

Abstract
Background
G protein coupled receptors (GPCRs) are seven helical transmembrane proteins that function as signal transducers. They bind ligands in their extracellular and transmembrane regions and activate cognate G proteins at their intracellular surface at the other side of the membrane. The relay of allosteric communication between the ligand binding site and the distant G protein binding site is poorly understood. In this study, GREMLIN [1], a recently developed method that identifies networks of co-evolving residues from multiple sequence alignments, was used to identify those that may be involved in communicating the activation signal across the membrane. The GREMLIN-predicted long-range interactions between amino acids were analyzed with respect to the seven GPCR structures that have been crystallized at the time this study was undertaken.
Results
GREMLIN significantly enriches the edges containing residues that are part of the ligand binding pocket, when compared to a control distribution of edges drawn from a random graph. An analysis of these edges reveals a minimal GPCR binding pocket containing four residues (T1183.33, M2075.42, Y2686.51 and A2927.39). Additionally, of the ten residues predicted to have the most long-range interactions (A1173.32, A2726.55, E1133.28, H2115.46, S186EC2, A2927.39, E1223.37, G902.57, G1143.29 and M2075.42), nine are part of the ligand binding pocket.
Conclusions
We demonstrate the use of GREMLIN to reveal a network of statistically correlated and functionally important residues in class A GPCRs. GREMLIN identified that ligand binding pocket residues are extensively correlated with distal residues. An analysis of the GREMLIN edges across multiple structures suggests that there may be a minimal binding pocket common to the seven known GPCRs. Further, the activation of rhodopsin involves these long-range interactions between extracellular and intracellular domain residues mediated by the retinal domain.
Keywords: GPCR, GREMLIN, Long-range interactions, Ligand binding pocket, Graphical model
Background
G-protein coupled receptors (GPCRs) are an important class of proteins initiating major biochemical pathways sensing environmental stimuli. They are the largest protein superfamily with an estimated 1000 genes in the human genome alone [2]. An estimated 30% of known drug compounds target these receptors [3]. Around 500 of GPCRs are odorant or taste receptors and the remaining bind endogenous ligands. The GPCR family is divided into five distinct classes, class A – E [4]. The class A family is the largest class and includes rhodopsin, the prototypical GPCR, for which the first crystal structure of any GPCR was solved [5]. Its ligand is 11-cis retinal (RT), covalently attached to the protein. 11-cis RT isomerizes to alltrans RT upon light incidence, resulting in activation of the receptor. As of 2011, several additional GPCR structures have been deposited in the PDB increasing the total number of structures to 43 representing seven distinct GPCRs (Table 1). All GPCR structures are characterized by a transmembrane (TM) region consisting of seven helices, the G-protein interacting intracellular (IC) domain and an extracellular (EC) domain.
Table 1.
GPCR summary table
| Receptor | PDB IDs [number of structures] | Ligands |
|---|---|---|
| Bovine Rhodopsin (BR) |
1 F88, 1GZM, 1HZX, 1JFP, 1L9H, 1LN6, 1U19, 2 G87, 2HPY, 2I35, 2I36, 2I37, 2J4Y, 2PED, 3C9L, 3C9M, 3CAP, 3DQB
[18] |
RT, Ligand free |
| Squid Rhodopsin (SR) |
2Z73, 2ZIY
[2] |
RT |
| Turkey β1 adrenergic receptor (β1AR) |
2VT4, 2Y00, 2Y01, 2Y02, 2Y03, 2Y04
[6] |
Cyanopindilol, Dobutamine Carmoterol, Isoprenaline Salbutamol |
| Human β2 adrenergic receptor (β2AR) |
2R4R, 2R4S, 2RH1, 3D4S, 3KJ6, 3NY8, 3NY9, 3NYA, 3P0G, 3PDS
[10] |
Carazalol, Timolol, ICI 118,551, (molecule from Kolb et al., 2009), Alprenolol, BI-167107, FAUC50 |
| Human A2A adenosine receptor (A2A) |
3EML
[1] |
ZM241385 |
| Human chemokine receptor (CXCR4) |
3ODU, 3OE0, 3OE6, 3OE8, 3OE9
[5] |
IT1t, CVX15 |
| Human dopamine D3 receptor (D3R) | 3PBL [1] | Eticlopride |
Summary of structural information available on GPCRs as of January 2011. While all ligands are small molecules, CVX15 (in bold) is a peptide.
In GPCRs, the binding of a ligand in the EC or TM domain is the signal that is propagated to the IC domain wherein different effectors bind, in particular the G protein heterotrimer, GPCR receptor kinases (GRK) and β-arrestin. Thus, receptor activation is an inherently allosteric process where the ligand binding signal is communicated to a distant site. The activation of rhodopsin and other class A GPCRs is thought to be conserved and involves rearrangements in structural microdomains [6]. Conformational changes of multiple ‘switches’ in tandem activate the receptor [7]. These long-range interactions between distant residues are important for the function of the receptors and are also closely involved in their folding and structural stability [8,9]. Identifying the residues involved in the propagation of signals within the protein is important to understand the mechanism of activation. While much information can be directly extracted from crystal structures, allosteric interactions are dynamic and implicit in nature and thus are not directly observable in static crystal structures. Experimental methods for investigating dynamics, such as nuclear magnetic resonance, are presently incapable of resolving allosteric interactions in large membrane proteins, such as GPCRs.
Due to the limitations of experimental methods, statistical analysis of GPCR sequences is an alternative in identifying residues that may be involved in allosteric communication. Here, considerable effort has been directed towards identifying networks of co-evolving residues from multiple sequence alignments (MSA), i.e. residues that are statistically correlated in the MSA. Such correlations are thought to be necessary for function, and may provide insights into how signals are propagated between different domains. A number of computational methods have been developed to identify such couplings from MSAs, including Hidden Markov Models (HMMs) [10], Statistical Coupling Analysis (SCA) [11,12], Explicit Likelihood of Subset Co-variation (ELSC) [13], Graphical Models for Residue Coupling (GMRC) [14], and Generative REgularized ModeLs of proteINs (GREMLIN) [1]. Like the GMRC method, GREMLIN learns an undirected probabilistic graphical model known as a Markov Random Field (MRF). Unlike HMMs, which are also graphical models, MRFs are well suited to modelling long-range couplings (i.e., between non-sequential residues). The SCA and ELSC methods return a set of residue couplings (which may include long-range couplings), but unlike MRFs, they do not distinguish between direct (conditionally dependent) and indirect (conditionally independent) correlations. This distinction is crucial in determining whether an observed correlation between two residues can be explained in terms of a network of correlations involving other residues. The key difference between the GMRC and GREMLIN methods is that GREMLIN is statistically consistent and guaranteed to learn an optimal MRF, whereas the GMRC uses heuristics to learn the MRF. We have previously reported detailed comparisons of the GMRC and GREMLIN methods [1] and found that GREMLIN achieved higher accuracy and superior scalability.
Multiple sequence alignments of class A GPCRs have previously been examined by the SCA [12] and GMRC [14] methods. In the SCA study, the authors focused on the critical residue at position 296 corresponding to a lysine (K2967.43), which is the covalent attachment site for RT in bovine rhodopsin [6,15]. Several networks of residues were proposed to mediate the signal flow from the ligand binding pocket to the G protein coupling site. This focus overlooked the important contribution of the EC domain to GPCR structure and dynamics [8]. In contrast to SCA, there were no statistically coupled residues involving K2967.43 in the GMRC study, rendering a comparison of SCA and GMRC results impossible. Only 5 edges in GMRC were considered statistically significant, limiting the interpretability of the results. At the time of the above studies, the rhodopsin crystal structure was the only GPCR structure available. The now larger number of structures published (Table 1) provides us with an opportunity to investigate the generality of the roles of individual residues for allostery in different GPCRs. Furthermore, we re-examine the communication across the entire membrane, not only from a single RT residue to the IC side, but considering all possible communication points.
Because of the demonstrated advantages of GREMLIN over other methods [1], we applied GREMLIN to the same GPCR sequence alignment previously investigated by SCA and GMRC studies for comparability [12,14]. Using GREMLIN we identified statistically significant long-range couplings in class A GPCRs and analyzed the results with respect to all seven GPCRs that had been crystallized at the time of our study. Our findings indicate that the ligand binding residues are significantly enriched in these long-range couplings, mediating not only communication to the IC, but also to the EC side of the membrane. 9 out of the 10 residues with the largest number of long-range couplings belong to the ligand binding domain. There a total of 34 statistically significant long-range couplings involving these 10 residues, involving experimentally determined microdomains and activation switches in GPCRs. Our study describes a comprehensive view of the network of statistical couplings across the membrane in class A GPCRs. The details of this network are consistent with the hypothesis that the ligand-binding pocket mediates allosteric communication. The independent identification of a crucial role of the ligand binding pocket in mediating this communication provides the first sequence-based support for the early notion that all three domains in GPCRs are structurally coupled [16]. Finally, the extent of enrichment of edges in different GPCR structures allowed us to propose a novel minimal binding pocket predicted to represent the common core of ligand contact residues crucial for activation of all class A GPCRs.
Results and discussion
GREMLIN [1] was used to identify a network of correlated mutations in class A GPCRs. We first used bovine rhodopsin as a template to map the edges (correlations) to the structure. We defined the set of residues involved in interaction with the RT ligand based on the structure of rhodopsin, and first analysed the results with respect to these residues. Subsequently, we identified the ligand binding pockets of all GPCRs with known structure to consider generality of our findings. Finally, we identified minimal binding pockets that capture the most general aspects of ligand binding across all GPCRs we examined.
Mapping of GREMLIN edges to the structure of bovine Rhodopsin
Our preliminary analysis (at regularization penalty λ = 38, see Methods) revealed that most edges involve residues in the RT ligand pocket, as compared to those between or within the residues belonging to EC, IC and TM domains outside of the RT pocket (Figure 1). The RT pocket is located in the TM domain, at the interface with the EC domain. To quantify the observation that there were differences in the number of edges connecting EC, IC, TM domains and RT pocket, we enumerated the GREMLIN edges and compared them to a control set, which included all possible edges (a total of 60,378 edges) involving all the 348 amino acids in rhodopsin. The results are summarized in Table 2. Assuming a significance level of α = 0.05, we find that there is a significant enrichment of edges involving RT residues compared to the control set (46.48% for GREMLIN vs. 13.87% for control; p-value of ~0). Similar enrichment was observed in the relative distributions of EC-EC (23.8% for GREMLIN vs. 6.78% for control; p-value of ~0) and IC-IC (14.93% vs. 7.24%, p-value ~0) edges. There was significant under-representation of edges in EC-IC (7.89% versus 14.17%, p-value ~ 0), EC-TM (21.55% versus 24.57%, p-value ~0.026) and IC-TM (11.41% versus 25.38%, p-value ~0). There was no significant difference in TM-TM contacts (20.42% versus 21.87%, p-value ~0.16).
Figure 1.

Distribution of GREMLIN edges between different domains. Mapping of (A) all (B) RT and (C) non RT edges identified by GREMLIN (at λ = 38) mapped onto the bovine rhodopsin structure (PDB ID: 1U19). The edges are EC-EC (red), EC-TM (green), EC-IC (blue), TM-TM (cyan), IC-TM (orange), IC-IC (grey40), EC-RT (red), RT-TM (blue), IC-RT (green) and RT-RT (orange) where EC, IC, TM and RT represent residues in extracellular, intracellular, transmembrane and RT (ligand binding) domains. In (D) The percentage of edges for GREMLIN (squares) and null set (diamonds) are plotted against the common ligand binding pockets sorted by their size. The bars indicate fold enrichment (values on secondary y-axis) of edges in GREMLIN over the null set
Table 2.
Comparison of edge distribution from control set and GREMLIN
| Categories |
Control set (Null Distribution) |
GREMLIN (at penaltyλ = 38) |
GREMLIN > Null |
GREMLIN < Null |
||
|---|---|---|---|---|---|---|
| Total edges | % of edges | Total edges | % of edges | p value | p value | |
|
EC-EC |
4095 |
6.78 |
169 |
23.80 |
0 |
1 |
|
EC-TM |
14833 |
24.57 |
153 |
21.55 |
0.97 |
0.03 |
|
EC-IC |
8554 |
14.17 |
56 |
7.89 |
1 |
0 |
|
TM-TM |
13203 |
21.87 |
145 |
20.42 |
0.84 |
0.16 |
|
IC-TM |
15322 |
25.38 |
81 |
11.41 |
1 |
0 |
|
IC-IC |
4371 |
7.24 |
106 |
14.93 |
0 |
1 |
|
TOTAL |
60378 |
100.00 |
710 |
100.00 |
|
|
|
EC-RT |
2125 |
3.52 |
114 |
16.06 |
0 |
1 |
|
RT-TM |
3600 |
5.96 |
98 |
13.80 |
0 |
1 |
|
IC-RT |
2350 |
3.89 |
67 |
9.44 |
0 |
1 |
|
RT-RT |
300 |
0.50 |
51 |
7.18 |
0 |
1 |
| SUB-TOTAL | 8375 | 13.87 | 330 | 46.48 | ||
The finding that there is significant enrichment in the EC-EC and IC-IC contacts and that there is an under-representation of EC-IC domain contacts is biologically meaningful, because EC-IC interactions would structurally be mediated via the TM domain. Interestingly, there is a lack of significant enrichment of edges within the TM domain and a slight under-representation of EC-TM and TM-IC edges. A lack of TM enrichment is in line with the general view of the TM helices as rigid bodies in the GPCR field [17-19]. Furthermore, an important evolutionary pressure experienced by the amino acids in the TM region is to ensure that hydrophobic residues in the helices face the lipid bilayer. This pressure may override the importance of specific TM-TM contacts. However, it was puzzling that EC-TM and TM-IC contacts are under-represented since we would expect to find long-range couplings between EC and IC domains to be mediated via the intermediate TM domain. We therefore hypothesized that the EC-IC long-range contacts are more specifically mediated through a subset of TM and EC residues, namely those participating in binding RT. Indeed, 20 residues out of 27 in the RT pocket are in TM regions. We therefore analyzed the edges involving RT binding pocket residues in more detail.
Long-range couplings involving the ligand binding pockets
The RT edges were further classified into EC-RT, RT-TM, IC-RT and RT-RT groups and were compared with the respective distributions in the control set. There is significant enrichment in EC-RT, IC-RT and all other groups compared to the control set (Table 2). This finding supports the hypothesis that the EC-IC long-range couplings are mediated via RT. This is in line with our current understanding of rhodopsin activation, as the initial conformational changes triggered on activation of the receptor are in the ligand binding domain which is ultimately propagated to the IC domain.
Mapping of GREMLIN edges to the structure of other GPCRs
To extend this observation to other GPCRs, we defined a common binding pocket for each GPCR with known structure by taking the union of all residues in proximity to the ligands in cases where the same receptor was crystallized in the presence of multiple ligands (see Methods; Table 3). We compared the percentage of edges formed by the residues in these common binding pockets to that of the null distribution and against each other. As expected, the percentage of edges for the null set decreased linearly from 21% to 8% with decreasing number of residues in the pocket, i.e. pocket size (Figure 1). In contrast, the percentage of edges for the receptor binding pockets plateaus between 47% - 51%, independent of pocket size except for A2A, which had a lower value of 20% (Figure 1). The fold enrichment of edges for receptor binding pockets over the null set varied between 2.4 to 3.8. These results are statistically significant at significance level 0.05 with p-value ~ 0. Thus, GREMLIN significantly enriches edges containing ligand binding pocket residues compared to the control set. Importantly, the plateau observed in the percentage of edges for CXCR4, β2AR, β1AR, BR, SR and D3R suggests that there is a conserved ligand binding pocket shared between these receptors. The most probable explanation for the lower percentage of edges for A2A is that the ligand ZM241385 binds more towards the EC side compared to the position of ligands of other GPCRs (Figure 2). Additionally, it is oriented parallel to the TM helical bundle unlike ligands in other receptors in which a relatively perpendicular orientation is found. Nonetheless, the ligand binding pocket still contains an overlapping set of residue contacts with the other GPCRs (Figure 2). These findings suggest that there is a minimal binding pocket common to all GPCRs crystallized to date.
Table 3.
Common ligand binding pockets defined for GPCRs with structural information
| CXCR4 | β2AR | β1AR | BR | SR | D3R | A2A |
|---|---|---|---|---|---|---|
| M1, G3, L31, Q36, F37, M44, T93, T94, T97, S98, F103, E113, G114, A117, T118, P171, L172, Y178, I179, P180, T193, P194, H195, E196, E197, N200, F203, V204, M207, Y268, A272, I275, H278, Q279, S281, P285, M288, T289, A292 |
M86, T94, T97, S98, F103, E113, G114, A117, T118, G121, E122, I179, P180, I189, Y191, F203, V204, M207, F208, H211, W265, Y268, A269, A272, P285, M288, T289, A292, F293, K296 |
T94, T97, S98, E113, G114, A117, T118, G121, E122, I179, P180, I189, Y191, F203, V204, M207, F208, H211, W265, Y268, A269, A272, M288, T289, A292, F293, K296 |
E113, G114, A117, T118, G121, E122, L125, Y178, E181, S186, C187, G188, I189, Y191, M207, F208, H211, F212, F261, W265, Y268, A269, A272, A292, F293, A295, K296 |
M86, G90, E113, G114, A117, T118, G121, E122, L125, Y178, E181, S186, C187, G188, I189, M207, F208, H211, F212, F261, W265, Y268, A269, A292, K296 |
T94, E113, G114, A117, T118, G121, E122, P180, G188, I189, V204, M207, F208, H211, W265, Y268, A269, A272, F273, M288, A292, K296 |
T118, P180, E181, F203, M207, W265, Y268, A269, A272, F283, P285, M288, T289, A292 |
| 39 | 30 | 27 | 27 | 25 | 22 | 14 |
The residues listed are analogous binding pockets mapped onto the rhodopsin structure (1U19). The binding pockets are arranged in the order of decreasing size of the binding pocket (left to right). The numbers in the last row represent the number of residues in the binding pocket.
Figure 2.

Overlay of all ligands obtained from GPCR crystal structures mapped onto the structure of rhodopsin. A total of 18 ligands from available GPCR structures are super positioned onto rhodopsin by structure alignment. The alignment and images were generated using PyMOL (Version 0.99rc6; http://pymol.org/pymol). For clarity loop regions and parts of TM regions of rhodopsin (in grey; PDB ID 1U19) are removed in the images. The ligands are colored as follows: BR (green), SR (pale green), β1AR (shades of red), β2AR (shades of yellow), A2A (magenta), D3R (cyan) and CXCR4 (CVX15 in blue and IT1t in marine blue). The PDB structures used in the figure are 1U19, 2Z73, 2VT4, 2Y00, 2Y02, 2Y03, 2Y04, 2RH1, 3D4S, 3NY8, 3NY9, 3NYA, 3P0G, 3PDS, 3EML, 3PBL, 3ODU and 3OE0. All the ligands show high overlap in their positions with RT, except CVX15, ZM241385 and IT1t, which only partially overlap with RT and other ligands
A minimal ligand binding pocket
We hypothesized that if there is a minimal binding pocket common to the seven known GPCRs, then GREMLIN would significantly enrich the percentage edges for this pocket of residues compared to the null distribution set. To test this hypothesis we first defined ligand binding pockets B1, B2, B3, B4, B5, B6 and B7 representing residues common to at least one, two, three, four, five, six and seven receptor ligand binding pockets, respectively (Table 4). We compared the percentage of edges formed by the residues in these pockets to that of the null distribution set and against each other. The percentage of edges for the null set decreased linearly from 32% to 2% with decreasing pocket size (Figure 3). The percentage edges over the same range for GREMLIN decreased 69% to 10% as expected because of the decreasing pocket size. However, the fold enrichment of edges for GREMLIN over the null set increased from 2.2 – 5.2 for pockets B1 – B6. These results are statistically significant at a significance level of 0.05 with p-value ~ 0. The fold enrichment for B7 slightly decreased to 4.3 because the pocket is small with only 4 residues.
Table 4.
Defining a minimal GPCR pocket
| B1 | B2 | B3 | B4 | B5 | B6 | B7 |
|---|---|---|---|---|---|---|
| M1, G3, L31, Q36, F37, M44, M86, G90, T93, T94, T97, S98, F103, E113, G114, A117, T118, G121, E122, L125, P171, L172, Y178, I179, P180, E181, S186, C187, G188, I189, Y191, T193, P194, H195, E196, E197, N200, F203, V204, M207, F208, H211, F212, F261, W265, Y268, A269, A272, F273, I275, H278, Q279, S281, F283, P285, M288, T289, A292, F293, A295, K296 |
M86, T94, T97, S98, F103, E113, G114, A117, T118, G121, E122, L125, Y178, I179, P180, E181, S186, C187, G188, I189, Y191, F203, V204, M207, F208, H211, F212, F261, W265, Y268, A269, A272, P285, M288, T289, A292, F293, K296 |
T94, T97, S98, E113, G114, A117, T118, G121, E122, Y178, I179, P180, E181, G188, I189, Y191, F203, V204, M207, F208, H211, W265, Y268, A269, A272, P285, M288, T289, A292, F293, K296 |
T94, E113, G114, A117, T118, G121, E122, P180, I189, F203, V204, M207, F208, H211, W265, Y268, A269, A272, M288, T289, A292, K296 |
E113, G114, A117, T118, G121, E122, P180, I189, M207, F208, H211, W265, Y268, A269, A272, M288, A292, K296 |
E113, G114, A117, T118, M207, W265, Y268, A269, A272, A292 |
T118, M207, Y268, A292 |
| 61 | 38 | 31 | 22 | 18 | 10 | 4 |
The residues listed are analogous binding pockets mapped onto the rhodopsin structure (1U19). The binding pockets are arranged in the order of decreasing size of the binding pocket (left to right). The numbers in the last row represent the number residues in the binding pocket. B1, B2, B3, B4, B5, B6 and B7 represent common sets of residues present in at least one, two, three, four, five, six and seven known receptor ligand binding pockets, respectively.
Figure 3.
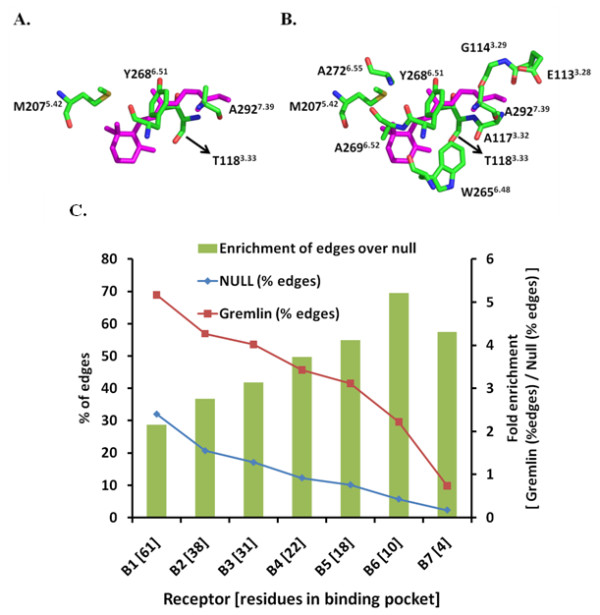
Edge distributions in the minimal ligand binding pockets (GREMLIN vs. null set) and Location of minimal ligand binding pocket residues in rhodopsin structure. The spatial organization of residues in the minimal binding pocket (A) B7 and the larger pocket (B) B6 as present in the rhodopsin structure (PDB id 1U19). Rhodopsin numbering along with Ballesteros-Weinstein numbering (superscript) is given for comparison with other GPCRs. For clarity only the binding pocket residues are shown along with bound RT (in magenta). The images were generated using PyMOL (Version 0.99rc6; http://pymol.org/pymol). In (C), the percentage of edges for GREMLIN (squares) and null set (diamonds) are plotted against the minimal ligand binding pockets sorted by their size. The bars indicate fold enrichment (values on secondary y-axis) of edges in GREMLIN over the null set
The four residues in B7 are T1183.33, M2075.42, Y2686.51 and A2927.39. These residues are uniquely positioned around the ligand (RT in rhodopsin; Figure 3) and make key interactions that stabilize RT [5,20]. On the other hand, the B6 pocket has the maximum enrichment of GREMLIN edges over the control set. There are 6 additional residues in B6 (for a total of 10 residues; E1133.28, G1143.29, A1173.32, T1183.33, M2075.42, W2656.48, Y2686.51, A2696.52, A2726.55, A2927.39) compared to B7 that seem to make contacts with RT towards the EC and IC side. These residues are known to stabilize ligand binding and are part of micro-domains that are involved in rhodopsin activation [5-7,20]. Thus, residues in B7 (T1183.33, M2075.42, Y2686.51 and A2927.39) form the minimal GPCR pocket but the expanded set of residues in B6 also represents a meaningful pocket for many GPCRs. Shown in Table 5 are all the edges formed by the residues in the minimal GPCR pocket, B7.
Table 5.
GREMLIN edges (λ = 38) involving residues from the B7 pocket
| T118 | M207 | Y268 | A292 |
|---|---|---|---|
| G90, T94, P171, E197, T198, H211, A269, A272, F293, M309, C316 | G90, S98, G114, G121, E122, P171, E181, C185, D190, E196, A233, A269, I275, H278, G284, M288, T289, A292, F293, C316, K325, N326 | NONE | A26, Y29, H65, L72, G90, T93, T94, V104, N111, A117, G121, N145, F148, S176, Y178, S186, D190, N199, N200, V204, M207, Q237, T243, A269, A272, I275, F276, Q312 |
Underlined residues form edges with at least two of the B7 pocket residues.
Identification of the most frequently observed residues involved in long-range interactions in rhodopsin
The previous section showed that GREMLIN is able to shed light on the biological and structural properties of the GPCR family. In this section we present a strategy for ranking GREMLIN edges. This strategy can be used for exploratory purposes in order to discover novel couplings and residues that might play a key role in structure and function of the GPCR protein family.
The strategy is based on the following two key insights. The first insight is that the residues that have high degree in the graph of GREMLIN couplings could be considered as hubs that lie on the communication pathways in GPCRs. This is motivated by the graphical model since a mutation/perturbation in the hub residue could affect a number of other residues. The second insight is based on the persistence of certain couplings even under stringent model complexity constraints. The larger the regularization parameter, λ, the sparser the Markov Random Field (MRF), see Methods. Thus, each edge in the MRF can be assigned a persistence score equal to the maximum λ until which the coupling was retained. The persistence score is an indicator of the importance of the couplings and the corresponding residues.
We ranked the residues based on the number of edges at a penalty of λ=38. The number of edges shown in the set of top 10 residues most frequently involved in an edge is shown in Table 6. Nine of these top ten residues (A1173.32, A2726.55, E1133.28, H2115.46, S186EC2, A2927.39, E1223.37, G902.57, G1143.29 and M2075.42) are part of the RT pocket and are involved in packing and stabilizing of RT [5,20]. Of these nine residues, eight are from the TM domain while S186EC2 is from the EC region. S186EC2 is involved in EC2 loop movement and its mutation to alanine alters the kinetics of activation [21,22]. The remaining residue G902.57 that is not part of the RT pocket as defined by a 5 Å distance cut-off is nonetheless an important residue. The naturally occurring mutation G902.57D in the RT degeneration disease, Retinitis pigmentosa, results in the constitutive activity of the receptor [23].
Table 6.
List of top ranked residues and the most persistent edges
| Rank | Position | Number of edges (at λ = 38) | Most persistent pair position (edges at penalty λ = 140) |
|---|---|---|---|
| 1 |
A1173.32 |
41 |
G902.57, E247 IC3, F2937.40, K2967.43 |
| 2 |
A2726.55 |
30 |
L72IC1, G1143.29, S176EC2, Y178EC2 |
| 3 |
E1133.28 |
29 |
M441.39, L72IC1, W1263.41, Q237IC3, F2937.40 |
| 4 |
H2115.46 |
29 |
F912.58, C140 IC2, F148 IC2 |
| 5 |
A2927.39 |
28 |
Y29EC (N-terminus) |
| 6 |
S186EC2 |
27 |
K67IC1, Q244IC3, P2917.38 |
| 7 |
E1223.37 |
26 |
I481.43, G902.57, E196EC3, M2075.42, A2696.52, F2937.40, C316IC (C-terminus) |
| 8 |
G902.57 |
23 |
A1173.32, G1203.35, E1223.37, M2075.42, Q237IC3, A2696.52, F2937.40 |
| 9 |
G1143.29 |
22 |
S176EC2, A2726.55, Y178EC2 |
| 10 | M2075.42 | 22 | G902.57, E1223.37, C316IC (C-terminus) |
Residues in bold are part of the RT binding pocket extracted from the rhodopsin structure (PDB ID: 1U19). The Ballesteros-Weinstein numbering (superscript) is given for comparison with other GPCRs. Only long-range edges are reported i.e. the edges formed with neighboring residues (8 amino acids on either side) are filtered out.
Involvement of long-range interactions in activation of rhodopsin
The above analysis indicated that RT forms a central hub for long-range edges. This can also be seen intuitively from a plot of the edges at penalty 140 in the rhodopsin structure (Figure 4). For clarity, we discuss these residues in two groups, those involving the EC and TM domain and those involving the distant IC domain, separately.
Figure 4.
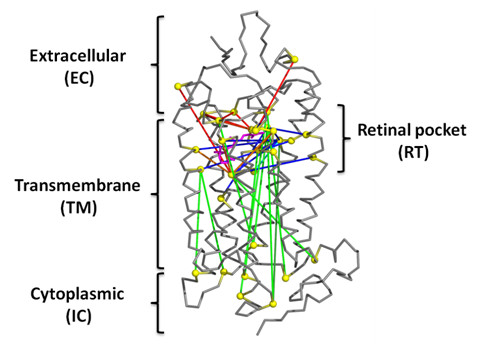
Persistent long-range contacts mapped onto structure of rhodopsin. Persistent edges at penalty 140 for the top 10 residues are mapped onto the rhodopsin structure (PDB id 1U19). The residues forming the edges are represented as yellow spheres. The edges are TM-TM (cyan), IC-TM (dark green), EC-RT (red), RT-TM (blue), IC-RT (green) and RT-RT (orange), where EC, IC, TM and RT represent residues in extracellular, intracellular, transmembrane and RT (ligand binding) domains, respectively. The image was generated using PyMOL (Version 0.99rc6; http://pymol.org/pymol)
Edges involving the EC and TM domains
The RT attachment site, K2967.43, to which RT is covalently linked via a Schiff base with the amino group of this lysine, has 15 edges at λ = 38 and the most persistent edge is A1173.32 - K2967.43, the only long-range edge at λ = 280. K2967.43 is also a key determinant for ligand specificity in different GPCRs [6,15]. The counter-ion [24] for the Schiff base is E1133.28, also a top-ranked GREMLIN edge residue. The imine moiety of the RT Schiff base is surrounded by several amino acids of which M441.39 and F2937.40 are identified in the edge list [19]. The major event on light-incidence is the isomerization of 11-cis-RT to all-trans-RT which results in the rotation of the C20 methyl group towards the EC2 loop [25]. This rotation triggers movements of the EC2 loop and rotation of the Schiff base to a more hydrophobic interior [7]. The EC2 loop displacement is one of the molecular switches in rhodopsin activation [7]. Three important residues that are part of this loop, namely S176EC2, Y178EC2 and S186EC2, are identified as top-ranked edges here.
Movement of EC2 is coupled to the outward rotation of the EC end of TM5. The shift in the RT β-ionone ring towards M2075.42 on TM5 results in a rearrangement of the hydrogen bonding network between this helix and TM3 [7]. Residue H2115.46 interacts with E1223.37 and W1263.41 and these interactions are important for receptor activation to form the Meta II state [26,27]. Other residues that are important for Meta II stability on TM3 and identified by GREMLIN are E1133.28, G1143.29, A1173.32, G1203.35, E1223.37 and W1263.41.
In addition to the rearrangement of the hydrogen bonding network between TM3 and TM5, RT isomerization in rhodopsin and ligand binding in GPCRs results in two major activation switches, the so-called rotamer toggle switch and the breakage of the ionic lock. Rotamer toggle switch refers to the rotation of W2656.48, a residue which is part of the conserved CWxP motif [28] causing reorientation of Y2235.58, M2576.40 and Y2686.51 on TM6 [7,29]. The conserved ionic lock involves the (E/D3.49)R3.50Y3.51 motif, Y2235.58 and E247IC3 at the IC side [30-32]. Note that R1353.50, Y2235.58 and W2656.48 did not appear in our edge lists because highly conserved residues naturally do not vary, and thus cannot co-vary, and so GREMLIN does not learn edges to/from such residues (see Methods). For the same reason, absent from our lists are residues from the highly conserved NPxxY motif [33] that are involved in the TM6 motions on the IC side. However, E247IC3 from the ionic lock which is not highly conserved is present in our list forming an edge with A1173.32. Other important residues that are present in our edge list are A2696.52, A2726.55 on TM6 and A2927.39 on TM7 which contribute to RT binding [5,20]. In addition, A2696.52 in rhodopsin is usually substituted by F6.52 in other GPCRs and is considered an extension of the conserved aromatic cluster on TM6. F6.52 is thought to act as ‘ligand-sensor’ in concert with the CWxP motif [34].
Edges involving the IC domain
The IC domain is the domain that interacts with the G protein and other proteins of the signal transduction cascade and the communication of RT with this distant domain is thus of particular functional significance. Several IC residues (K67IC1, L72IC1, C140IC2, F148 IC2, Q237IC3, Q244IC3, E247IC3, and C316IC (C-terminus)) form edges with the top ten residues that have the highest number of edges (Table 7). The conformational changes in the IC domain of rhodopsin that ensure receptor activation have been extensively investigated by cysteine mutagenesis coupled with biophysical studies of the cysteine mutants [35-39]. In rhodopsin, K67IC1C, F148IC2C, Q244IC3C display decreased G protein, in rhodospin called transducin (Gt), activation compared to wild-type while L72IC1C has no effect on activation [38,40-42]. Moreover, solvent accessibility studies have shown that L72IC1C undergoes the largest conformational change in IC1 upon activation whereby it becomes more solvent exposed than in the dark state [37,41-43]. L72IC1 in the crystal structure of opsin makes Van der Waals contacts with the Gt peptide [30]. EPR studies show an increase in mobility of C140IC2, Q244IC3C on photoactivation while no such changes are seen for Q237IC3C and E247IC3C [40,43]. E247IC3 is a critical residue that forms a salt bridge with the conserved ionic lock motif and undergoes major conformational changes during activation leading to the formation of the Gt binding pocket [43]. C316 IC (C-terminus) is identified as a persistent edge and displays increased mobility upon activation by EPR studies [36,40].
Table 7.
Persistent edges categorized based on the long-range contacts between different domains
| Edge category | Subset containing RT residues |
|---|---|
|
EC – TM [7]* |
EC – RT[7]: |
| A2726.55 - S176EC2, A2726.55 - Y178EC2, A2927.39 - Y29EC (N-terminus), S186EC2 - P2917.38, E1223.37 - E196EC3, G1143.29 - S176EC2, G1143.29 - Y178EC2 | |
|
TM – TM [17] |
TM(not RT) – TM(not RT) [1]: |
| G902.57 - G1203.35 | |
|
RT – TM [10]: | |
| A1173.32 - G902.57, E1133.28 - M441.39, E1133.28 - W1263.41, H2115.46 - F912.58, E1223.37 - I481.43, E1223.37 - G902.57, E1223.37 - M2075.42, G902.57 - M2075.42, G902.57 - A2696.52, G902.57 - F2937.40 | |
|
RT – RT [6]: | |
| A1173.32 - F2937.40, A1173.32 - K2967.43, A2726.55 - G1143.29, E1133.28 - F2937.40, E1223.37 - A2696.52, E1223.37 - F2937.40 | |
|
TM – IC [8] |
TM(not RT) – IC [1]: |
| G902.57 - Q237IC3 | |
|
RT – IC [7]: | |
| A1173.32 - E247 IC3, A2726.55 - L72IC1, E1133.28 - L72IC1, H2115.46 - C140 IC2, H2115.46 - F148 IC2, E1223.37 - C316IC (C-terminus), M2075.42 - C316IC (C-terminus) | |
|
EC – IC [2] |
RT – IC [2]: |
| S186EC2 - K67IC1, S186EC2 - Q244IC3 |
*The number of edges in category is given in brackets.
Categorization of edges based on the long-range contacts between the EC, IC and TM domains. The number of edges are in each category are given in brackets. There are a total of 34 edges formed by top 10 ranking residues at penalty λ = 140. There are no edges in the EC – EC and IC – IC categories. In the second column, the edges are sub-categorized to include the RT domain. Ballesteros-Weinstein numbering (superscript) is given for comparison with other GPCRs. Only long-range edges are reported. These are edges where neighbouring residues (8 amino acids on either side) are filtered out. *The number of total edges is given in brackets.
Involvement of long-range interaction residues identified by GREMLIN in ligand binding and function of angiotensin II type I receptor (AT1R)
To validate our findings using a GPCR not used in the present analysis and for which no structure is yet known, we chose the rat angiotensin II type I receptor (AT1R). AT1R is a class A GPCR which plays a vital role in cardiovascular physiology. Unlike rhodopsin, there is no full length structural or extensive biophysical data available for AT1R. However, pharmacological and structure-function properties of this receptor have been well studied by mutagenesis experiments [44].
Residues in AT1R that are homologous top ranking edge forming residues in rhodopsin were extracted based on the MSA used in GREMLIN analysis (Table 8; AT1R residues in the table and in the following text are highlighted by underlining to differentiate them from rhodopsin). Although rhodopsin and AT1R share only 20% sequence identity, general GPCR motifs such as the ionic lock and NPxxY on TM7 are conserved. In addition to these general features, we find that the subset of edges we discovered in this study have been independently shown by previous experiments to be important for ligand binding and the function of AT1R as discussed below.
Table 8.
Residues in AT1R that are homologous top ranking edge forming residues in rhodopsin
| Ballesteros-Weinstein numbering | Rhodopsin residues | ATR1 residues |
|---|---|---|
|
EC (N-Terminus) |
Y29 |
K20 |
|
1.39 |
M44 |
Y35 |
|
1.43 |
I48 |
F39 |
|
IC1 |
K67 |
M57 |
|
IC1 |
L72 |
A63 |
|
2.57 |
G90 |
L81 |
|
2.58 |
F91 |
P82 |
|
3.28 |
E113 |
A104 |
|
3.29 |
G114 |
S105 |
|
3.32 |
A117 |
V108 |
|
3.35 |
G120 |
N111 |
|
3.37 |
E122 |
Y113 |
|
3.41 |
W126 |
F117 |
|
IC2 |
C140 |
V131 |
|
IC2 |
F148 |
R140 |
|
EC2 |
S176 |
N168 |
|
EC2 |
Y178 |
F170 |
|
EC2 |
S186 |
V179 |
|
EC2 |
E196 |
S189 |
|
5.42 |
M207 |
K199 |
|
5.46 |
H211 |
G203 |
|
IC3 |
Q237 |
-- |
|
IC3 |
Q244 |
K232 |
|
IC3 |
E247 |
N235 |
|
6.52 |
A269 |
Q257 |
|
6.55 |
A272 |
T260 |
|
7.38 |
P291 |
T287 |
|
7.39 |
A292 |
I288 |
|
7.40 |
F293 |
C289 |
|
7.43 |
K296 |
Y292 |
| IC (C-Terminus) | C316 | Y312 |
Experimental and computational docking studies suggest that AT1R receptor agonist (angiotensin II [Ang II]) and antagonist (losartan) bind in the homologous RT binding site [44,45], thus hinting that many of the residues in the top ranking edge list may play a role in ligand binding in AT1R. Interestingly, the first step in AngII binding is thought to be the insertion of the C-terminus of the peptide in the receptor followed by the interaction of N-terminus residues of the peptide with EC and TM ends on the EC face [44]. AngII binding is supposed to extend from the EC face of the protein to the homologous RT binding site buried in the TM similar to peptide bound chemokine structure [46]. The carboxylate group on the C-terminus of AngII forms a salt bridge with K1995.42 on TM5 [47-49]. In addition, K1995.42 is also involved in insurmountable antagonism with carboxylate containing ligands [50]. Similar to K1995.42, Q2576.52 is also shown to be involved in insurmountable antagonism [51]. The C-terminal residue of AngII (F8) makes critical stacking interactions with the minimal binding pocket residue H2566.51 and the aromaticity of F8 and H2566.51 is important for receptor activation [49,52]. A N1113.35G mutation on TM3 results in constitutive activation of AT1R [53]. The mechanism of constitutive activation of the N1113.35G mutation is due its steric effects involving Y2927.43 on TM7 [54]. N111 is also required for discriminating AT1R specific ligands [55]. Other residues such as V179EC2 in the EC loop are also important for Ang II binding [56]. Residues like Y2927.43 in TM7, N235IC3 and Y312IC(C-terminus) in the IC face are critical for G-protein coupling and second messenger generation in cells [57-59]. Thus, AT1R residues identified to be important by empirically performed mutagenesis experiments represent the bulk of the edges identified by GREMLIN, thus validating the applicability of our approach to other GPCRs, including those for which structural information is lacking.
Comparison of results from GREMLIN with SCA and GMRC
Since we applied GREMLIN to the same MSA previously studied by the SCA [12] and GMRC [14] methods, we can directly compare, the residues found statistically coupled by the three methods, listed in Table 9. The GREMLIN residues correspond to those obtained at a penalty of λ = 38. In the SCA study, the authors focused on K2967.43, since this is a moderately conserved residue and a key determinant of ligand interaction in GPCRs [12]. The common residues between GREMLIN and SCA forming edges with K2967.43 are T932.60, A1173.32, G1213.36 and F2937.40. There are no statistically coupled residues involving K2967.43 in the GMRC study (Table 9). There are only 5 edges in GMRC that are identified to be statistically significant and none of the residues that are identified have any edges in GREMLIN at a penalty of λ = 38. GMRC also shares no common edges with SCA. Only two out of five edges in the GMRC study qualify as long-range and the residues involved (A822.49, C2646.47 and A2997.46) are strategically located in the middle of TM helices. This might be an artefact of the topology learning heuristic used by GMRC when compared with the other methods. It is important to note that in the GMRC study, the authors considered a sub-class of the original MSA [12] involving only amine (196 sequences), peptide (333 sequences) and rhodopsin (143 sequences) that represents the bulk of the sequences (672 out of a total of 948 sequences) [14].
Table 9.
Comparison of edges reported in SCA and GMRC studies with GREMLIN
| GREMLIN | SCA [12] | GMRC [14] |
|---|---|---|
| Residues involved in edges with K296 (at λ = 38) |
Residues that are statistically coupled to K296 perturbation |
Statistically coupled residues in amine + peptide + rhodopsin model |
| M44, L72, N73, G90, T93, G114, A117, G121, W175, Y178, C185, D190, S202, H211, A269, P291, A292,F293 |
I54, T58, N73, N78, F91, T92, T93, E113, A117, G121, E122, I123, L125, V129, E134, Y136, F148, A164, F212, I213, I219, M257, F261, W265, Y268, F293, F294, A295, S298, A299,N302,F313, M317 |
L57 – A82, F313 – R314, I305 – Y306, N302 – I304, C264 – A299 |
| Note: None of the above residues have any edges in GREMLIN (at λ =38) |
Short range edges are italicized while bold residues are common edges between SCA and GREMLIN. Edges from GRMC are not shared by SCA or GREMLIN.
In the SCA study, the residues statistically coupled to K2967.43 were classified further into three classes: (1) Immediate neighbours - F2937.40, L2947.41, A2957.42, A2997.46, F912.56, E1133.28, (2) Linked network - F2616.44, W2656.48, Y2686.51, F2125.47 and (3) Sparse but contiguous network: G1213.36, I1233.38, L1253.40, I2195.54, F2616.44, S2987.45, A2997.46, N3027.49. These categories were formulated on mapping the residues onto the rhodopsin structure. Residues in the immediate neighbour category are in the vicinity of K2967.43 and are mainly involved in helix packing interactions except for E1133.28. E1133.28 forms a salt bridge interaction with the protonated Schiff base on K2967.43 and is an important interaction identified by SCA. In the GREMLIN model, E1133.28 and K2967.43 aren’t connected by an edge, but they do share three common neighbours: M44, L72, and F293, and are thus indirectly correlated. The linked network residues in SCA are parallel to the membrane and form an aromatic cluster around the β-ionone ring of RT in rhodopsin. The residues in the sparse but contiguous network are distant from K2967.43 and form helix packing interactions toward the IC side. There are critical residues identified in the SCA study, most importantly W2656.48 which is part of the CWxP motif [28] and N3027.49 which is part of the NPxxY motif [33]. The SCA method performs a perturbation on a particular amino acid only if the corresponding sub-alignment size is beyond a certain cutoff in order to calculate ΔΔGstat values. GREMLIN on the other hand makes no such distinction. Hence it is possible that SCA detects edges even if a position is fairly conserved whereas GREMLIN ignores them. This could be a source of difference between GREMLIN and SCA edge couplings. Overall, compared to SCA and GMRC, GREMLIN seems to identify couplings that are more extensive (i.e., involving EC, TM, RT and IC) and are part of experimentally functional switches and structural micro-domains that are critical for activation as discussed above.
Limitations of the GREMLIN approach
GREMLIN is subject to the same kinds of limitations that all MSA-based analyses face. We briefly discuss these limitations here so that readers can better understand the nature of the results of our study.
GREMLIN is very sensitive to the size and contents of the MSA. A small, and/or poorly constructed MSA may result in subpar models. However, GREMLIN does attempt to deal with small MSAs (i.e., those with relatively few sequences) through regularization. As described in the Methods section, GREMLIN selects a value for the regularization parameter, λ, via a permutation of the columns of the MSA. Specifically, it selects a λ value that minimizes the expected number of false positive edges. It does this at the expense of an increase in the number of false negative edges. The value of λ is expected to be roughly inversely proportional to the number of sequences in the MSA. Likewise, the number of edges in the resulting model will be roughly inversely proportional to λ. So, small MSAs will inherently produce sparse graphs which will probably contain many “missing” edges that don’t have strong statistical support. Users must therefore consider the size of the MSA when interpreting the set of edges in the graph returned by GREMLIN.
In addition to the size of the MSA, the contents of the MSA are also important, especially if the MSA contains functionally heterogeneous sequences (as is the case in our study). In particular, weak signals in the MSA (e.g., due to sampling imbalances between different functional groups) are very likely to be missed. This is especially true for GREMLIN since it is biased towards minimizing false positive edges. Conversely, the GREMLIN algorithm will learn the conservation and correlation statistics for two (or more) divergent subclasses, provided that they are well represented in the MSA. Additionally, some of the edges learned by GREMLIN are due to correlations that distinguish functionally divergent sequences, while others are due to other constraints (e.g., conservation of charge). GREMLIN cannot distinguish between these two kinds of couplings. Naturally, one may attempt to compare the set of edges learned from functionally homogeneous MSAs to those learned from heterogeneous MSAs, but differences in the sizes of the MSAs can make it difficult to compare models, as discussed above. Addressing this limitation is one of our goals as part of on-going research.
As noted by an anonymous reviewer, GREMLIN is not well-suited to learning couplings between one residue and a cluster of functionally redundant residues (e.g., a cluster of Glu residues, any one of which could form a salt bridge with a nearby Lys), unless, the MSA contains examples of each possible clustering. Thus, care must be taken if the MSA contains such clusters.
Finally, the results presented in this paper are limited to GPCRs where the binding pocket is at or near the corresponding binding pocket of rhodopsin. Our MSA did not contain a significant number of GPCRs with binding pockets substantially different than rhodopsin, such as A2A.
Conclusions
In this study we demonstrated the use of GREMLIN to identify a network of statistically correlated and functionally important residues in class A GPCRs. Based on sequence only, GREMLIN identified that ligand binding pocket residues are extensively correlated with distal residues, compared to those that are not part of the ligand pocket. An analysis of the GREMLIN edges across multiple structures suggests that there is a minimal binding pocket common to the seven known GPCRs. Statistically significant long-range couplings identified here were previously identified experimentally to be critical for activation of rhodopsin. Further, the activation of rhodopsin involves these long-range interactions between EC and IC residues mediated by RT. Compared to previously applied methods SCA and GMRC, GREMLIN identifies edges that span the entire protein and are functionally important. Based on our findings here with the GPCR family and our earlier studies with several soluble protein families [1], GREMLIN can be used to identify functionally important residue couplings in both soluble and membrane proteins. Future work can include validating the functional importance of novel residues and couplings identified by GREMLIN using molecular modelling tools such as GOBLIN [60] or via Molecular Dynamic Simulations and ultimately wet-lab experiments.
Methods
GREMLIN methodology
We employed GREMLIN [1] to learn a Markov Random Field (MRF) model (Figure 5) from a MSA of class A GPCRs (see details below). MRFs are undirected probabilistic graphical models. In this paper, MRFs are used to model the conservation and coupling statistics observed in the MSA. In particular, each node in the MRF corresponds to a column in the MSA. An edge between two nodes indicates that they are coupled. Conversely, the absence of an edge between two nodes means that they are conditionally independent. The conservation and coupling statistics in a MRF are encoded via node (ϕ) and edge potentials (ψ). Informally, these potentials can be thought of as un-normalized probabilities. Collectively, these potentials encode the joint probability distribution over protein sequences such that the probability of any given length p sequence x = (x1, x2, …, xp) can be computed as:
Figure 5.

Cartoon of a multiple sequence alignment and its mapping to a Markov random field. Shown in the figure is a cartoon figure of a multiple sequence alignment (MSA) and a corresponding Markov random field (MRF). There is one node in the MRF for each column in the MSA. The column-wise conservation statistics in the MSA are encoded by node potentials (Φi). Similarly, the co-variation statistics in the MSA (e.g., between columns 1 and 4) are encoded by edge potentials (φ1,4) in the MRF. The lack of an edge between two nodes means that the corresponding columns are conditionally independent.
| (1) |
Here, Z is the normalization constant, V and E are the nodes and edges in the MRF, respectively. We note MRFs are generative and can thus be used to sample new sequences (as in protein design).
Figure 5 shows a toy example of the relationship between the input MSA and the MRF that GREMLIN learns. Here, a 7-column MSA is shown. Column 2 is completely conserved, and is therefore statistically independent of the remaining columns. This independence is encoded in the MRF by the absence of an edge to the variable corresponding to the second column. On the other hand, columns 1 and 4 co-vary such that whenever there is an ‘S’ in column 1, there is a ‘H’ in column 4, and whenever there is an ‘F’ in column 1, there is a ‘W’ in column 4. This coupling is represented in the MRF by an edge between the variables corresponding to columns 1 and 4. In this paper, we examine the topology of the learned MRF to gain insights into the network of correlated mutations. Specifically, we are most interested in correlations that are observed between spatially distant residues from different domains of GPCRs.
Multiple sequence alignment (MSA) of class A GPCRs
The authors of the SCA study [12] obtained the class A GPCR alignment from GPCRDB [61] and TinyGRAP [62] databases and manually adjusted the sequences using structure-based sequence alignments. The final MSA has 940 sequences and 348 residue positions covering the entire length of bovine rhodopsin without any gaps (Figure 6). We used this MSA here. As a pre-processing step, we selected the top 1000 candidate edges using a mutual information metric on which the structure learning approach would be subsequently run. This pre-processing step was done purely for computational reasons. Later versions of GREMLIN can avoid this pre-processing step by scaling up to larger sized proteins by parallelizing the computations using a Map-Reduce framework [63].
Figure 6.
Multiple sequence alignment of class A GPCRs. For easy visualization the logo of the MSA alignment is generated using Weblogo ( http://weblogo.berkeley.edu). The amino acids numbering is based on the positions of bovine opsin (NCBI Reference Sequence: NP_001014890.1). The individual letter height of amino acid(s) at each position indicates their relative frequencies and conservation in the alignment. The TM helices are indicated as lines above the sequence. Most of the conserved regions are restricted to TM regions.
Model selection
GREMLIN uses a single parameter, λ, which determines the sparsity of the MRF (i.e., the number of edges) and the likelihood of the sequences in the MSA under the model. Higher values of λ will produce sparser models. In general, a dense graph will yield higher likelihoods than a sparse graph. However, maximizing the likelihood of the MSA is likely to over-fit the data. Thus, the regularization parameter, λ, controls the trade-off between goodness-of-fit to the data and the tendency to over-fit. As in previous work, we used a permutation-based method to select λ. Briefly, we randomly permute the columns of the MSA in order to destroy all correlations between columns while retaining the column-wise distribution of amino acids. We then run GREMLIN on the permutated MSA using different values of λ. The smallest λ yielding zero edges on the permuted MSA is selected. This is a conservative estimate designed to minimize the number of false positive edges. A comparison of the number of edges versus λ for the permuted and the original alignment are shown in Figure 7A and 7B, respectively. In our experiments the optimal λ value was 38 (Figure 7A). We used GREMLIN to learn models from the un-permuted MSA using penalties of 38, or higher (Figure 7B). We consider such edges as the most “robust”. The analysis of GPCRs described here is based on these robust edges unless otherwise stated.
Figure 7.
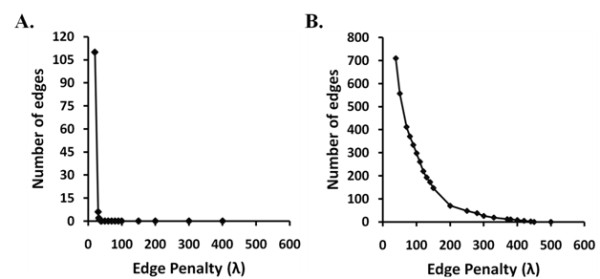
Model Complexity Curve. This figure shows a plot of the number of edges learned by the MRF as a function of the model complexity parameter, λ (A) on the permuted GPCR MSA (Null model) and (B) on the GPCR MSA. This exercise was carried out to define ‘robust’ edges or edges which are selected at a zero false positive rate. The penalty for which the number of edges goes to zero is λ = 38 for the permuted GPCR MSA and this is used as the parameter for defining ‘robust’ edges. The number of edges goes to zero at around λ = 450 for GPCR MSA. The smaller the parameter the denser the model and larger the parameter the sparser the model.
GPCR structures files
As of January 2011, there were a total of 43 structures representing seven different GPCRs deposited in the PDB (Table 1). Only class A GPCRs have been crystallized so far. The GPCRs for which structural information is available are bovine rhodopsin (BR; 18 structures including opsin), squid rhodopsin (SR; 2 structures) turkey β1 adrenergic receptor (β1AR; 6 structures), human β2 adrenergic receptor (β2AR; 10 structures), human A2A adenosine receptor (A2A; 1 structure), human chemokine receptor CXCR4 (5 structures) and human dopamine D3 receptor (D3R; 1 structure).
Residue numbering scheme
The amino acids of the bovine rhodopsin sequence were used as position references (NCBI Reference Sequence [64]: NP_001014890.1). The positions of amino acids are represented by the single letter amino acid code followed by the sequence number in rhodopsin. To allow easier comparison with other GPCRs, given in superscript is the generic numbering proposed by Ballesteros and Weinstein [65].
Description of ligand binding pockets in GPCR structures
The residues in the ligand pocket of the different GPCR crystal structures available to date were defined as those which have at least one atom within 5 Å of the respective ligand. Python scripts were written to extract residues within a ligand binding pocket using this cut-off distance from crystal structures.
We mapped the ligand binding pockets of the different GPCRs onto bovine rhodopsin for comparison. Pair-wise sequence/structure based alignments between rhodopsin (PDB ID: 1U19) and other GPCR structures were generated using the ‘salign’ module in the MODELLER [66] software. All ligand binding pockets discussed in this paper are mapped onto the structure of bovine rhodopsin.
In addition to comparing ligand binding pockets directly (i.e. extracting 5 Å residues in PDB ID: 1F88 for rhodopsin to identify the RT ligand binding pocket), we also generated the following combined sets of pocket residues to investigate similarities and differences between ligand binding pockets of different GPCRs (Table 1). For each of the 7 GPCRs, we defined a common ligand binding pocket by combining the ligand binding pockets from all available crystal structures for the respective receptor (Table 3). Thus, for bovine rhodopsin, the common ligand pocket is the combination of all RT binding pockets of 12 different structures. [Note: Rhodopsin PDBs excluded are 1JFP and 1LN6, because these represent structure models from NMR structures of protein fragments. 2I36, 2I37, 3CAP and 3DQB were also excluded because these are opsin structures and have no RT in them.] In analogous fashion, common pockets were created for squid rhodopsin (SR), turkey β1 adrenergic receptor (β1AR), human β2 adrenergic receptor (β2AR), human A2A adenosine receptor (A2A), human chemokine receptor CXCR4 and human dopamine D3 receptor (D3R).
Finally, to generalize across different GPCRs, we derived additional ligand pockets B1, B2, B3, B4, B5, B6 and B7 representing common sets of residues present in at least one, two, three, four, five, six and seven receptor ligand binding pockets, respectively. These combined ligand binding pockets are listed in Table 4.
Definition of long-range interactions
A long-range interaction is defined as a statistical coupling between two amino acids that are separated by at least 8 amino acids in the sequence (a definition used in CASP [13]).
Control dataset and statistical significance tests
GREMLIN derived robust edges were checked for statistically over- or under-represented patterns amongst couplings observed. These tests were not done to validate the efficacy of GREMLIN in terms of modeling the protein family, but to get structural and biological insights into the nature of couplings that the model learns. For this purpose we compared the edges that GREMLIN returns against a control distribution of edges. The control distribution is created by drawing edges from a random graph. We classified the edges into one of the following categories: EC-EC, EC-IC, EC-TM, EC-RT, IC-IC, IC-RT, IC-TM, TM-TM, RT-TM and RT-RT. Here, RT stands for the ligand binding domain in rhodopsin (PDB ID: 1F88). To define the control distribution, we enumerated all possible edges coupling any two amino acids in rhodopsin (PDB ID: 1U19) and assigned these edges into the previously defined categories. We defined a control distribution of a category as the probability of randomly picking an edge in that category from the control dataset. To check for statistical significance, we enumerated the edges returned by GREMLIN in each category and compared the fraction of edges in this category against the control distribution. A p-value was calculated by a one-sided binomial test for statistical significance of GREMLIN categories against categories of the control distribution.
Abbreviations
GREMLIN: Generative REgularized ModeLs of proteINs; GPCR: G protein coupled receptors; SCA: Statistical Coupling Analysis; GMRC: Graphical Models for Residue Coupling; HMM: Hidden Markov Model; MRF: Markov Random Field; MSA: Multiple sequence analysis; TM: Transmembrane; EC: Extracellular; IC: Intracellular; RT: Retinal; PDB: Protein data bank.
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
SM ran the GREMLIN experiments, participated in the design and performed the statistical analysis, and drafted the manuscript. KCT performed the structural analysis, participated in the design and statistical analysis of the study and drafted the manuscript. JKS and CJL conceived of the study, directed its design, coordinated all work and edited the manuscript. All authors read and approved the final manuscript.
Contributor Information
Subhodeep Moitra, Email: subho@cmu.edu.
Kalyan C Tirupula, Email: tirupuk@ccf.org.
Judith Klein-Seetharaman, Email: jks33@pitt.edu.
Christopher James Langmead, Email: cjl@cs.cmu.edu.
Acknowledgements
This study was supported by NSF grants 1144281 and IIS-0905193, and NIH grant R01 LM007994-07.
References
- Balakrishnan S, Kamisetty H, Carbonell JG, Lee SI, Langmead CJ. Learning generative models for protein fold families. Proteins. 2011;79:1061–1078. doi: 10.1002/prot.22934. [DOI] [PubMed] [Google Scholar]
- Takeda S, Kadowaki S, Haga T, Takaesu H, Mitaku S. Identification of G protein-coupled receptor genes from the human genome sequence. FEBS letters. 2002;520:97–101. doi: 10.1016/S0014-5793(02)02775-8. [DOI] [PubMed] [Google Scholar]
- Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there? Nat Rev Drug Discov. 2006;5:993–996. doi: 10.1038/nrd2199. [DOI] [PubMed] [Google Scholar]
- Fredriksson R, Lagerstrom MC, Lundin LG, Schioth HB. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol Pharmacol. 2003;63:1256–1272. doi: 10.1124/mol.63.6.1256. [DOI] [PubMed] [Google Scholar]
- Palczewski K, Kumasaka T, Hori T, Behnke CA, Motoshima H, Fox BA, Le Trong I, Teller DC, Okada T, Stenkamp RE. et al. Crystal structure of rhodopsin: A G protein-coupled receptor. Science. 2000;289:739–745. doi: 10.1126/science.289.5480.739. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Shi L, Javitch JA. Structural mimicry in G protein-coupled receptors: implications of the high-resolution structure of rhodopsin for structure-function analysis of rhodopsin-like receptors. Mol Pharmacol. 2001;60:1–19. [PubMed] [Google Scholar]
- Ahuja S, Smith SO. Multiple switches in G protein-coupled receptor activation. Trends Pharmacol Sci. 2009;30:494–502. doi: 10.1016/j.tips.2009.06.003. [DOI] [PubMed] [Google Scholar]
- Rader AJ, Anderson G, Isin B, Khorana HG, Bahar I, Klein-Seetharaman J. Identification of core amino acids stabilizing rhodopsin. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:7246–7251. doi: 10.1073/pnas.0401429101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein-Seetharaman J. Dual role of interactions between membranous and soluble portions of helical membrane receptors for folding and signaling. Trends Pharmacol Sci. 2005;26:183–189. doi: 10.1016/j.tips.2005.02.009. [DOI] [PubMed] [Google Scholar]
- Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K. et al. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockless SW, Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science. 1999;286:295–299. doi: 10.1126/science.286.5438.295. [DOI] [PubMed] [Google Scholar]
- Suel GM, Lockless SW, Wall MA, Ranganathan R. Evolutionarily conserved networks of residues mediate allosteric communication in proteins. Nature structural biology. 2003;10:59–69. doi: 10.1038/nsb881. [DOI] [PubMed] [Google Scholar]
- Dekker JP, Fodor A, Aldrich RW, Yellen G. A perturbation-based method for calculating explicit likelihood of evolutionary co-variance in multiple sequence alignments. Bioinformatics (Oxford, England) 2004;20:1565–1572. doi: 10.1093/bioinformatics/bth128. [DOI] [PubMed] [Google Scholar]
- Thomas J, Ramakrishnan N, Bailey-Kellogg C. Graphical models of residue coupling in protein families. IEEE/ACM Trans Comput Biol Bioinform. 2008;5:183–197. doi: 10.1109/TCBB.2007.70225. [DOI] [PubMed] [Google Scholar]
- Gether U. Uncovering molecular mechanisms involved in activation of G protein-coupled receptors. Endocrine reviews. 2000;21:90–113. doi: 10.1210/er.21.1.90. [DOI] [PubMed] [Google Scholar]
- Hwa J, Garriga P, Liu X, Khorana HG. Structure and function in rhodopsin: packing of the helices in the transmembrane domain and folding to a tertiary structure in the intradiscal domain are coupled. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:10571–10576. doi: 10.1073/pnas.94.20.10571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenbach C, Yang K, Farrens DL, Farahbakhsh ZT, Khorana HG, Hubbell WL. Structural features and light-dependent changes in the cytoplasmic interhelical E-F loop region of rhodopsin: a site-directed spin-labeling study. Biochemistry. 1996;35:12470–12478. doi: 10.1021/bi960849l. [DOI] [PubMed] [Google Scholar]
- Farrens DL, Altenbach C, Yang K, Hubbell WL, Khorana HG. Requirement of rigid-body motion of transmembrane helices for light activation of rhodopsin. Science. 1996;274:768–770. doi: 10.1126/science.274.5288.768. [DOI] [PubMed] [Google Scholar]
- Sakmar TP, Menon ST, Marin EP, Awad ES. Rhodopsin: insights from recent structural studies. Annu Rev Biophys Biomol Struct. 2002;31:443–484. doi: 10.1146/annurev.biophys.31.082901.134348. [DOI] [PubMed] [Google Scholar]
- Okada T, Sugihara M, Bondar AN, Elstner M, Entel P, Buss V. The retinal conformation and its environment in rhodopsin in light of a new 2.2 A crystal structure. J Mol Biol. 2004;342:571–583. doi: 10.1016/j.jmb.2004.07.044. [DOI] [PubMed] [Google Scholar]
- Ahuja S, Hornak V, Yan EC, Syrett N, Goncalves JA, Hirshfeld A, Ziliox M, Sakmar TP, Sheves M, Reeves PJ. et al. Helix movement is coupled to displacement of the second extracellular loop in rhodopsin activation. Nat Struct Mol Biol. 2009;16:168–175. doi: 10.1038/nsmb.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan ECY, Epps J, Lewis JW, Szundi I, Bhagat A, Sakmar TP, Kliger DS. Photointermediates of the Rhodopsin S186A Mutant as a Probe of the Hydrogen-Bond Network in the Chromophore Pocket and the Mechanism of Counterion Switch†. The Journal of Physical Chemistry C. 2007;111:8843–8848. doi: 10.1021/jp067172o. [DOI] [Google Scholar]
- Rao VR, Cohen GB, Oprian DD. Rhodopsin mutation G90D and a molecular mechanism for congenital night blindness. Nature. 1994;367:639–642. doi: 10.1038/367639a0. [DOI] [PubMed] [Google Scholar]
- Sakmar TP, Franke RR, Khorana HG. Glutamic acid-113 serves as the retinylidene Schiff base counterion in bovine rhodopsin. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:8309–8313. doi: 10.1073/pnas.86.21.8309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamichi H, Okada T. Crystallographic analysis of primary visual photochemistry. Angew Chem Int Ed Engl. 2006;45:4270–4273. doi: 10.1002/anie.200600595. [DOI] [PubMed] [Google Scholar]
- Lewis JW, Szundi I, Kazmi MA, Sakmar TP, Kliger DS. Proton movement and photointermediate kinetics in rhodopsin mutants. Biochemistry. 2006;45:5430–5439. doi: 10.1021/bi0525775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SW, Sakmar TP. Specific tryptophan UV-absorbance changes are probes of the transition of rhodopsin to its active state. Biochemistry. 1996;35:11149–11159. doi: 10.1021/bi960858u. [DOI] [PubMed] [Google Scholar]
- Shi L, Liapakis G, Xu R, Guarnieri F, Ballesteros JA, Javitch JA. Beta2 adrenergic receptor activation. Modulation of the proline kink in transmembrane 6 by a rotamer toggle switch. J Biol Chem. 2002;277:40989–40996. doi: 10.1074/jbc.M206801200. [DOI] [PubMed] [Google Scholar]
- Patel AB, Crocker E, Reeves PJ, Getmanova EV, Eilers M, Khorana HG, Smith SO. Changes in interhelical hydrogen bonding upon rhodopsin activation. J Mol Biol. 2005;347:803–812. doi: 10.1016/j.jmb.2005.01.069. [DOI] [PubMed] [Google Scholar]
- Scheerer P, Park JH, Hildebrand PW, Kim YJ, Krauss N, Choe HW, Hofmann KP, Ernst OP. Crystal structure of opsin in its G-protein-interacting conformation. Nature. 2008;455:497–502. doi: 10.1038/nature07330. [DOI] [PubMed] [Google Scholar]
- Park JH, Scheerer P, Hofmann KP, Choe HW, Ernst OP. Crystal structure of the ligand-free G-protein-coupled receptor opsin. Nature. 2008;454:183–187. doi: 10.1038/nature07063. [DOI] [PubMed] [Google Scholar]
- Ballesteros JA, Jensen AD, Liapakis G, Rasmussen SG, Shi L, Gether U, Javitch JA. Activation of the beta 2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J Biol Chem. 2001;276:29171–29177. doi: 10.1074/jbc.M103747200. [DOI] [PubMed] [Google Scholar]
- Fritze O, Filipek S, Kuksa V, Palczewski K, Hofmann KP, Ernst OP. Role of the conserved NPxxY(x)5,6 F motif in the rhodopsin ground state and during activation. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:2290–2295. doi: 10.1073/pnas.0435715100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein H. Hallucinogen actions on 5-HT receptors reveal distinct mechanisms of activation and signaling by G protein-coupled receptors. AAPS J. 2005;7:E871–884. doi: 10.1208/aapsj070485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai K, Klein-Seetharaman J, Farrens D, Zhang C, Altenbach C, Hubbell WL, Khorana HG. Single-cysteine substitution mutants at amino acid positions 306–321 in rhodopsin, the sequence between the cytoplasmic end of helix VII and the palmitoylation sites: sulfhydryl reactivity and transducin activation reveal a tertiary structure. Biochemistry. 1999;38:7925–7930. doi: 10.1021/bi9900119. [DOI] [PubMed] [Google Scholar]
- Cai K, Klein-Seetharaman J, Hwa J, Hubbell WL, Khorana HG. Structure and function in rhodopsin: effects of disulfide cross-links in the cytoplasmic face of rhodopsin on transducin activation and phosphorylation by rhodopsin kinase. Biochemistry. 1999;38:12893–12898. doi: 10.1021/bi9912443. [DOI] [PubMed] [Google Scholar]
- Klein-Seetharaman J, Hwa J, Cai K, Altenbach C, Hubbell WL, Khorana HG. Single-cysteine substitution mutants at amino acid positions 55–75, the sequence connecting the cytoplasmic ends of helices I and II in rhodopsin: reactivity of the sulfhydryl groups and their derivatives identifies a tertiary structure that changes upon light-activation. Biochemistry. 1999;38:7938–7944. doi: 10.1021/bi990013t. [DOI] [PubMed] [Google Scholar]
- Altenbach C, Cai K, Klein-Seetharaman J, Khorana HG, Hubbell WL. Structure and function in rhodopsin: mapping light-dependent changes in distance between residue 65 in helix TM1 and residues in the sequence 306–319 at the cytoplasmic end of helix TM7 and in helix H8. Biochemistry. 2001;40:15483–15492. doi: 10.1021/bi011546g. [DOI] [PubMed] [Google Scholar]
- Altenbach C, Klein-Seetharaman J, Cai K, Khorana HG, Hubbell WL. Structure and function in rhodopsin: mapping light-dependent changes in distance between residue 316 in helix 8 and residues in the sequence 60–75, covering the cytoplasmic end of helices TM1 and TM2 and their connection loop CL1. Biochemistry. 2001;40:15493–15500. doi: 10.1021/bi011545o. [DOI] [PubMed] [Google Scholar]
- Altenbach C, Cai K, Khorana HG, Hubbell WL. Structural features and light-dependent changes in the sequence 306–322 extending from helix VII to the palmitoylation sites in rhodopsin: a site-directed spin-labeling study. Biochemistry. 1999;38:7931–7937. doi: 10.1021/bi9900121. [DOI] [PubMed] [Google Scholar]
- Ridge KD, Zhang C, Khorana HG. Mapping of the amino acids in the cytoplasmic loop connecting helices C and D in rhodopsin. Chemical reactivity in the dark state following single cysteine replacements. Biochemistry. 1995;34:8804–8811. doi: 10.1021/bi00027a032. [DOI] [PubMed] [Google Scholar]
- Yang K. Farrens DL, Hubbell WL. Khorana HG: Structure and function in rhodopsin. Single cysteine substitution mutants in the cytoplasmic interhelical E-F loop region show position-specific effects in transducin activation. Biochemistry. 1996;35:12464–12469. doi: 10.1021/bi960848t. [DOI] [PubMed] [Google Scholar]
- Farahbakhsh ZT, Ridge KD, Khorana HG, Hubbell WL. Mapping light-dependent structural changes in the cytoplasmic loop connecting helices C and D in rhodopsin: a site-directed spin labeling study. Biochemistry. 1995;34:8812–8819. doi: 10.1021/bi00027a033. [DOI] [PubMed] [Google Scholar]
- Oliveira L, Costa-Neto CM, Nakaie CR, Schreier S, Shimuta SI, Paiva AC. The angiotensin II AT1 receptor structure-activity correlations in the light of rhodopsin structure. Physiological reviews. 2007;87:565–592. doi: 10.1152/physrev.00040.2005. [DOI] [PubMed] [Google Scholar]
- Baleanu-Gogonea C, Karnik S. Model of the whole rat AT1 receptor and the ligand-binding site. Journal of molecular modeling. 2006;12:325–337. doi: 10.1007/s00894-005-0049-z. [DOI] [PubMed] [Google Scholar]
- Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, Abagyan R, Brooun A, Wells P, Bi FC. et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science (New York, NY. 2010;330:1066–1071. doi: 10.1126/science.1194396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda K, Saad Y, Kinoshita A, Boyle TP, Graham RM, Husain A, Karnik SS. Tetrazole and carboxylate groups of angiotensin receptor antagonists bind to the same subsite by different mechanisms. The Journal of biological chemistry. 1995;270:2284–2289. doi: 10.1074/jbc.270.5.2284. [DOI] [PubMed] [Google Scholar]
- Yamano Y, Ohyama K, Chaki S, Guo DF, Inagami T. Identification of amino acid residues of rat angiotensin II receptor for ligand binding by site directed mutagenesis. Biochemical and biophysical research communications. 1992;187:1426–1431. doi: 10.1016/0006-291X(92)90461-S. [DOI] [PubMed] [Google Scholar]
- Noda K, Saad Y, Karnik SS. Interaction of Phe8 of angiotensin II with Lys199 and His256 of AT1 receptor in agonist activation. The Journal of biological chemistry. 1995;270:28511–28514. doi: 10.1074/jbc.270.48.28511. [DOI] [PubMed] [Google Scholar]
- Fierens FL, Vanderheyden PM, Gaborik Z, Minh TL, Backer JP, Hunyady L, Ijzerman A, Vauquelin G. Lys(199) mutation of the human angiotensin type 1 receptor differentially affects the binding of surmountable and insurmountable non-peptide antagonists. Journal of the renin-angiotensin-aldosterone system : JRAAS. 2000;1:283–288. doi: 10.3317/jraas.2000.044. [DOI] [PubMed] [Google Scholar]
- Takezako T, Gogonea C, Saad Y, Noda K, Karnik SS. "Network leaning" as a mechanism of insurmountable antagonism of the angiotensin II type 1 receptor by non-peptide antagonists. The Journal of biological chemistry. 2004;279:15248–15257. doi: 10.1074/jbc.M312728200. [DOI] [PubMed] [Google Scholar]
- Miura S, Feng YH, Husain A, Karnik SS. Role of aromaticity of agonist switches of angiotensin II in the activation of the AT1 receptor. The Journal of biological chemistry. 1999;274:7103–7110. doi: 10.1074/jbc.274.11.7103. [DOI] [PubMed] [Google Scholar]
- Noda K, Feng YH, Liu XP, Saad Y, Husain A, Karnik SS. The active state of the AT1 angiotensin receptor is generated by angiotensin II induction. Biochemistry. 1996;35:16435–16442. doi: 10.1021/bi961593m. [DOI] [PubMed] [Google Scholar]
- Feng YH, Miura S, Husain A, Karnik SS. Mechanism of constitutive activation of the AT1 receptor: influence of the size of the agonist switch binding residue Asn(111) Biochemistry. 1998;37:15791–15798. doi: 10.1021/bi980863t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monnot C, Bihoreau C, Conchon S. Curnow KM, Corvol P, Clauser E: Polar residues in the transmembrane domains of the type 1 angiotensin II receptor are required for binding and coupling. Reconstitution of the binding site by co-expression of two deficient mutants. The Journal of biological chemistry. 1996;271:1507–1513. doi: 10.1074/jbc.271.3.1507. [DOI] [PubMed] [Google Scholar]
- Hjorth SA, Schambye HT, Greenlee WJ, Schwartz TW. Identification of peptide binding residues in the extracellular domains of the AT1 receptor. The Journal of biological chemistry. 1994;269:30953–30959. [PubMed] [Google Scholar]
- Zhang M, Zhao X, Chen HC, Catt KJ, Hunyady L. Activation of the AT1 angiotensin receptor is dependent on adjacent apolar residues in the carboxyl terminus of the third cytoplasmic loop. The Journal of biological chemistry. 2000;275:15782–15788. doi: 10.1074/jbc.M000198200. [DOI] [PubMed] [Google Scholar]
- Sano T, Ohyama K, Yamano Y, Nakagomi Y, Nakazawa S, Kikyo M, Shirai H, Blank JS, Exton JH, Inagami T. A domain for G protein coupling in carboxyl-terminal tail of rat angiotensin II receptor type 1A. The Journal of biological chemistry. 1997;272:23631–23636. doi: 10.1074/jbc.272.38.23631. [DOI] [PubMed] [Google Scholar]
- Marie J, Maigret B, Joseph MP, Larguier R, Nouet S, Lombard C, Bonnafous JC. Tyr292 in the seventh transmembrane domain of the AT1A angiotensin II receptor is essential for its coupling to phospholipase C. The Journal of biological chemistry. 1994;269:20815–20818. [PubMed] [Google Scholar]
- Kamisetty H, Ramanathan A, Bailey-Kellogg C, Langmead CJ. Accounting for conformational entropy in predicting binding free energies of protein-protein interactions. Proteins. 2011;79:444–462. doi: 10.1002/prot.22894. [DOI] [PubMed] [Google Scholar]
- Horn F, Weare J, Beukers MW, Horsch S, Bairoch A, Chen W, Edvardsen O, Campagne F, Vriend G. GPCRDB: an information system for G protein-coupled receptors. Nucleic Acids Res. 1998;26:275–279. doi: 10.1093/nar/26.1.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beukers MW, Kristiansen I. AP IJ, Edvardsen I: TinyGRAP database: a bioinformatics tool to mine G-protein-coupled receptor mutant data. Trends Pharmacol Sci. 1999;20:475–477. doi: 10.1016/S0165-6147(99)01403-0. [DOI] [PubMed] [Google Scholar]
- Kamisetty H. Structured Probabilistic Models of Proteins across Spatial and Fitness Landscape. Carnegie Mellon, Computer Science; 2011. [Google Scholar]
- Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods in Neurosciences. 1995;25:366–428. [Google Scholar]
- Eswar N, Eramian D, Webb B, Shen MY, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol. 2008;426:145–159. doi: 10.1007/978-1-60327-058-8_8. [DOI] [PubMed] [Google Scholar]