B) Flow cytometric analysis (%) of cytokine-secreting cells from transduced T cells after activation with α-CD3/CD28 antibodies. Data in (A) and (B) represent the average (mean ± SEM) of three independent experiments. (C) Suppression assays of wildtype Foxp3 or the 3KR mutant. Naive mouse CD4+ T cells were infected with retroviral vectors expressing wildtype Foxp3 or the 3KR mutant along with control retrovirus after activation by α-CD3/CD28 antibody for 24 hrs as above. Infected cells were sorted and co-cultured with CFSE-labeled responder T cells for 3 days. Proliferation of responder T cells was assessed by CFSE dilution within the responder T cell population. The left panel shows one representative FACS plot. The proliferating cells were presented as cells having divided more than 3 cycles. The right panel shows the data represented as the mean ± SEM of five independent experiments.
To test the suppressive activity of these cells directly, Thy1.1+ cells were sorted from infected T-cell cultures and were co-cultured with CFSE-labeled CD4+ effector T cells. Cells expressing the 3KR mutant inhibited the proliferation of responder T cells more potently than cells expressing wildtype Foxp3 or vector-transduced cells as shown by a lack of dilution of the CFSE fluorescent marker (Figure 5C). These data indicate that enhanced expression of 3KR mutant Foxp3 correlates with better suppressive function of T cells expressing this protein.
Hyperacetylation of Foxp3 promotes differentiation of T cells into Tregs
We speculated that these observations may have therapeutic relevance and that hyperacetylation of Foxp3 may increase the yield and stability of Foxp3-expressing T cells generated ex vivo. Naive CD4+ T cells were isolated from mouse spleen and lymph nodes and were activated with α-CD3/CD28 antibodies in the presence of IL-2 and increasing amounts of TGF-β to differentiate them into iTregs. Addition of Ex-527 increased the number of Foxp3-expressing iTregs even in the absence of exogenous TGF-β (Figure 6A). Furthermore, Ex-527 synergized with low concentrations of exogenous TGF-β (0.1 and 0.5 ng/ml) to induce differentiation of iTregs. A synergy with Ex-527 was no longer observed at higher concentrations of TGF-β. No effect of Ex-527 was observed on cell proliferation as measured by CFSE labeling except in cells co-treated with TGF-β at concentrations higher than 1 ng/ml (Figure 6A) Similar results were obtained when iTreg differentiation was induced in the absence of IL-2 (data not shown).
Figure 6. Inhibition of SIRT1 activity by Ex-527 promotes differentiation of iTregs.
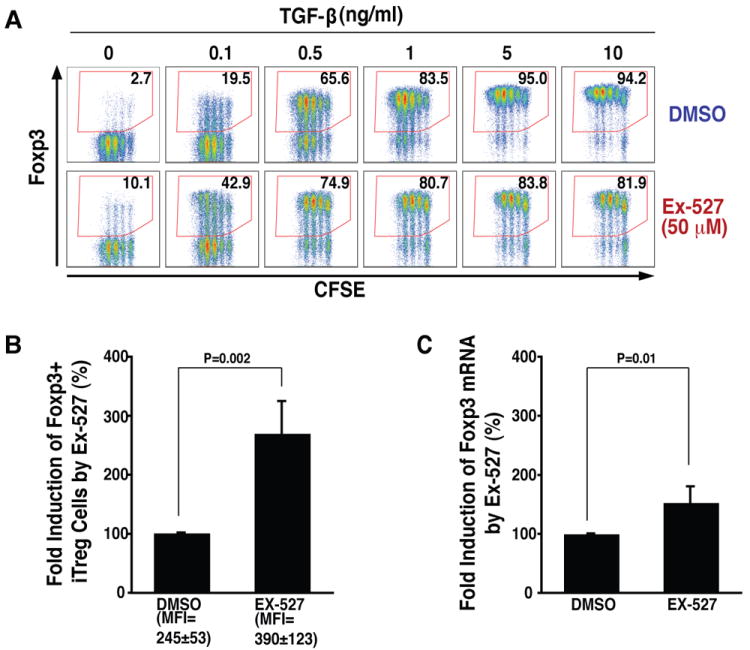
(A) Intracellular flow cytometric analysis of Foxp3 expression in iTregs differentiated at indicated concentrations of TGF-β in the presence and the absence of Ex-527 (50 μM). One experiment performed in triplicate is shown. (B) Intracellular flow cytometric analysis of Foxp3+ cells and MFI of intracellular Foxp3 in iTregs after treatment with Ex-527 (50 μM) or DMSO control during differentiation from naive T cells. (C) Real-time RT-PCR analysis of Foxp3 mRNA levels in iTregs described in (B). Data in (B) and (C) are presented as fold induction by Ex-527 treatment relative to DMSO vehicle (100%). The averages (mean ± SD) of three independent experiments are shown.
The increased yield of Foxp3-expressing T cells was accompanied by an increase in the expression level of Foxp3 (MFI), supporting the model that hyperacetylation induced by Ex-527 stabilizes Foxp3 protein in iTregs (Figure 6B). However, we also measured a slight increase in Foxp3 mRNA levels, pointing to an additional transcriptional effect of Ex-527 during iTreg differentiation (Figure 6C).
Ex-527 treatment stabilizes Foxp3 in iTregs and nTregs
To examine the effect of Ex-527 in vivo, we performed adoptive transfer experiments of iTregs into mice. iTregs were generated from CD4+ T cells isolated from Foxp3-GFP knock-in mice. This mouse line expresses a fusion protein composed of Foxp3 and GFP from the endogenous Foxp3 promoter, and Foxp3 expression can be monitored by flow cytometry of GFP in live cells (25). GFP+ cells were sorted after differentiation in vitro and transferred into Boy/J recipient mice by retro-orbital injection. Mice were treated with either Ex-527 or carrier control by intraperitoneal injection for two weeks. CD4+ T cells were isolated from pancreatic and mesenteric lymph nodes of Boy/J mice, and Foxp3 expression was analyzed by intracellular Foxp3 staining in recipient and donor Treg cell populations with different CD45 allele markers. After two weeks, only 50% of transferred iTregs still expressed Foxp3 in control mice as compared to 90% in mice treated with Ex-527 (Figure 7A, lower panel). The MFI of Foxp3 was markedly higher in Treg isolated from Ex-527-treated mice as compared to control-treated littermates (MFI=463 versus 122). Ex-527 also increased the MFI of Foxp3 in Tregs isolated from recipient Boy/J mice (MFI=419 versus 205); however, no change in total Treg numbers was noted in recipient mice (Figure 7A, upper panel). These results indicate that inhibition of SIRT1 deacetylase activity by Ex-527 prevents the loss of Foxp3 protein and increases the stability of iTregs in vivo.
Figure 7. Ex-527 promotes the stability of iTregs in vivo and nTregs ex vivo.
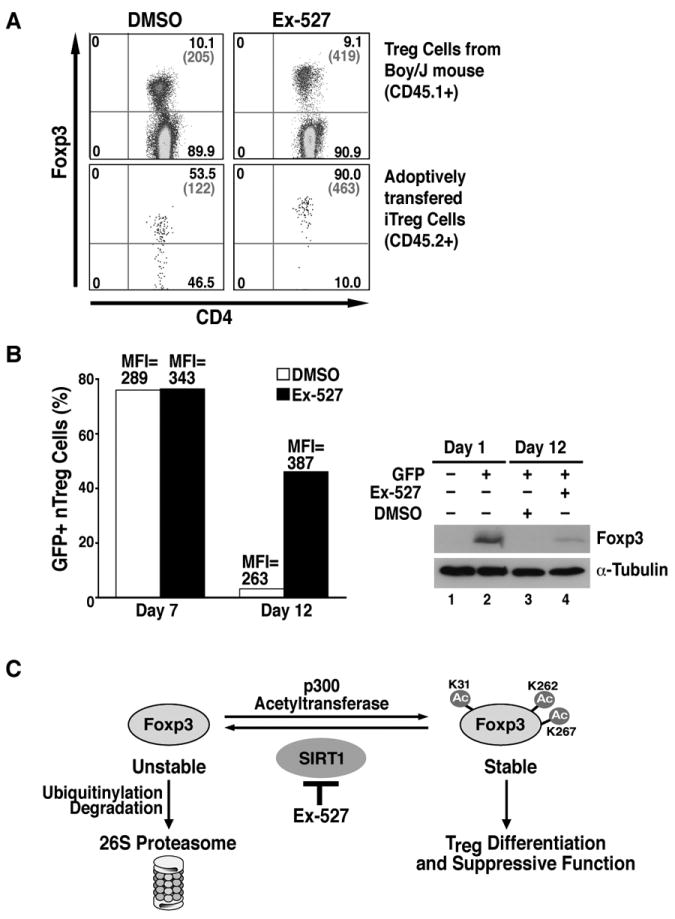
(A) Flow cytometric analysis of CD4 and Foxp3 (% in black and MFI in gray) in Tregs isolated from the draining lymph nodes of Boy/J mice 2 weeks after adoptive transfer. Highly sorted iTregs differentiated in vitro from naive CD4+ T cells were transferred into Boy/J mice by retro-orbital injection. DMSO and Ex-527 were injected intraperitoneally into these mice every day for 2 weeks. The data shows one representative experiment of three independent experiments with similar results. (B) The left panel shows flow cytometric analysis of GFP in nTregs isolated from GFP-Foxp3 knock-in mice after ex vivo expansion with or without DMSO. The right panel shows western blot analysis of Foxp3 in cell lysates from GFP- and GFP+ cultures before and after expansion. We show one representative experiment of two independent experiments with similar results. (C) Model of how inhibition of SIRT1 deacetylase activity by Ex-527 increases the acetylation at three newly identified lysine residues in Foxp3 and promotes Treg differentiation and stability. See text for details.
Finally, we tested the effect of Ex-527 on the stability of nTregs during ex vivo expansion. Total CD4+ T cells were isolated from spleen and lymph nodes of GFP-Foxp3 knock-in mice, and GFP+ nTregs were sorted with >95% purity. GFP+ cells were cultured in the presence of α-CD3/CD28 beads and IL-2 for 5 days when Ex-527 or carrier control was added. Two days later, 75% of cells were GFP+ regardless of whether Ex-527 was added or not; however, the MFI of GFP was increased in Ex-527-treated cells, pointing to stabilization of Foxp3 protein levels in the absence of SIRT1 activity (Figure 7B, day 7). At day 12 post-isolation, only 5% of cells remained GFP+ in control-treated cells versus 45% in Ex-527-treated cultures (Figure 7B, day 12). The level of Foxp3 expression remained higher in Ex-527-treated versus control-treated cells by flow cytometry (MFI=387 versus 263) and in western blot analysis (Figure 7B, right panel). These data underscore the negative role of the SIRT1 deacetylase activity on Foxp3 expression in the natural context of induced or natural Treg cells.
Discussion
Our study identifies three novel acetylation sites in Foxp3 that regulate expression of the Foxp3 protein in induced and natural Treg cells. The three sites are targeted by the SIRT1 deacetylase, a process that destabilizes Foxp3 expression (Figure 7C). Paradoxically, 3KR mutant Foxp3, which cannot be acetylated, accumulates to higher levels than wildtype Foxp3 in CD4+ T cells and confers better suppressive activity to T cells than the wildtype protein. These data fit with a proposed acetylation/ubiquitinylation switch in Foxp3, where acetylation and mutation of the acetylated residues both exert a positive effect on protein stability and function (13,28-30). Accordingly, hyperacetylation induced by Ex-527, a small-molecule inhibitor of SIRT1, stabilizes cellular Foxp3 expression and promotes in vitro differentiation and in vivo stability of iTregs as well as maintenance of Foxp3 in nTregs during ex vivo expansion (Figure 7C).
One of our most striking findings is that Ex-527 can replace or synergize with low doses of TGF-β in the in vitro generation and in the in vivo maintenance of iTregs (Figure 6A). These data uncover a novel role of SIRT1 in iTreg differentiation. The cooperative effects of Ex-527 with TCR/TGF-β signaling on iTreg differentiation is comparable to the effects of all-trans retinoic acid (RA), a well-established enhancer of iTreg generation (31-37). While all-trans RA enhances Foxp3 expression in iTregs through multiple mechanisms including transcriptional regulation of the Foxp3 promoter, enhancement of TCR/TGF-β signaling, and indirect effects by modulating the function of antigen presenting cells, our combined data point to a major effect of Ex-527 in the stabilization of Foxp3 protein through hyperacetylation at the three newly defined lysines.
However, we also observed a small but reproducible increase in Foxp3 mRNA levels in response to Ex-527 in iTregs. This may result from hyperacetylation of other transcriptional regulators involved in Foxp3 gene activation in these cells. p65/RelA, a subunit of the NF-κB transcription factor as well as Foxo transcription factors and Smad3 proteins are substrates of SIRT1 (38-41) and activate Foxp3 transcription in response to TCR/TGF-β signaling (5,42,43). Therefore, Ex-527 may activate Foxp3 transcription through p65/RelA, Foxo, or Smad3 acetylation during in vitro differentiation of iTregs. In addition, Foxp3 protein stabilized by Ex-527 may promote its own transcription through a positive feed-forward regulatory loop (5,44,45).
Published results indicate that acetylation regulates the transcriptional activity of Foxp3 (i.e., by regulating the DNA-binding affinity of Foxp3) (10-12). Our results point to a major function of acetylation in regulating the stability of the Foxp3 protein. It is striking that both the hyperacetylated and acetylation-deficient Foxp3 proteins accumulate to higher levels in cells, and that the 3KR mutant shows enhanced transcriptional activity on target genes, such as IL-2 and IL-10. Our data showing that mutant Foxp3 has higher suppressive activity than wildtype protein argues against an additional positive transcriptional effect of Foxp3 acetylation at these three sites. A positive transcriptional effect would likely be abrogated by the lysine-to-arginine mutations and would result in a decrease in Treg suppressive functions. Instead, suppressive functions were enhanced, indicating that stabilization of Foxp3 may be the major effect of lysine acetylation at the three sites.
Although many reports have demonstrated that Tregs expressing low levels of Foxp3 protein or Ex-Tregs have pathogenic effects in the immune system (6,9,46,47,48,49), molecular mechanisms underlying the down-regulation of Foxp3 expression are poorly understood. Interestingly, Foxp3 mRNA is expressed at similar levels in human Treg cells expressing Foxp3 at low and high levels, indicating that regulation of FoxP3 expression may occur at the post-transcriptional level (48). Our findings that SIRT1 negatively regulates the stability of Foxp3 expression through deacetylation of three lysine residues support a model whereby downregulation of Foxp3 expression is regulated in part at the post-transcriptional level. Elevated cellular expression or selective activation of the deacetylase activity of SIRT1 during inflammatory responses could lead to enhanced deacetylation and destabilization of Foxp3 followed by conversion of Foxp3+ Treg cells into Foxp3− or Foxp3low Treg cells.
While our data provide robust evidence for the presence and functional relevance of the three newly described acetylation sites in Foxp3, it remains unclear how many other acetylation sites exist in Foxp3. Tip60 acetylates K8 in human FoxP3 (50). In addition, TSA treatment was shown to change the acetylation levels of K8, K382 and K393 in Foxp3 (10). Our analysis did not recover any peptides containing these acetylated lysines, indicating that different acetyltransferases may target different lysines in Foxp3 possibly under different biological circumstances.
Our observation that Ex-527 enhances Foxp3 expression during ex vivo Treg expansion may be relevant to ongoing efforts to enhance Foxp3 expression in therapeutic ex vivo approaches to allograft rejection or autoimmune diseases (2,7,47,51,52). A recent publication showed that intraperitoneal injection of Ex-527 in mice prolonged allograft survival, supporting the model that Ex-527 treatment is enhancing Treg activity in vivo (15). In addition, administration of Ex-527 may render infused Treg cells more resistant to conversion into effector T cells during adoptive Treg cell therapy and may boost endogenous iTreg cell generation by synergizing with natural TGF-β present within chronically inflamed tissues or transplanted tissues (4). Future studies will evaluate the potential of SIRT1 inhibitors in in vivo and ex vivo Treg-based therapeutic applications in autoimmune diseases or graft rejections.
Supplementary Material
Acknowledgments
We thank Drs. S. Sakaguchi, T. Jacks, and J. Bluestone for sharing important reagents, Ruth Getachew for technical assistance, Lukas Jerker for helpful discussions and Veronica Fonseca for administrative assistance.
We thank Andy, Sara, Ken, and Meg Kurtzig and Leslie Mulholland for generous support. M. Ott is supported by grants from the Gladstone Institute, Sandler foundation/UCSF, and the NIH (R01 AI081651).
References
- 1.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Cell. 2008;133:775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 2.Littman DR, Rudensky AY. Cell. 2010;140:845–858. doi: 10.1016/j.cell.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 3.Shevach EM. Immunity. 2009;30:636–645. doi: 10.1016/j.immuni.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 4.Curotto de Lafaille MA, Lafaille JJ. Immunity. 2009;30:626–635. doi: 10.1016/j.immuni.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 5.Josefowicz SZ, Rudensky A. Immunity. 2009;30:616–625. doi: 10.1016/j.immuni.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou L, Chong MM, Littman DR. Immunity. 2009;30:646–655. doi: 10.1016/j.immuni.2009.05.001. [DOI] [PubMed] [Google Scholar]
- 7.Zhou X, Bailey-Bucktrout S, Jeker LT, Bluestone JA. Curr Opin Immunol. 2009;21:281–285. doi: 10.1016/j.coi.2009.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lal G, Bromberg JS. Blood. 2009;114:3727–3735. doi: 10.1182/blood-2009-05-219584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou X, Bailey-Bucktrout SL, Jeker LT, Penaranda C, Martinez-Llordella M, Ashby M, Nakayama M, Rosenthal W, Bluestone JA. Nat Immunol. 2009;10:1000–1007. doi: 10.1038/ni.1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tao R, de Zoeten EF, Ozkaynak E, Chen C, Wang L, Porrett PM, Li B, Turka LA, Olson EN, Greene MI, Wells AD, Hancock WW. Nat Med. 2007;13:1299–1307. doi: 10.1038/nm1652. [DOI] [PubMed] [Google Scholar]
- 11.Li B, Samanta A, Song X, Iacono KT, Bembas K, Tao R, Basu S, Riley JL, Hancock WW, Shen Y, Saouaf SJ, Greene MI. Proc Natl Acad Sci U S A. 2007;104:4571–4576. doi: 10.1073/pnas.0700298104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Samanta A, Li B, Song X, Bembas K, Zhang G, Katsumata M, Saouaf SJ, Wang Q, Hancock WW, Shen Y, Greene MI. Proc Natl Acad Sci U S A. 2008;105:14023–14027. doi: 10.1073/pnas.0806726105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Loosdregt J, Vercoulen Y, Guichelaar T, Gent YY, Beekman JM, van Beekum O, Brenkman AB, Hijnen DJ, Mutis T, Kalkhoven E, Prakken BJ, Coffer PJ. Blood. 2010;115:965–974. doi: 10.1182/blood-2009-02-207118. [DOI] [PubMed] [Google Scholar]
- 14.van Loosdregt J, Brunen D, Fleskens V, Pals CE, Lam EW, Coffer PJ. PLoS One. 2011;6:e19047. doi: 10.1371/journal.pone.0019047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beier UH, Wang L, Bhatti TR, Liu Y, Han R, Ge G, Hancock WW. Mol Cell Biol. 2011;31:1022–1029. doi: 10.1128/MCB.01206-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sauve AA, Wolberger C, Schramm VL, Boeke JD. Annu Rev Biochem. 2006;75:435–465. doi: 10.1146/annurev.biochem.74.082803.133500. [DOI] [PubMed] [Google Scholar]
- 17.Yu J, Auwerx J. Pharmacol Res. 2010;62:35–41. doi: 10.1016/j.phrs.2009.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kwon HS, Ott M. Trends Biochem Sci. 2008;33:517–525. doi: 10.1016/j.tibs.2008.08.001. [DOI] [PubMed] [Google Scholar]
- 19.Kwon HS, Brent MM, Getachew R, Jayakumar P, Chen LF, Schnolzer M, McBurney MW, Marmorstein R, Greene WC, Ott M. Cell Host Microbe. 2008;3:158–167. doi: 10.1016/j.chom.2008.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang J, Lee SM, Shannon S, Gao B, Chen W, Chen A, Divekar R, McBurney MW, Braley-Mullen H, Zaghouani H, Fang D. J Clin Invest. 2009;119:3048–3058. doi: 10.1172/JCI38902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shevchenko A, Tomas H, Havlis J, Olsen JV, Mann M. Nat Protoc. 2006;1:2856–2860. doi: 10.1038/nprot.2006.468. [DOI] [PubMed] [Google Scholar]
- 22.Pagans S, Sakane N, Schnoelzer M, Ott M. Methods. 2011;53:91–96. doi: 10.1016/j.ymeth.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ventura A, Meissner A, Dillon CP, McManus M, Sharp PA, Van Parijs L, Jaenisch R, Jacks T. Proc Natl Acad Sci U S A. 2004;101:10380–10385. doi: 10.1073/pnas.0403954101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pagans S, Pedal A, North BJ, Kaehlcke K, Marshall BL, Dorr A, Hetzer-Eger C, Henklein P, Frye R, McBurney MW, Hruby H, Jung M, Verdin E, Ott M. PLoS Biol. 2005;3:e41. doi: 10.1371/journal.pbio.0030041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fontenot JD, Rasmussen JP, Williams LM, Dooley JL, Farr AG, Rudensky AY. Immunity. 2005;22:329–341. doi: 10.1016/j.immuni.2005.01.016. [DOI] [PubMed] [Google Scholar]
- 26.Napper AD, Hixon J, McDonagh T, Keavey K, Pons JF, Barker J, Yau WT, Amouzegh P, Flegg A, Hamelin E, Thomas RJ, Kates M, Jones S, Navia MA, Saunders JO, DiStefano PS, Curtis R. J Med Chem. 2005;48:8045–8054. doi: 10.1021/jm050522v. [DOI] [PubMed] [Google Scholar]
- 27.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Asher G, Gatfield D, Stratmann M, Reinke H, Dibner C, Kreppel F, Mostoslavsky R, Alt FW, Schibler U. Cell. 2008;134:317–328. doi: 10.1016/j.cell.2008.06.050. [DOI] [PubMed] [Google Scholar]
- 29.Nakahata Y, Kaluzova M, Grimaldi B, Sahar S, Hirayama J, Chen D, Guarente LP, Sassone-Corsi P. Cell. 2008;134:329–340. doi: 10.1016/j.cell.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Min SW, Cho SH, Zhou Y, Schroeder S, Haroutunian V, Seeley WW, Huang EJ, Shen Y, Masliah E, Mukherjee C, Meyers D, Cole PA, Ott M, Gan L. Neuron. 2010;67:953–966. doi: 10.1016/j.neuron.2010.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benson MJ, Pino-Lagos K, Rosemblatt M, Noelle RJ. J Exp Med. 2007;204:1765–1774. doi: 10.1084/jem.20070719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, Belkaid Y. J Exp Med. 2007;204:1775–1785. doi: 10.1084/jem.20070602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, Cheroutre H. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 34.Kang SG, Lim HW, Andrisani OM, Broxmeyer HE, Kim CH. J Immunol. 2007;179:3724–3733. doi: 10.4049/jimmunol.179.6.3724. [DOI] [PubMed] [Google Scholar]
- 35.Mucida D, Pino-Lagos K, Kim G, Nowak E, Benson MJ, Kronenberg M, Noelle RJ, Cheroutre H. Immunity. 2009;30:471–472. doi: 10.1016/j.immuni.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu L, Kitani A, Stuelten C, McGrady G, Fuss I, Strober W. Immunity. 2010;33:313–325. doi: 10.1016/j.immuni.2010.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hill JA, Hall JA, Sun CM, Cai Q, Ghyselinck N, Chambon P, Belkaid Y, Mathis D, Benoist C. Immunity. 2008;29:758–770. doi: 10.1016/j.immuni.2008.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW. EMBO J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giannakou ME, Partridge L. Trends Cell Biol. 2004;14:408–412. doi: 10.1016/j.tcb.2004.07.006. [DOI] [PubMed] [Google Scholar]
- 40.Huang H, Tindall DJ. J Cell Sci. 2007;120:2479–2487. doi: 10.1242/jcs.001222. [DOI] [PubMed] [Google Scholar]
- 41.Li J, Qu X, Ricardo SD, Bertram JF, Nikolic-Paterson DJ. Am J Path. 2010;177:1065–1071. doi: 10.2353/ajpath.2010.090923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruan Q, Kameswaran V, Tone Y, Li L, Liou HC, Greene MI, Tone M, Chen YH. Immunity. 2009;31:932–940. doi: 10.1016/j.immuni.2009.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ouyang W, Li MO. Trends Immunol. 2010;32:26–33. doi: 10.1016/j.it.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Williams LM, Rudensky AY. Nat Immunol. 2007;8:277–284. doi: 10.1038/ni1437. [DOI] [PubMed] [Google Scholar]
- 45.Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, Rudensky AY. Nature. 2007;445:771–775. doi: 10.1038/nature05543. [DOI] [PubMed] [Google Scholar]
- 46.Wan YY, Flavell RA. Nature. 2007;445:766–770. doi: 10.1038/nature05479. [DOI] [PubMed] [Google Scholar]
- 47.Komatsu N, Mariotti-Ferrandiz ME, Wang Y, Malissen B, Waldmann H, Hori S. Proc Natl Acad Sci U S A. 2009;106:1903–1908. doi: 10.1073/pnas.0811556106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miyara M, Yoshioka Y, Kitoh A, Shima T, Wing K, Niwa A, Parizot C, Taflin C, Heike T, Valeyre D, Mathian A, Nakahata T, Yamaguchi T, Nomura T, Ono M, Amoura Z, Gorochov G, Sakaguchi S. Immunity. 2009;30:899–911. doi: 10.1016/j.immuni.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 49.Lee YK, Mukasa R, Hatton RD, Weaver CT. Curr Opin Immunol. 2009;21:274–280. doi: 10.1016/j.coi.2009.05.021. [DOI] [PubMed] [Google Scholar]
- 50.Xiao Y, Li B, Zhou Z, Hancock WW, Zhang H, Greene MI. Curr Opin Immunol. 2010;22:583–591. doi: 10.1016/j.coi.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Riley JL, June CH, Blazar BR. Immunity. 2009;30:656–665. doi: 10.1016/j.immuni.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Miyara M, Wing K, Sakaguchi S. J Allergy Clin Immunol. 2009;123:749–75. doi: 10.1016/j.jaci.2009.03.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.