

Abstract
Prelamin A processing impairment is a common feature of a restricted group of rare genetic alterations/disorders associated with a wide range of clinical phenotypes. Changes in histone posttranslational modifications, alterations in non-histone chromatin proteins and chromatin disorganization have been specifically linked to impairment of specific, distinct prelamin A processing steps, but the molecular mechanism involved in these processes is not yet understood . In this study, we show that the accumulation of wild-type prelamin A detected in restrictive dermopathy (RD), as well as the accumulation of mutated forms of prelamin A identified in familial partial lipodystrophy (FPLD) and mandibuloacral dysplasia (MADA), affect the nuclear localization of barrier-to-autointegration factor (BAF), a protein able to link lamin A precursor to chromatin remodeling functions. Our findings, in accordance with previously described results, support the hypothesis of a prelamin A involvement in BAF nuclear recruitment and suggest BAF-prelamin A complex as a protein platform usually activated in prelamin A-accumulating diseases. Finally, we demonstrate the involvement of the inner nuclear membrane protein emerin in the proper localization of BAF-prelamin A complex.
Keywords: BAF, BANF1, prelamin A, lamin A/C, laminopathies, emerin, EDMD1
Introduction
The ability to store and translate instructions from the genetic code is essential to maintain life in all cells. Since the human genetic code is composed of over 3 billion nucleotides, tight packaging and an accurate spatial organization is necessary in order to fit it within each micron-sized nuclei. Packaging of the human genome includes folding the DNA into chromatin fibers, chromosome domains and ultimately chromosomes. This higher-order organization is known to contribute to gene regulation, and, therefore, it is not surprising that defects in chromatin and chromosome organization cause different diseases, including cancer and aging.1 Recently, chromatin alterations and epigenetic changes have been identified as common features in a group of rare genetic disorders (laminopathies) due to mutations in LMNA gene, a DNA sequence codifying for two of major nuclear lamina components: lamin A and lamin C.2 In accordance with the nuclear lamina, proposed functions including supporting transcription, replication, genome organization, development and DNA repair; the nuclear lamina defects cause a wide range of clinical phenotypes that can be associated to a specific tissue failure (muscular dystrophy, lipodystrophy, cerebellar disorders) or to multisystem disorders (mandibuloacral dysplasia, premature aging).3 Among laminopathies themselves, chromatin and nuclear structure alterations appear to be more severe in the prelamin A-accumulating forms of these diseases.4 Prelamin A is a protein precursor of lamina A, which is posttranslationally modified at its C terminal region, where the CaaX motif triggers a sequence of modifications, including farnesylation, carboxymethylation and proteolytic cleavage by ZMPSTE 24 metalloproteinase.5,6 The prelamin A maturation pathway has been found altered in Hutchinson-Gilford progeria syndrome (HGPS),7,8 Mandibuloacral dysplasia (MAD),9 Dunnigan-type familial partial lipodystrophy (FPLD), atypical Werner syndrome (WS)10 and restrictive dermopathy (RD.)11 These diseases are characterized by loss of the key architectural chromatin protein HP1 and alterations in the amount of heterochromatin-associated histones H3K9me3 and H3K27me3.4 Interestingly, similar effects have been obtained in human control fibroblasts induced to accumulate different prelamin A forms through the exogenous expression of uncleavable prelamin A mutant constructs or by treatments with prelamin A interfering drugs.12,13
How can prelamin A affect chromatin organization is unclear, but we recently described its interaction with barrier-to-autointegration factor (BAF), a DNA-binding protein involved in both chromatin dynamics and epigenetic modifications.14 BAF is a soluble protein localized in both cytoplasm and nucleus, where it interacts with LEM-domain proteins (LAP2, emerin, MAN1, Lem2/NET25) and affects chromatin silencing. BAF can condense DNA in vitro;15 it influences higher-order chromatin structure and represses transcription at specific promoters.16,17 In addition, the previously described BAF interactions with histone H3 and retinoblastoma binding protein 4 (RBBP4) suggest its association with nucleosomes and nucleosome remodeling mechanisms.18Interestingly, BAF has been identified as a protein responsible for a progeroid syndrome featuring dysmorphic cell nuclei, atrophic skin, generalized lipoatrophy, severe osteoporosis and marked osteolysis features that overlap with those described in prelamin A-accumulating diseases.19,20
Since experimentally accumulated prelamin A varieties, as well as progerin (a mutated prelamin A form lacking 50 aa), accumulated in HGPS cells, affect BAF distribution,21 we wondered if a similar event occurs in FPLD, MADA and RD cells which are known to accumulate point-mutated or wild-type prelamin A forms, respectively. In this study, we show that in human skin fibroblasts from RD, MADA and FPLD patients, BAF is prevalently located in the nucleus, where it colocalizes with accumulated prelamin A. Moreover, we demonstrate that LMNA gene mutations occurring in MADA and FPLD cells do not interfere with in vivo BAF-prelamin A interaction. Finally we show that emerin, a BAF and prelamin A binding partners, is involved in BAF-prelamin A complex localization in the nucleus.
Our observations strongly suggest that BAF-prelamin A complex could be considered as a chromatin-regulator element involved in the pathophysiological mechanism of prelamin A-accumulating diseases.
Results
Barrier-to-autointegration factor localization and expression in FPLD, MADA and RD cells
BAF cellular localization was evaluated in control and laminopathic cells. Since FPLD and MADA cells were obtained from adult patients (aged 20 to 30 y) and RD cells were obtained from neonatal patients, we used two different age matched groups of healthy donors referred to in the Figure 1 as control 1 (cont.1, adult healthy donor) and control 2 (cont.2, neonatal healthy donor).

Figure 1. Barrier-to-autointegration factor nuclear localization in prelamin A-accumulating diseases. (A) BAF and prelamin A distribution in control (cont.1 adult healthy donor, cont.2 neonatal healthy donor) Familial partial lipodystrophy (FPLD, LMNA-R482Q), Mandibuloacral dysplasia (MADA, LMNA-R527H) and Restrictive dermopathy (RD, ZMPSTE24 c.1085_1086InsT). Control and pathological cells at early passage (passage 5–10) were used for each experiment. BAF was evaluated on methanol-fixed cells by a rabbit-polyclonal anti-BAF antibody visualized by FITC-conjugated secondary antibody (green). Prelamin A was evaluated using a goat-polyclonal anti-prelamin A antibody visualized by TRITC-conjugated secondary antibody (red). In merge, arrow, arrowhead and asterisk indicate double-stained nuclei. DNA was detected using DAPI. Bar, 10 μm. (B) Higher magnification of prelamin A-BAF-positive nuclei indicated by arrow, arrowhead and asterisk in panel A. Prelamin A (red), BAF (green) merge (merge) and merge plus DAPI staining (merge + DAPI) are shown. (C) The percentage of nuclei showing prelamin A staining is reported in the graph as means ± SD of three different counts (100 nuclei per count). Examined samples are control fibroblast from adult healthy donors (cont.1), familial partial lipodystrophy fibroblasts (FPLD), mandibuloacral dysplasia fibroblasts (MADA), control fibroblast from neonate healthy donors (cont.2) and restrictive dermopathy fibroblasts (RD). Asterisks indicate statistically significant differences at the Student’s t-test (p < 0.05), with respect to control cells values. (D) Western blotting evaluation of BAF protein levels in control (cont.1 and cont.2), FPLD, MADA and RD cells. Eighty micrograms of total cell lysates were subjected to western blot detection of prelamin A (prelamin A), lamin A/C (lamin A/C), BAF (BAF) and actin (actin). Immunolabeled bands visualized by ECL chemiluminescence are shown.
BAF immunofluorescence evaluation performed in control cells bearing undetectable levels of prelamin A showed BAF ubiquitously distributed between cytoplasm and nucleus in 80% of the cells, while in less than 15% of the cells, BAF were prevalently located in the nucleus (Fig. 1A and C). In FPLD, MADA and RD cells, the distribution of BAF changed, all prelamin A-positive nuclei showing BAF nuclear recruitment (Fig. 1A).
Prelamin A accumulation at the nuclear rim or at the intranuclear aggregates was observed in 45% of FPLD cells.22 In these cells, BAF was predominantly located in the nucleus, where it was distributed in the nuclear lamina and nucleoplasm as well as at prelamin A-positive structures (Fig. 1A and B).
Similar results were obtained in two different MADA cell lines in which a previously described R527H LMNA mutation9 leads to prelamin A accumulation in 55% of the cells (Fig. 1C); this was detected at the rim of normally shaped nuclei or, alternatively, as intranuclear aggregates (Fig. 1A). BAF nuclear localization was observed in the MADA prelamin A-positive cells, where it perfectly colocalized with prelamin A-labeled structures (Fig. 1A and B).
Finally, prelamin A and BAF localization were evaluated in two RD cell lines bearing the same ZMPSTE24 gene mutation (c.1085_1086InsT) (Fig. 1A), which causes complete elimination of the last proteolytic cleavage step of prelamin A processing and accumulation of different non-mutated prelamin A intermediates.23Using an anti-prelamin A antibody, we detected prelamin A in the nucleus (Fig. 1A and C). BAF nuclear labeling was observed in 90% of RD cells (Fig. 1A and C), where it localized in the nucleoplasm of normally shaped nuclei or colocalized with prelamin A at the nuclear lamina of dysmorphic nuclei (Fig. 1A and B, asterisk). These findings agree with our previous results reporting different BAF nuclear distribution following experimental accumulation of full-length or farnesylated-carboxymethylated prelamin A.21 Western blotting analysis confirmed that the total amount of BAF was not altered (Fig. 1D) by the high levels of prelamin A, which is accumulated and clearly detectable in FPLD, MADA and RD cells.
Different prelamin A mutated forms interact with BAF and affect its cellular distribution
The above described results were confirmed in HEK293 cells expressing processable prelamin A (LA-WT), laminopathic, uncleaved prelamin A (LA-L647R) or laminopathic processable prelamin A mutants (LA-R482Q and LA-R527H) (Fig. 2). FLAG-tagged prelamin A forms were efficiently expressed in HEK293 cells, and the nuclear distribution of each pathological mutant resembled that observed respectively in human fibroblasts from laminopathic patients. Moreover, in accordance with our previously described results,21,24 we observed that all FLAG-tagged prelamin A constructs (including LA-WT) were able to induce accumulation of prelamin A (Fig. S1). FLAG immunological detection showed that LA-WT and uncleaved prelamin A localized at the nuclear lamina and, in addition, nucleoplasmic distribution of LA-L647R was also detected.21 LA-R527H localized both at the nuclear lamina and at the intranuclear foci (Fig. 2, arrowhead), while LA-R482Q was observed over the aggregates located at the nuclear lamina (Fig. 2, arrow). BAF nuclear staining was observed in all transfected cells where it colocalized with FLAG-tagged prelamin A forms (Fig. 2A); on the contrary, a prevalent cytoplasmic BAF localization was observed in untransfected cells (Fig. 2A, asterisk). Western blotting analysis confirmed the expression of FLAG-tagged proteins and showed the same BAF protein amount in untransfected and transfected cells (Fig. 2B). In order to evaluate BAF nuclear translocation in response to prelamin A accumulation, HEK293 cells were transfected with GFP-BAF construct alone or in combination with LA-WT or each prelamin A mutant (LA-R527H, LA-R482Q, LA-L647R). Total extracts, cytoplasmic fractions and isolated nuclei from single and cotransfected cells were subjected to FLAG and GFP western blotting evaluation. Immunolabeled bands showed that nuclear GFP-BAF level was definitely higher in GFP-BAF/FLAG-prelamin A-expressing cells than in GFP-BAF single transfected cells (Fig. 3). GFP-BAF bands staining was decreased in cytosolic fractions from cotransfected cells (Fig. 3). Nuclear fraction purity and equal protein loading was demonstrated by the exclusive detection of caveolin 1 in whole lysates and cytosolic fractions. On the contrary, lamin B1 staining was used as a nuclear fractions marker and nuclear loading control (Fig. 3).

Figure 2. Barrier-to-autointegration factor localization and expression in prelamin A mutants HEK293 transfected cells. (A) BAF localization in HEK293 cells expressing prelamin A mutants. Cells were transfected with wild-type processable prelamin A (LA-WT), MADA processable prelamin A mutant (LA-R527H), FPLD processable prelamin A mutant (LA-R482Q) or uncleavable farnesylated-carboxymethylated prelamin A mutant (LA-L647R). Immunofluorescence detection of overexpressed proteins was performed using a mouse monoclonal anti FLAG-Cy3 conjugated antibody (red). Arrowhead and arrow indicate LA-R527H and LA-R482Q distribution, respectively. Endogenous BAF localization was evaluated by a rabbit polyclonal anti-BAF antibody visualized by FITC-conjugated secondary antibody (green). Asterisk indicates BAF distribution in untransfected cells. Nuclei were stained with DAPI. Bar, 10 μm. (B) BAF protein levels in prelamin A mutants expressing cells. BAF protein levels were determined in HEK293 cells expressing FLAG-tagged prelamin A constructs (LA-WT, LA-R527H, LA-R482Q, LA-L647R) as well as in untransfected cells (unt.). Thirty micrograms of total cell lysates were separated by SDS-PAGE (5–20%) and subjected to western blot. FLAG-tagged protein and endogenous BAF detection was performed using a mouse-monoclonal and a rabbit-polyclonal, respectively. Actin (Actin) was detected as a protein-loading control.

Figure 3. Barrier-to-autointegration factor nuclear translocation in HEK293 cells transfected with prelamin A mutants. Western blotting analysis of whole cellular lysates, cytosol and isolated nuclei from HeK293 cells transfected with GFP-BAF (GFP-BAF), alone or in combination with: wild-type processable prelamin A (LA-WT + GFP-BAF), MADA processable prelamin A mutant (LA-R527H+GFP-BAF), FPLD processable prelamin A mutant (LA-R482Q+GFP-BAF) or uncleavable prelamin A mutant (LA-L647R+GFP-BAF). Immunolabeled bands of FLAG, GFP, lamin B1 and caveolin 1 are shown.
Since BAF nuclear localization in response to prelamin A (WT or mutated forms) accumulation suggested a possible reciprocal protein interaction,21 a coimmunoprecipitation study was performed (Fig. 4). Total lysates from HEK293 cells expressing GFP-BAF in combination with FLAG-tagged prelamin A constructs (LA-WT, LA-R527H, LA-R482Q, LA-L647R) were subjected to anti-FLAG immunoprecipitation. Western blotting detection showed FLAG and GFP immunolabeled bands in both total cellular extracts and in FLAG-IP samples (Fig. 4A) while no immunolabeled bands were observed in protein A/G lanes. In the same transfected cells, the interaction of prelamin A mutated or WT forms with BAF was also evaluated. GFP-coimmunoprecipitation experiment was performed, and the obtained protein complexes were subject to GFP, FLAG, prelamin A and lamin A western blotting detection (Fig. 4B). GFP, FLAG, prelamin A and lamin A immunolabeled bands were observed in all total lysates as well as in all GFP-IP samples (Fig. 4B), demonstrating that LMNA gene mutations occurring in MADA and FPLD cells do not interfere, per se, with BAF-prelamin A interaction. In addition, comparison of 70 kD lamin A bands with the 74 kD prelamin A bands suggested that BAF interacts preferentially with prelamin A rather than with mature lamin A.
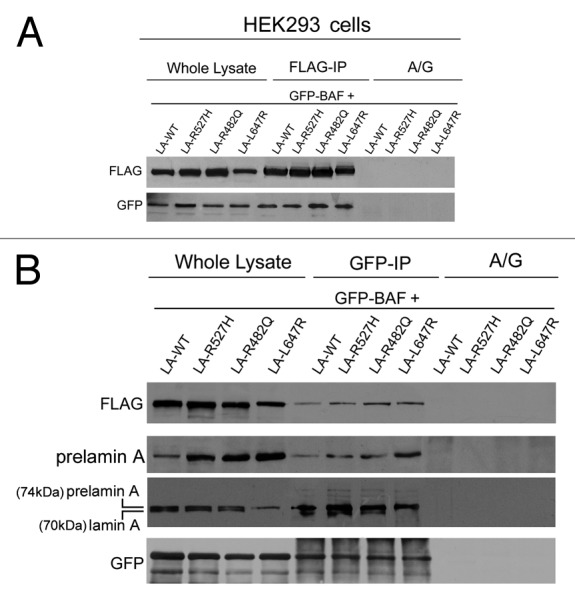
Figure 4. Barrier-to-autointegration factor interaction with laminopathic forms of prelamin A. HEK293 cells were transfected with FLAG-tagged prelamin A constructs (LA-WT, LA-R527H, LA-R482Q, LA-L647R) in combination with GFP-BAF (GFP-BAF) construct. Cotransfected cells were lysed and subjected to coimmunoprecipitation experiments. Total lysates (whole lysate) and immunoprecipitated proteins, FLAG-IP in panel (A) and GFP-IP in panel (B), were subjected to western blotting analysis. FLAG and GFP immunolabeled bands were observed in whole lysates as well as in immunoprecipitated samples, while no protein staining was observed in coimmunoprecipitation samples obtained in absence of anti-FLAG antibody or anti GFP- antibody (A/G) (A and B). In (B), prelamin A staining was observed in whole lysates and in GFP-IP lanes subjected to specific anti-prelamin A detection (prelamin A) and to anti-lamin A detection performed with Abcam antibody, which detects both mature lamin A (70kDa) and prelamin A (74kDa) (prelamin A, lamin A).
Emerin affects BAF-prelamin A complex localization
Finally, we wondered if the BAF and prelamin A-binding protein emerin, involved in the proper localization of lamin A precursor,24,25 could affect BAF nuclear localization during prelamin A accumulation. Thus, we evaluated prelamin A and BAF localization in mevinolin-treated skin fibroblasts obtained from control and emerin-null patients affected by Emery Dreifuss Muscular Dystrophy type 1 (EDMD1)26 (Fig. 5). Mevinolin is a prelamin A-interfering drug able to induce the accumulation of non-farnesylated prelamin A through the inhibition of 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the enzyme responsible for the farnesyl-moiety biosynthesis. After 18 h of pharmacological treatment, BAF and prelamin A were evaluated. In control and EDMD1 untreated cells, prelamin A was undetectable and BAF localized normally throughout the cell compartments (Fig. 5A and B). After mevinolin treatment, both control and EDMD1 cells showed non-farnesylated prelamin A accumulation and BAF nuclear recruitment (Fig. 5B): control cells showed non-farnesylated prelamin A accumulation at intranuclear aggregates, which colocalized with BAF fluorescence (Fig. 5A, arrow and square), while EDMD1 cells showed an altered nuclear distribution of non-farnesylated prelamin A and, unexpectedly, of BAF (Fig. 5A). In accordance with our previously described results, we observed that non-farnesylated prelamin A aggregates were retained at the nuclear periphery in emerin-null cells24(Fig. 5A, square). Interestingly, BAF colocalized with these prelamin A-containing delocalized structures (Fig. 5A, arrowhead), suggesting that emerin does not affect BAF cellular distribution or its prelamin A-dependent nuclear translocation, but it could be necessary for the proper localization of the whole prelamin A-BAF protein complex inside the nucleus. Western blotting analysis confirmed a quantitatively comparable BAF protein amount in all samples (Fig. 5C). In order to confirm these results, we evaluated whether the restoration of emerin expression in EDMD1 cells was able to induce the proper localization of BAF. In this regard, GFP-emerin fusion protein was expressed in emerin-null EDMD1 fibroblasts, and prelamin A was accumulated by mevinolin treatment. We observed that emerin expression in EDMD1 did not affect prelamin A processing or BAF localization (Fig. 6A). On the contrary, mevinolin treatment on GFP-emerin transfected cells showed that the restoring of emerin expression was able to recover prelamin A and BAF nuclear aggregates distribution (Fig. 6B, arrow).

Figure 5. Localization and expression of barrier-to-autointegration factor and prelamin A in EDMD1 cells. (A) BAF and prelamin A were localized in control cells (control) treated with 10 μM mevinolin (+mevinolin) and in Emery Dreifuss Muscular Dystrophy type 1 (EDMD1) cells untreated (untreated) or treated (+mevinolin) with 10 μM mevinolin. After pharmacological treatment, BAF (green) and prelamin A staining (red) were performed. BAF and prelamin A were detected using a rabbit-polyclonal antibody and a goat-polyclonal antibody, respectively. Prelamin A and BAF colocalization is indicated by arrow and arrowhead in merge (merge). Squares indicate BAF/prelamin A aggregates localization. In the lower panels, BAF/prelamin A aggregates localization is reported at higher magnification, omitting DAPI staining. DNA was stained by DAPI. Bar, 10 μm. (B) Proportions of cell populations with different BAF localization: Nuc > Cyto, Nuc = Cyto and Nuc < Cyto represent cells with prevailing nuclear staining, uniform or mostly cytoplasmic staining, respectively. The proportion of cell populations was determined in control and EDMD1 cells at the same population doubling untreated (untreated) or mevinolin-treated cells (mevinolin). Data are means ± s.d. of at least 100 cells for each of three independent experiments. Asterisks indicate statistically significant differences at the Student’s t-test (p < 0.05), with respect to control and EDMD1 mevinolin untreated cells values. (C) Western blotting evaluation of BAF in prelamin A EDMD1-accumulating cells. Western blotting evaluation of prelamin A, emerin, BAF and actin were performed in total lysates from control cells (control) or Emery Dreifuss Muscular Dystrophy type 1 cells (EDMD1) untreated (unt.) or treated with mevinolin (+mev.). Immunolabeled bands are shown.

Figure 6. Emerin exogenous expression rescues BAF intranuclear distribution in EDMD1 cells accumulating prelamin A. Prelamin A and BAF evaluation in EDMD1 GFP-emerin transfected cells with or without mevinolin treatment. In panel A, untreated EDMD1 GFP-emerin transfected cells (green) were subjected to prelamin A (prelamin A) and BAF (BAF) detection, both protein were visualized by TRITC-conjugated secondary antibody (red). In merge (merge+DAPI) DNA was detected using DAPI. Bar, 10 μm. In panel B, mevinolin-treated EDMD1 GFP-emerin transfected cells (green). Prelamin A (prelamin A) and BAF (BAF) localization are shown. Arrow indicates recover of intranuclear BAF distribution in GFP-emerin transfected cells. Arrowhead indicates the altered intranuclear BAF distribution in the absence of emerin.
Discussion
Nuclear structure alteration and chromatin disorganization are considered common features of prelamin A accumulating diseases, whose mechanism and precise molecular processes remain largely unknown. Expanding on our previous work that demonstrated in vivo BAF-prelamin A interaction, this study provides further insights into a possible BAF-prelamin A interaction also in all known diseases involving an accumulation of lamin A precursor. In particular, we demonstrate that prelamin A pathogenic forms identified in FPLD, MADA and RD cells interact with BAF and affect its intracellular distribution. As previously described in HGPS cells, we observe an evident BAF nuclear localization and its colocalization with prelamin A in FPLD, MADA and RD nuclei. Finally, a possible implication of the inner nuclear membrane protein emerin in the proper localization of the BAF-prelamin A complex is proposed.
Since BAF has been recently identified as a protein that interacts with mononucleosomes and is also able to affect the epigenetic organization of chromatin,14 the prelamin A interaction with this DNA-binding protein we describe here may suggest through what kind of molecular platforms the known lamin A precursors could affect chromatin organization. Indeed, nuclear structure alteration and chromatin disorganization are considered common features of different prelamin A-accumulating diseases.27
In FPLD cells, where farnesylated lamin A precursor is accumulated, nuclei show an altered heterochromatin arrangement with areas characterized by heterochromatin loss.10 In MADA, nuclei heterochromatin domains are disorganized or completely lost, in accordance with the delocalization of both heterochromatin-associated protein (HP1β) and histone H3 methylated at lysine 9 (Tri-H3K9) and with a lamin B receptor increased solubility.9 In RD cells, in which the impairment of ZMPSTE24 function profoundly affects the prelamin A maturation pathway at different steps, preventing the production of mature lamina A, heterochromatin is lost, and a generalized nuclear enlargement is detected. Interestingly, the severity of such alterations has been recently linked to the combination of different prelamin A forms accumulated in the nucleus.23
Thus, in RD normally shaped nuclei, where both unprocessed prelamin A and farnesylated prelamin A are detected, an evident clustering of tri-H3K9 and tri-H4K20 is observed. On the contrary, in highly dysmorphic RD nuclei, where a combination of different prelamin A forms (unprocessed and uncleaved-prelamin A) is detected, a disorganization of tri-H4K20 and a decrease of tri-H3K9 are observed.23 In this regard, a BAF direct interaction with histones H3 and H4 has been recently demonstrated, and also it has been described how posttranslational modifications of such nucleosome components may change in BAF-overexpressing cells.14
Thus, it is conceivable that BAF nuclear recruitment observed in prelamin A-accumulating diseases could resemble, in some aspects, a BAF-overexpression condition. In accordance with these data, a significant decrease in H4K16 acetylation has been recently described in BAF-overexpressing HEK293 cells and in Zmpste24−/− mouse embryonic fibroblasts.14,28 The acetylation of H4 plays an important role in several processes such as transcriptional activation, chromatin architecture maintenance and DNA repair.29 Interestingly, it has been demonstrated that prelamin A accumulation inhibits H4 acetylation by interfering with the nuclear retention of histone acetyltransferase Mof.28 In addition, BAF nuclear accumulation and its direct interaction with histones could prevent epigenetic changes by interfering with the histone-modifying enzymes that bind to the nucleosomes.14 Finally, it must be considered that the chromatin disorganization observed in prelamin A-accumulating disease could also be due to an inhibition of specific BAF physiological interactions caused by its actual prelamin A binding.
This hypothesis is enforced by the decreased level of retinoblastoma binding protein 4 (RBBP4) observed in HGPS cells.30 RBBP4 is a BAF-binding protein with a role in chromatin organization whose downregulation in HGPS cells may appear in contrast with BAF nuclear recruitment in progerin-accumulating cells.21 Nevertheless, its downregulation is consistent with our hypothesis, which suggests that the persistence of progerin, as well as of other prelamin A forms in a BAF containing complex, could compete with, reduce or eventually abolish BAF interactions with other binding partners. Thus, prelamin A interaction with BAF could block specific BAF protein domains necessary for interaction with DNA remodeling enzymes, like the recently identified endonuclease Ankle1, or with inner nuclear membrane proteins (lem3 in C. elegans) involved in the BAF-mediate DNA damage response.31,32 In accordance, prelamin A-accumulating diseases are characterized by DNA damage increase.33,34
Among the described BAF-binding partners, some proteins have been recently identified that not only may help to understand prelamin A chromatin remodeling effects, but may also suggest how prelamin A accumulation causes tissue-specific symptoms.14 In this regard, an in vivo BAF interaction has been detected with the histone methyltransferase G9a, a chromatin remodeling enzyme involved in the transition of preadipocytes in adipocytes.14,35 Lipodystrophy is a common phenotypic sign observed in FPLD, MADA and HGPS patients, and we previously demonstrated that prelamin A accumulation interferes with the adipogenic program affecting SREBP1 nuclear translocation.10 The identification of G9a methyltransferase as a BAF-binding protein suggests an alternative pathological mechanism that could be involved in prelamin A-mediated lipodystrophy.14 It has been demonstrated that G9a methyltransferase inhibits adipogenic differentiation silencing PPAR-gamma gene expression. In preadipocytes, G9a activity increases the methylation status of the PPAR-gamma gene promoter, reducing its accessibility at transcription machinery.35 Thus, it is conceivable that BAF-G9a interaction could interfere with the dissociation of the repressor-complexes at the PPAR-gamma regulatory site, leading to the suppression of the late-stage adipogenesis. In accordance, in prelamin A-accumulating mouse preadipocytes induced to differentiate PPAR-gamma, expression is impaired and the adipogenic differentiation can’t occur.10
In MADA and HGPS patients, the accumulation of prelamin A causes premature aging. It has been proposed that premature aging may be a consequence of premature stem cell exhaustion.36 Mutations in A-type lamins could perturb the balance between proliferation and differentiation in adult stem cells, leading to less efficient tissue regeneration37,38 and differentiation.39 Interestingly, BAF interacts with, and seems to regulate the function of, Sox2, a transcription factor involved in maintaining the pluripotency in embryonic stem cells.14 In accordance, BAF depletion decreases cell survival and cloning efficiency in mouse and human stem cells and promotes the differentiation of both cellular types. We propose that the accumulation of prelamin A could interfere with BAF-mediated gene activation/inactivation function by affecting BAF exchange between cytosol and nucleus.40 Since BAF downregulation increased the expression of specific cell lineage genes, the forced BAF nuclear retention could abolish BAF nuclear quantitative changes necessary for chromatin remodeling events, which, in turn, should actuate gene expression.14 Interestingly, a new progeroid syndrome has been recently identified, which is due to a point mutation in BAF encoding gene (BANF1).19 Néstor-Guillermo progeria syndrome (NGPS) is a chronic progeria syndrome with early onset and slow clinical course. As observed in HGPS, NGPS patients are characterized by aged appearance, growth retardation, decreased subcutaneous fat, thin limbs and stiff joints, but they have no signs of cardiovascular impairment, diabetes mellitus or hypertriglyceridemia in early adulthood.41 In addition, NGPS shows a severe osteolysis and osteoporosis. At the cellular level, the BAF p.Ala12Thr mutation reduces BAF protein amount, probably affecting the protein stability and induces nuclear abnormalities.19 Interestingly, NGPS and HGPS cells show similar nuclear morphological defects, suggesting that BAF downregulation, as well as progerin interaction, affects nuclear structure in the same way. Thus, it is conceivable that BAF interaction with progerin could abolish BAF binding with the nuclear envelope and/or the nuclear lamina proteins involved in BAF structural functions.14,19
Since prelamin A expression is linked to chromatin remodeling, it is not surprising that this protein precursor is accumulated during skeletal muscle differentiation, which is characterized by a large reorganization of chromatin packaging.42
We demonstrated that prelamin A governs nuclear positioning in human skeletal muscle cells, and that LMNA gene mutations causing EDMD2 reduce prelamin A protein level and induce nuclear clustering.43 Interestingly, LMNA gene mutations leading to EDMD2 can impair the lamin A/C-emerin interaction, which has a role in prelamin A localization as underscored by prelamin A delocalization observed in EDMD1 cells.24 Since prelamin A, emerin and BAF may be considered as components of the same protein complexes, our finding on BAF-prelamin A delocalization in emerin-null cells is not surprising and suggests the involvement of this molecular platform in other muscular laminopathies. In particular, the impairment of BAF-prelamin A complex functions could be influenced by disruption of the known structural link between nucleus and cytoplasm (LINC complex).44 In fact, mutations in LINC complex proteins nesprin 1 and nesprin 2 affect lamin A/C and emerin-localization and cause EDMD4.45 Unfortunately, in order to better define prelamin A-BAF complex role in muscular dystrophy pathogenesis, important elements are still lacking. In particular, BAF expression, localization and protein interactions during skeletal muscle differentiation have never been described.
In conclusion, in this study we demonstrate that prelamin A accumulated in FPLD, MADA and RD cells interacts with BAF and affects its cellular distribution. This finding is in accordance with the rescue of BAF cellular distribution following lowering of progerin amount in HGPS cells treated with rapamycin.46 Also, our results strongly suggest prelamin A as a BAF-binding protein is able to recruit BAF in the nucleus. Moreover, our findings suggest that prelamin A requires specific protein-binding partners, like BAF, to affect nuclear function not only during disease, but probably also during physiological events requiring important reorganization of genome.
Materials and Methods
Cell cultures and transfection
Skin fibroblast cultures were obtained from skin biopsies of healthy, FPLD, MADA, RD and EDMD1 patients following a written consent. Cultures were established and cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum (FCS) and antibiotics. Control and pathological cells at early-passage (passage 5–10) were used for each experiment.
For immunoprecipitation assays, HEK293 cells were used. GFP-BAF, full-length FLAG-tagged rat prelamin A (LA-WT, pCI mammalian expression vector) and the mutated constructs LA-R482Q, LA-R527H and LA-L647R were transiently transfected into HEK293 cells using FuGene reagent (Roche).21 GFP-emerin was transiently transfected in human skin fibroblast as previously described.24 Biochemical and immunofluorescence analyses were performed 24 h after transfection. In human skin fibroblast cells, the accumulation of prelamin A intermediate was obtained using 25 μM mevinolin (Sigma) in growth medium for 18 h.24
Western blotting and immunoprecipitation
Human fibroblasts were lysed in lysis buffer containing 20 mM Tris–HCl, pH 7.5, 1% SDS, 1 mM Na3VO4, 1 mM PMSF, 5% β-mercaptoethanol and protease inhibitors. Proteins were subjected to SDS gradient gel (5–20%) electrophoresis and transferred to nitrocellulose membrane overnight at 4°C. Incubation with primary antibodies was performed for the indicated time. Bands were revealed by the Amersham ECL detection system. To isolate cytoplasms and nuclei of transfected cells, HEK293 cells pellets were resuspended in a lysis buffer containing 10 mM Tris pH 7.8, 1% Nonidet P-40 (NP-40), 2 mM MgCl2 and protease inhibitors. The separation was obtained by hypotonic shock; nuclei and cytoplasms were obtained by centrifugation at 1,000 g at 4°C. Cytoplasmic fractions and isolated nuclei were subject to SDS gradient gel (5–20%).
For immunoprecipitation experiments, HEK293 transfected cells were lysed in a buffer containing 50 mM TRIS-HCl, pH 8.0, 150 mM NaCl, 1% NP40, 0.1% SDS, 1 mM DTT and protease inhibitors (IP-buffer).21 Five hundred micrograms of total cell lysates were incubated with specific antibodies overnight at 4°C. Control immunoprecipitations were performed in the presence of aspecific immunoglobulins. After the addition of 30 μl of protein A/G (Santa Cruz Biotechnology) for 60 min at 4°C, immunoprecipitated protein complexes were washed, added to Laemmli’s buffer, boiled and subjected to western blot analysis.
Immunofluorescence
Human fibroblasts and HEK293 transfected cells grown on coverslips were fixed in methanol at –20°C for 7 min. Samples were incubated with PBS containing 4% BSA to saturate non-specific binding and incubated with primary antibodies and secondary antibodies. The nuclei were then counterstained with 4,6-diamino-2-phenylindole (DAPI). The slides were mounted with an anti-fade reagent in glycerol and observed. Immunofluorescence microscopy was performed using a Nikon E600 epifluorescence microscope and a Nikon oil-immersion objective [100x magnification, 1,3 NA (numerical aperture)]. Photographs were taken using a Nikon digital camera (DXm) and NIS-Element BR2.20 software. All images were taken at similar exposures within an experiment for each antibody. Images were processed using Adobe Photoshop (Adobe Systems).
Antibodies
The antibodies employed for western blot analysis or immunofluorescence labeling were: anti-FLAG, mouse monoclonal (Sigma M2, diluted 1:300, 1 h, for the immunofluorescence analysis and 1:1,000, 1 h, for the western blot analysis); anti-BAF, rabbit polyclonal (Santa Cruz Biotechnology FL-89, diluted 1:10, overnight at 4°C for immunofluorescence analysis and 1:100 overnight at 4°C, for the western blot analysis); anti-GFP rabbit polyclonal (Santa Cruz Biotechnology FL, diluted 1:1,000 for 1 h, for the western blot analysis) anti-emerin, mouse monoclonal (Monosan, diluted 1:200 for 1 h, for the western blot analysis); anti-prelamin A, goat polyclonal (Santa Cruz Biotechnology SC-6214, used 1:100 for 1 h at room temperature for the immunofluorescence analysis and 1:500 for 1 h, for the western blot analysis); anti-lamin A/C, goat polyclonal (Santa Cruz Biotechnology N-18, used 1:100 for 1 h for the western blot analysis); anti-lamin A rabbit polyclonal (Abcam ab-26300 diluted 1:500 for 1 h, for western blot analysis) anti-actin, goat polyclonal (Santa Cruz Biotechnology I-19, diluted 1:1,000 for 1 h, for the western blot analysis).
Supplementary Material
Acknowledgments
We thank A. Valmori, S. Grasso and D. Zini for the technical support. This work was supported by Italian Ministry for Education, University and Research Prin 2008 to G.L., FIRB 2010 to N.M.M., the “Fondazione Carisbo,” Italy and A.I.Pro.Sa.B., Italy. The authors are grateful to colleagues of Italian Network for Laminopathies for the helpful discussion.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/21869
References
- 1.Misteli T. Higher-order genome organization in human disease. Cold Spring Harb Perspect Biol. 2010;2:a000794. doi: 10.1101/cshperspect.a000794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Worman HJ. Nuclear lamins and laminopathies. J Pathol. 2012;226:316–25. doi: 10.1002/path.2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maraldi NM, Lattanzi G, Capanni C, Columbaro M, Merlini L, Mattioli E, et al. Nuclear envelope proteins and chromatin arrangement: a pathogenic mechanism for laminopathies. Eur J Histochem. 2006;50:1–8. [PubMed] [Google Scholar]
- 4.Maraldi NM, Lattanzi G. Involvement of prelamin A in laminopathies. Crit Rev Eukaryot Gene Expr. 2007;17:317–34. doi: 10.1615/CritRevEukarGeneExpr.v17.i4.50. [DOI] [PubMed] [Google Scholar]
- 5.Sinensky M, Fantle K, Trujillo M, McLain T, Kupfer A, Dalton M. The processing pathway of prelamin A. J Cell Sci. 1994;107:61–7. doi: 10.1242/jcs.107.1.61. [DOI] [PubMed] [Google Scholar]
- 6.Corrigan DP, Kuszczak D, Rusinol AE, Thewke DP, Hrycyna CA, Michaelis S, et al. Prelamin A endoproteolytic processing in vitro by recombinant Zmpste24. Biochem J. 2005;387:129–38. doi: 10.1042/BJ20041359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, et al. Lamin a truncation in Hutchinson-Gilford progeria. Science. 2003;300:2055. doi: 10.1126/science.1084125. [DOI] [PubMed] [Google Scholar]
- 8.Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature. 2003;423:293–8. doi: 10.1038/nature01629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Filesi I, Gullotta F, Lattanzi G, D’Apice MR, Capanni C, Nardone AM, et al. Alterations of nuclear envelope and chromatin organization in mandibuloacral dysplasia, a rare form of laminopathy. Physiol Genomics. 2005;23:150–8. doi: 10.1152/physiolgenomics.00060.2005. [DOI] [PubMed] [Google Scholar]
- 10.Capanni C, Mattioli E, Columbaro M, Lucarelli E, Parnaik VK, Novelli G, et al. Altered pre-lamin A processing is a common mechanism leading to lipodystrophy. Hum Mol Genet. 2005;14:1489–502. doi: 10.1093/hmg/ddi158. [DOI] [PubMed] [Google Scholar]
- 11.Navarro CL, De Sandre-Giovannoli A, Bernard R, Boccaccio I, Boyer A, Geneviève D, et al. Lamin A and ZMPSTE24 (FACE-1) defects cause nuclear disorganization and identify restrictive dermopathy as a lethal neonatal laminopathy. Hum Mol Genet. 2004;13:2493–503. doi: 10.1093/hmg/ddh265. [DOI] [PubMed] [Google Scholar]
- 12.Lattanzi G, Columbaro M, Mattioli E, Cenni V, Camozzi D, Wehnert M, et al. Pre-Lamin A processing is linked to heterochromatin organization. J Cell Biochem. 2007;102:1149–59. doi: 10.1002/jcb.21467. [DOI] [PubMed] [Google Scholar]
- 13.Mattioli E, Columbaro M, Capanni C, Santi S, Maraldi NM, D’Apice MR, et al. Drugs affecting prelamin A processing: effects on heterochromatin organization. Exp Cell Res. 2008;314:453–62. doi: 10.1016/j.yexcr.2007.11.012. [DOI] [PubMed] [Google Scholar]
- 14.Montes de Oca R, Andreassen PR, Wilson KL. Barrier-to-Autointegration Factor influences specific histone modifications. Nucleus. 2011;2:580–90. doi: 10.4161/nucl.2.6.17960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Skoko D, Li M, Huang Y, Mizuuchi M, Cai M, Bradley CM, et al. Barrier-to-autointegration factor (BAF) condenses DNA by looping. Proc Natl Acad Sci USA. 2009;106:16610–5. doi: 10.1073/pnas.0909077106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Margalit A, Neufeld E, Feinstein N, Wilson KL, Podbilewicz B, Gruenbaum Y. Barrier to autointegration factor blocks premature cell fusion and maintains adult muscle integrity in C. elegans. J Cell Biol. 2007;178:661–73. doi: 10.1083/jcb.200704049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Xu S, Rivolta C, Li LY, Peng GH, Swain PK, et al. Barrier to autointegration factor interacts with the cone-rod homeobox and represses its transactivation function. J Biol Chem. 2002;277:43288–300. doi: 10.1074/jbc.M207952200. [DOI] [PubMed] [Google Scholar]
- 18.Montes de Oca R, Shoemaker CJ, Gucek M, Cole RN, Wilson KL. Barrier-to-autointegration factor proteome reveals chromatin-regulatory partners. PLoS One. 2009;4:e7050. doi: 10.1371/journal.pone.0007050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Puente XS, Quesada V, Osorio FG, Cabanillas R, Cadiñanos J, Fraile JM, et al. Exome sequencing and functional analysis identifies BANF1 mutation as the cause of a hereditary progeroid syndrome. Am J Hum Genet. 2011;88:650–6. doi: 10.1016/j.ajhg.2011.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cabanillas R, Cadiñanos J, Villameytide JA, Pérez M, Longo J, Richard JM, et al. Néstor-Guillermo progeria syndrome: a novel premature aging condition with early onset and chronic development caused by BANF1 mutations. Am J Med Genet A. 2011;155A:2617–25. doi: 10.1002/ajmg.a.34249. [DOI] [PubMed] [Google Scholar]
- 21.Capanni C, Cenni V, Haraguchi T, Squarzoni S, Schüchner S, Ogris E, et al. Lamin A precursor induces barrier-to-autointegration factor nuclear localization. Cell Cycle. 2010;9:2600–10. doi: 10.4161/cc.9.13.12080. [DOI] [PubMed] [Google Scholar]
- 22.Gambineri A, Semple RK, Forlani G, Genghini S, Grassi I, Hyden CS, et al. Monogenic polycystic ovary syndrome due to a mutation in the lamin A/C gene is sensitive to thiazolidinediones but not to metformin. Eur J Endocrinol. 2008;159:347–53. doi: 10.1530/EJE-08-0272. [DOI] [PubMed] [Google Scholar]
- 23.Columbaro M, Mattioli E, Schena E, Capanni C, Cenni V, Levy N, et al. Prelamin A processing and functional effects in restrictive dermopathy. Cell Cycle. 2010;9:4766–8. doi: 10.4161/cc.9.23.14210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Capanni C, Del Coco R, Mattioli E, Camozzi D, Columbaro M, Schena E, et al. Emerin-prelamin A interplay in human fibroblasts. Biol Cell. 2009;101:541–54. doi: 10.1042/BC20080175. [DOI] [PubMed] [Google Scholar]
- 25.Bengtsson L, Wilson KL. Barrier-to-autointegration factor phosphorylation on Ser-4 regulates emerin binding to lamin A in vitro and emerin localization in vivo. Mol Biol Cell. 2006;17:1154–63. doi: 10.1091/mbc.E05-04-0356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bione S, Maestrini E, Rivella S, Mancini M, Regis S, Romeo G, et al. Identification of a novel X-linked gene responsible for Emery-Dreifuss muscular dystrophy. Nat Genet. 1994;8:323–7. doi: 10.1038/ng1294-323. [DOI] [PubMed] [Google Scholar]
- 27.Maraldi NM, Lattanzi G, Capanni C, Columbaro M, Mattioli E, Sabatelli P, et al. Laminopathies: a chromatin affair. Adv Enzyme Regul. 2006;46:33–49. doi: 10.1016/j.advenzreg.2006.01.001. [DOI] [PubMed] [Google Scholar]
- 28.Krishnan V, Chow MZ, Wang Z, Zhang L, Liu B, Liu X, et al. Histone H4 lysine 16 hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24-deficient mice. Proc Natl Acad Sci USA. 2011;108:12325–30. doi: 10.1073/pnas.1102789108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li X, Corsa CA, Pan PW, Wu L, Ferguson D, Yu X, et al. MOF and H4 K16 acetylation play important roles in DNA damage repair by modulating recruitment of DNA damage repair protein Mdc1. Mol Cell Biol. 2010;30:5335–47. doi: 10.1128/MCB.00350-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pegoraro G, Kubben N, Wickert U, Göhler H, Hoffmann K, Misteli T. Ageing-related chromatin defects through loss of the NURD complex. Nat Cell Biol. 2009;11:1261–7. doi: 10.1038/ncb1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brachner A, Braun J, Ghodgaonkar M, Castor D, Zlopasa L, Ehrlich V, et al. The endonuclease Ankle1 requires its LEM and GIY-YIG motifs for DNA cleavage in vivo. J Cell Sci. 2012;125:1048–57. doi: 10.1242/jcs.098392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dittrich CM, Kratz K, Sendoel A, Gruenbaum Y, Jiricny J, Hengartner MO. LEM-3 - A LEM domain containing nuclease involved in the DNA damage response in C. elegans. PLoS One. 2012;7:e24555. doi: 10.1371/journal.pone.0024555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Musich PR, Zou Y. Genomic instability and DNA damage responses in progeria arising from defective maturation of prelamin A. Aging (Albany NY) 2009;1:28–37. doi: 10.18632/aging.100012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Musich PR, Zou Y. DNA-damage accumulation and replicative arrest in Hutchinson-Gilford progeria syndrome. Biochem Soc Trans. 2011;39:1764–9. doi: 10.1042/BST20110687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gulbagci NT, Li L, Ling B, Gopinadhan S, Walsh M, Rossner M, et al. SHARP1/DEC2 inhibits adipogenic differentiation by regulating the activity of C/EBP. EMBO Rep. 2009;10:79–86. doi: 10.1038/embor.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Halaschek-Wiener J, Brooks-Wilson A. Progeria of stem cells: stem cell exhaustion in Hutchinson-Gilford progeria syndrome. J Gerontol A Biol Sci Med Sci. 2007;62:3–8. doi: 10.1093/gerona/62.1.3. [DOI] [PubMed] [Google Scholar]
- 37.Gotzmann J, Foisner R. A-type lamin complexes and regenerative potential: a step towards understanding laminopathic diseases? Histochem Cell Biol. 2006;125:33–41. doi: 10.1007/s00418-005-0050-8. [DOI] [PubMed] [Google Scholar]
- 38.Vlcek S, Foisner R. Lamins and lamin-associated proteins in aging and disease. Curr Opin Cell Biol. 2007;19:298–304. doi: 10.1016/j.ceb.2007.04.001. [DOI] [PubMed] [Google Scholar]
- 39.Scaffidi P, Misteli T. Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat Cell Biol. 2008;10:452–9. doi: 10.1038/ncb1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haraguchi T, Koujin T, Osakada H, Kojidani T, Mori C, Masuda H, et al. Nuclear localization of barrier-to-autointegration factor is correlated with progression of S phase in human cells. J Cell Sci. 2007;120:1967–77. doi: 10.1242/jcs.03461. [DOI] [PubMed] [Google Scholar]
- 41.Cabanillas R, Cadiñanos J, Villameytide JA, Pérez M, Longo J, Richard JM, et al. Néstor-Guillermo progeria syndrome: a novel premature aging condition with early onset and chronic development caused by BANF1 mutations. Am J Med Genet A. 2011;155A:2617–25. doi: 10.1002/ajmg.a.34249. [DOI] [PubMed] [Google Scholar]
- 42.Brero A, Easwaran HP, Nowak D, Grunewald I, Cremer T, Leonhardt H, et al. Methyl CpG-binding proteins induce large-scale chromatin reorganization during terminal differentiation. J Cell Biol. 2005;169:733–43. doi: 10.1083/jcb.200502062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mattioli E, Columbaro M, Capanni C, Maraldi NM, Cenni V, Scotlandi K, et al. Prelamin A-mediated recruitment of SUN1 to the nuclear envelope directs nuclear positioning in human muscle. Cell Death Differ. 2011;18:1305–15. doi: 10.1038/cdd.2010.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Méjat A, Misteli T. LINC complexes in health and disease. Nucleus. 2010;1:40–52. doi: 10.4161/nucl.1.1.10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Q, Bethmann C, Worth NF, Davies JD, Wasner C, Feuer A, et al. Nesprin-1 and -2 are involved in the pathogenesis of Emery Dreifuss muscular dystrophy and are critical for nuclear envelope integrity. Hum Mol Genet. 2007;16:2816–33. doi: 10.1093/hmg/ddm238. [DOI] [PubMed] [Google Scholar]
- 46.Cenni V, Bavelloni A, Beretti F, Tagliavini F, Manzoli L, Lattanzi G, et al. Ankrd2/ARPP is a novel Akt2 specific substrate and regulates myogenic differentiation upon cellular exposure to H(2)O(2) Mol Biol Cell. 2011;22:2946–56. doi: 10.1091/mbc.E10-11-0928. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.