

Abstract
There is general agreement that many cancers are associated with aberrant phosphotyrosine signaling, which can be caused by the inappropriate activities of tyrosine kinases or tyrosine phosphatases. Furthermore, incorrect activation of signaling pathways has been often linked to changes in adhesion events mediated by cell surface receptors. Among these receptors, receptor protein tyrosine phosphatases (RPTPs) both antagonize tyrosine kinases as well as engage extracellular ligands. A recent wealth of data on this intriguing family indicates that its members can fulfill either tumor suppressing or oncogenic roles. The interpretation of these results at a molecular level has been greatly facilitated by the recent availability of structural information on the extra- and intracellular regions of RPTPs. These structures provide a molecular framework to understand how alterations in extracellular interactions can inactivate RPTPs in cancers or why the overexpression of certain RPTPs may also participate in tumor progression.
Keywords: cell adhesion, phosphorylation, phosphatase, inactivating somatic mutations, tumor suppressor, oncogene, receptor overexpression, crystal structure, receptor protein tyrosine phosphatase
Introduction
Tumor development and progression is associated with changes in cell proliferation, adhesion and migration, which are themselves tightly regulated by manifold signaling pathways. Tyrosine phosphorylation is a key molecular mechanism for most of these regulatory pathways. Mutations in protein tyrosine kinases have often been associated with aberrant cellular functions that lead to cancers1 because their altered activities create an imbalance in the phosphorylation states of key signaling molecules. On the other hand, changes in adhesive properties due to the loss of expression of cell adhesion molecules (CAMs) (see LeBras, Taubenslag and Andl, in this issue) are required for cancer cell migration and invasion. The loss of CAM expression is also associated with alterations in downstream signaling pathways. Kiefel et al. in this special focus discuss how altering the expression of L1CAM alters intracellular signaling. Hence, it is perhaps no surprise that molecules such as receptor protein tyrosine phosphatases (RPTPs), which combine CAM-like extracellular regions and intracellular tyrosine phosphatase domains, have been associated with human cancers2,3 (Fig. 1).
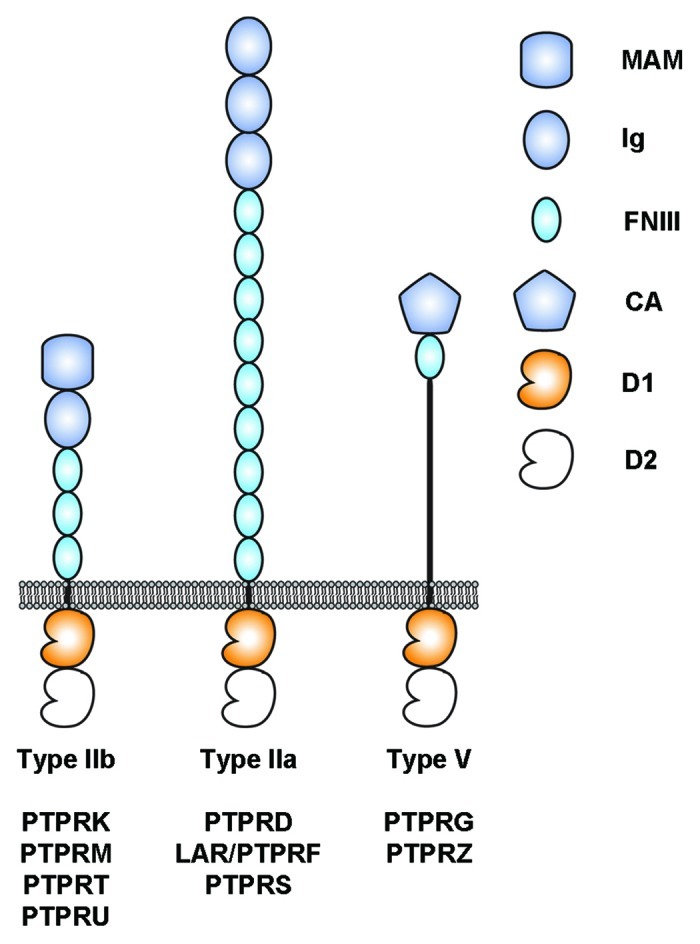
Figure 1. Architecture of selected human receptor protein tyrosine phosphatases considered in this review. The intracellular regions are composed of two tyrosine phosphatase domains called D1 and D2. D1 is catalytically active whereas D2 is not. MAM, Meprin-A5-RPTPμ domain; Ig, Immunoglobulin-like domain; FNIII, fibronectin type III domain; CA, Carbonic anhydrase domain; D1, active protein tyrosine phosphatase domain; D2, inactive protein tyrosine phosphatase domain.
Twenty-one RPTPs have been identified in humans since the initial characterization of CD454,5 and those receptors are involved in critical processes including immune regulation, nervous system development and vascular development.5-7 Unfortunately, RPTPs are not as well understood as receptor tyrosine kinases. A general mechanism that would account for the control of their catalytic activities by extracellular cues is lacking and most RPTPs remain orphan receptors.6,8 Nevertheless, their potential roles in cancer were described soon after their discovery when it was shown that the gene encoding a family member called PTPRG mapped to a chromosomal region deleted in lung and kidney cancer cell lines and thus could be a potential tumor suppressor.9 At first glance it appears logical that the absence or inactivation of a protein that antagonizes tyrosine kinase activity would be linked to tumor growth and indeed, many RPTPs are tumor suppressors.3 Yet, it quickly appeared that overexpression of RPTPs may play a role in tumor progression as well when Krueger and colleagues discovered that a PTPRG homolog called PTPRZ was overexpressed in glioblastoma cells.10 Data accumulated in the past few years now indicates clearly that RPTPs can fulfill both tumor suppressing and oncogenic roles.3
In this review, we will focus on the three classes of RPTPs for which structural information is available, which will in turn help us understand their functions in cell adhesion and cancer progression (Fig. 1). We will consider the importance of the some of the somatic mutations identified in type IIa and IIb RPTPs to illustrate the roles of RPTPs as tumor suppressors, particularly in the case of type IIb receptors. We will also focus on the potential oncogenic effect of type V receptors, especially in the case of PTPRZ. However, we will not discuss comprehensively the alterations of RPTP function that occur in cancer as these have already been reviewed elsewhere.2,3
Type IIb RPTPs and Homophilic Interactions
The prototypical member of the type IIb subgroup of RPTP, PTPRM, was identified in 1991 from a mouse brain library.11 This subgroup now includes four members in vertebrates (PTPRK/RPTPκ, PTPRM/RPTPμ, PTPRT/RPTPρ and PTPRU/PCP2/RPTPλ), but lacks a clear ortholog in Drosophila. The four family members share a common domain topology including an N-terminal Meprin-A5-RPTPμ (MAM) domain, an Ig repeat and four FNIII domains in their respective extracellular regions while the intracellular segment includes two tyrosine phosphatase domains (Fig. 1). These RPTPs represent a particularly interesting subgroup because three of the four family members (PTPRM, PTPRK and PTPRT) “act as their own ligands” and mediate homophilic interactions between two apposing cells.12-15 Despite strong homology in their extracellular regions (typically more than 50% amino acid sequence identity), the homophilic interactions between type IIb family members are specific. For example, both PTPRM and PTPRK interact homophilically to form dimers yet are unable to interact with one another16 and similar results have been obtained for PTPRM and PTPRT.15 In contrast with other type IIb RPTPs, PTPRU is not a homophilic binding protein as it does not mediate cell-cell aggregation and the identities of its potential extracellular binding partners are as of yet unknown.17
One of the most interesting aspects of type IIb RPTPs is their relationship with cadherins and intracellular components of cadherin adhesion complexes called catenins.18 Cadherins are Ca2+-dependent homophilic-binding molecules that mediate adhesive interactions between adjacent cells and are central to the development and maintenance of stable tissues.18 Critically, alterations in the assembly of cadherin-catenin adhesion complexes are linked to the disassociation of adherent epithelial cells and their subsequent migration, a process that can be part of the normal aspect of tissue development, but is also a hallmark of cancer growth and metastasis18,19 (the effects of E- and N-cadherin in cancer progression and epithelial-mesenchymal transitions are discussed by LeBras, Taubenslag and Andl in this issue). These transitions can be brought about by enhanced tyrosine phosphorylation of β-catenin,20,21 which explains why the association of type IIb RPTPs with components of cadherin-catenin adhesion complexes has been of particular interest.22-28 For example, PTPRM associates with E-cadherin23 and dephosphorylates p120 catenin.26 Yet, the role played by type IIb RPTPs in cadherin-mediated cell adhesion may not be solely attributed to their catalytic activity. Indeed, expression of PTPRM in a prostate cancer cell line could restore cadherin-dependent adhesion of these cells, but so did a catalytically inactive form of PTPRM,29 highlighting the importance of additional intracellular effectors of PTPRM signaling.
Consistent with their association with cadherins and catenins, type IIb RPTPs have been linked to the progression of human cancers. Downregulation of PTPRM in glioma cells due to proteolysis correlates with increased cell movement and tumor invasiveness.30 Similarly, cell surface cleavage of PTPRK favors the dispersion of colon cancer cells.31,32 These findings strongly suggest that shedding of these RPTPs is involved in cancer progression and recent results indicate that this process may not be limited to type IIb family members.33 On the other hand, PTPRT associates with E-cadherin and is linked to the stability of cadherin-catenin adhesion complexes27 so that a decrease of PTPRT adhesiveness due to mutations in its ectodomain found in cancer cells15,34 may in turn promote the migration of these cells. Hence, the picture that emerges is that impairing type IIb-mediated cell adhesion can favor the dispersion of cells expressing these receptors and thus promote tumor progression. Because of the increased interest in the roles of RPTPs in cancer, several groups have worked to identify mutations in genes encoding RPTPs in several human cancers. In particular, mutational analysis of colorectal cancer cells has highlighted PTPRT has a frequently mutated gene in this cancer35 and mice lacking PTPRT are more susceptible to cancer development.36
The recent availability of the crystal structure of the PTPRM ectodomain37 provides us with an opportunity to evaluate the effects of mutations in the ectodomains of type IIb receptors that have been identified in cancer patients. Given the sequence identity between type IIb RPTPs, this structure likely recapitulates the structures of all members of this family. PTPRM adopts an extended, rod-like conformation and the short linker segments between the individual modules of the extracellular region presumably contributes to the rigidity of this structure.37 The MAM-Ig segment forms a single structural unit in which the MAM and Ig domains interact extensively.38 Importantly, the homophilic contacts between PTPRM molecules expressed on apposing cells involve the four N-terminal domains of PTPRM37 (MAM, Ig, FN1 and FN2) and analysis of the contact residues accounted for the absence of heterophilic contacts between type IIb family members.
Visualization of the homophilic PTPRM interface allowed mapping of the cancer mutations found in type IIb family members that have been identified in multiple human cancers39-49 and can be accessed via the COSMIC database.50 Missense mutations locate at the homophilic interface between two opposing type IIb molecules,15,34,37 suggesting that disruption in these contacts may be linked to tumor growth, in line with the notion that loss of adhesiveness can favor the progression of cancers (Fig. 2). A second set of mutations is localized at interfaces between modules of PTPRM. The potential effect of these mutations could be to reduce the rigidity of the entire extracellular region, which would in turn impair homophilic interactions in much the same way that removal of the Ca2+ ions that rigidify the interfaces of successive cadherin modules inhibits homophilic interactions between cadherins.51 Finally, a third set of mutated amino acid residues were found in buried portions of the PTPRM ectodomain and may impair its folding or processing, thus reducing or even preventing receptor presentation at the cell surface52 (Fig. 2).
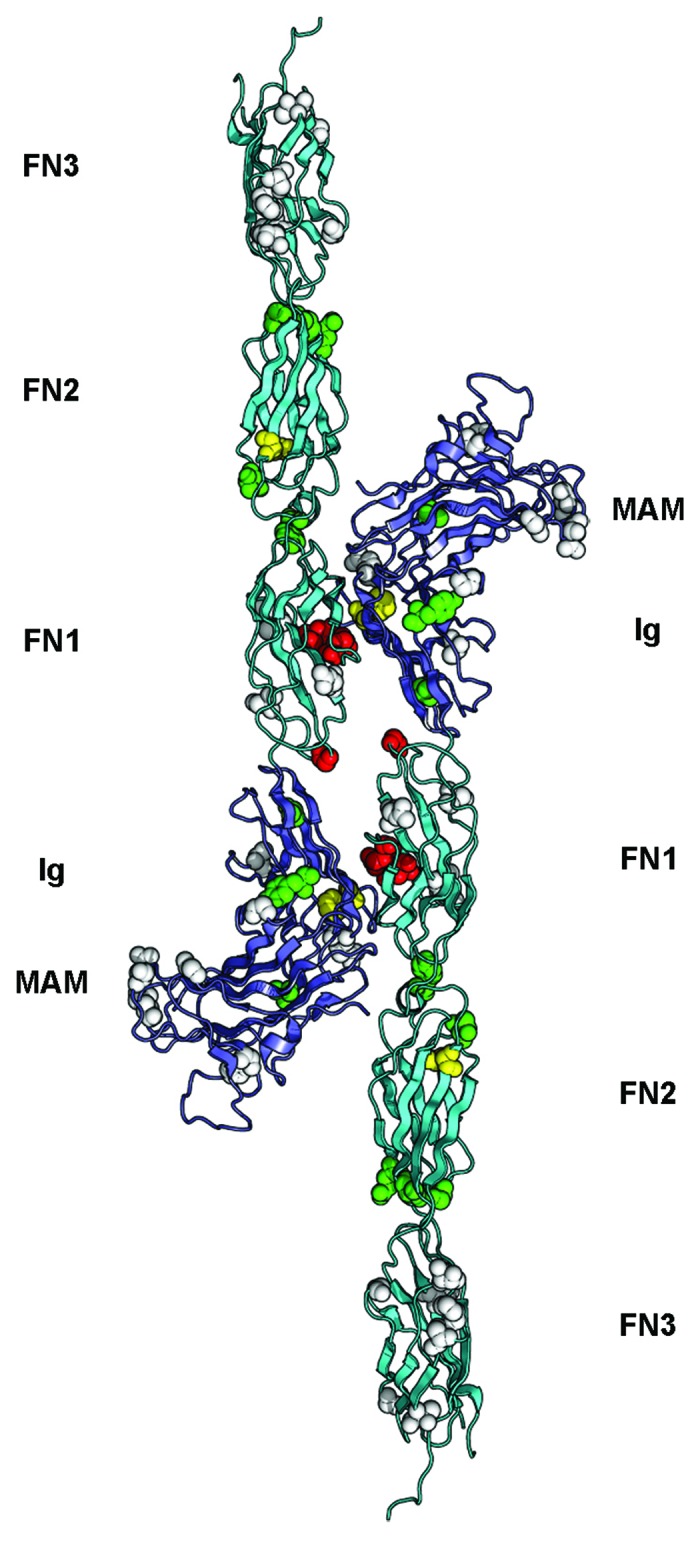
Figure 2. Overview of somatic mutations in type IIb RPTPs shown in the PTPRM homophilic dimer. The structure of the PTPRM homodimer was obtained from PDB ID 2V5Y determined by Aricescu et al.37 and is shown as a ribbon diagram. The MAM and Ig region are colored slate and the FNIII modules are colored cyan. The residues that are mutated in cancers are shown as spheres colored based on the predicted effects of the location of the mutations (yellow, buried; green, interdomain interface; red, homophilic interface; white, solvent exposed). The expected effect of the mutations in yellow, green or red would be to impair homophilic interactions whereas the expected effects of mutating residues shown as white spheres are unknown.
Importantly, the common effect of the changes found in cancers would be to inhibit the formation of homophilic contacts. This notion was confirmed by assessing the ability of non-adherent Spodoptera frugiperda Sf9 cells to form clusters when transfected with wild-type or mutated PTPRT.15,34 In these experiments, the aggregation of PTPRT-transfected Sf9 cells was impaired when cancer missense mutations were introduced in the PTPRT ectodomain, thus providing a clear relationship between amino acid changes at these positions and reduced cellular adhesion. However, little can be inferred from a fourth group of mutations include amino acid residues at the protein surface that are located away from the homophilic interface (Fig. 2). It is tempting to speculate that these may affect the association of a given type IIb RPTP with itself on the same cell surface53 or with another cell surface receptor such as cadherins,24 or may render the receptor ectodomain more susceptible to proteolysis, but evidence of this is lacking.
Type IIa RPTPs
Leukocyte common antigen related or LAR was first identified in 1988 from a human placental genomic DNA library54 and is the founding member of the type IIa subgroup of RPTPs, represented by Dlar in Drosophila55 and by PTPRF/LAR, PTPRD/RPTPδ and PTPRS/RPTPσ in vertebrates7 (Fig. 1). Historically, this family of receptors is particularly important because it was studies on the development of the Drosophila nervous system that demonstrated that Dlar plays an important role in the guidance of motor axons,56,57 which paved the way for investigating the specific physiological functions of RPTPs during neural development.7 Since then, vertebrate type IIa members have been linked to neurogenesis (PTPRS58), axon guidance (PTPRS59) and synaptogenesis (LAR, PTPRD and PTPRS60-63).
The physiological roles of type IIa RPTPs in neural development are mediated, at least in part, by interactions with chondroitin sulfate proteoglycans (CSPGs) and heparan sulfate proteoglycans (HSPGs). HSPGs bind to PTPRS in chick retina64 and the sole type IIa Drosophila RPTP Dlar binds to heparan sulfate (HS) chains on syndecan and dally-like protein to drive the formation of synapses at neuromuscular junctions.65,66 The interactions between these receptors and proteoglycans depend on their N-terminal Ig module and in particular on the presence of a cluster of basic residues in a loop region,64 which is conserved in Dlar and its vertebrate orthologs.67 On the other hand, this region interacts functionally with CSPGs, at least in the case of PTPRS.68,69 Critically, the physiological outcomes of the interactions with these two classes of ligands are distinct since interactions with HSPGs promote axonal outgrowth whereas interactions with CSPGs impair outgrowth.68,69 These radically different outcomes are linked to the different oligomeric state of PTPRS in the presence of HS or CS chains. The current model proposes that PTPRS aggregates in the presence of HS chains expressed on the same cell leading to an uneven distribution of phosphatase activity at the cell surface because of the presence of PTPRS clusters. This redistribution would be reversed when PTPRS interacts with CS chains expressed on a different cell as PTPRS would revert to its monomeric state.69 The molecular basis accounting for the distinct oligomeric state of PTPRS in the presence of its glycosaminoglycan ligands remains unclear, however.
Evidence for the involvement of type IIa RPTPs in cancer came in 2004 when somatic mutations were identified in the PTPRF gene in colorectal tumors.35 Soon after, deletions in the PTPRD gene were found in human cancer cell lines70 and more recent data indicate that this gene is inactivated in glioblastoma multiforme, malignant melanoma and lung carcinoma as well as head and neck carcinoma.71,72 Similarly, PTPRS is frequently deleted in head and neck squamous cell carcinoma.73 Whole genome studies aiming to document the genetic variations existing in human cancers uncovered several missense mutations in the genes encoding type IIa family members.39,40,42,44,45,48,49,71,74-77 Although most of these mutations are located in the phosphatase region where they presumably lead to a loss of phosphatase activity, some of the changes localize to the extracellular region and in particular to the N-terminal Ig region for which structural data were obtained recently67,69 (Figs. 1 and3).
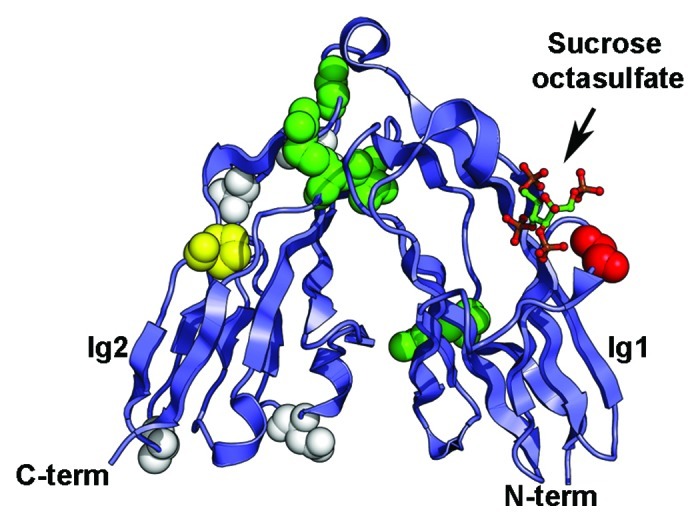
Figure 3. Somatic mutations identified in the Ig1-Ig2 tandem repeats of type IIa RPTPs. Mutations in LAR, PTPRD and PTPRS are shown on the structure of human LAR bound to the glycosaminoglycan mimic sucrose octasulfate (PDB ID 2YD869). The sucrose octasulfate is shown in ball-and-stick representation. The residues that are mutated in cancers are shown as spheres colored based on the predicted effects of the location of the mutations (yellow, buried; green, interdomain interface; red, ligand interface; white, solvent exposed). The expected effect of the mutations in yellow would be to disrupt the folding of the Ig domain while mutation of the residues colored green is expected to disrupt the interface between Ig1 and Ig2. Introducing a glycine to glutamate change at position 61 (shown in red in the structure) is expected to impair interactions with the ligand. The potential effects of mutating residues colored white are unknown.
The structures of the first two Ig repeats of LAR, PTPRD and PTPRS revealed unusual features. Although there are little differences between the topologies of each Ig domain and Ig modules found in other cell surface receptors,67 the Ig1-Ig2 pair adopts an antiparallel arrangement giving this region the appearance of a horseshoe. This conformation is conserved in the Drosophila family member Dlar67,69 and resembles the structure of FNIII tandem repeats found in the extracellular region of the Drosophila hedgehog co-receptor iHog.67 It is however distinct from horseshoe conformations recently identified in contactins, Dscams and neurofascin.78-84 These structural analyses also revealed the nature of the glycosaminoglycan-binding site and confirmed earlier biochemical experiments that had mapped it to the first Ig domain.64,68
Interestingly, several of the cancer somatic mutations map to this region and in particular to residues at the interface between Ig1 and Ig2 in both PTPRF (G123E in malignant melanoma50 and R206H in ovarian tumor44) and PTPRS (V224M in colorectal cancer76) and may disrupt the interdomain interface (Fig. 3, residues are shown as green spheres). Furthermore, a glutamate residue is found in place of a conserved glycine residue in the PTPRD-glycosaminoglycan binding site in malignant melanomas.71 This change (G61E, colored red on Fig. 3) introduces a negative charge and would be expected to weaken the interaction between this usually positively-charged region conserved in all type IIa receptors and the negative charge on the glycosaminoglycan chains.67 Finally, a proline to serine change at position 141 in PTPRS50 is found in malignant melanomas. This proline is buried and conserved in type IIa family members so that this change may impair the folding of this region altogether (Fig. 3, residue is depicted as yellow spheres). Inferring the potential roles of these mutations in tumor progression is more challenging, however, because the effect these changes have on protein function have not been assessed. In particular, binding of HS chains to the Ig1-Ig2 region of Dlar appears to be solely mediated by Ig1 and is seemingly unaffected by mutations that are expected to impair the formation of the Ig1-Ig2 interface,67 although this particular set of experiments did not examine the roles of the mutations within the context of the full-length protein nor if the physiological properties were altered after mutations. Assessment of the biochemical properties of changes in the Ig region of type IIa RPTPs is warranted and may provide yet another clear link between control of cell adhesion by phosphatases and regulation of intracellular signal transduction pathways.
The potential mechanisms by which the absence of type IIa RPTPs would promote tumor progression have been studied in more detail. Inactivation of PTPRD results in activation of the oncoprotein signal transducer and activator of transcription 3 (STAT3) and in turn expression of STAT3-target genes that promote tumorigenesis.72 The implication of STAT3 in this pathway is particularly interesting because it had already been identified as a substrate of the type IIb receptor PTPRT by Zhang and colleagues36 and because inactivation of PTPRT leads to enhanced expression of STAT3 target genes as is now the case for PTPRD. Activation of STAT3 is not the only change associated with type IIa inactivation. Indeed, deletion of PTPRS in head and neck squamous cell carcinomas results in activation of the PI3K/epidermal growth factor receptor pathway presumably because PTPRS is required to antagonize the epidermal growth factor receptor kinase activity.73,85 Although speculative, an additional mechanism for type IIa-dependent tumor progression may be associated with cadherin-catenin adhesion complexes as both LAR and PTPRS associate with cadherins and dephosphorylate β-catenin.86-90 Moreover, inducing expression of LAR in migrating epithelial cells, in which tyrosine phosphorylation of β-catenin is enhanced, inhibits their migration.88 The structural basis for the association of type IIa receptors with cadherins and β-catenin is not known, but it will be very interesting to determine if some of the somatic mutations identified in LAR or PTPRS map to these potential binding interfaces.
Overexpression of Type V RPTPs
The receptors PTPRG and PTPRZ can be considered somewhat like oddities among RPTPs. Instead of the combination of multiple Ig and FNIII modules that are found in the ectodomains of most RPTPs, PTPRG and PTPRZ include an N-terminal inactive carbonic anhydrase (CA), a single FNIII repeat and a long spacer region that is presumably heavily glycosylated. PTPRG and PTPRZ were initially cloned in the early 1990s10,91-93 and were quickly linked to human tumors as PTPRG was mapped to a chromosomal region frequently deleted in kidney and lung cancers9 whereas a glioblastoma cell line (U373MG) was found to express high levels of PTPRZ.10 Both receptors are expressed mostly in the nervous system during embryogenesis and adulthood, but their respective localization differs as PTPRG is expressed mostly on neurons and PTPRZ on glial cells.94,95 Despite their similar architectures, PTPRG and PTPRZ appear to have distinct roles in neural development as PTPRG inhibited nerve growth factor-induced neurite outgrowth when expressed recombinantly in PC12D cells whereas PTPRZ had no effect on this process.96 However, PTPRZ mediates the outgrowth of neurites when bound to its ligand contactin-1 (CNTN1) expressed on neurons97,98 and this complex also plays an important role in the maturation of oligodendrocytes.83 The role of PTPRG in vivo is less well described although it has recently been shown to be involved in spinal cord neurogenesis in chicken,99 which is somewhat in contrast with the results obtained in PC12D cells and underscores the potentially complex role that this receptor may have in neural development.
Although the search for an extracellular binding partner for PTPRG was not initially successful, several ligands for PTPRZ have been found, including the neural CAM contactin-1 (CNTN1) mentioned above and the growth factor pleiotrophin100 (PTN). The latter proved instrumental in our understanding of how tyrosine phosphatase activity can be regulated by extracellular cues and also shed light into the role of PTPRZ in tumor progression. Initial insights on the effect of PTN on PTPRZ activity came in 2000 when Meng and colleagues demonstrated that PTN binds to and inactivates PTPRZ expressed on the surface of the glioblastoma cells U373MG,101 leading to an increase in the tyrosine phosphorylation of β-catenin. Furthermore, another series of experiments extended those initial findings by demonstrating that PTPRZ is catalytically active in its monomeric state, but is inactivated after it dimerizes following treatment with PTN resulting in the enhanced phosphorylation of its substrates.102 In parallel experiments, the role played by PTPRZ in glioblastoma was examined because it is typically overexpressed in these tumors.103 PTN stimulates migration of glioblastoma cells. When PTPRZ expression is reduced by siRNA, PTN no longer stimulates the migration of these cells. This demonstrates that PTPRZ mediates the stimulatory effect of PTN on glioblastoma cell migration.103 These migratory properties were explained in part by enhanced tyrosine phosphorylation of β-catenin after PTPRZ is inactivated by PTN, which then promotes disassembly of cadherin-catenin adhesion complexes and thus a loss of cell adhesiveness.104 Consistent with this notion, aberrant β-catenin signaling has recently been linked to the etiology of astrocytomas.21
The previous observations hinged on the fact that PTN was able to induce dimerization of PTPRZ and that this change of oligomeric state inactivated the intracellular phosphatase activity. A model implicating the dimerization/inhibition of RPTPs has been favored since the crystal structure of a dimeric form of the D1 domain of PTPRA was determined and showed that a helix-turn-helix motif (called a wedge) of one protomer was inserted into the active site of a second protomer and blocked its active site.105 The influence of RPTP dimerization on phosphatase activity had already been suggested by experiments demonstrating that the phosphatase activity of an EGFR-CD45 chimera could be inhibited upon EGF binding106 so that it was hypothesized that the formation of dimers implicating the wedge region could be an attractive model to explain the control of phosphatase activity. However, the crystal structures of the active phosphatase domain of PTPRM107 and of the tandem phosphatase domains of LAR108 and CD45109 did not show any hint of dimerization for these proteins, casting doubts that wedge-mediated dimerization/inhibition of RPTPs represented a general mechanism for control of RPTP activity. Therefore it was a significant result when it was shown that the tandem phosphatase region of PTPRG dimerizes in solution and that the phosphatase domains are arranged in an antiparallel fashion so that the two active sites are occluded in the dimer crystal structure110 (Fig. 4). The high sequence identity between PTPRG and PTPRZ as well as the conservation of the residues at the dimer interface strongly suggests that two PTPRZ phosphatase regions can assemble in similar fashion, thus providing structural insights into the mechanism by which PTN binding inactivates the tyrosine phosphatase activity of PTPRZ and ultimately promotes tumor progression.
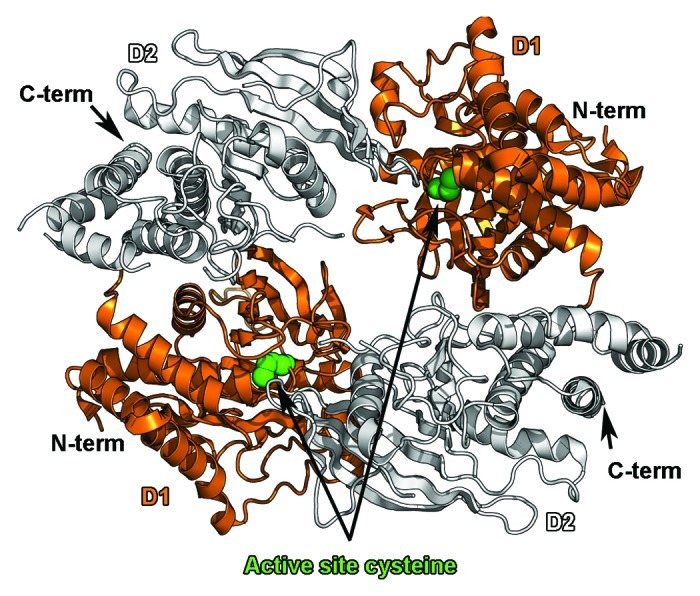
Figure 4. Dimerization of the tandem phosphatase regions of PTPRG. The antiparallel dimer of the tandem phosphatase region of PTPRG (PDB ID 2NLK110) is shown as a ribbon diagram. The catalytically active D1 domains are colored orange, while the inactive D2 domains are colored white. Dimerization blocks access to a key cysteine residue in the active site (depicted as green spheres).
In addition, the work on PTN/PTPRZ interactions paved the way for understanding how ligands of type V RPTPs could regulate the intracellular phosphatase activity of their cognate receptors. Indeed, inactivation of the catalytic activity of PTPRZ could be induced after incubation with an antibody raised against the extracellular region of PTPRZ102 suggesting that other binding partners of PTPRZ could inhibit its phosphatase activity. CNTN1 is probably the best understood of these ligands since formation of a complex of PTPRZ and CNTN1 is critical for the proper maturation of oligodendrocytes.83 Interestingly, reduced expression of CNTN1 correlates with impaired invasiveness of lung adenocarcinoma cells111 and more recently, increased expression of CNTN1 has been identified in melanoma,112 indicating that CNTN1 could play a role in the etiology of some human cancers. Consistent with this notion, CNTN1 is expressed in astrocytic tumors with increasing malignancies correlating with increasing CNTN1 expression.113 In particular, it was demonstrated that PTPRZ-expressing cells adhered less strongly to CNTN1-expressing cells than control cells and that this decreased adhesion was due to the presence of PTPRZ. The sum of these experiments suggested that interactions between CNTN1 and PTPRZ could have a repulsive effect113 and thus would promote the migration of the PTPRZ-expressing glioma cells. Keeping in mind the notion that interactions between PTN and PTPRZ result in loss of adhesiveness in part because of enhanced tyrosine phosphorylation of β-catenin and subsequent loss of cadherin-mediated adhesion, one is tempted to speculate that formation of CNTN1/PTPRZ complexes may also inactivate PTPRZ, leading to enhanced phosphorylation of PTPRZ substrates such as β-catenin and ultimately promote cell migration.
In this context, the recent identification of CNTN1 homologs as potential binding partners for PTPRG is particularly interesting. Indeed, four of the six members of the CNTN family of neural recognition molecules called CNTN3, 4, 5 and 6 were recently shown to interact with PTPRG82 and a crystal structure of a PTPRG/CNTN4 complex showed that it is closely related to the one formed by CNTN1 and PTPRZ.83 It is not yet known what the precise physiological functions of CNTN/PTPRG interactions are, but based on their localization in the nervous system it is speculated that these proteins could be involved in the development and function of the nervous system.82 Interestingly, as is the case for PTPRZ, PTPRG is overexpressed in gliomas,114 hinting that PTPRG/CNTN signaling could play a role in tumor progression in much the same way that PTPRZ/CNTN1 may especially since β-catenin is an in vitro PTPRG substrate.99
Concluding Remarks
The architecture of RPTPs has long suggested that these receptors are able to mediate both cell adhesion and cell signaling. Since the progression of tumors is often characterized by changes in both adhesion and signaling properties, recent work has been aimed at characterizing the alterations in RPTP activity that are associated with human cancers. Even though it is now clear that RPTPs can both suppress or activate the growth of tumors, our understanding is still limited by the fact that the in vivo substrates of RPTPs are not well described and the extracellular cues to which they respond remain poorly defined. A significant progress in the RPTP field will undoubtedly come from the availability of crystal structures of full RPTP ectodomains in the absence and presence of their ligands, which will explain how RPTP ligands control receptor dimerization and phosphatase activity. Until then, renewed efforts to identify RPTP ligands and to obtain structural information on RPTPs have already helped bridge the gap between the description of RPTP alterations in cancer and the role these changes may have at a molecular level. This trend will continue and will likely set the stage for the development of new cancer therapies based on RPTP biology.
Acknowledgments
This work was supported by the National Institute of General Medical Sciences (award number R01GM088806).
Footnotes
Previously published online: www.landesbioscience.com/journals/celladhesion/article/21242
References
- 1.Hunter T, Sefton BM. Transforming gene product of Rous sarcoma virus phosphorylates tyrosine. Proc Natl Acad Sci U S A. 1980;77:1311–5. doi: 10.1073/pnas.77.3.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ostman A, Hellberg C, Böhmer FD. Protein-tyrosine phosphatases and cancer. Nat Rev Cancer. 2006;6:307–20. doi: 10.1038/nrc1837. [DOI] [PubMed] [Google Scholar]
- 3.Julien SG, Dubé N, Hardy S, Tremblay ML. Inside the human cancer tyrosine phosphatome. Nat Rev Cancer. 2011;11:35–49. doi: 10.1038/nrc2980. [DOI] [PubMed] [Google Scholar]
- 4.Charbonneau H, Tonks NK, Walsh KA, Fischer EH. The leukocyte common antigen (CD45): a putative receptor-linked protein tyrosine phosphatase. Proc Natl Acad Sci U S A. 1988;85:7182–6. doi: 10.1073/pnas.85.19.7182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, et al. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 6.Hendriks WJAJ, Elson A, Harroch S, Stoker AW. Protein tyrosine phosphatases: functional inferences from mouse models and human diseases. FEBS J. 2008;275:816–30. doi: 10.1111/j.1742-4658.2008.06249.x. [DOI] [PubMed] [Google Scholar]
- 7.Johnson KG, Van Vactor D. Receptor protein tyrosine phosphatases in nervous system development. Physiol Rev. 2003;83:1–24. doi: 10.1152/physrev.00016.2002. [DOI] [PubMed] [Google Scholar]
- 8.Stoker A. Methods for identifying extracellular ligands of RPTPs. Methods. 2005;35:80–9. doi: 10.1016/j.ymeth.2004.07.011. [DOI] [PubMed] [Google Scholar]
- 9.LaForgia S, Morse B, Levy J, Barnea G, Cannizzaro LA, Li F, et al. Receptor protein-tyrosine phosphatase gamma is a candidate tumor suppressor gene at human chromosome region 3p21. Proc Natl Acad Sci U S A. 1991;88:5036–40. doi: 10.1073/pnas.88.11.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krueger NX, Saito H. A human transmembrane protein-tyrosine-phosphatase, PTP zeta, is expressed in brain and has an N-terminal receptor domain homologous to carbonic anhydrases. Proc Natl Acad Sci U S A. 1992;89:7417–21. doi: 10.1073/pnas.89.16.7417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gebbink MF, van Etten I, Hateboer G, Suijkerbuijk R, Beijersbergen RL, Geurts van Kessel A, et al. Cloning, expression and chromosomal localization of a new putative receptor-like protein tyrosine phosphatase. FEBS Lett. 1991;290:123–30. doi: 10.1016/0014-5793(91)81241-Y. [DOI] [PubMed] [Google Scholar]
- 12.Brady-Kalnay SM, Flint AJ, Tonks NK. Homophilic binding of PTP mu, a receptor-type protein tyrosine phosphatase, can mediate cell-cell aggregation. J Cell Biol. 1993;122:961–72. doi: 10.1083/jcb.122.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gebbink MF, Zondag GC, Wubbolts RW, Beijersbergen RL, van Etten I, Moolenaar WH. Cell-cell adhesion mediated by a receptor-like protein tyrosine phosphatase. J Biol Chem. 1993;268:16101–4. [PubMed] [Google Scholar]
- 14.Sap J, Jiang YP, Friedlander D, Grumet M, Schlessinger J. Receptor tyrosine phosphatase R-PTP-kappa mediates homophilic binding. Mol Cell Biol. 1994;14:1–9. doi: 10.1128/mcb.14.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu J, Becka S, Zhang P, Zhang X, Brady-Kalnay SM, Wang Z. Tumor-derived extracellular mutations of PTPRT /PTPrho are defective in cell adhesion. Mol Cancer Res. 2008;6:1106–13. doi: 10.1158/1541-7786.MCR-07-2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zondag GC, Koningstein GM, Jiang YP, Sap J, Moolenaar WH, Gebbink MF. Homophilic interactions mediated by receptor tyrosine phosphatases mu and kappa. A critical role for the novel extracellular MAM domain. J Biol Chem. 1995;270:14247–50. doi: 10.1074/jbc.270.24.14247. [DOI] [PubMed] [Google Scholar]
- 17.Becka S, Zhang P, Craig SEL, Lodowski DT, Wang Z, Brady-Kalnay SM. Characterization of the adhesive properties of the type IIb subfamily receptor protein tyrosine phosphatases. Cell Commun Adhes. 2010;17:34–47. doi: 10.3109/15419061.2010.487957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gumbiner BM. Regulation of cadherin-mediated adhesion in morphogenesis. Nat Rev Mol Cell Biol. 2005;6:622–34. doi: 10.1038/nrm1699. [DOI] [PubMed] [Google Scholar]
- 19.Berx G, van Roy F. Involvement of members of the cadherin superfamily in cancer. Cold Spring Harb Perspect Biol. 2009;1:a003129. doi: 10.1101/cshperspect.a003129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roura S, Miravet S, Piedra J, García de Herreros A, Duñach M. Regulation of E-cadherin/Catenin association by tyrosine phosphorylation. J Biol Chem. 1999;274:36734–40. doi: 10.1074/jbc.274.51.36734. [DOI] [PubMed] [Google Scholar]
- 21.Yang C, Iyer RR, Yu ACH, Yong RL, Park DM, Weil RJ, et al. β-Catenin signaling initiates the activation of astrocytes and its dysregulation contributes to the pathogenesis of astrocytomas. Proc Natl Acad Sci U S A. 2012;109:6963–8. doi: 10.1073/pnas.1118754109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fuchs M, Müller T, Lerch MM, Ullrich A. Association of human protein-tyrosine phosphatase kappa with members of the armadillo family. J Biol Chem. 1996;271:16712–9. doi: 10.1074/jbc.271.28.16712. [DOI] [PubMed] [Google Scholar]
- 23.Brady-Kalnay SM, Mourton T, Nixon JP, Pietz GE, Kinch M, Chen H, et al. Dynamic interaction of PTPmu with multiple cadherins in vivo. J Cell Biol. 1998;141:287–96. doi: 10.1083/jcb.141.1.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Del Vecchio RL, Tonks NK. The conserved immunoglobulin domain controls the subcellular localization of the homophilic adhesion receptor protein-tyrosine phosphatase mu. J Biol Chem. 2005;280:1603–12. doi: 10.1074/jbc.M410181200. [DOI] [PubMed] [Google Scholar]
- 25.Oblander SA, Ensslen-Craig SE, Longo FM, Brady-Kalnay SM. E-cadherin promotes retinal ganglion cell neurite outgrowth in a protein tyrosine phosphatase-mu-dependent manner. Mol Cell Neurosci. 2007;34:481–92. doi: 10.1016/j.mcn.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zondag GC, Reynolds AB, Moolenaar WH. Receptor protein-tyrosine phosphatase RPTPmu binds to and dephosphorylates the catenin p120(ctn) J Biol Chem. 2000;275:11264–9. doi: 10.1074/jbc.275.15.11264. [DOI] [PubMed] [Google Scholar]
- 27.Besco JA, Hooft van Huijsduijnen R, Frostholm A, Rotter A. Intracellular substrates of brain-enriched receptor protein tyrosine phosphatase rho (RPTPrho/PTPRT) Brain Res. 2006;1116:50–7. doi: 10.1016/j.brainres.2006.07.122. [DOI] [PubMed] [Google Scholar]
- 28.Yan H-X, Yang W, Zhang R, Chen L, Tang L, Zhai B, et al. Protein-tyrosine phosphatase PCP-2 inhibits beta-catenin signaling and increases E-cadherin-dependent cell adhesion. J Biol Chem. 2006;281:15423–33. doi: 10.1074/jbc.M602607200. [DOI] [PubMed] [Google Scholar]
- 29.Hellberg CB, Burden-Gulley SM, Pietz GE, Brady-Kalnay SM. Expression of the receptor protein-tyrosine phosphatase, PTPmu, restores E-cadherin-dependent adhesion in human prostate carcinoma cells. J Biol Chem. 2002;277:11165–73. doi: 10.1074/jbc.M112157200. [DOI] [PubMed] [Google Scholar]
- 30.Burgoyne AM, Phillips-Mason PJ, Burden-Gulley SM, Robinson S, Sloan AE, Miller RH, et al. Proteolytic cleavage of protein tyrosine phosphatase μ regulates glioblastoma cell migration. Cancer Res. 2009;69:6960–8. doi: 10.1158/0008-5472.CAN-09-0863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim Y-S, Kang H-Y, Kim J-Y, Oh S, Kim C-H, Ryu CJ, et al. Identification of target proteins of N-acetylglucosaminyl transferase V in human colon cancer and implications of protein tyrosine phosphatase kappa in enhanced cancer cell migration. Proteomics. 2006;6:1187–91. doi: 10.1002/pmic.200500400. [DOI] [PubMed] [Google Scholar]
- 32.Kim Y-S, Jung J-A, Kim H-J, Ahn YH, Yoo JS, Oh S, et al. Galectin-3 binding protein promotes cell motility in colon cancer by stimulating the shedding of protein tyrosine phosphatase kappa by proprotein convertase 5. Biochem Biophys Res Commun. 2011;404:96–102. doi: 10.1016/j.bbrc.2010.11.071. [DOI] [PubMed] [Google Scholar]
- 33.Craig SEL, Brady-Kalnay SM. Tumor-derived extracellular fragments of receptor protein tyrosine phosphatases (RPTPs) as cancer molecular diagnostic tools. Anticancer Agents Med Chem. 2011;11:133–40. doi: 10.2174/187152011794941244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang P, Becka S, Craig SE, Lodowski DT, Brady-Kalnay SM, Wang Z. Cancer-derived mutations in the fibronectin III repeats of PTPRT/PTPrho inhibit cell-cell aggregation. Cell Commun Adhes. 2009;16:146–53. doi: 10.3109/15419061003653771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Z, Shen D, Parsons DW, Bardelli A, Sager J, Szabo S, et al. Mutational analysis of the tyrosine phosphatome in colorectal cancers. Science. 2004;304:1164–6. doi: 10.1126/science.1096096. [DOI] [PubMed] [Google Scholar]
- 36.Zhang X, Guo A, Yu J, Possemato A, Chen Y, Zheng W, et al. Identification of STAT3 as a substrate of receptor protein tyrosine phosphatase T. Proc Natl Acad Sci U S A. 2007;104:4060–4. doi: 10.1073/pnas.0611665104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aricescu AR, Siebold C, Choudhuri K, Chang VT, Lu W, Davis SJ, et al. Structure of a tyrosine phosphatase adhesive interaction reveals a spacer-clamp mechanism. Science. 2007;317:1217–20. doi: 10.1126/science.1144646. [DOI] [PubMed] [Google Scholar]
- 38.Aricescu AR, Hon W-C, Siebold C, Lu W, van der Merwe PA, Jones EY. Molecular analysis of receptor protein tyrosine phosphatase mu-mediated cell adhesion. EMBO J. 2006;25:701–12. doi: 10.1038/sj.emboj.7600974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Parsons DW, Jones S, Zhang X, Lin JC-H, Leary RJ, Angenendt P, et al. An integrated genomic analysis of human glioblastoma multiforme. Science. 2008;321:1807–12. doi: 10.1126/science.1164382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333:1157–60. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Parsons DW, Li M, Zhang X, Jones S, Leary RJ, Lin JC-H, et al. The genetic landscape of the childhood cancer medulloblastoma. Science. 2011;331:435–9. doi: 10.1126/science.1198056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360–3. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones S, Zhang X, Parsons DW, Lin JC-H, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cancer Genome Atlas Research Network Integrated genomic analyses of ovarian carcinoma. Nature. 2011;474:609–15. doi: 10.1038/nature10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Durinck S, Ho C, Wang NJ, Liao W, Jakkula LR, Collisson EA, et al. Temporal Dissection of Tumorigenesis in Primary Cancers. Cancer Discov. 2011;1:137–43. doi: 10.1158/2159-8290.CD-11-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Quesada V, Conde L, Villamor N, Ordóñez GR, Jares P, Bassaganyas L, et al. Exome sequencing identifies recurrent mutations of the splicing factor SF3B1 gene in chronic lymphocytic leukemia. Nat Genet. 2012;44:47–52. doi: 10.1038/ng.1032. [DOI] [PubMed] [Google Scholar]
- 47.Jones S, Wang T-L, Shih IeM, Mao T-L, Nakayama K, Roden R, et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science. 2010;330:228–31. doi: 10.1126/science.1196333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei X, Walia V, Lin JC, Teer JK, Prickett TD, Gartner J, et al. NISC Comparative Sequencing Program Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat Genet. 2011;43:442–6. doi: 10.1038/ng.810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sjöblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–74. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 50.Forbes SA, Bhamra G, Bamford S, Dawson E, Kok C, Clements J, et al. The Catalogue of Somatic Mutations in Cancer (COSMIC). Curr Protoc Hum Genet 2008; Chapter 10:Unit 10.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Boggon TJ, Murray J, Chappuis-Flament S, Wong E, Gumbiner BM, Shapiro L. C-cadherin ectodomain structure and implications for cell adhesion mechanisms. Science. 2002;296:1308–13. doi: 10.1126/science.1071559. [DOI] [PubMed] [Google Scholar]
- 52.De Angelis E, Watkins A, Schäfer M, Brümmendorf T, Kenwrick S. Disease-associated mutations in L1 CAM interfere with ligand interactions and cell-surface expression. Hum Mol Genet. 2002;11:1–12. doi: 10.1093/hmg/11.1.1. [DOI] [PubMed] [Google Scholar]
- 53.Cismasiu VB, Denes SA, Reiländer H, Michel H, Szedlacsek SE. The MAM (meprin/A5-protein/PTPmu) domain is a homophilic binding site promoting the lateral dimerization of receptor-like protein-tyrosine phosphatase mu. J Biol Chem. 2004;279:26922–31. doi: 10.1074/jbc.M313115200. [DOI] [PubMed] [Google Scholar]
- 54.Streuli M, Krueger NX, Hall LR, Schlossman SF, Saito H. A new member of the immunoglobulin superfamily that has a cytoplasmic region homologous to the leukocyte common antigen. J Exp Med. 1988;168:1523–30. doi: 10.1084/jem.168.5.1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tian SS, Tsoulfas P, Zinn K. Three receptor-linked protein-tyrosine phosphatases are selectively expressed on central nervous system axons in the Drosophila embryo. Cell. 1991;67:675–85. doi: 10.1016/0092-8674(91)90063-5. [DOI] [PubMed] [Google Scholar]
- 56.Desai CJ, Gindhart JG, Jr., Goldstein LS, Zinn K. Receptor tyrosine phosphatases are required for motor axon guidance in the Drosophila embryo. Cell. 1996;84:599–609. doi: 10.1016/S0092-8674(00)81035-1. [DOI] [PubMed] [Google Scholar]
- 57.Krueger NX, Van Vactor D, Wan HI, Gelbart WM, Goodman CS, Saito H. The transmembrane tyrosine phosphatase DLAR controls motor axon guidance in Drosophila. Cell. 1996;84:611–22. doi: 10.1016/S0092-8674(00)81036-3. [DOI] [PubMed] [Google Scholar]
- 58.Meathrel K, Adamek T, Batt J, Rotin D, Doering LC. Protein tyrosine phosphatase sigma-deficient mice show aberrant cytoarchitecture and structural abnormalities in the central nervous system. J Neurosci Res. 2002;70:24–35. doi: 10.1002/jnr.10382. [DOI] [PubMed] [Google Scholar]
- 59.Rashid-Doubell F, McKinnell I, Aricescu AR, Sajnani G, Stoker A. Chick PTPsigma regulates the targeting of retinal axons within the optic tectum. J Neurosci. 2002;22:5024–33. doi: 10.1523/JNEUROSCI.22-12-05024.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Uetani N, Chagnon MJ, Kennedy TE, Iwakura Y, Tremblay ML. Mammalian motoneuron axon targeting requires receptor protein tyrosine phosphatases sigma and delta. J Neurosci. 2006;26:5872–80. doi: 10.1523/JNEUROSCI.0386-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Woo J, Kwon S-K, Choi S, Kim S, Lee J-R, Dunah AW, et al. Trans-synaptic adhesion between NGL-3 and LAR regulates the formation of excitatory synapses. Nat Neurosci. 2009;12:428–37. doi: 10.1038/nn.2279. [DOI] [PubMed] [Google Scholar]
- 62.Takahashi H, Arstikaitis P, Prasad T, Bartlett TE, Wang YT, Murphy TH, et al. Postsynaptic TrkC and presynaptic PTPσ function as a bidirectional excitatory synaptic organizing complex. Neuron. 2011;69:287–303. doi: 10.1016/j.neuron.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kwon S-K, Woo J, Kim S-Y, Kim H, Kim E. Trans-synaptic adhesions between netrin-G ligand-3 (NGL-3) and receptor tyrosine phosphatases LAR, protein-tyrosine phosphatase delta (PTPdelta), and PTPsigma via specific domains regulate excitatory synapse formation. J Biol Chem. 2010;285:13966–78. doi: 10.1074/jbc.M109.061127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Aricescu AR, McKinnell IW, Halfter W, Stoker AW. Heparan sulfate proteoglycans are ligands for receptor protein tyrosine phosphatase sigma. Mol Cell Biol. 2002;22:1881–92. doi: 10.1128/MCB.22.6.1881-1892.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fox AN, Zinn K. The heparan sulfate proteoglycan syndecan is an in vivo ligand for the Drosophila LAR receptor tyrosine phosphatase. Curr Biol. 2005;15:1701–11. doi: 10.1016/j.cub.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 66.Johnson KG, Tenney AP, Ghose A, Duckworth AM, Higashi ME, Parfitt K, et al. The HSPGs Syndecan and Dallylike bind the receptor phosphatase LAR and exert distinct effects on synaptic development. Neuron. 2006;49:517–31. doi: 10.1016/j.neuron.2006.01.026. [DOI] [PubMed] [Google Scholar]
- 67.Biersmith BH, Hammel M, Geisbrecht ER, Bouyain S. The immunoglobulin-like domains 1 and 2 of the protein tyrosine phosphatase LAR adopt an unusual horseshoe-like conformation. J Mol Biol. 2011;408:616–27. doi: 10.1016/j.jmb.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shen Y, Tenney AP, Busch SA, Horn KP, Cuascut FX, Liu K, et al. PTPsigma is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Science. 2009;326:592–6. doi: 10.1126/science.1178310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Coles CH, Shen Y, Tenney AP, Siebold C, Sutton GC, Lu W, et al. Proteoglycan-specific molecular switch for RPTPσ clustering and neuronal extension. Science. 2011;332:484–8. doi: 10.1126/science.1200840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cox C, Bignell G, Greenman C, Stabenau A, Warren W, Stephens P, et al. A survey of homozygous deletions in human cancer genomes. Proc Natl Acad Sci U S A. 2005;102:4542–7. doi: 10.1073/pnas.0408593102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Solomon DA, Kim J-S, Cronin JC, Sibenaller Z, Ryken T, Rosenberg SA, et al. Mutational inactivation of PTPRD in glioblastoma multiforme and malignant melanoma. Cancer Res. 2008;68:10300–6. doi: 10.1158/0008-5472.CAN-08-3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Veeriah S, Brennan C, Meng S, Singh B, Fagin JA, Solit DB, et al. The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers. Proc Natl Acad Sci U S A. 2009;106:9435–40. doi: 10.1073/pnas.0900571106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Morris LGT, Taylor BS, Bivona TG, Gong Y, Eng S, Brennan CW, et al. Genomic dissection of the epidermal growth factor receptor (EGFR)/PI3K pathway reveals frequent deletion of the EGFR phosphatase PTPRS in head and neck cancers. Proc Natl Acad Sci U S A. 2011;108:19024–9. doi: 10.1073/pnas.1111963108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–73. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- 75.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–75. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wood LD, Parsons DW, Jones S, Lin J, Sjöblom T, Leary RJ, et al. The genomic landscapes of human breast and colorectal cancers. Science. 2007;318:1108–13. doi: 10.1126/science.1145720. [DOI] [PubMed] [Google Scholar]
- 77.Li M, Zhao H, Zhang X, Wood LD, Anders RA, Choti MA, et al. Inactivating mutations of the chromatin remodeling gene ARID2 in hepatocellular carcinoma. Nat Genet. 2011;43:828–9. doi: 10.1038/ng.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Freigang J, Proba K, Leder L, Diederichs K, Sonderegger P, Welte W. The crystal structure of the ligand binding module of axonin-1/TAG-1 suggests a zipper mechanism for neural cell adhesion. Cell. 2000;101:425–33. doi: 10.1016/S0092-8674(00)80852-1. [DOI] [PubMed] [Google Scholar]
- 79.Mörtl M, Sonderegger P, Diederichs K, Welte W. The crystal structure of the ligand-binding module of human TAG-1 suggests a new mode of homophilic interaction. Protein Sci. 2007;16:2174–83. doi: 10.1110/ps.072802707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sawaya MR, Wojtowicz WM, Andre I, Qian B, Wu W, Baker D, et al. A double S shape provides the structural basis for the extraordinary binding specificity of Dscam isoforms. Cell. 2008;134:1007–18. doi: 10.1016/j.cell.2008.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Meijers R, Puettmann-Holgado R, Skiniotis G, Liu J-H, Walz T, Wang J-H, et al. Structural basis of Dscam isoform specificity. Nature. 2007;449:487–91. doi: 10.1038/nature06147. [DOI] [PubMed] [Google Scholar]
- 82.Bouyain S, Watkins DJ. The protein tyrosine phosphatases PTPRZ and PTPRG bind to distinct members of the contactin family of neural recognition molecules. Proc Natl Acad Sci U S A. 2010;107:2443–8. doi: 10.1073/pnas.0911235107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lamprianou S, Chatzopoulou E, Thomas J-L, Bouyain S, Harroch S. A complex between contactin-1 and the protein tyrosine phosphatase PTPRZ controls the development of oligodendrocyte precursor cells. Proc Natl Acad Sci U S A. 2011;108:17498–503. doi: 10.1073/pnas.1108774108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Liu H, Focia PJ, He X. Homophilic adhesion mechanism of neurofascin, a member of the L1 family of neural cell adhesion molecules. J Biol Chem. 2011;286:797–805. doi: 10.1074/jbc.M110.180281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Súrez Pestana E, Tenev T, Gross S, Stoyanov B, Ogata M, Böhmer FD. The transmembrane protein tyrosine phosphatase RPTPsigma modulates signaling of the epidermal growth factor receptor in A431 cells. Oncogene. 1999;18:4069–79. doi: 10.1038/sj.onc.1202794. [DOI] [PubMed] [Google Scholar]
- 86.Aicher B, Lerch MM, Müller T, Schilling J, Ullrich A. Cellular redistribution of protein tyrosine phosphatases LAR and PTPsigma by inducible proteolytic processing. J Cell Biol. 1997;138:681–96. doi: 10.1083/jcb.138.3.681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kypta RM, Su H, Reichardt LF. Association between a transmembrane protein tyrosine phosphatase and the cadherin-catenin complex. J Cell Biol. 1996;134:1519–29. doi: 10.1083/jcb.134.6.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Müller T, Choidas A, Reichmann E, Ullrich A. Phosphorylation and free pool of beta-catenin are regulated by tyrosine kinases and tyrosine phosphatases during epithelial cell migration. J Biol Chem. 1999;274:10173–83. doi: 10.1074/jbc.274.15.10173. [DOI] [PubMed] [Google Scholar]
- 89.Siu R, Fladd C, Rotin D. N-cadherin is an in vivo substrate for protein tyrosine phosphatase sigma (PTPsigma) and participates in PTPsigma-mediated inhibition of axon growth. Mol Cell Biol. 2007;27:208–19. doi: 10.1128/MCB.00707-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Muise AM, Walters T, Wine E, Griffiths AM, Turner D, Duerr RH, et al. Protein-tyrosine phosphatase sigma is associated with ulcerative colitis. Curr Biol. 2007;17:1212–8. doi: 10.1016/j.cub.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 91.Krueger NX, Streuli M, Saito H. Structural diversity and evolution of human receptor-like protein tyrosine phosphatases. EMBO J. 1990;9:3241–52. doi: 10.1002/j.1460-2075.1990.tb07523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Kaplan R, Morse B, Huebner K, Croce C, Howk R, Ravera M, et al. Cloning of three human tyrosine phosphatases reveals a multigene family of receptor-linked protein-tyrosine-phosphatases expressed in brain. Proc Natl Acad Sci U S A. 1990;87:7000–4. doi: 10.1073/pnas.87.18.7000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Barnea G, Silvennoinen O, Shaanan B, Honegger AM, Canoll PD, D’Eustachio P, et al. Identification of a carbonic anhydrase-like domain in the extracellular region of RPTP gamma defines a new subfamily of receptor tyrosine phosphatases. Mol Cell Biol. 1993;13:1497–506. doi: 10.1128/mcb.13.3.1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Harroch S, Palmeri M, Rosenbluth J, Custer A, Okigaki M, Shrager P, et al. No obvious abnormality in mice deficient in receptor protein tyrosine phosphatase beta. Mol Cell Biol. 2000;20:7706–15. doi: 10.1128/MCB.20.20.7706-7715.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lamprianou S, Vacaresse N, Suzuki Y, Meziane H, Buxbaum JD, Schlessinger J, et al. Receptor protein tyrosine phosphatase gamma is a marker for pyramidal cells and sensory neurons in the nervous system and is not necessary for normal development. Mol Cell Biol. 2006;26:5106–19. doi: 10.1128/MCB.00101-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Shintani T, Maeda N, Noda M. Receptor-like protein tyrosine phosphatase gamma (RPTPgamma), but not PTPzeta/RPTPbeta, inhibits nerve-growth-factor-induced neurite outgrowth in PC12D cells. Dev Neurosci. 2001;23:55–69. doi: 10.1159/000048696. [DOI] [PubMed] [Google Scholar]
- 97.Peles E, Nativ M, Campbell PL, Sakurai T, Martinez R, Lev S, et al. The carbonic anhydrase domain of receptor tyrosine phosphatase beta is a functional ligand for the axonal cell recognition molecule contactin. Cell. 1995;82:251–60. doi: 10.1016/0092-8674(95)90312-7. [DOI] [PubMed] [Google Scholar]
- 98.Sakurai T, Lustig M, Nativ M, Hemperly JJ, Schlessinger J, Peles E, et al. Induction of neurite outgrowth through contactin and Nr-CAM by extracellular regions of glial receptor tyrosine phosphatase beta. J Cell Biol. 1997;136:907–18. doi: 10.1083/jcb.136.4.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hashemi H, Hurley M, Gibson A, Panova V, Tchetchelnitski V, Barr A, et al. Receptor tyrosine phosphatase PTPγ is a regulator of spinal cord neurogenesis. Mol Cell Neurosci. 2011;46:469–82. doi: 10.1016/j.mcn.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Maeda N, Nishiwaki T, Shintani T, Hamanaka H, Noda M. 6B4 proteoglycan/phosphacan, an extracellular variant of receptor-like protein-tyrosine phosphatase zeta/RPTPbeta, binds pleiotrophin/heparin-binding growth-associated molecule (HB-GAM) J Biol Chem. 1996;271:21446–52. doi: 10.1074/jbc.271.35.21446. [DOI] [PubMed] [Google Scholar]
- 101.Meng K, Rodriguez-Peña A, Dimitrov T, Chen W, Yamin M, Noda M, et al. Pleiotrophin signals increased tyrosine phosphorylation of beta beta-catenin through inactivation of the intrinsic catalytic activity of the receptor-type protein tyrosine phosphatase beta/zeta. Proc Natl Acad Sci U S A. 2000;97:2603–8. doi: 10.1073/pnas.020487997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fukada M, Fujikawa A, Chow JPH, Ikematsu S, Sakuma S, Noda M. Protein tyrosine phosphatase receptor type Z is inactivated by ligand-induced oligomerization. FEBS Lett. 2006;580:4051–6. doi: 10.1016/j.febslet.2006.06.041. [DOI] [PubMed] [Google Scholar]
- 103.Müller S, Kunkel P, Lamszus K, Ulbricht U, Lorente GA, Nelson AM, et al. A role for receptor tyrosine phosphatase zeta in glioma cell migration. Oncogene. 2003;22:6661–8. doi: 10.1038/sj.onc.1206763. [DOI] [PubMed] [Google Scholar]
- 104.Perez-Pinera P, Alcantara S, Dimitrov T, Vega JA, Deuel TF. Pleiotrophin disrupts calcium-dependent homophilic cell-cell adhesion and initiates an epithelial-mesenchymal transition. Proc Natl Acad Sci U S A. 2006;103:17795–800. doi: 10.1073/pnas.0607299103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bilwes AM, den Hertog J, Hunter T, Noel JP. Structural basis for inhibition of receptor protein-tyrosine phosphatase-alpha by dimerization. Nature. 1996;382:555–9. doi: 10.1038/382555a0. [DOI] [PubMed] [Google Scholar]
- 106.Desai DM, Sap J, Schlessinger J, Weiss A. Ligand-mediated negative regulation of a chimeric transmembrane receptor tyrosine phosphatase. Cell. 1993;73:541–54. doi: 10.1016/0092-8674(93)90141-C. [DOI] [PubMed] [Google Scholar]
- 107.Hoffmann KM, Tonks NK, Barford D. The crystal structure of domain 1 of receptor protein-tyrosine phosphatase mu. J Biol Chem. 1997;272:27505–8. doi: 10.1074/jbc.272.44.27505. [DOI] [PubMed] [Google Scholar]
- 108.Nam HJ, Poy F, Krueger NX, Saito H, Frederick CA. Crystal structure of the tandem phosphatase domains of RPTP LAR. Cell. 1999;97:449–57. doi: 10.1016/S0092-8674(00)80755-2. [DOI] [PubMed] [Google Scholar]
- 109.Nam H-J, Poy F, Saito H, Frederick CA. Structural basis for the function and regulation of the receptor protein tyrosine phosphatase CD45. J Exp Med. 2005;201:441–52. doi: 10.1084/jem.20041890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Barr AJ, Ugochukwu E, Lee WH, King ONF, Filippakopoulos P, Alfano I, et al. Large-scale structural analysis of the classical human protein tyrosine phosphatome. Cell. 2009;136:352–63. doi: 10.1016/j.cell.2008.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Su J-L, Yang C-Y, Shih J-Y, Wei L-H, Hsieh C-Y, Jeng Y-M, et al. Knockdown of contactin-1 expression suppresses invasion and metastasis of lung adenocarcinoma. Cancer Res. 2006;66:2553–61. doi: 10.1158/0008-5472.CAN-05-2645. [DOI] [PubMed] [Google Scholar]
- 112.Mauerer A, Roesch A, Hafner C, Stempfl T, Wild P, Meyer S, et al. Identification of new genes associated with melanoma. Exp Dermatol. 2011;20:502–7. doi: 10.1111/j.1600-0625.2011.01254.x. [DOI] [PubMed] [Google Scholar]
- 113.Eckerich C, Zapf S, Ulbricht U, Müller S, Fillbrandt R, Westphal M, et al. Contactin is expressed in human astrocytic gliomas and mediates repulsive effects. Glia. 2006;53:1–12. doi: 10.1002/glia.20254. [DOI] [PubMed] [Google Scholar]
- 114.Vezzalini M, Mombello A, Menestrina F, Mafficini A, Della Peruta M, van Niekerk C, et al. Expression of transmembrane protein tyrosine phosphatase gamma (PTPgamma) in normal and neoplastic human tissues. Histopathology. 2007;50:615–28. doi: 10.1111/j.1365-2559.2007.02661.x. [DOI] [PubMed] [Google Scholar]