

SUMMARY
Aldosterone has been shown to mediate p21-dependent cellular senescence in rat kidney proximal tubules in vivo and in cultured human proximal tubular cells. p21-induced senescent cells expressed higher levels of apoptotic cytokines, such as tumour necrosis factor-α (TNF-α), compared to non-senescent cells. The aim of this study was to investigate the hypothesis that aldosterone increases proximal tubular apoptosis by increasing the secretion of apoptosis-inducing factors through a p21-dependent mechanism.
Human proximal tubular cells were incubated with aldosterone (10 nmol/L) and cell senescence was detected by senescence-associated β-galactosidase staining and expression of p21. Apoptosis was analysed by terminal nick-end labelling and annexin/propidium iodide staining. p21 localisation was observed by immunofluorescence.
Aldosterone for 3 or 5 days increased senescence-associated β-galactosidase staining, expression of p21 and TNF-α mRNAs, and secretion of TNF-α into the culture medium. These changes were abolished by gene silencing of p21. Aldosterone failed to increase apoptotic cells at day 3, but did increase them at day 5. Neutralising antibody against TNF-α prevented the aldosterone-induced apoptotic changes. Aldosterone did not affect the localisation of p21.
These findings indicate that aldosterone increases TNF-α synthesis and secretion in proximal tubular cells via p21/senescence-dependent cell phenotypic changes, and that secreted TNF-α plays an important role as a paracrine factor in mediating cell apoptosis, indicating a possible involvement in aldosterone-induced renal damage.
Keywords: p21, apoptosis, aldosterone, proximal tubular cells
INTRODUCTION
Cells in the human body are known to undergo several fates in response to exogenous and endogenous stresses. Although the triggers determining cell fate are still under investigation, senescence, represented by termination of mitosis, is a recognised cellular response to stresses such as tumourigenesis,1, 2 irradiation,3, 4 and aging.5, 6 Considerable recent evidence has demonstrated the physiological importance of the accumulation of senescent cells on organ function. Cyclin-dependent kinase (CDK) inhibitors (CDKI), such as p16 and p21, induce cell senescence by binding to CDK in the nucleus,7 Baker et al.8 revealed that targeting apoptosis induction in p16-positive cells attenuated age-associated pathological changes in adipose tissue, skeletal muscle, and the eye, indicating that the accumulation of CDKI-positive senescent cells in organs accelerates the aging phenotype. In addition, the transfusion of plasma from aged mice (supposed to have more senescent cells than young mice) into young mice impaired their cognitive function and neurogenesis,9 indicating that soluble factors in the circulation were also responsible for inducing an aging phenotype. Changes in secreted molecules from senescent cells (senescence-associate secreting phenotype, SASP) are considered to be a possible mechanism whereby cell senescence can affect the function of other organs through the release of paracrine and/or hormonal factors.10
Renal proximal tubular cells (PTCs) play several important physiological roles, such as controlling electrolyte balance,11, 12 glucose reabsorption,13, 14 uric acid transport,15, 16 and filtered protein reuptake.17, 18 Damage to PTCs, either acutely or chronically, can thus affect renal function. We recently demonstrated that aldosterone increased cellular senescence in vivo and in vitro, especially in PTCs, through an MR/oxidative stress/Sirt-1/p53/p21-dependent pathway.19 The senescent cells expressed higher levels of tumour necrosis factor-α (TNF-α) mRNA than non-senescent cells; this expression was shown to be totally dependent on p21 by its abolition by small interfering RNA for p21. These results suggest that aldosterone-induced p21 up-regulation may induce not only cell senescence, but also SASP as a secondary response. However, this has not yet been investigated in detail. We therefore examined the effect of the aldosterone-induced increase in p21 on SASP and apoptosis, and the involvement of newly-synthesised cytokines such as TNF-α, in human PTCs (HPTCs).
MATERIALS AND METHODS
Cell culture and experimental protocol
We used HPTCs immortalised by temperature-sensitive SV40 large T antigen-containing adenovirus. We previously demonstrated that HPTCs maintained proximal tubular characteristics, including higher sodium-glucose transporter 2 expression and alkaline phosphatase activity compared to other nephron cells, such as mesangial cells. They also approached the Hayflick limit (a limitation of cell mitosis that is not observed in immortalised cells at 37°C), indicating that they were not immortalised at this temperature.19 HPTCs were maintained in growth medium comprising Click’s medium:RPMI-1640 (1:1; Quality Biological, Gaithersburg, MD, USA) containing 1% insulin-transferrin-selenium, 40 ng/mL dexamethasone, 10 ng/mL epidermal growth factor, 2% foetal bovine serum (FBS) and 2% penicillin in a humidified atmosphere of 5% CO2 at 33°C. After reaching 30% confluence in growth medium, the cells were transferred to 0.2% FBS Click’s medium:RPMI-1640 containing 1% insulin-transferrin-selenium and 40 ng/mL dexamethasone for 24 h at 37°C, then incubated with vehicle or aldosterone (10 scrambled siRNA. In another group of experiments, 10 μg/mL TNF-α neutralising antibody (Thermo Scientific, Waltham, MA, USA) was added to cells treated with aldosterone, with normal rabbit IgG (MBL, Nagoya, Japan) used as a control. Cells were analysed 3 or 5 days after incubation with aldosterone. The experimental duration and aldosterone dose were determined based on our previous results.19
RNA interference
siRNA targeting p21 (Invitrogen, San Diego, CA, USA) or its scrambled control were transfected using Lipofectamine 2000 (Invitrogen). Subconfluent (40–50%) HPTCs in growth medium without antibiotics were transfected in Click’s medium:RPMI-1640-free containing 5 μL Lipofectamine 2000 with 100 pmol siRNA per well (6-well plate) for 24 h, and the medium was then replaced with growth medium.
Senescence-associated β-galactosidase (SAβ-Gal) staining
SAβ-Gal activity was detected using a senescence detection kit (BioVision, Mountain View, CA, USA) according to the manufacturer’s protocol. Briefly, cells were fixed for 15 min at room temperature in fixative solution, washed twice with phosphate-buffered saline (PBS), and incubated for 12 h at 37°C in freshly prepared X-Gal staining solution mix. Thereafter, the staining solution was removed and the samples were overlaid with 70% glycerol and the development of blue colour was assessed at ×100 magnification.
Real-time reverse transcription-polymerase chain reaction (RT-PCR)
mRNA was extracted from cells using the phenol-chloroform extraction method. The levels of β-actin, p21 and TNF-α mRNAs in HPTCs were analysed by RT-PCR using an ABI Prism 7000 with Power SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA, USA). The primers have been described previously.19 All data are shown as relative differences between vehicle- and aldosterone-treated cells after normalisation to β-actin expression.
Western blotting
Expression of p21 protein was measured by western blotting. Protein (50 μg) was separated by 15% sodium dodecyl sulphate-polyacrylamide gel electrophoresis, transferred to a nitrocellulose membrane, and immunoblotted with antibodies specific for p21 (1:1000; Millipore, Temecula, CA, USA). Equal loading was confirmed by reprobing the membranes with an antibody against β-actin (1:10,000 dilution; Sigma, St Louis, MO, USA). IRDye®-labelled anti-mouse IgG antibody (1:15,000, Li-Cor, Lincoln, NE, USA) was used for the detection of p21 and β-actin. Detection was performed using an Odyssey System (Li-Cor). Data were normalised to β-actin protein expression levels.
TNF-α secretion
TNF-α secretion was evaluated by measuring TNF-α levels in the medium of aldosterone-treated cells using an enzyme-linked immunosorbent assay kit (Quantikine®, R&D systems, Minneapolis, MN, USA) according to the manufacturer’s protocol.
Detection of apoptosis
Apoptosis was detected by terminal deoxynucleotidyl transferase dUTP nick-end labelling (TUNEL) and annexin V/propidium iodide (PI) staining using commercial kits (Apop-Tag Red in situ Apoptosis Detection Kit; Chemicon International, Temecula, CA, USA, and Annexin V-FITC Apoptosis Detection Kit; BioVision, respectively). The numbers of apoptotic cells positive for TUNEL or annexin V/PI double staining were counted in at least five randomly selected regions. Hoechst 33342 (Invitrogen) was used to identify nuclei in TUNEL staining.
Immunofluorescence staining for p21 localisation
HPTCs were treated with vehicle or aldosterone for 3 days, washed with PBS, then fixed with 4% formaldehyde for 10 min. After washing with PBS, the cells were blocked with 10% normal goat serum (Nitirei Bioscience, Tokyo, Japan) for 15 min, and incubated at 37°C for 1 h with mouse anti-human p21 (F-5) antibody (1:100, Santa Cruz min at 37°C with Alexa Fluor 594 goat anti-mouse antibody (1:1000, Molecular Probes, CA, USA). After washing, the nuclei were counterstained with Hoechst 33342 (Invitrogen). The cells were washed, mounted, and examined under a fluorescence microscope.
Statistical analysis
All values are expressed as mean ± S.E.M. Relevant data were processed using InStat (Graph-PAD Software for Science, San Diego, CA). Statistical analysis was performed using one-way analysis of variance followed by Dunnett’s multiple comparison tests. Differences were considered significant at two-sided P < 0.05.
RESULTS
SAβ-Gal staining and p21 expression in aldosterone-treated HPTCs
Aldosterone treatment for 3 days clearly increased SAβ-Gal staining and p21 mRNA and protein expression levels. These changes were markedly suppressed by knockdown of p21 with siRNA (Fig. 1A–C, G, H). Aldosterone treatment for 5 days also increased the number of SAβ-Gal-positive cells compared to vehicle treatment, with no clear difference in the numbers of SAβ-Gal-positive cells between 3-day and 5-day aldosterone treatments (Fig. 1A–F). Aldosterone treatment for 5 days induced increases in p21 mRNA and protein other hand, cells treated with vehicle for 5 days showed higher p21 protein levels than cells treated with vehicle for 3 days, with no apparent change in SAβ-Gal staining (Fig. 1D, H), possibly as a result of contact inhibition induced by spontaneous cell proliferation.20
Fig. 1.
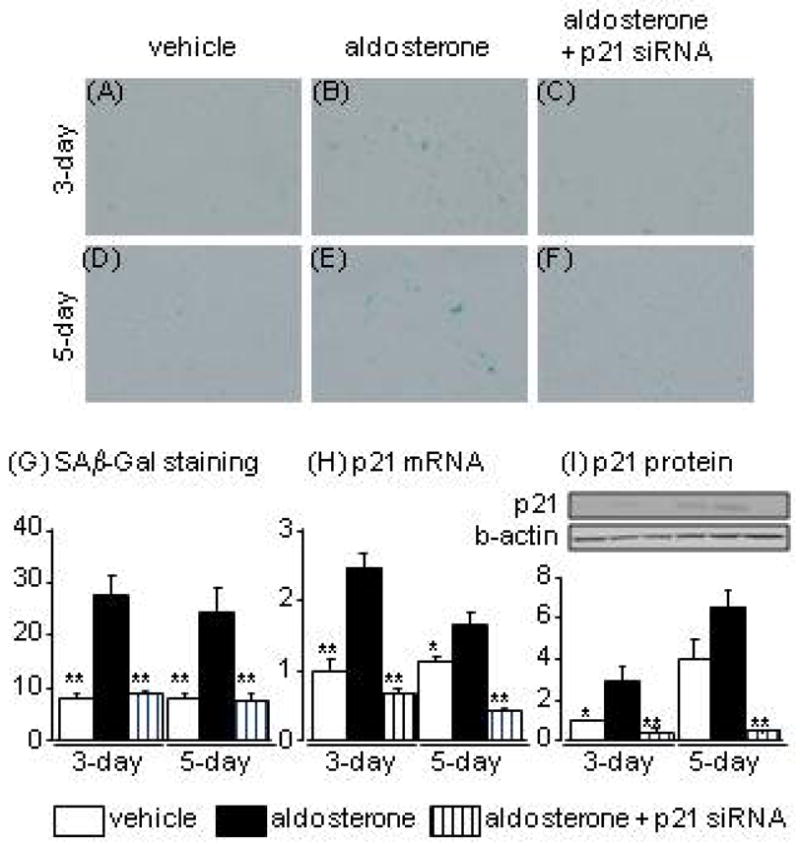
Effects of aldosterone and p21 siRNA on senescence-associated β-galactosidase staining (A-G), and levels of p21 mRNA (H) and protein (I) in human proximal tubular cells at 3 and 5 days after aldosterone treatment. Data are expressed as means ± S.E.M.; n = 6 per group. *P < 0.05, **P < 0.01, compared to the same-day aldosterone-treatment group.
Apoptosis in aldosterone-treated HPTCs
Apoptosis was analysed by TUNEL staining and annexin V/PI-double staining, which are the established methods for the detection of apoptosis. In contrast to SAβ-Gal staining, aldosterone treatment for 3 days resulted in no significant changes in either annexin V/PI or TUNEL staining (Fig. 2A, B, F, G, K, L). Meanwhile, 5-day aldosterone treatment markedly increased numbers of both annexin V/PI- and TUNEL-positive cells (Fig. 2C, D, H, I, K, L). This increase in apoptotic cells at 5 days after aldosterone treatment was significantly suppressed in p21-knockdown HPTCs (Fig. 2E, J, K, L).
Fig. 2.

Effects of aldosterone and p21 siRNA on apoptosis evaluated by annexin-V (green)/propidium iodide (PI; red) (A–F) and terminal deoxynucleotidyl transferase dUTP nick-end labelling (TUNEL; red) staining (G–L) in human proximal tubular cells at 3 and 5 days after aldosterone treatment (arrows indicate the fluorophore-positive cells). Hoechst 33342 (blue) was used to identify nuclei in each staining (left pictures). Data are expressed as means ± S.E.M.; n = 3 or 4 per group. **P < 0.01, compared to the same-day aldosterone-treatment group.
TNF-α expression and secretion in aldosterone-treated HPTCs
Cell senescence occurred earlier than apoptosis and p21 contributed to aldosterone-induced cell apoptosis. We therefore examined the possible target of SASP. We focused on the involvement of TNF-α in aldosterone-induced apoptosis, because we previously reported that aldosterone increased TNF-α mRNA expression via a p21-dependent pathway,19 and because TNF-α is a well known paracrine factor.21 Aldosterone significantly increased TNF-α mRNA in cells at day-3, and aldosterone treatment for 5 days tended to increase TNF-α mRNA. Aldosterone treatment increased TNF-α levels in the culture medium, and levels were significantly higher at day 5 than at day 3. These changes in mRNA and secreted protein levels were suppressed by p21 siRNA (Fig. 3A, B).
Fig. 3.

Effects of aldosterone and p21 siRNA on tumour necrosis factor (TNF)-α mRNA levels in human proximal tubular cells (A) and protein levels in the culture medium (B) at 3 and 5 days after aldosterone treatment. Data are expressed as means ± S.E.M.; n = 5 or 6 per group. *P < 0.05, **P < 0.01, compared to the same-day aldosterone-treatment group.
Effects of TNF-α neutralising antibody on aldosterone-induced apoptosis
We examined the involvement of TNF-α in the culture medium on aldosterone-induced apoptosis using TNF-α neutralising antibody, to clarify the contribution of p21/senescent cell-induced TNF-α secretion to aldosterone-induced apoptosis. Co-administration of TNF-α neutralising antibody obviously blocked aldosterone-induced increases in annexin-V/PI- and TUNEL-positive cell numbers at day 5 (Fig. 4A–H).
Fig. 4.

Effects of tumour necrosis factor (TNF)-α neutralising antibody on 5-day aldosterone-induced apoptosis evaluated by annexin-V (green)/propidium iodide (PI; red) (A–D) and terminal deoxynucleotidyl transferase dUTP nick-end labelling (TUNEL; red) staining (E–H) in human proximal tubular cells at 3 and 5 days after aldosterone treatment (arrows indicate the fluorophore-positive cells). Hoechst 33342 (blue) was used to identify nuclei in each staining (left pictures). Data are expressed as means ± S.E.M.; n = 3 or 4 per group. *P < 0.05, **P < 0.01, compared to the same-day aldosterone-treatment group.
Localisation of p21 in HPTCs
Numbers of p21-immunostained HPTCs were increased by aldosterone-treatment (Fig. 5A, B). p21 immunostaining was mainly localised in the nuclei, though a few vehicle-treated and aldosterone-treated cells showed detectable cytosolic p21 immunostaining (Fig. 5C). Cells expressing cytosolic p21 showed saturated levels of nuclear expression and were located in areas of higher HPTC density.
Fig. 5.
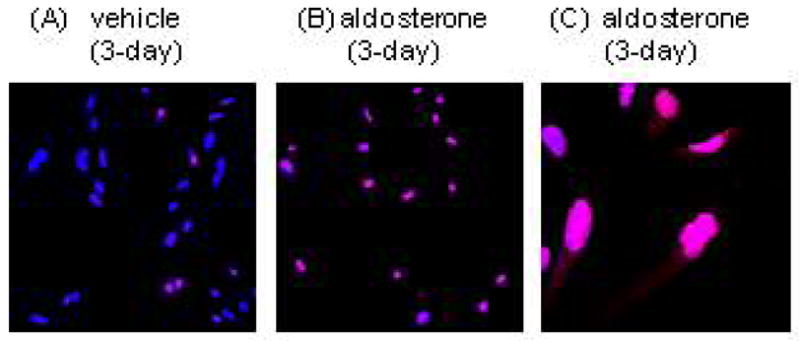
Localisation of p21 in vehicle- (A) and aldosterone (B)-treated human proximal tubular cells. Aldosterone treatment increased p21 expression in nuclei. Cytosolic staining of p21 was rarely found (C). Red; p21, blue; Hoechst 33342.
DISCUSSION
Aldosterone is an important hormone in regulating electrolyte balance. The role of aldosterone in tissues has become an important issue since clinical studies (RALES and EPHESUS) demonstrated beneficial effects of aldosterone blockers on outcomes of heart failure patient compared with placebo controls.22, 23 Chronic aldosterone infusion induced glomerulosclerosis and tubulointerstitial fibrosis in rats,24, 25 but the precise mechanisms involved in aldosterone-induced tissue injury are still under investigation. The current study demonstrated that aldosterone enhanced TNF-α expression and secretion in HPTCs, and increased SAβ-Gal-positive HPTCs. Importantly, the increase in TNF-α was abolished by knockdown of p21, an important molecule involved in cellular senescence, indicating that the aldosterone-induced increase in TNF-α might be mediated through p21-dependent cellular senescence. Experiments using TNF-α neutralising antibody revealed that TNF-α secretion was involved in apoptosis through autocrine/paracrine action of TNF-α. Overall, the results of the present study thus demonstrate that aldosterone elicits cell senescence and SASP in PTCs via a p21-dependent pathway. SASP in PTCs may accelerate apoptotic changes in surrounding cells and induce nephron damage. This hypothesis may be supported by an earlier study showing that mice lacking the p21 gene developed less interstitial fibrosis, with increased cell proliferation, in a renal ablation model, compared to normal mice.26
Cells exposed to stress are maintained in cell cycle arrest to allow repair of DNA damage.27 Cells exposed to excessive stress that cannot be managed by the endogenous repair system are driven into senescence, dedifferentiate into other phenotypes, and ultimately undergo apoptosis.27, 28 Patni et al.29 revealed that aldosterone (at 100 times greater concentration than the current study) induced tubular cell apoptosis by activating the mitochondrial pathway and generating reactive oxygen species in human proximal tubular HK2 cells. Our findings that the aldosterone-induced increase in p21 changes cell phenotype and elicits TNF-α-induced apoptosis via an autocrine/paracrine pathway support this earlier study.
The present results may also indicate that senescent PTCs may induce apoptosis/inflammation by secreting cytokines, such as TNF-α, and may try to remove themselves from tubules to protect or maintain proximal tubule function as a self-defence system. However, secreted molecules such as TNF-α could stimulate other neighbouring cells through paracrine action and induce apoptosis and inflammation, resulting in the decline of tubular (nephron) function. It would be interesting to isolate nephrons containing senescent cells and measure tubular function, such as electrolyte-transport ability; however, to the best of our knowledge, there is currently no reliable means of identifying such nephrons in vivo. Recent advances in technology, such as drug-induced removal of CDKI-positive cells developed by Baker et al.8, may be suitable for examining this issue.
Recent reports have indicated that soluble factors in the plasma of aged animals (i.e. CCL11) impaired neurogenesis and cognitive function in young animals, indicating the involvement of systemic circulating factors in aging-associated changes in the body.9 However, how these soluble molecules are secreted into the systemic circulation, and whether molecules secreted by senescent PTCs could spill over into the circulation, remain unclear. This situation may not apply in aldosterone-induced SASP of PTCs, because patients with primary aldosteronism have low or similar plasma TNF-α levels to patients with essential hypertension.30, 31
Clinical studies have shown poor graft survival of transplanted kidneys from elder donors,32–34 and the increase in p21 expression in the kidney of allograft nephropathy patients.35 Such evidence may reflect the hypothesis that the p21-up-regulated/senescent kidney is more susceptible to various diseases, particularly diseases affecting tubular function, such as acute kidney injury and proteinuria during the development of chronic kidney disease because of reduced tubular repair activity by senescence/SASP/apoptosis. Thus, considering our present findings, patients with excessive aldosterone/MR levels in their kidney may show unfavorable outcomes, although this has not yet been evaluated in clinical studies.
Some studies have reported the involvement of cytosolic p21 in apoptosis.36–38 p21 in the cytoplasm forms a complex with pro-apoptotic factors, such as apoptosis signal-regulating kinase 136 and proliferating cell nuclear antigen bound to procaspases,37 and attenuates the activity of these molecules. We therefore expected that aldosterone might accelerate p21 translocation from the cytosol to the nucleus, and that the released pro-apoptotic factors might induce apoptosis. However, the present study detected much less immunoreactivity for p21 in the cytosol compared to the nuclei in both vehicle- and aldosterone-treated HPTCs. This suggests that an involvement of p21 localisation in aldosterone-induced SASP is unlikely.
In conclusion, aldosterone-induced increases in p21 and cell senescence were accompanied by increased expression and secretion of TNF-α in HPTCs. This p21-depedent SASP plays an important role in aldosterone-induced apoptosis in HPTCs. An important issue that has not been addressed in our study is whether patients with hyperaldosteronism exhibit senescent changes in the kidney and whether those changes can be treated or prevented by pharmacotherapy with MR blockers.
Acknowledgments
This work was supported in part by the Fund for Kagawa University Young Scientists 2010 and the Fund for Priority Research Areas from Kagawa University 2011 to Daisuke Nakano.
Footnotes
None of the authors have any conflicts of interest to declare.
References
- 1.Blagoev KB. Organ aging and susceptibility to cancer may be related to the geometry of the stem cell niche. Proc Natl Acad Sci USA. 2011;108:19216–21. doi: 10.1073/pnas.1106105108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Burgess DJ. Senescence: Tumorigenesis under surveillance. Nat Rev Cancer. 2011;12:6–7. doi: 10.1038/nrc3188. [DOI] [PubMed] [Google Scholar]
- 3.Wang M, Morsbach F, Sander D, et al. EGF receptor inhibition radiosensitizes NSCLC cells by inducing senescence in cells sustaining DNA double-strand breaks. Cancer Res. 2011;71:6261–9. doi: 10.1158/0008-5472.CAN-11-0213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hovest MG, Krieg T, Herrmann G. Differential roles for Chk1 and FANCD2 in ATR-mediated signalling for psoralen photoactivation-induced senescence. Exp Dermatol. 2011;20:883–9. doi: 10.1111/j.1600-0625.2011.01365.x. [DOI] [PubMed] [Google Scholar]
- 5.Kume S, Uzu T, Horiike K, et al. Calorie restriction enhances cell adaptation to hypoxia through Sirt1-dependent mitochondrial autophagy in mouse aged kidney. J Clin Invest. 2010;120:1043–55. doi: 10.1172/JCI41376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ikeda Y, Inagi R, Miyata T, et al. Glyoxalase I retards renal senescence. Am J Pathol. 2011;179:2810–21. doi: 10.1016/j.ajpath.2011.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Erol A. Deciphering the intricate regulatory mechanisms for the cellular choice between cell repair, apoptosis or senescence in response to damaging signals. Cell Signal. 2011;23:1076–81. doi: 10.1016/j.cellsig.2010.11.023. [DOI] [PubMed] [Google Scholar]
- 8.Baker DJ, Wijshake T, Tchkonia T, et al. Clearance of p16(Ink4a)-positive senescent cells delays ageing-associated disorders. Nature. 2011 doi: 10.1038/nature10600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Villeda SA, Luo J, Mosher KI, et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature. 2011;477:90–4. doi: 10.1038/nature10357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fumagalli M, d’Adda di Fagagna F. SASPense and DDRama in cancer and ageing. Nat Cell Biol. 2009;11:921–3. doi: 10.1038/ncb0809-921. [DOI] [PubMed] [Google Scholar]
- 11.Panchapakesan U, Pollock C, Saad S. Renal epidermal growth factor receptor: its role in sodium and water homeostasis in diabetic nephropathy. Clin Exp Pharmacol Physiol. 2011;38:84–8. doi: 10.1111/j.1440-1681.2010.05472.x. [DOI] [PubMed] [Google Scholar]
- 12.Dantzler WH. Regulation of renal proximal and distal tubule transport: sodium, chloride and organic anions. Comp Biochem Physiol A Mol Integr Physiol. 2003;136:453–78. doi: 10.1016/s1095-6433(03)00135-1. [DOI] [PubMed] [Google Scholar]
- 13.Vallon V, Platt KA, Cunard R, et al. SGLT2 mediates glucose reabsorption in the early proximal tubule. J Am Soc Nephrol. 2011;22:104–12. doi: 10.1681/ASN.2010030246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mather A, Pollock C. Glucose handling by the kidney. Kidney Int Suppl. 2011:S1–6. doi: 10.1038/ki.2010.509. [DOI] [PubMed] [Google Scholar]
- 15.Miura D, Anzai N, Jutabha P, et al. Human urate transporter 1 (hURAT1) mediates the transport of orotate. J Physiol Sci. 2011;61:253–7. doi: 10.1007/s12576-011-0136-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cunningham R, Brazie M, Kanumuru S, et al. Sodium-hydrogen exchanger regulatory factor-1 interacts with mouse urate transporter 1 to regulate renal proximal tubule uric acid transport. J Am Soc Nephrol. 2007;18:1419–25. doi: 10.1681/ASN.2006090980. [DOI] [PubMed] [Google Scholar]
- 17.Amsellem S, Gburek J, Hamard G, et al. Cubilin is essential for albumin reabsorption in the renal proximal tubule. J Am Soc Nephrol. 2010;21:1859–67. doi: 10.1681/ASN.2010050492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Desmond MJ, Lee D, Fraser SA, et al. Tubular proteinuria in mice and humans lacking the intrinsic lysosomal protein SCARB2/Limp-2. Am J Physiol Renal Physiol. 2011;300:F1437–47. doi: 10.1152/ajprenal.00015.2011. [DOI] [PubMed] [Google Scholar]
- 19.Fan YY, Kohno M, Hitomi H, et al. Aldosterone/Mineralocorticoid receptor stimulation induces cellular senescence in the kidney. Endocrinology. 2011;152:680–8. doi: 10.1210/en.2010-0829. [DOI] [PubMed] [Google Scholar]
- 20.Puliafito A, Hufnagel L, Neveu P, et al. Collective and single cell behavior in epithelial contact inhibition. Proc Natl Acad Sci USA. 2012;109:739–44. doi: 10.1073/pnas.1007809109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Crescenzi E, Pacifico F, Lavorgna A, et al. NF-kappaB-dependent cytokine secretion controls Fas expression on chemotherapy-induced premature senescent tumor cells. Oncogene. 2011;30:2707–17. doi: 10.1038/onc.2011.1. [DOI] [PubMed] [Google Scholar]
- 22.Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med. 1999;341:709–17. doi: 10.1056/NEJM199909023411001. [DOI] [PubMed] [Google Scholar]
- 23.Pitt B, Remme W, Zannad F, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348:1309–21. doi: 10.1056/NEJMoa030207. [DOI] [PubMed] [Google Scholar]
- 24.Nishiyama A, Yao L, Nagai Y, et al. Possible contributions of reactive oxygen species and mitogen-activated protein kinase to renal injury in aldosterone/salt-induced hypertensive rats. Hypertension. 2004;43:841–8. doi: 10.1161/01.HYP.0000118519.66430.22. [DOI] [PubMed] [Google Scholar]
- 25.Fan YY, Baba R, Nagai Y, et al. Augmentation of intrarenal angiotensin II levels in uninephrectomized aldosterone/salt-treated hypertensive rats; renoprotective effects of an ultrahigh dose of olmesartan. Hypertens Res. 2006;29:169–78. doi: 10.1291/hypres.29.169. [DOI] [PubMed] [Google Scholar]
- 26.Megyesi J, Price PM, Tamayo E, Safirstein RL. The lack of a functional p21(WAF1/CIP1) gene ameliorates progression to chronic renal failure. Proc Natl Acad Sci USA. 1999;96:10830–5. doi: 10.1073/pnas.96.19.10830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hayflick L. Living forever and dying in the attempt. Exp Gerontol. 2003;38:1231–41. doi: 10.1016/j.exger.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 28.Minamino T, Komuro I. Vascular aging: insights from studies on cellular senescence, stem cell aging, and progeroid syndromes. Nat Clin Pract Cardiovasc Med. 2008;5:637–48. doi: 10.1038/ncpcardio1324. [DOI] [PubMed] [Google Scholar]
- 29.Patni H, Mathew JT, Luan L, Franki N, Chander PN, Singhal PC. Aldosterone promotes proximal tubular cell apoptosis: role of oxidative stress. Am J Physiol Renal Physiol. 2007;293:F1065–71. doi: 10.1152/ajprenal.00147.2007. [DOI] [PubMed] [Google Scholar]
- 30.Irita J, Okura T, Manabe S, et al. Plasma osteopontin levels are higher in patients with primary aldosteronism than in patients with essential hypertension. Am J Hypertens. 2006;19:293–7. doi: 10.1016/j.amjhyper.2005.08.019. [DOI] [PubMed] [Google Scholar]
- 31.Carvajal CA, Herrada AA, Castillo CR, et al. Primary aldosteronism can alter peripheral levels of transforming growth factor beta and tumor necrosis factor alpha. J Endocrinol Invest. 2009;32:759–65. doi: 10.1007/BF03346533. [DOI] [PubMed] [Google Scholar]
- 32.Rigotti P, Ekser B, Furian L, et al. Outcome of renal transplantation from very old donors. N Engl J Med. 2009;360:1464–5. doi: 10.1056/NEJMc0900169. [DOI] [PubMed] [Google Scholar]
- 33.Melk A, Schmidt BM, Braun H, et al. Effects of donor age and cell senescence on kidney allograft survival. Am J Transplant. 2009;9:114–23. doi: 10.1111/j.1600-6143.2008.02500.x. [DOI] [PubMed] [Google Scholar]
- 34.Braun H, Schmidt BM, Raiss M, et al. Cellular Senescence Limits Regenerative Capacity and Allograft Survival. J Am Soc Nephrol. 2012 doi: 10.1681/ASN.2011100967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chkhotua AB, Altimari A, Gabusi E, et al. Increased expression of P21((WAF1/CIP1)) CDKI gene in chronic allograft nephropathy correlating with the number of acute rejection episodes. Transplant Proc. 2003;35:655–8. doi: 10.1016/s0041-1345(03)00025-3. [DOI] [PubMed] [Google Scholar]
- 36.Asada M, Yamada T, Ichijo H, et al. Apoptosis inhibitory activity of cytoplasmic p21(Cip1/WAF1) in monocytic differentiation. EMBO J. 1999;18:1223–34. doi: 10.1093/emboj/18.5.1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Witko-Sarsat V, Mocek J, Bouayad D, et al. Proliferating cell nuclear antigen acts as a cytoplasmic platform controlling human neutrophil survival. J Exp Med. 2010;207:2631–45. doi: 10.1084/jem.20092241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koster R, di Pietro A, Timmer-Bosscha H, et al. Cytoplasmic p21 expression levels determine cisplatin resistance in human testicular cancer. J Clin Invest. 2010;120:3594–605. doi: 10.1172/JCI41939. [DOI] [PMC free article] [PubMed] [Google Scholar]