

Abstract
Here, we investigated the compartment-specific role of cell cycle arrest and senescence in breast cancer tumor growth. For this purpose, we generated a number of hTERT-immortalized senescent fibroblast cell lines overexpressing CDK inhibitors, such as p16(INK4A), p19(ARF) or p21(WAF1/CIP1). Interestingly, all these senescent fibroblast cell lines showed evidence of increased susceptibility toward the induction of autophagy (either at baseline or after starvation), as well as significant mitochondrial dysfunction. Most importantly, these senescent fibroblasts also dramatically promoted tumor growth (up to ~2-fold), without any comparable increases in tumor angiogenesis. Conversely, we generated human breast cancer cells (MDA-MB-231 cells) overexpressing CDK inhibitors, namely p16(INK4A) or p21(WAF1/CIP1). Senescent MDA-MB-231 cells also showed increased expression of markers of cell cycle arrest and autophagy, including β-galactosidase, as predicted. Senescent MDA-MB-231 cells had retarded tumor growth, with up to a near 2-fold reduction in tumor volume. Thus, the effects of CDK inhibitors are compartment-specific and are related to their metabolic effects, which results in the induction of autophagy and mitochondrial dysfunction. Finally, induction of cell cycle arrest with specific inhibitors (PD0332991) or cellular stressors [hydrogen peroxide (H₂O₂) or starvation] indicated that the onset of autophagy and senescence are inextricably linked biological processes. The compartment-specific induction of senescence (and hence autophagy) may be a new therapeutic target that could be exploited for the successful treatment of human breast cancer patients.
Keywords: CDK inhibitors, PD0332991, aging, autophagy, cancer metabolism, cancer-associated fibroblast, cell cycle arrest, glycolysis, mitophagy, senescence, tumor initiation, tumor stroma
Introduction
The exact functional relationship between cell cycle arrest, senescence and autophagy remains unknown.1-9 Both senescence and autophagy are thought to play key mechanistic roles in the development of multiple aging-associated diseases, and especially human cancers.10,11 Thus, understanding senescence and autophagy has wide clinical implications, for both regenerative medicine and cancer prevention. Two relevant mechanistic questions are: (1) Is autophagy sufficient to induce senescence? and, conversely, (2) Is senescence sufficient to induce autophagy?
Recent studies have shown that senescence and autophagy may be part of the same metabolic program, known as the autophagy-senescence transition (AST).10-21 In support of this notion, recombinant expression of autophagy genes (BNIP3, Cathepsin B or ATG16L1) in stromal fibroblasts is sufficient to induce the onset of constitutive autophagy as well as the development of senescence.12,13 In this model, “leaky” lysosomes drive the onset of aging and senescence in response to cellular stress.22,23 Thus, autophagy is indeed sufficient to induce senescence.22,23 The AST also leads to mitophagy, due to the onset of mitochondrial dysfunction, resulting in a cellular shift toward aerobic glycolysis and ketone production.12,13 Importantly, these autophagic-senescent fibroblasts undergo catabolism and locally produce high-energy mitochondrial fuels (such as L-lactate, ketone bodies and glutamine).12,13 These mitochondrial fuels can then act as onco-catabolites, driving anabolic tumor growth and cancer cell metastasis.24-36 This simple energy-transfer mechanism may also be very important for tumor initiation, especially under conditions where tumor angiogenesis has yet to occur, providing a “fertile ground” for tumor growth.37-42
However, it remains unknown if cell cycle arrest and senescence are also sufficient to drive the onset of autophagy, resulting in the senescence-autophagy transition (SAT). If this was indeed the case, then the SAT would explain why chronological aging is one of the single most important risk factors for the development of human cancers. It would establish, unequivocally, that chronological aging directly generates high-energy nutrients (onco-catabolites) to “feed” cancer cells.
Here, we used a genetic approach to test the hypothesis that cell cycle arrest and senescence were sufficient to induce an autophagic phenotype in cancer-associated fibroblasts. To test this idea, we created a panel of hTERT-immortalized fibroblast cell lines that recombinantly overexpress well-known CDK inhibitors, such as p16(INK4A), p19(ARF), smARF and p21(WAF1/CIP1). Interestingly, overexpression of CDK-inhibitor proteins was sufficient to induce autophagy and to drive the onset of mitophagy, as well as mitochondrial dysfunction. Thus, we validated the existence of the senescence-autophagy transition (SAT). Importantly, these CDK-overexpressing fibroblasts also significantly promoted tumor growth, without an increase in angiogenesis.
As such, our current results provide a new genetically tractable model for understanding the metabolic role of “host aging” in promoting tumor growth and metastasis by providing a “fertile,” local microenvironment.
Conversely, overexpression of CDK-inhibitor proteins in breast cancer cells resulted in reduced tumor growth, secondary to the induction of autophagy in tumor cells. This may provide an innovative targeted approach for new avenues of therapeutic intervention.
Finally, using a pharmacological approach relying on specific inhibitors (PD0332991) and cellular stressors (hydrogen peroxide or starvation), we show that autophagy and senescence are two closely linked biological phenomena, implying that they are part of the same coordinated metabolic program.
Results
Cell cycle arrest, autophagy and senescence are three closely linked biological processes
To experimentally assess the relationship between cell cycle arrest, senescence and autophagy, we first used a pharmacological approach to induce cell cycle arrest with the CDK4/6 chemical inhibitor, PD0332991.43,44 For this purpose, hTERT-immortalized fibroblasts were treated with PD0332991 (0.1, 0.5 and 1 μM) for a period of 36 h, and then the cells were subjected to biochemical analysis.
Figure 1 shows that treatment with PD0332991 dramatically inhibits RB-phosphorylation, as expected, resulting in the upregulation of markers of cell cycle arrest, such as CDK inhibitors (p16 and p21), as well as markers of senescence (β-galactosidase) and autophagy (LC3-I/II). Thus, the induction of cell cycle arrest is closely linked to the onset of senescence and autophagy.
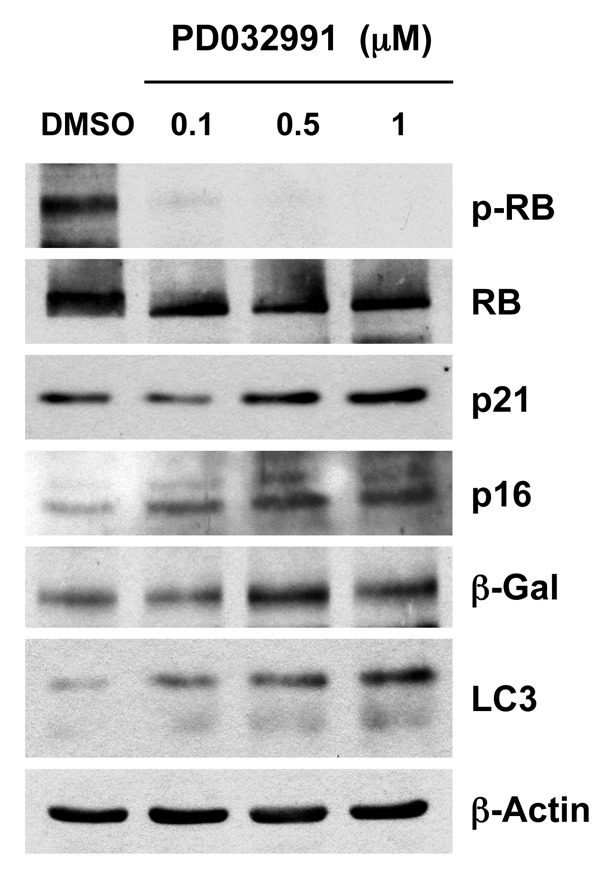
Figure 1. Treatment with PD0332991, a CDK4/6-inhibitor, induces markers of senescence and autophagy. hTERT-BJ1 fibroblasts were incubated with PD0332991 (0.1, 0.5 and 1 μM) for 36 h before the cells were harvested and subjected to immunoblot analysis with specific antibody probes. Note that PD0332991 effectively inhibits the phosphorylation of RB, as predicted. In addition, PD0332991 induces the upregulation of three markers of senescence [p21(WAF1/CIP1), p16(INK4A) and β-galactosidase]. Similarly, LC3 expression (an autophagy marker) is progressively upregulated. Blotting with β-actin is shown as a control for equal protein loading.
Next, we assessed the response to a well-established cellular stress, hydrogen peroxide (H2O2; 300 μM), which is known to induce oxidative stress, senescence and autophagy. Then, we followed changes in markers of the cell cycle and autophagy, over a time course of 8 d post-treatment. Figure 2A shows that H202-treatment induces the upregulation of markers of cell cycle arrest and senescence (p53, p21, β-galactosidase), as well the downregulation of markers of cell proliferation (phospho-RB, PCNA), within the first 2–3 d post-treatment. Figure 2B indicates that H2O2 treatment induces a panel of lysosomal (Lamp-1, cathepsin B) and autophagy/mitophagy markers (Beclin-1, BNIP3, LC3-I/II) within 4–6 d post-treatment.

Figure 2. Acute treatment with hydrogen peroxide (H2O2) induces markers of senescence and autophagy. hTERT-BJ1 fibroblasts were treated for 1 h with H2O2 (300 μM) and then cultured for 0–8 d. Then, at each time point, the cells were harvested and subjected to immunoblot analysis. Blotting with β-actin is shown as a control for equal protein loading. (A) Markers of the cell cycle and senescence. Note that several markers of cell cycle arrest and senescence were increased at days 2 and 3 post-treatment [p53, p21(WAF1/CIP1) and β-galactosidase], while markers of cell cycle progression are decreased (phospho-RB and PCNA). (B) Markers of autophagy and mitophagy. Note that several markers of autophagy, mitophagy and lysosomes (Lamp-1, Beclin-1, BNIP3, cathepsin B and LC3) were increased, mainly at days 4–6 post-treatment.
Finally, we determined the response to another cellular stress closely associated with autophagy, i.e., amino acid starvation. A preliminary time course study indicated that autophagy peaked at 3 h and cell cycle arrest peaked at 4 h. Figure 3 (left panel) shows that at 3 h post-starvation, several markers of autophagy (Lamp-1, BNIP3 and LC3) are upregulated. Similarly, Figure 3 (right panel) illustrates that at 4 h post-starvation, a panel of markers associated with cell cycle arrest and senescence (p16, p19, p21, p53, β-galactosidase) are all upregulated. Conversely, at the same time point, markers of cell proliferation are downregulated (phospho-RB and cyclin D1).
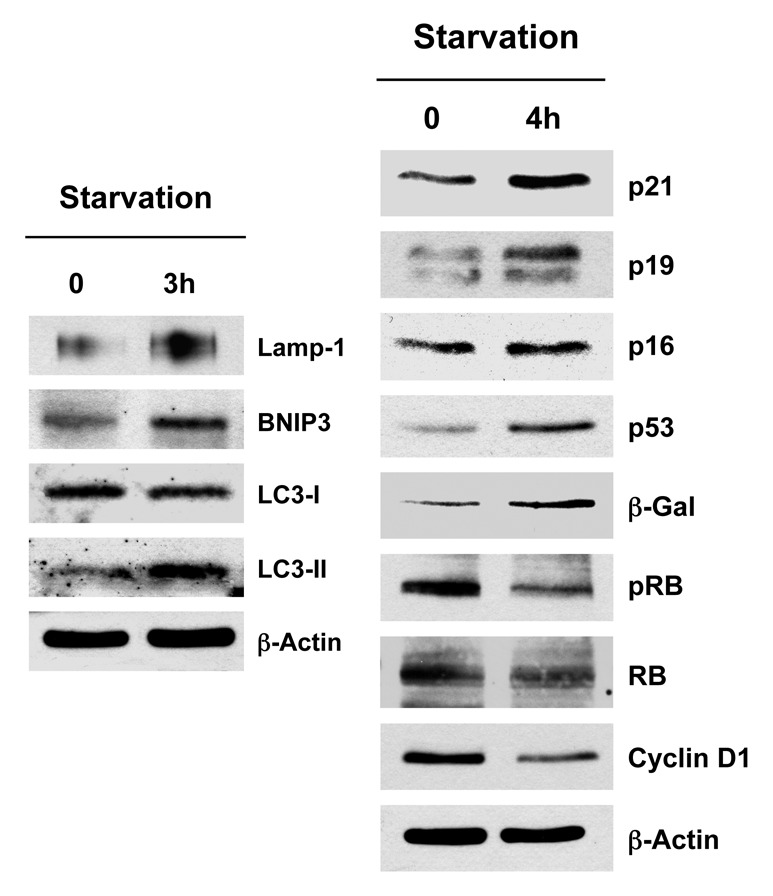
Figure 3. Acute starvation induces markers of autophagy and senescence. After 3 h of acute starvation with Hepes-buffered HBSS, several markers of autophagy were increased (Lamp-1, BNIP3, LC3). Similarly, after 4 h of acute starvation with HBSS, a panel of markers for cell cycle arrest [p21(WAF1/CIP1), p19(ARF), p16(INK4A), p53, β-galactosidase] were all increased. Conversely, markers of cell cycle progression were significantly decreased (phospho-RB and cyclin D1). Blotting with β-actin is shown as a control for equal protein loading.
In summary, based on this analysis, we conclude that cell cycle arrest, autophagy and senescence are three closely linked conserved biological processes, such that autophagy induces senescence and, conversely, senescence induces autophagy.
p19(ARF) and smARF drive the onset of senescence and autophagy in cancer-associated fibroblasts, metabolically promoting tumor growth, without increased angiogenesis
p19(ARF) and its closely related truncated isoform, smARF, are best known to function as CDK inhibitors.45-51 To study the potential metabolic effects of p19(ARF) and smARF, they were both stably expressed in hTERT-immortalized fibroblasts. Figure 4A indicates that we successfully generated stable cell lines expressing either p19(ARF) or smARF. Since they are both continuously degraded by the proteasomal machinery, we used treatment with the proteasomal-inhibitor MG-132 (10 μM for 16 h) to facilitate their detection. In addition, Figure 4B indicates that p19(ARF)- and smARF-expressing fibroblasts are more susceptible to the induction of p16 and p21 under conditions of H2O2 exposure, as expected. Under these conditions (Fig. 4C), we also observed a loss of Cav-1 expression, suggesting its lysosomal degradation by autophagy.
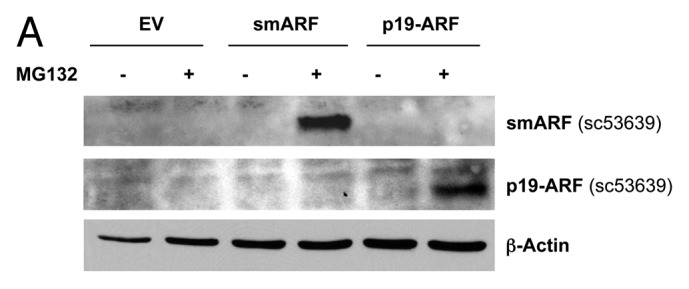
Figure 4. Generating stable fibroblast cell lines which constitutively express smARF and p19(ARF). hTERT-BJ1 fibroblasts were transduced with lenti-viral vectors encoding smARF or p19(ARF) and then selected for puromycin-resistance. Control fibroblasts transduced with the empty vector (EV; Lv-105) were produced in parallel. Then, these stable cell lines were subjected to biochemical analysis. Blotting with β-actin is shown as a control for equal protein loading. (A) Treatment with the proteasome inhibitor MG-132. After overnight treatment (16 h) with MG-132 (10 μM), the cells were harvested and subjected to immunoblot analysis. Note that both smARF and p19(ARF) were stably expressed. (B) Acute treatment with H202. After acute treatment with H2O2 (300 μM), the fibroblasts were cultured for an additional 5 d to allow the onset of senescence. Note that fibroblasts transduced with smARF or p19(ARF) show the upregulation of p16(INK4A) and/or p21 (WAF1/CIP1), other CDK inhibitors. (C) As in (B), except the expression of Cav-1 was assessed. Note the 2-fold reduction in Cav-1 expression.
To validate the hypothesis that both smARF and p19(ARF) expression increase the susceptibility to autophagy, these stable fibroblast cell lines were acutely treated with H202 and cultured for an additional 5 d to allow the onset of senescence. Figure 5 shows that the expression of either smARF or p19(ARF) increases the levels of a panel of autophagy markers (Lamp-1, Beclin-1, BNIP3, cathepsin B, LC3 and legumain), in accordance with our hypothesis.
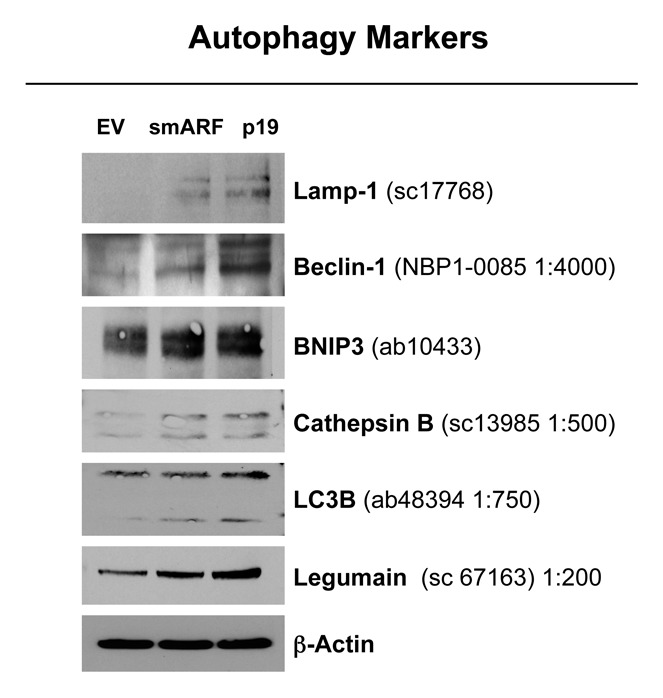
Figure 5. Both smARF and p19(ARF) expression increases the susceptibility of fibroblasts to the onset of autophagy. Stable fibroblast cell lines were acutely treated with H202 and cultured for an additional 5 d. Then, the expression of autophagy markers was assessed. Note that the expression of either smARF or p19(ARF) increases the levels of a panel of autophagy markers (Lamp-1, Beclin-1, BNIP3, cathepsin B, LC3 and legumain). Blotting with β-actin is shown as a control for equal protein loading.
We further validated that that both smARF- and p19(ARF)-expressing fibroblasts are more susceptible to the onset of H2O2-induced senescence, using β-galactosidase as a marker. Figure 6 shows representative images and corresponding quantitation, indicating that smARF and p19(ARF) expression increase β-galactosidase positivity by > 3-fold. In addition, under conditions of hypoxia, p19(ARF) also induces a > 2-fold increase in L-lactate production, consistent with the onset of mitophagy and mitochondrial dysfunction (Fig. 7). Thus, p19(ARF)-induced cell cycle arrest increases the susceptibility of cells toward the onset of stress-induced cellular senescence, resulting in the activation of autophagic processes and a metabolic shift toward glycolysis.
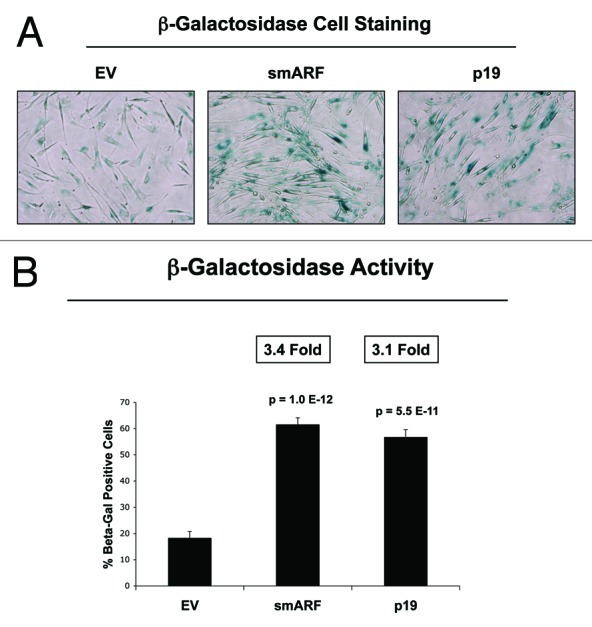
Figure 6. Both smARF and p19(ARF) expression increases the susceptibility of fibroblasts to the onset of senescence. Stable fibroblast cell lines were acutely treated with H202 and cultured for an additional 5 d. Then, the expression of β-galactosidase activity was assessed. Note that the expression of either smARF or p19(ARF) increases β-galactosidase activity by ~3-fold. Representative images are shown in (A), and quantitation is presented in (B).
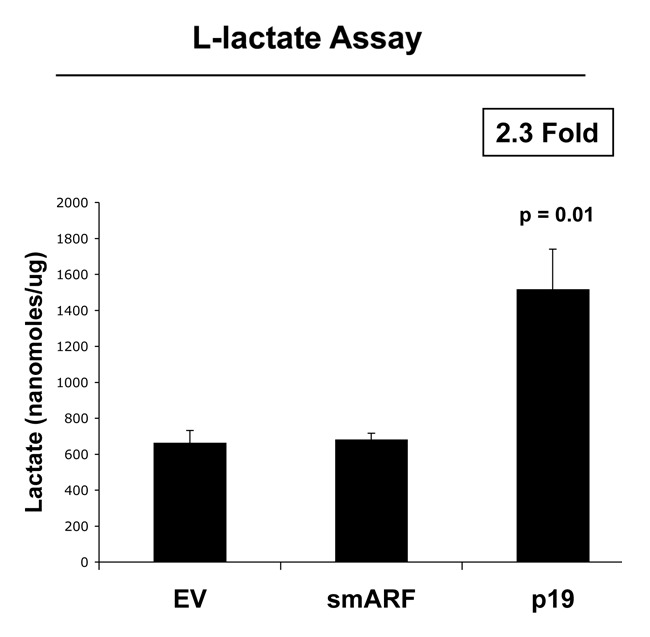
Figure 7. p19(ARF) increases L-lactate production. Stable fibroblast cell lines were subjected to chronic hypoxia for 48 h. Then, the levels of L-lactate that accumulated in the tissue-culture media were assessed and normalized to the amount of total cellular protein. Note that p(19)ARF expression increases L-lactate accumulation by > 2-fold.
Most importantly, fibroblasts overexpressing smARF or p19(ARF) effectively promoted tumor growth (Fig. 8). Stable fibroblast cell lines were co-injected with MDA-MB-231(GFP) breast cancer cells into the flanks of immunodeficient mice. Then, tumor growth was followed for a period of 30 d post-injection. Figure 8A and B show that fibroblasts overexpressing smARF or p19(ARF) resulted in a near 2-fold increase in tumor growth. However, CD31 immunostaining indicated that angiogenesis could not account for the observed increase in tumor growth (Fig. 8C).

Figure 8. Fibroblasts overexpressing smARF or p19(ARF) promote tumor growth. Stable fibroblast cell lines were co-injected with MDA-MB-231(GFP) breast cancer cells into the flanks of immunodeficient mice. Then, tumor growth was followed for a period of 30 d post-injection. (A) shows the time course of tumor growth and (B) shows the final tumor volume at sacrifice, which resulted in a near 2-fold increase in tumor growth. (C) shows the results CD31 immunostaining, indicating that angiogenesis cannot account for the observed increase in tumor growth.
p16(INK4A) and p21(WAF1/CIP1) induce senescence and autophagy in cancer-associated fibroblasts, metabolically promoting tumor growth, without increased angiogenesis
To further assess the relationship between cell cycle arrest, autophagy and senescence, we next focused on the metabolic effects of two other well-known CDK inhibitors, namely p16(INK4A) and p21(WAF1/CIP1). For this purpose, we generated hTERT fibroblast cell lines overexpressing p16(INK4A) and p21(WAF1/CIP1). Figure 9 directly shows that p16(INK4A) and p21(WAF1/CIP1) were effectively stably expressed in hTERT-BJ1 cells. As predicted, this resulted in the corresponding upregulation of a panel of markers of cell cycle arrest and senescence (p16, p21, p53 and β-galactosidase).
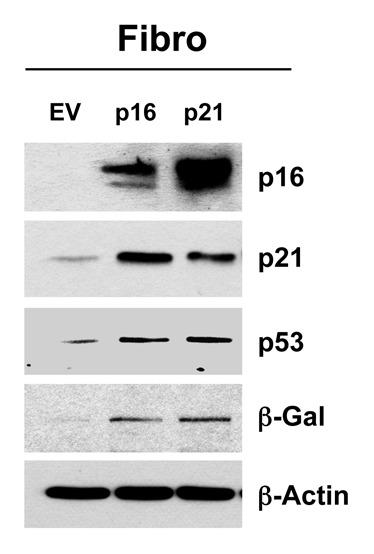
Figure 9. Generating stable fibroblast cell lines, which constitutively express p16(INK4A) and p21(WAF1/CIP1). hTERT-BJ1 fibroblasts were transduced with lenti-viral vectors encoding p16(INK4A) and p21(WAF1/CIP1) and then selected for puromycin-resistance. Control fibroblasts transduced with the empty vector (EV; Lv-105) were produced in parallel. Then, these stable cell lines were subjected to biochemical analysis. Note that after 6 d of culture, the overexpression of markers of cell cycle arrest and senescence [p16(INK4A, p21(WAF1/CIP1), p53 and β-galactosidase] is observed. Blotting with β-actin is shown as a control for equal protein loading.
In further support of the notion that senescence and autophagy are part of the same metabolic program, Figure 10 shows that p16(INK4A)- and p21(WAF1/CIP1)-expressing fibroblasts are more autophagic, both at baseline and under conditions of starvation (Fig. 10A and B), and they also show a tendency toward cell hypertrophy, consistent with a senescent phenotype (Fig. 10C). Overexpression of markers of autophagy (Lamp-1 and BNIP3) is observed, especially in cells overexpressing p21(WAF1/CIP1). Similarly, after 12 h of starvation in Hepes-buffered HBSS, the overexpression of markers of autophagy (LC3-I/II) is detected, especially in cells overexpressing p21(WAF1/CIP1). However, cell hypertrophy is observed most significantly in fibroblasts overexpressing p16(INK4A).
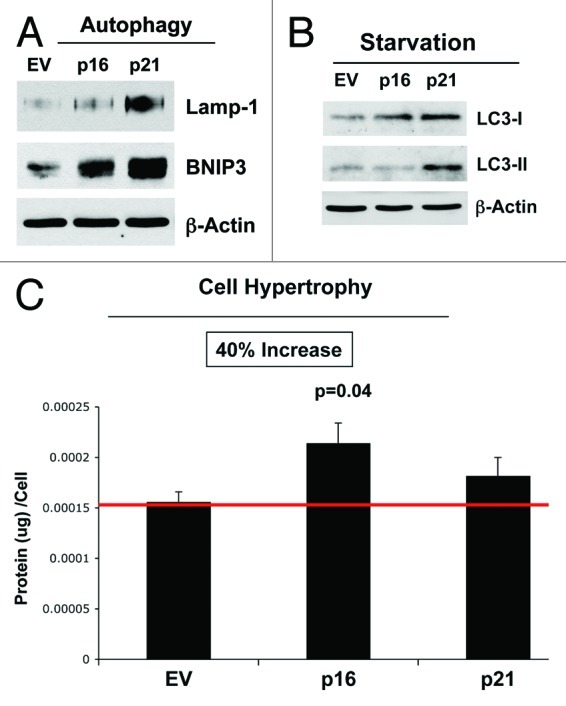
Figure 10. Stable fibroblast cell lines, which constitutively express p16(INK4A) and p21(WAF1/CIP1), show the onset of increased autophagy and/or cell hypertrophy. (A) Autophagy at baseline. Note that after 6 d of culture, the overexpression of markers of autophagy (Lamp-1 and BNIP3) is observed, especially in cells overexpressing p21(WAF1/CIP1). Blotting with β-actin is shown as a control for equal protein loading. (B) Autophagy after starvation. Note that after 12 h of starvation in Hepes-buffered HBSS, the overexpression of markers of autophagy (LC3-I/II) is observed, especially in cells overexpressing p21(WAF1/CIP1). Blotting with β-actin is shown as a control for equal protein loading. (C) Cell hypertrophy. Note that after 2 d of culture, cell hypertrophy is observed, especially in cells overexpressing p16(INK4A).
As strong BNIP3 overexpression was observed in p16- and p21-overexpressing fibroblasts, and BNIP334,52-55 is associated with mitophagy and mitochondrial dysfunction, we assessed the status of mitochondrial oxidative phosphorylation (OXPHOS). Figure 11 shows that fibroblasts overexpressing p16(INK4A) and p21(WAF1/CIP1) both show significant changes in mitochondrial OXPHOS components. Importantly, dramatic reductions in several OXPHOS complex components was observed, especially complex I, III and V. Similarly, reductions were also observed in complex II and IV, but to a lesser extent. This is consistent with mitochondrial dysfunction and a shift toward glycolysis.
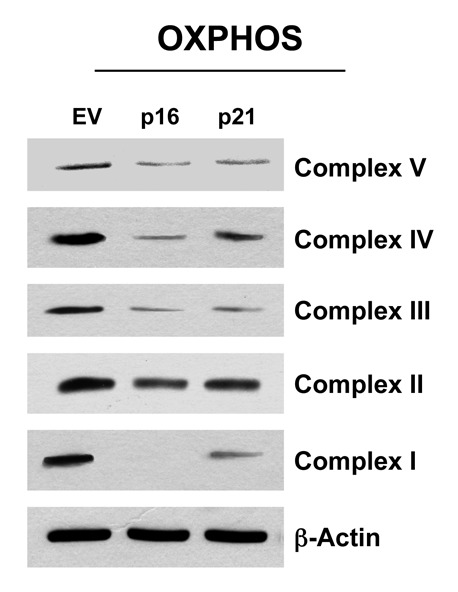
Figure 11. Fibroblast cell lines, which constitutively express p16(INK4A) and p21(WAF1/CIP1), show significant reductions in OXPHOS complex components. Note that after 6 d of culture, dramatic reductions in OXPHOS complex components is observed, especially complex I, III and V. Reductions were also observed in complex II and IV, but to a lesser extent. Blotting with β-actin is shown as a control for equal protein loading.
We also determined the tumor-promoting activity of p16- and p21-senescent fibroblasts. These stable fibroblast cell lines were co-injected with MDA-MB-231(GFP) breast cancer cells into the flanks of immunodeficient mice. Then, tumor growth was followed for a period of 4 wk post-injection. Figure 12A shows the final tumor volume at sacrifice, which resulted in a near 2-fold increase in tumor growth. Figure 12B illustrates that tumor-associated CD31 immunostaining is not elevated, indicating that angiogenesis does account for the increased tumor growth. Thus, senescent-autophagic fibroblasts promote tumor growth, independently of neo-angiogenesis.

Figure 12. Fibroblasts overexpressing p16(INK4A) and p21(WAF1/CIP1) promote tumor growth in a paracrine fashion. Stable fibroblast cell lines were co-injected with MDA-MB-231(GFP) breast cancer cells into the flanks of immunodeficient mice. Then, tumor growth was followed for a period of 4 wk post-injection. (A) shows the final tumor volume at sacrifice, which resulted in a near 2-fold increase in tumor growth. (B) shows the results CD31 immunostaining, indicating that angiogenesis cannot account for the observed increase in tumor growth.
p16(INK4A) and p21(WAF1/CIP1) induce senescence and autophagy in breast cancer cells, metabolically inhibiting tumor growth
To examine the compartment-specific effects of senescence and autophagy on tumor growth, we also created MDA-MB-231 breast cancer cell lines overexpressing p16(INK4A) and p21(WAF1/CIP1). Figure 13 shows that we successfully generated MDA-MB-231 breast cancer cells overexpressing p16 or p21, which resulted in the induction of β-galactosidase, an established marker of cell cycle arrest and senescence.
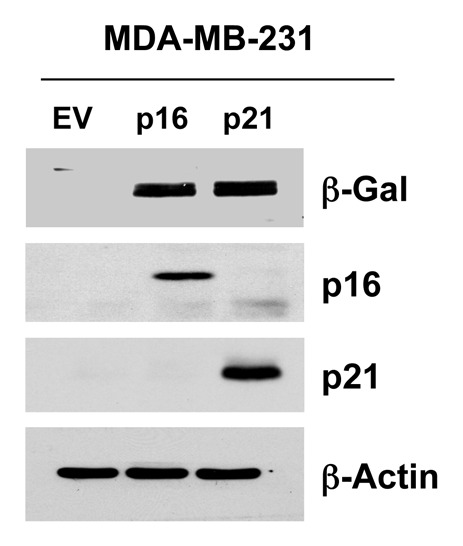
Figure 13. Generating MDA-MB-231 breast cancer cell lines, which overexpress p16(INK4A) and p21(WAF1/CIP1). MDA-MB-231(GFP) cells were transduced with lenti-viral vectors encoding p16(INK4A) and p21(WAF1/CIP1), and then selected for puromycin-resistance. Control cells transduced with the empty vector (EV; Lv-105) were produced in parallel. Then, these stable cell lines were subjected to biochemical analysis. Note the overexpression of markers of cell cycle arrest and senescence [p16(INK4A, p21(WAF1/CIP1) and β-galactosidase] is observed. Blotting with β-actin is shown as a control for equal protein loading.
Morphologically, MDA-MB-231 cells overexpressing p16(INK4A) and p21(WAF1/CIP1) also appeared more hypertrophic and expressed increased β-galactosidase activity, consistent with a senescence phenotype (Fig. 14).

Figure 14. MDA-MB-231 breast cancer cell lines, which overexpress p16(INK4A) and p21(WAF1/CIP1), show signs of senescence. (A) Cell hypertrophy. MDA-MB-231 cells, which overexpress p16(INK4A) and p21(WAF1/CIP1), morphologically appear flatter or hypertrophic. Representative phase images are shown. (B) Beta-galactosidase. MDA-MB-231 cells, which overexpress p16(INK4A) and p21(WAF1/CIP1), also appear to have increased β-galactosidase activity.
Figure 15 illustrates that MDA-MB-231 cells, which overexpress p16(INK4A) and p21(WAF1/CIP1), undergo increased authophagy, either at baseline (Lamp-1, BNIP3, LC3-I/II) or under conditions of starvation for 12 h with HBSS (Beclin-1, and cathepsin B). Therefore, cell cycle arrest, senescence and autophagy in cancer cells are all part of the same metabolic program.
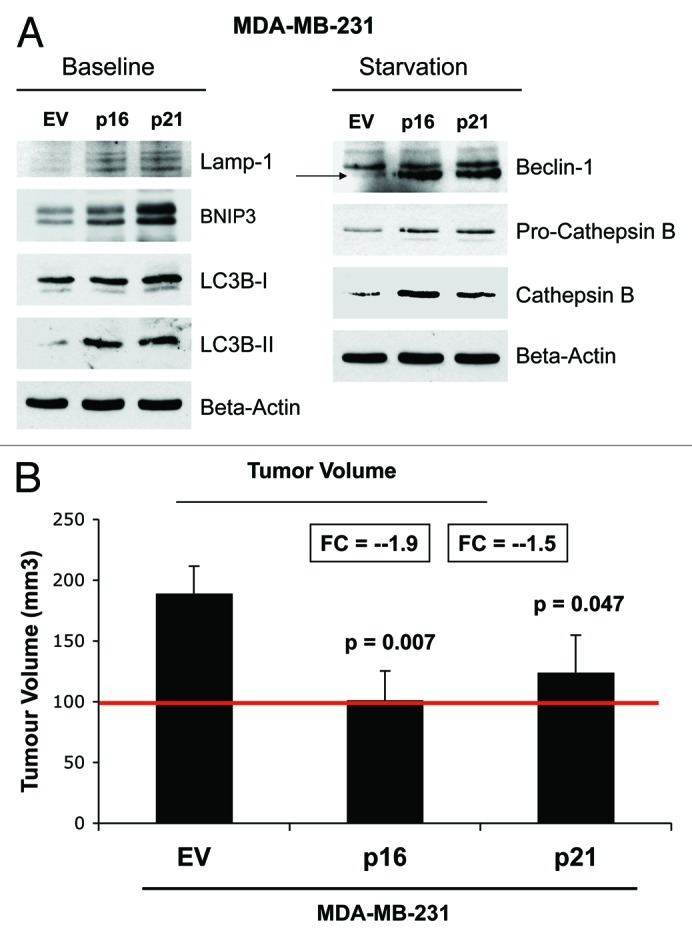
Figure 15. MDA-MB-231 breast cancer cell lines, which overexpress p16(INK4A) and p21(WAF1/CIP1), undergo autophagy, and show dramatically reduced tumor growth. (A) Autophagy status. MDA-MB-231 cells, which overexpress p16(INK4A) and p21(WAF1/CIP1), undergo increased authophagy, either at baseline (Lamp-1, BNIP3, LC3-I/II) or under conditions of starvation for 12 h with HBSS (Beclin-1 and cathepsin B). (B) Tumor growth. MDA-MB-231 cells, which overexpress p16(INK4A) and p21(WAF1/CIP1), show a near 2-fold reduction in tumor volume.
Most importantly, MDA-MB-231 cells, which overexpress p16(INK4A) and p21(WAF1/CIP1), show a near 2-fold reduction in tumor volume. Thus, senescence and autophagy in cancer cells metabolically inhibits tumor growth.
Discussion
Here, we investigated the existence of the senescence-autophagy transition (SAT), using a molecular genetic approach (Fig. 16). More specifically, we tested the hypothesis that chronological aging may facilitate the growth of tumors via the onset of cell cycle arrest and senescence, which, in turn, drives the induction of an autophagic program in stromal fibroblasts. Senescence-induced autophagy would provide a “fertilized” microenvironment to facilitate the growth of cancer cells via the paracrine production of high-energy mitochondrial fuels (such L-lactate) that can then function as onco-catabolites.41,42,56-60 These recycled nutrients promote anabolic tumor growth by increasing mitochondrial metabolism (OXPHOS) in adjacent cancer cells.31-34,37-40,61-63
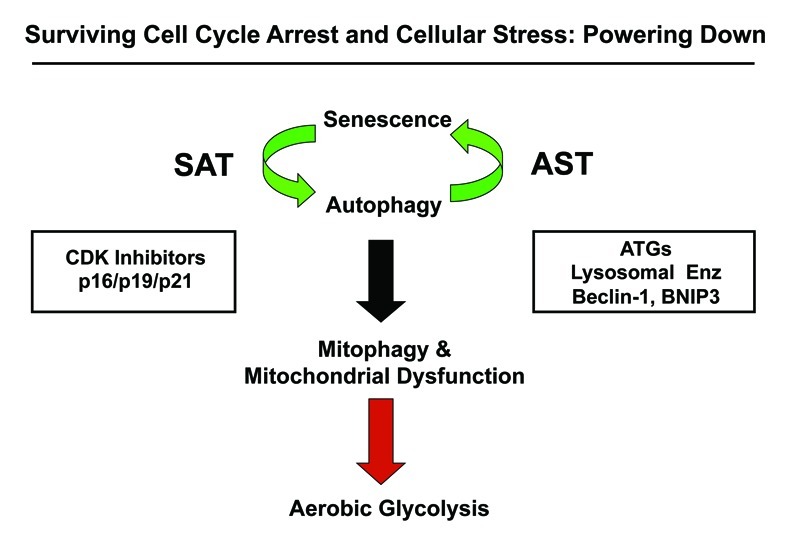
Figure 16. Understanding cell cycle arrest, senescence and autophagy: The sensescence-autophagy transition (SAT). Previously, we showed that recombinant expression of autophagy-associated genes (BNIP3, cathepsin B or ATG16L1) is sufficient to induce senescence, driving the autophagy-senescence transition (AST). Here, we show that recombinant expression of CDK inhibitors (p16/p19/p21) is sufficient to induce autophagy, driving the senescence-autophagy transition (SAT). Both SAT and AST result in mitochondrial dysfunction and a metabolic shift toward glycolysis, “powering down” cells during cell cycle arrest. Thus, cell cycle arrest, autophagy and senescence are all part of the same metabolic program that occurs in response to cellular stress.
To test this hypothesis, we created a variety of hTERT-immortalized fibroblast cell lines stably overexpressing established CDK-inhibitor proteins, such as p16(INK4A), p19(ARF), smARF and p21(WAF1/CIP1). Importantly, these cell lines overexpressing CDK inhibitors were clearly more sensitive to the induction of senescence and autophagy, especially under conditions of stress, such as hydrogen peroxide exposure and/or starvation. These CDK-inhibitor overexpressing cell lines also showed evidence of mitochondrial dysfunction, such as either increased L-lactate production or reductions in mitochondrial OXPHOS complexes as well as the upregulation of mitophagy markers, such as BNIP3. Thus, it appears that senescence and autophagy are two related faces of the same metabolic aging-associated program.
Interestingly, these CDK-inhibitor-overexpressing fibroblast cell lines also significantly promoted tumor growth, by up to 2-fold, without any significant increases in neo-angiogenesis. Thus, senescent-autophagic fibroblasts have tumor growth-promoting properties. Conversely, CDK-inhibitor protein expression in breast cancer cells significantly retarded tumor growth and was associated with the induction of markers of senescence, autophagy and mitophagy in these epithelial cancer cells.
Pharmacological induction of cell cycle arrest, by using a chemical CDK4/6 inhibitor (PD0332991) or use of other relevant cellular stressors (hydrogen peroxide and starvation), all had the same net effect, resulting in the induction of molecules associated both with senescence and autophagy. Thus, we have unequivocally established that these two biological processes are tightly linked.
Several previous studies have also highlighted the tumor-promoting properties of senescent fibroblasts.9,64-66 For example, Dr. Judy Campisi’s group has shown that senescent fibroblasts directly promote tumor growth via the increased secretion of cytokines and chemokines.9,64-66 This is known as SASP, the senescence-associated secretory phenotype. However, Dr. Campisi’s group has also shown that SASP is actually not required for the tumor-promoting activity of senescent fibroblasts,64 implying the existence of another related or independent paracrine mechanism. Thus, the senescence-autophagy transition (SAT) may represent another mechanism by which senescent fibroblasts effectively promote tumor growth.
These two seemingly unrelated mechanisms may also be intimately linked, as NFκB is both a master regulator of cytokine production, and its activation is required for the induction of autophagy. Thus, activation of NFκB in cancer-associated fibroblasts could account for both the SASP and SAT. In fact, previous studies have shown that tumor cells induce the activation of NFκB in cancer-associated fibroblasts via oxidative stress, resulting in cytokine production and a shift toward autophagy and glycolytic metabolism.24,30,34,35,67,68 This is essentially the same phenotype as the SASP and SAT, as oxidative stress is a strong inducer of both senescence and autophagy. In fact, increased protein synthesis and the secretion of cytokines are known to occur during autophagy as a protective response against excessive protein degradation and apoptosis.11,69
Our current studies on the tumor-promoting effects of p16(INK4A) fibroblasts may also explain recent biomarker data showing that an increase in stromal p16(INK4A) levels in human DCIS lesions (a precursor to full-blown breast cancer) dramatically increases the hazard ratio for recurrence by nearly 9-fold (p < 0.0001).70 Thus, senescent-autophagic fibroblasts may be critical for “fueling” tumor initiation, progression and cancer recurrence. As such, our findings have important clinical and translational implication for future biomarker discovery and treatment stratification in DCIS and breast cancer patients.
Materials and Methods
Materials
Commercially available antibodies used were as follows: β-actin (Sigma-Aldrich, #A5441); Cav-1 mAb 2297 (BD Transduction Laboratories, #610406); Beclin1 (Novus Biologicals, #NBP1-00085); BNIP3 (Abcam, #ab10433); BNIP3L (Abcam, #ab8399); Beta-galactosidase (GLB1; Abcam, #ab55176); p53 (Ab-6; Calbiochem, #OP43); Cathepsin B (FL-339) (Santa Cruz, #sc-13985); Lamp-1 (E-5) (Santa Cruz, #sc-17768); LC3B (Abcam, #ab48394); legumain (Santa Cruz, #sc-67163); p21 (H-164 or F-5) (Santa Cruz, #sc-756 or #sc-6246); p19 (DCS-240) (Santa Cruz, #sc-53639); p16 (H-156) (Santa Cruz, #sc-759); cyclin D1 (Thermo Scientific, #MS-210) and OXPHOS (MitoSciences, #MS601). Lenti-viral vectors containing full-length cDNAs encoding various CDK-inhibitor genes [p16(INK4A), p19(ARF) and p21(WAF1/CIP1)] were all obtained commercially from GeneCopoeia. PD0332991 (the specific CDK4/6-inhibitor; #S1116) was obtained from Selleck Chemicals, Inc.
Cell culture
Immortalized human fibroblasts (hTERT-BJ1 cells) and human breast cancer cells (MDA-MB-231-GFP) were cultured in DMEM implemented with 10% fetal bovine serum (FBS) and penicillin (100 units/mL)/streptomycin (100 μg/mL) (P/S). Hank’s balanced salt solution (HBSS), supplemented with 40 mM Hepes and 1% P/S, was used to initiate cell starvation. For co-culture studies, hTERT fibroblasts and MDA-MB-231-GFP cells were plated onto glass coverslips in 12-well plates in complete media at a cellular ratio of 5:1. The next day, the media was switched to DMEM supplemented with 10% NuSerum. Cells were maintained in co-culture for 3 d.
Lenti-virus production and cell line derivation
For lenti-virus production, 293Ta cells were transfected with different plasmids, in particular, EX-neg-Lv105 (the empty vector control), EX-H0288-Lv105 (encoding p19), EX-T2276-Lv105 (encoding smARF), EX-T1759-Lv105 (encoding p16) and EX-G0313-Lv105 (encoding p21). All plasmids also contained a puromycin-resistance marker. The resulting lenti-viruses were then used to infect hTERT fibroblasts or MDA-MB-231-GFP cells to obtain stable cell lines overexpressing the target gene of interest.
Immunoblot analysis
Immunoblotting was performed on samples obtained through OG buffer extraction. For this purpose, cells were scraped in OG lyses buffer (10 mM Tris, pH 7.5, 150 mM NaCl, 1% Triton X-100 and 60 mM n-octyl-glucoside), supplemented with a protease-inhibitor cocktail (Roche Diagnostics) and a phosphatase-inhibitor cocktail (Sigma). The samples were rotated for 40 min, then centrifuged for 10 min at 13,000 × g at 4°C. Finally, supernatants were collected. To determine the expression of cell cycle-related proteins, the cells were scraped into RIPA buffer (50 mM Tris pH 7.5, 150 mM NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 0.1% SDS, plus protease-inhibitor and phosphatase-inhibitor cocktails) and sonicated for 20 sec, then the samples were centrifuge 13,000 × g for 10 min at 4°C, and the supernatants were collected. The protein concentrations were determined using the BCA protein assay kit (Thermo scientific, #23225). Protein lysates were then separated by SDS-PAGE (using a 10-to-15% acrylamide gel) and transferred to nitrocellulose membranes. To detect expression of the protein of interest, specific primary antibodies and peroxidase-conjugated secondary antibodies were used. Bound antibodies were revealed using enhanced chemiluminescence (ECL) substrates (Thermo Scientific).
L-lactate assays
L-lactate levels were assessed according to the manufacturer’s instructions, using the EnzyChromTM L-Lactate assay kit (cat #ECLC-100, BioAssay Systems). For this purpose, cells were seeded in 12-well plates in complete media. The next day, the media was switched to DMEM containing 2% FBS. After 48 h, the media was collected, and the concentration of L-lactate was measured. Results were normalized for total cell number.
β-galactosidase staining assay
β-galactosidase activity was also detected by using a Senescence β-galactosidase staining kit (Cell Signaling, #9860). For this purpose, cells were seeded into 6-well plates in complete media. After 24 h, the media was changed to DMEM supplemented with 10% Nu-serum. After 48 h, the cells were fixed and incubated overnight at 37°C in a dry incubator without CO2 with the β-galactosidase Staining solution. Afterwards, cells were observed under the microscope.
Cellular hypertrophy assay
Approximately 100,000 cells were seeded per well in 12-well plates in DMEM containing 10% FBS. The next day, the media was switched to DMEM supplemented with 10% Nu-serum. After 24 h, cells were counted and protein lysates were prepared with OG buffer. Protein quantification was performed using the BCA protein assay kit (Thermo scientific, #23225). The values obtained were expressed as a ratio between the total amount of protein per well and the total number of cells per well (protein/cell number). An increased protein/cell number ratio would be indicative of cellular hypertrophy.
Animal studies
To evaluate the in vivo tumor-promoting effects of CDK-inhibitor proteins, flank injections were performed on athymic NCr nude mice (NCRNU; Taconic Farms; at 6–8 wk of age). Experiments were performed according to the National Institutes of Health (NIH) guidelines, with the consensus of the Institutional Animal Care and Use Committee (IACUC) of Thomas Jefferson University. MDA-MB-231-GFP cells (alone or plus hTERT-fibroblasts) were injected into the flanks of nude mice. More specifically, MDA-MB-231-GFP cancer cells (1 million cells) plus hTERT-BJ1 fibroblasts (300,000 cells) were re-suspended in 100 μl of sterile PBS, just prior to co-injection into the flank. After 4 wk post-injection, the tumors were collected and tumor growth was quantitatively measured.
Quantitation of tumor angiogenesis
Six-μm tumor frozen sections were fixed with 4% paraformaldehyde in PBS for 10 min at 4°C and washed 3x with PBS. A three-step biotin-streptavidin-horseradish peroxidase method was used for antibody detection. After fixation, the sections were blocked with 10% rabbit serum and incubated overnight at 4°C with rat monoclonal CD31 antibody (550274, BD Biosciences). The sections were then incubated with biotinylated rabbit anti-rat IgG (Vector Labs) and streptavidin-HRP (Dako). Immunoreactivity was revealed with 3, 3′ diaminobenzidine. For quantitation of vessels, CD31-positive vessels were counted in 6–8 fields within the central area of each tumor using a 20x objective lens and an ocular grid (0.25 mm2 per field). The total numbers of vessel per unit area was calculated and the data was represented graphically.
Acknowledgments
F.S. and her laboratory were supported by grants from the Breast Cancer Alliance (BCA) and the American Cancer Society (ACS). U.E.M. was supported by a Young Investigator Award from the Margaret Q. Landenberger Research Foundation. M.P.L. was supported by grants from the NIH/NCI (R01-CA-080250; R01-CA-098779; R01-CA-120876; R01-AR-055660) and the Susan G. Komen Breast Cancer Foundation. R.G.P. was supported by grants from the NIH/NCI (R01-CA-70896, R01-CA-75503, R01-CA-86072 and R01-CA-107382) and the Dr. Ralph and Marian C. Falk Medical Research Trust. The Kimmel Cancer Center was supported by the NIH/NCI Cancer Center Core grant P30-CA-56036 (to R.G.P.). Funds were also contributed by the Margaret Q. Landenberger Research Foundation (to M.P.L. and U.E.M.O.). This project is funded, in part, under a grant with the Pennsylvania Department of Health (to M.P.L. and F.S.). The department specifically disclaims responsibility for any analyses, interpretations or conclusions. This work was also supported, in part, by a centre grant in Manchester from Breakthrough Breast Cancer in the UK (to A.H.) and an Advanced ERC Grant from the European Research Council.
Footnotes
Previously published online: www.landesbioscience.com/journals/cc/article/21884
References
- 1.Blagosklonny MV. Calorie restriction: decelerating mTOR-driven aging from cells to organisms (including humans) Cell Cycle. 2010;9:683–8. doi: 10.4161/cc.9.4.10766. [DOI] [PubMed] [Google Scholar]
- 2.Blagosklonny MV. Revisiting the antagonistic pleiotropy theory of aging: TOR-driven program and quasi-program. Cell Cycle. 2010;9:3151–6. doi: 10.4161/cc.9.16.13120. [DOI] [PubMed] [Google Scholar]
- 3.Blagosklonny MV. Linking calorie restriction to longevity through sirtuins and autophagy: any role for TOR. Cell Death Dis. 2010;1:e12. doi: 10.1038/cddis.2009.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Blagosklonny MV. Prevention of cancer by inhibiting aging. Cancer Biol Ther. 2008;7:1520–4. doi: 10.4161/cbt.7.10.6663. [DOI] [PubMed] [Google Scholar]
- 5.Blagosklonny MV. Aging, stem cells, and mammalian target of rapamycin: a prospect of pharmacologic rejuvenation of aging stem cells. Rejuvenation Res. 2008;11:801–8. doi: 10.1089/rej.2008.0722. [DOI] [PubMed] [Google Scholar]
- 6.Blagosklonny MV. Inhibition of S6K by resveratrol: in search of the purpose. Aging (Albany NY) 2009;1:511–4. doi: 10.18632/aging.100059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blagosklonny MV. TOR-driven aging: speeding car without brakes. Cell Cycle. 2009;8:4055–9. doi: 10.4161/cc.8.24.10310. [DOI] [PubMed] [Google Scholar]
- 8.Blagosklonny MV. Rapamycin and quasi-programmed aging: four years later. Cell Cycle. 2010;9:1859–62. doi: 10.4161/cc.9.10.11872. [DOI] [PubMed] [Google Scholar]
- 9.Blagosklonny MV, Campisi J, Sinclair DA. Aging: past, present and future. Aging (Albany NY) 2009;1:1–5. doi: 10.18632/aging.100009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Narita M, Young AR, Narita M. Autophagy facilitates oncogene-induced senescence. Autophagy. 2009;5:1046–7. doi: 10.4161/auto.5.7.9444. [DOI] [PubMed] [Google Scholar]
- 11.Narita M, Young AR, Arakawa S, Samarajiwa SA, Nakashima T, Yoshida S, et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science. 2011;332:966–70. doi: 10.1126/science.1205407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Capparelli C, Guido C, Whitaker-Menezes D, Bonuccelli G, Balliet R, Pestell TG, et al. Autophagy and senescence in cancer-associated fibroblasts metabolically supports tumor growth and metastasis via glycolysis and ketone production. Cell Cycle. 2012;11:2285–302. doi: 10.4161/cc.20718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Capparelli C, Whitaker-Menezes D, Guido C, Balliet R, Pestell TG, Howell A, et al. CTGF drives autophagy, glycolysis and senescence in cancer-associated fibroblasts via HIF1 activation, metabolically promoting tumor growth. Cell Cycle. 2012;11:2272–84. doi: 10.4161/cc.20717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerland LM, Peyrol S, Lallemand C, Branche R, Magaud JP, Ffrench M. Association of increased autophagic inclusions labeled for beta-galactosidase with fibroblastic aging. Exp Gerontol. 2003;38:887–95. doi: 10.1016/S0531-5565(03)00132-3. [DOI] [PubMed] [Google Scholar]
- 15.Luo Y, Zou P, Zou J, Wang J, Zhou D, Liu L. Autophagy regulates ROS-induced cellular senescence via p21 in a p38 MAPKα dependent manner. Exp Gerontol. 2011;46:860–7. doi: 10.1016/j.exger.2011.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. Autophagy mediates the process of cellular senescence characterizing bile duct damages in primary biliary cirrhosis. Lab Invest. 2010;90:835–43. doi: 10.1038/labinvest.2010.56. [DOI] [PubMed] [Google Scholar]
- 17.Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. Autophagy may precede cellular senescence of bile ductular cells in ductular reaction in primary biliary cirrhosis. Dig Dis Sci. 2012;57:660–6. doi: 10.1007/s10620-011-1929-y. [DOI] [PubMed] [Google Scholar]
- 18.Sasaki M, Miyakoshi M, Sato Y, Nakanuma Y. A possible involvement of p62/sequestosome-1 in the process of biliary epithelial autophagy and senescence in primary biliary cirrhosis. Liver Int. 2012;32:487–99. doi: 10.1111/j.1478-3231.2011.02656.x. [DOI] [PubMed] [Google Scholar]
- 19.White E, Lowe SW. Eating to exit: autophagy-enabled senescence revealed. Genes Dev. 2009;23:784–7. doi: 10.1101/gad.1795309. [DOI] [PubMed] [Google Scholar]
- 20.Young AR, Narita M. Connecting autophagy to senescence in pathophysiology. Curr Opin Cell Biol. 2010;22:234–40. doi: 10.1016/j.ceb.2009.12.005. [DOI] [PubMed] [Google Scholar]
- 21.Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009;23:798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen J, Goligorsky MS. Premature senescence of endothelial cells: Methusaleh’s dilemma. Am J Physiol Heart Circ Physiol. 2006;290:H1729–39. doi: 10.1152/ajpheart.01103.2005. [DOI] [PubMed] [Google Scholar]
- 23.Chen J, Xavier S, Moskowitz-Kassai E, Chen R, Lu CY, Sanduski K, et al. Cathepsin cleavage of sirtuin 1 in endothelial progenitor cells mediates stress-induced premature senescence. Am J Pathol. 2012;180:973–83. doi: 10.1016/j.ajpath.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle. 2010;9:3256–76. doi: 10.4161/cc.9.16.12553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martinez-Outschoorn UE, Goldberg A, Lin Z, Ko YH, Flomenberg N, Wang C, et al. Anti-estrogen resistance in breast cancer is induced by the tumor microenvironment and can be overcome by inhibiting mitochondrial function in epithelial cancer cells. Cancer Biol Ther. 2011;12:924–38. doi: 10.4161/cbt.12.10.17780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Martinez-Outschoorn UE, Lin Z, Ko YH, Goldberg AF, Flomenberg N, Wang C, et al. Understanding the metabolic basis of drug resistance: therapeutic induction of the Warburg effect kills cancer cells. Cell Cycle. 2011;10:2521–8. doi: 10.4161/cc.10.15.16584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martinez-Outschoorn UE, Lin Z, Trimmer C, Flomenberg N, Wang C, Pavlides S, et al. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect: implications for PET imaging of human tumors. Cell Cycle. 2011;10:2504–20. doi: 10.4161/cc.10.15.16585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Tanowitz HB, Sotgia F, et al. Stromal-epithelial metabolic coupling in cancer: integrating autophagy and metabolism in the tumor microenvironment. Int J Biochem Cell Biol. 2011;43:1045–51. doi: 10.1016/j.biocel.2011.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Martinez-Outschoorn UE, Pavlides S, Sotgia F, Lisanti MP. Mitochondrial biogenesis drives tumor cell proliferation. Am J Pathol. 2011;178:1949–52. doi: 10.1016/j.ajpath.2011.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Daumer KM, Milliman JN, Chiavarina B, et al. Tumor cells induce the cancer associated fibroblast phenotype via caveolin-1 degradation: implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle. 2010;9:2423–33. doi: 10.4161/cc.9.12.12048. [DOI] [PubMed] [Google Scholar]
- 31.Martinez-Outschoorn UE, Pestell RG, Howell A, Tykocinski ML, Nagajyothi F, Machado FS, et al. Energy transfer in “parasitic” cancer metabolism: mitochondria are the powerhouse and Achilles’ heel of tumor cells. Cell Cycle. 2011;10:4208–16. doi: 10.4161/cc.10.24.18487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Martinez-Outschoorn UE, Prisco M, Ertel A, Tsirigos A, Lin Z, Pavlides S, et al. Ketones and lactate increase cancer cell “stemness,” driving recurrence, metastasis and poor clinical outcome in breast cancer: achieving personalized medicine via Metabolo-Genomics. Cell Cycle. 2011;10:1271–86. doi: 10.4161/cc.10.8.15330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martinez-Outschoorn UE, Sotgia F, Lisanti MP. Power surge: supporting cells “fuel” cancer cell mitochondria. Cell Metab. 2012;15:4–5. doi: 10.1016/j.cmet.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 34.Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: Role of hypoxia, HIF1 induction and NFκB activation in the tumor stromal microenvironment. Cell Cycle. 2010;9:3515–33. doi: 10.4161/cc.9.17.12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martinez-Outschoorn UE, Whitaker-Menezes D, Lin Z, Flomenberg N, Howell A, Pestell RG, et al. Cytokine production and inflammation drive autophagy in the tumor microenvironment: role of stromal caveolin-1 as a key regulator. Cell Cycle. 2011;10:1784–93. doi: 10.4161/cc.10.11.15674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinez-Outschoorn UE, Whitaker-Menezes D, Pavlides S, Chiavarina B, Bonuccelli G, Casey T, et al. The autophagic tumor stroma model of cancer or “battery-operated tumor growth”: A simple solution to the autophagy paradox. Cell Cycle. 2010;9:4297–306. doi: 10.4161/cc.9.21.13817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lisanti MP, Martinez-Outschoorn UE, Chiavarina B, Pavlides S, Whitaker-Menezes D, Tsirigos A, et al. Understanding the “lethal” drivers of tumor-stroma co-evolution: emerging role(s) for hypoxia, oxidative stress and autophagy/mitophagy in the tumor microenvironment. Cancer Biol Ther. 2010;10:537–42. doi: 10.4161/cbt.10.6.13370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lisanti MP, Martinez-Outschoorn UE, Lin Z, Pavlides S, Whitaker-Menezes D, Pestell RG, et al. Hydrogen peroxide fuels aging, inflammation, cancer metabolism and metastasis: the seed and soil also needs “fertilizer”. Cell Cycle. 2011;10:2440–9. doi: 10.4161/cc.10.15.16870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lisanti MP, Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Pestell RG, Howell A, et al. Accelerated aging in the tumor microenvironment: connecting aging, inflammation and cancer metabolism with personalized medicine. Cell Cycle. 2011;10:2059–63. doi: 10.4161/cc.10.13.16233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sotgia F, Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, et al. Mitochondrial metabolism in cancer metastasis: visualizing tumor cell mitochondria and the “reverse Warburg effect” in positive lymph node tissue. Cell Cycle. 2012;11:1445–54. doi: 10.4161/cc.19841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sotgia F, Martinez-Outschoorn UE, Howell A, Pestell RG, Pavlides S, Lisanti MP. Caveolin-1 and cancer metabolism in the tumor microenvironment: markers, models, and mechanisms. Annu Rev Pathol. 2012;7:423–67. doi: 10.1146/annurev-pathol-011811-120856. [DOI] [PubMed] [Google Scholar]
- 42.Sotgia F, Martinez-Outschoorn UE, Pavlides S, Howell A, Pestell RG, Lisanti MP. Understanding the Warburg effect and the prognostic value of stromal caveolin-1 as a marker of a lethal tumor microenvironment. Breast Cancer Res. 2011;13:213. doi: 10.1186/bcr2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Toogood PL, Harvey PJ, Repine JT, Sheehan DJ, VanderWel SN, Zhou H, et al. Discovery of a potent and selective inhibitor of cyclin-dependent kinase 4/6. J Med Chem. 2005;48:2388–406. doi: 10.1021/jm049354h. [DOI] [PubMed] [Google Scholar]
- 44.Fry DW, Harvey PJ, Keller PR, Elliott WL, Meade M, Trachet E, et al. Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther. 2004;3:1427–38. [PubMed] [Google Scholar]
- 45.Reef S, Kimchi A. Nucleolar p19ARF, unlike mitochondrial smARF, is incapable of inducing p53-independent autophagy. Autophagy. 2008;4:866–9. doi: 10.4161/auto.6691. [DOI] [PubMed] [Google Scholar]
- 46.Ueda Y, Koya T, Yoneda-Kato N, Kato JY. Small mitochondrial ARF (smARF) is located in both the nucleus and cytoplasm, induces cell death, and activates p53 in mouse fibroblasts. FEBS Lett. 2008;582:1459–64. doi: 10.1016/j.febslet.2008.03.032. [DOI] [PubMed] [Google Scholar]
- 47.Abida WM, Gu W. p53-Dependent and p53-independent activation of autophagy by ARF. Cancer Res. 2008;68:352–7. doi: 10.1158/0008-5472.CAN-07-2069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Reef S, Shifman O, Oren M, Kimchi A. The autophagic inducer smARF interacts with and is stabilized by the mitochondrial p32 protein. Oncogene. 2007;26:6677–83. doi: 10.1038/sj.onc.1210485. [DOI] [PubMed] [Google Scholar]
- 49.Reef S, Kimchi A. A smARF way to die: a novel short isoform of p19ARF is linked to autophagic cell death. Autophagy. 2006;2:328–30. doi: 10.4161/auto.3107. [DOI] [PubMed] [Google Scholar]
- 50.Codogno P. Autophagy and caspase-independent cell death: p19ARF enters the game. Dev Cell. 2006;10:688–9. doi: 10.1016/j.devcel.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 51.Reef S, Zalckvar E, Shifman O, Bialik S, Sabanay H, Oren M, et al. A short mitochondrial form of p19ARF induces autophagy and caspase-independent cell death. Mol Cell. 2006;22:463–75. doi: 10.1016/j.molcel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 52.Chinnadurai G, Vijayalingam S, Gibson SB. BNIP3 subfamily BH3-only proteins: mitochondrial stress sensors in normal and pathological functions. Oncogene. 2008;27(Suppl 1):S114–27. doi: 10.1038/onc.2009.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mazure NM, Pouysségur J. Atypical BH3-domains of BNIP3 and BNIP3L lead to autophagy in hypoxia. Autophagy. 2009;5:868–9. doi: 10.4161/auto.9042. [DOI] [PubMed] [Google Scholar]
- 54.Tan EY, Campo L, Han C, Turley H, Pezzella F, Gatter KC, et al. BNIP3 as a progression marker in primary human breast cancer; opposing functions in in situ versus invasive cancer. Clin Cancer Res. 2007;13:467–74. doi: 10.1158/1078-0432.CCR-06-1466. [DOI] [PubMed] [Google Scholar]
- 55.Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem. 2008;283:10892–903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 56.Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N, et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle. 2010;9:3485–505. doi: 10.4161/cc.9.17.12721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Loss of stromal caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the “reverse Warburg effect”: a transcriptional informatics analysis with validation. Cell Cycle. 2010;9:2201–19. doi: 10.4161/cc.9.11.11848. [DOI] [PubMed] [Google Scholar]
- 58.Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Transcriptional evidence for the “Reverse Warburg Effect” in human breast cancer tumor stroma and metastasis: similarities with oxidative stress, inflammation, Alzheimer’s disease, and “Neuron-Glia Metabolic Coupling”. Aging (Albany NY) 2010;2:185–99. doi: 10.18632/aging.100134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pavlides S, Vera I, Gandara R, Sneddon S, Pestell RG, Mercier I, et al. Warburg meets autophagy: cancer-associated fibroblasts accelerate tumor growth and metastasis via oxidative stress, mitophagy, and aerobic glycolysis. Antioxid Redox Signal. 2012;16:1264–84. doi: 10.1089/ars.2011.4243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle. 2009;8:3984–4001. doi: 10.4161/cc.8.23.10238. [DOI] [PubMed] [Google Scholar]
- 61.Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, et al. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle. 2011;10:4047–64. doi: 10.4161/cc.10.23.18151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, et al. Evidence for a stromal-epithelial “lactate shuttle” in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle. 2011;10:1772–83. doi: 10.4161/cc.10.11.15659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Witkiewicz AK, Whitaker-Menezes D, Dasgupta A, Philp NJ, Lin Z, Gandara R, et al. Using the “reverse Warburg effect” to identify high-risk breast cancer patients: stromal MCT4 predicts poor clinical outcome in triple-negative breast cancers. Cell Cycle. 2012;11:1108–17. doi: 10.4161/cc.11.6.19530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Coppé JP, Rodier F, Patil CK, Freund A, Desprez PY, Campisi J. Tumor suppressor and aging biomarker p16(INK4a) induces cellular senescence without the associated inflammatory secretory phenotype. J Biol Chem. 2011;286:36396–403. doi: 10.1074/jbc.M111.257071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Krtolica A, Campisi J. Cancer and aging: a model for the cancer promoting effects of the aging stroma. Int J Biochem Cell Biol. 2002;34:1401–14. doi: 10.1016/S1357-2725(02)00053-5. [DOI] [PubMed] [Google Scholar]
- 66.Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci USA. 2001;98:12072–7. doi: 10.1073/pnas.211053698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chiavarina B, Whitaker-Menezes D, Martinez-Outschoorn UE, Witkiewicz AK, Birbe RC, Howell A, et al. Pyruvate kinase expression (PKM1 and PKM2) in cancer-associated fibroblasts drives stromal nutrient production and tumor growth. Cancer Biol Ther. 2011;12:1101–13. doi: 10.4161/cbt.12.12.18703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chiavarina B, Whitaker-Menezes D, Migneco G, Martinez-Outschoorn UE, Pavlides S, Howell A, et al. HIF1-alpha functions as a tumor promoter in cancer associated fibroblasts, and as a tumor suppressor in breast cancer cells: Autophagy drives compartment-specific oncogenesis. Cell Cycle. 2010;9:3534–51. doi: 10.4161/cc.9.17.12908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zoncu R, Sabatini DM. Cell biology. The TASCC of secretion. Science. 2011;332:923–5. doi: 10.1126/science.1207552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Witkiewicz AK, Rivadeneira DB, Ertel A, Kline J, Hyslop T, Schwartz GF, et al. Association of RB/p16-pathway perturbations with DCIS recurrence: dependence on tumor versus tissue microenvironment. Am J Pathol. 2011;179:1171–8. doi: 10.1016/j.ajpath.2011.05.043. [DOI] [PMC free article] [PubMed] [Google Scholar]