

Abstract
Ligand specificity characterizes receptors for antibody and many other immune receptors, but the common use of the FcR-γ-chain as their signaling subunit challenges the concept that these receptors are functionally distinct. We hypothesized that elements for specificity might be determined by the unique cytoplasmic domain (CY) sequences of the ligand-binding α-chains of γ-chain associated receptors. Among Fcγ receptors (FcRs), a protein kinase C (PKC) phosphorylation consensus motif, [RSSTR], identified within the FcγRIIIa (CD16A) CY by in silico analysis, is specifically phosphorylated by PKCs, unlike other FcRs. Phosphorylated CD16A mediates a more robust calcium flux, tyrosine phosphorylation of Syk and pro-inflammatory cytokine production while non-phosphorylatable CD16A is more effective at activation of the Gab2/PI3K pathway, leading to enhanced degranulation. S100A4, a specific protein binding partner for CD16A-CY newly identified by yeast two-hybrid analysis, inhibits phosphorylation of CD16A-CY by PKC in vitro, and reduction of S100A4 levels in vivo enhances receptor phosphorylation upon cross-linking. Taken together, PKC-mediated phosphorylation of CD16A modulates distinct signaling pathways engaged by the receptor. Calcium activated binding of S100A4 to CD16A, promoted by the initial calcium flux, attenuates the phosphorylation of CY, and acting as a molecular switch, may both serve as a negative feedback on cytokine production pathways during sustained receptor engagement and favor a shift to degranulation, consistent with the importance of granule release following conjugate formation between CD16A+ effector cells and target cells. This switch mechanism points to new therapeutic targets and provides a frame for understanding novel receptor polymorphisms.
Introduction
Receptors for antibody play a crucial role in immune complex clearance and initiation of multiple cell programs in including cytokine synthesis (1) and antibody-dependent cell mediated cytotoxicity (ADCC) (2). Playing a prominent role in immune complex-mediated inflammatory and anaphylactic responses (3), these receptors engage different subclasses of antibody preferentially (4), and structural polymorphisms affecting receptor function in humans (5) are implicated in susceptibility to some autoimmune diseases, including SLE (6).
Multiple FcR associate with a homo- or hetero-dimer of FcεRI γ-chain or γ-chain both for expression and for signal transduction (7, 8), which engages tyrosine phosphorylation events mediated through a ITAM (immunoreceptor tyrosine-based activating motif) (9, 10). However, since studies using mouse models suggest that these Fc receptors are not simply functionally redundant (3, 11, 12), we hypothesized that the unique FcγR cytoplasmic sequences of the ligand-binding α-chains play a role in determining receptor specificity. Indeed, within a myeloid environment, total truncation mutants of the cytoplasmic domain of FcγRI suggest that the CY affects γ-chain tyrosine-based signaling, receptor-induced specific gene expression and phagocytosis (13, 14). Similarly, deletion of the CD16A cytoplasmic domain results in altered receptor/γ chain complex-mediated signaling in T cell lines (15).
In silico analysis indicates that, unlike the highly homologous extracellular domains, each α-chain cytoplasmic domain sequence is without homologues in the human genome, and phosphorylation of a PKC motif, unique to CD16A among FcγR within the cytoplasmic domain, plays a critical role in modulating cellular responses following receptor-specific activation. PKC specifically mediates phosphorylation of CD16A-CY in vitro. CD16A phosphorylation is induced in cells upon receptor cross-linking, concurrent with enhanced calcium flux, tyrosine phosphorylation of Syk and pro-inflammatory cytokine production. However, non-phosphorylatable CD16A mutants are more effective at activation of the Gab2/PI3K pathway, resulting in enhanced degranulation.
Recognizing that CD16A-CY domain binding partners might modulate receptor specific functions; we identified S100A4 as specifically interacting with CD16A–CY. The association is direct, specific and Ca2+-dependent. S100A4, a member of the S100 family of calcium-binding proteins (16), widely distributed in normal cells and tumor cells (reviewed by (17)), interacts with a number of intracellular protein partners (18-20). S100A4 exerts its biological effects by inhibition of the phosphorylation of its target proteins by PKC and/or casein kinase II (20-23), and we show that the presence of S100A4 inhibits phosphorylation of CD16A-CY by PKC in vitro. Reduction of S100A4 levels in vivo enhances CD16A phosphorylation upon cross-linking. Taken together, we propose a molecular switch that regulates the dual pathways of effector functions for CD16A. While initially activating cytokine production, CD16A undergoes a shift to more efficient granule release, favored by S100A4 mediated inhibition of receptor phosphorylation, as might occur during conjugate formation between CD16A+ effector cells and target cells. Distinct from other FcRs, this switch mechanism highlights new therapeutic targets and provides a frame for understanding novel receptor polymorphisms.
Materials and Methods
Antibodies and reagents
The following antibodies were used: 3G8 anti-CD16 (Abbott Biotech); FITC-3G8 (Invitrogen); F(ab’)2 3G8 (prepared by Rockland Labs); 32.2 anti-CD64 (Medarex); rabbit anti-CD16 ( from Dr. Howard Fleit); rabbit anti-CD16A cytoplasmic tail (CY) and anti-CD64 cytoplasmic tail (CY) (made by Research Genetics), rabbit anti-S100A4 (from Dr. Arthur Polans, Univ. Wisconsin, Madison); rabbit anti-S100A4 (made by Cocalico Biologicals); 5C7 anti-S100A4 and 7D3 anti-FcR γ-chain mAb (generated in our laboratory); F(ab’)2 mouse IgG and F(ab’)2 goat anti-mouse IgG (Jackson ImmunoResearch); IgE anti-TNP (BD Pharmingen); rabbit anti-GST, anti-Syk, and anti-Gab2 (Santa Cruz Biotechnology) and mouse anti-GST (Pharmingen). Mouse IL-12β, IL-6, and TNFα ELISA kits (BD Pharmingen); PhoshoPlus AKT Activation Kit (Cell Signaling); wortmannin (Sigma); lipofectamine 2000 (Invitrogen); and Alexa monoclonal antibody labeling kits (Molecular Probes) were purchased.
Construction of expression plasmids
The S100A4/pcDNA3 expression construct was created by cloning S100A4 cDNA from pGEX2T/S100A4 into pcDNA3 expression vector. The triple alanine mutant GST-fusion protein CD16A CY (RAAAR) was generated using Qiagen plasmid purification kits (Valencia, CA). To generate GST-fusion proteins, the DNA fragment encoding human CD16A-CY was sub cloned into pGEX-2T (Amersham Biosciences). DNA mutagenesis was performed using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). GST-CD89-CY, GST-CD32a-CY and GST-CD64-CY were constructed in our lab. The GST-S100A4 fusion was generated by cloning human S100A4 cDNA into the EcoRI site of pGEX2T.
Cell lines and mononuclear cell preparation
The mouse macrophage cell line P388D1 and rat mast cell line RBL-2H3 (ATCC) were stably transfected by electroporation and cultured in selective medium. Comparable quantitative receptor expression for individual constructs was established by FACS. The HEK cell line 293 was transiently transfected using Lipofectamine 2000 (Invitrogen) and harvested for analysis 48 hours after transfection. Mononuclear cells from peripheral blood of healthy donors were obtained by Ficoll-Hypaque density gradients after informed consent.
Cell activation and immunoblotting
Cells were resuspended in ice-cold HBSA++ (HBSS buffer, pH 7.3, with 20mM HEPES, 0.1% BSA, 1mM CaCl2 and 1mM MgCl2) at 40 × 106/ml. The cells were incubated with 10ug/ml F(ab’)2 3G8 or F(ab’)2 mouse IgG on ice for 15 minutes. The cells were washed and incubated with F(ab’)2 goat or rabbit anti-mouse at 20ug/mL in HBSA++ for various time points at RT. Cells were then lysed with ice cold Triton X-100 lysis buffer (24)on ice for 10 minutes. Cell lysates or immunoprecipitates were analyzed by immunoblotting.
Cytokine analysis
Cytokine production was measured as described previously (13)with modification. Cells were stimulated in 24-well tissue culture plates (Costar) with 1 ug/ml LPS, surface absorbed mAb 3G8 F(ab’)2 , mouse IgE, or F(ab’)2 mouse IgG . Wells were coated with absorbed F(ab’)2 antibody, mouse IgG (40 μg/ml), or mouse IgE (40 μg/ml), overnight at room temperature. After washing, cells (5 × 105 cells/ml) were added to the wells and cultured for varying periods of time. Levels of murine or rat cytokines in culture supernatants were quantitated by enzyme-linked immunosorbent assays following manufacturer’s protocols.
Degranulation assay
Degranulation of RBL-2H3 cells was determined by measuring the release of the granule enzyme β-hexosaminidase (25). Briefly, cells were plated on 24-well plates at 1×105/well overnight. Monolayer cells were washed with Tyrode buffer (130mM NaCl, 5mM KCl, 1.4mM CaCl2, 1mM MgCl2, 5.6mM glucose, 10mM HEPES, and 0.1% BSA, pH 7.4). The cells were incubated with 100 uL of 5ug/mL F(ab’)2 3G8, F(ab’)2 mouse IgG, or mouse IgE on ice for 45 minutes. After washing with Tyrode buffer cells were incubated with 100 uL of F(ab’)2 goat anti-mouse at 20ug/mL at 37°C for various time points. Cell supernatants (10uL) were incubated with 30 uL of the substrate (3mM p-nitrophenyl-N-acetyl-β-D-glucosaminide in 0.1M citrate, pH 4.5) at 37°C for 1 hour. The reaction was stopped with 150 uL of 100mM NaCO3-NaHCO3 (pH 10.0) and absorbance at 405 nm was measured. Results are expressed as percentages of the total β-hexosaminidase contents of the cells after subtracting the spontaneous release in the absence of receptor cross-linking.
Measurement of Intracellular Calcium
Intracellular free calcium [Ca +2]i was determined as described previously (26). Briefly, suspensions of cells at 107/ml in Ca+2 and Mg+2 –free Hanks’ phosphate buffered saline, pH 7.4, were incubated with 5 uM Indo-1/AM at 37°C for 40 minutes. Following incubation, cell suspensions were incubated another 25 minutes at room temperature, washed and resuspended to 107/ml in Hank’s PBS with 1 mM Ca+2 and Mg+2, 1 mg/ml BSA, and 10mM HEPES (Hank’s +/+). Cells were opsonized with 7.5 ug/ml mAb 3G8 F(ab’)2 for 40 min. at 4°C, washed once, resuspended in Hanks+/+ and 2 × 106 cells were immediately transferred to the SLM 8000. With excitation at 355 nm, the simultaneous fluorescence emission at 405 and 490nm was measured, integrated, and recorded each second. After establishing a baseline for 60s, goat anti-mouse F(ab’)2 was added (20 ug/ml final concentration), and data acquisition was continued for an additional 3.5 min. Each sample was individually calibrated by lysing cells in 1% Triton X-100 to determine the maximal emission ratio and by adding EDTA (20 mM final concentration) to determine the minimal ratio. Alternatively, 2.5 × 106 RBL-2H3 cells expressing [RSSTR] or [RAAAR] CD16A were loaded with 2 uM Fluo-4 and opsonized with 10 ug/ml murine IgE for 20 minutes on ice. After establishing a baseline, cells were stimulated with goat anti-mouse Ig (20 ug/ml) and further analyzed by FACS analysis for intracellular calcium release.
In vitro kinase assays
Active PKC isozymes (Upstate Biotechnology) in 50 uL kinase buffer (for PKC α, β, and γ: 4mM MOPS, pH 7.2, 5mM β-glycerophosphate, 0.2mM Na3VO4, 0.2mM dithiothreitol, 0.2mM CaCl2; for PKC δ, ε, η, and ı: 20mM HEPS, pH7.4, 0.03% Triton-X 100; for PKC γ: 8mM MOPS, pH 7.0, 0.2mM EDTA) with diacylglycerol (5 ug/mL), sonicated phosphatidyl serine (50 ug/mL), 50uM ATP, 10mM MgCl2, and 10-20 uCi of [32P ATP] (Amersham) were incubated with 6 ug of GST-fusion proteins for 10 minutes at 32°C. The proteins were precipitated with 20% tricloroacetic acid, separated by SDS-PAGE and analyzed by autoradiography. In all PKC assays, a second set of samples without [32P ATP] was run in parallel and stained with Coomassie blue for protein loading controls.
Induced CD16A phosphorylation
Cells stably expressing [RSSTR] or [RAAAR] CD16A were metabolically labeled with [32P] orthophosphate (27). Labeled cells were incubated with 3G8 F(ab’)2 at 10 ug/mL in phosphate-free DMEM at 4°C for 10 minutes. After wash and resuspend in ice cold phosphate-free DMEM, cells were incubated with 20 ug/mL F(ab’)2 rabbit anti-mouse at RT for 5 min. The control samples were treated the same but incubated on ice all the time, or without addition of F(ab’)2 rabbit anti-mouse. Cells were lysed in Triton X-100 lysis buffer and CD16A was precipitated with 3G8 overnight at 4°C. Alternatively, CD16A was precipitated with F(ab’)2 goat anti-mouse and F(ab’)2 goat anti-rabbit at 4°C overnight. Precipitates were analyzed by SDS-PAGE and autoradiography. Parallel samples without [32P] labeling were analyzed by western blotting to control protein loading.
Yeast two-hybrid studies
Yeast two-hybrid studies were performed as detailed in the manufacturer’s protocols (DupLEX-A Yeast Two-Hybrid System, OriGene Technologies). Briefly, the bait plasmid, pEG202-CD16A-CY was used to screen the human peripheral blood leukocyte library. Yeast cells were transformed first with the bait plasmid and selected on leucine-free plates to produce a stable LexA-CD16A-CY strain. The cells were then transformed with the library and selected on leucine-, tryptophan-, and histidine-free plates and media. Expression of LacZ was determined by the appearance of blue colonies within 24 hours. Interaction of CD16A-CY mutants with S100A4 was measured by cell growth in liquid medium lacking leucine, tryptophan, and histidine. Positive yeast clones from the two-hybrid system were screened for identity. The identity of target plasmid was determined by amplification using vector specific upper and lower primers provided in the DupLEX-A system. An S100A4 specific upper primer was designed to be compatible with the vector specific lower primer to screen all of the positive yeast clones. S100A4 primer was: - 5′ ggg caa aga ggg tga caa. The interaction of CD16A-CY with S100A4 was confirmed by co-transforming S100A4 and CD16A-CY expression plasmids into a less sensitive yeast strain, EGY 188.
In vitro binding assays
GST-CD16A-CY (12 ug) was incubated with 12 ug of GST-S100A4, GST-CD64-CY, or GST in 100 ul KTT buffer (28) with 1mM CaCl2 or 10mM EDTA at 4°C overnight. CD16A was immunoprecipitated with rabbit anti-CD16A-CY overnight at 4°C. The precipitates were washed five times with KTT buffer with 1mM CaCl2 or 10mM EDTA and analyzed by SDS-PAGE and western blotting. To pull down S100A4 in cell lysate, U937 cells were lysed in NP40 lysis buffer (29) containing no EDTA(Roche) with Ca2+ (1.09mM CaCl2) or without Ca2+ but containing 10mM EDTA. The supernatant was incubated with various GST fusion proteins on glutathione-Sepharose 4B beads overnight at 4°C. The beads were washed with the lysis buffer with or without Ca2+ for three times, and analyzed by SDS-PAGE and western blotting with anti-S100A4 mAb 5C7. Blots were then stripped and re-probed with an anti-GST mAb to control protein loading.
Co-immunoprecipitation assays
NK3.3 cells, 293 cells transiently expressing S100A4 and CD16A or CD64, or primary mononuclear cells were lysed in NP-40 lysis buffer with Ca2+ or without Ca2+ but containing 10mM EDTA as above. CD16A or CD64 were immunoprecipitated from the supernatants with 3G8 or 197, overnight at 4°C. Immunoprecipitates were analyzed by western blotting with anti-S100A4. Blots were then stripped and re-probed with polyclonal anti-CD16A-CY antibodies and/or a polyclonal anti-CD64-CY antibody.
Results
PKC phosphorylates CD16A-CY but not other FcR CYs
The α-chain CYs of FcγRIIIa (CD16A), FcγRIa (CD64) and FcαRI (CD89) each have unique sequences without homology in the human genome. ProfileScan (Prosite data base) suggested several putative PKC phosphorylation motifs in their α-chain CYs although in vitro kinase assays (IVK) showed essentially no phosphorylation of the CYs of CD89 or CD64. However, CD16A-CY, which contains a potential PKC phosphorylation motif [RSSTR] (Fig. 1C), was strongly phosphorylated by PKCs (Fig.1A, 1B and S1). Mutation of the CD16A [RSSTR] motif to [RAAAR] abolished the phosphorylation. Mutation of serine-219 and threonine-220 to A-219 and A-220 also abolished PKC phosphorylation, localizing the phosphorylation site to these two residues (Fig.1C).
FIGURE 1.
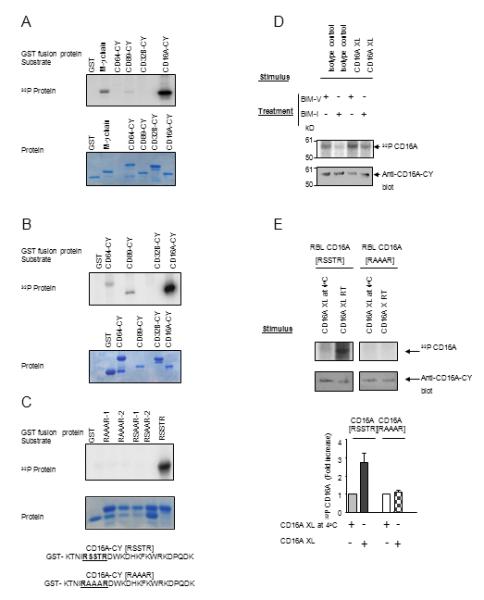
CD16A is specifically phosphorylated by PKC. (A) and (B) In vitro kinase assay using PKCı (A) and PKCαβγ (B) with various GST-Fc receptor CY domains (upper panel). Protein gel was stained with Coomassie Blue (lower panel). Data are representative of six experiments. (C) Cytoplasmic domain sequences of wild type ([RSSTR]) and the mutant ([RAAAR]) CD16A. IVK substrates include separate clones of CD16A-CY with S/T to A mutations within the [RSSTR] motif (RAAAR-1 and -2, RSAAR-1 and -2 represent duplicate assays). Protein gel was stained with Coomassie Blue (lower panel). Data are representative of three separate experiments using PKCδ. (D) CD16A-stably transfected P388D1 cells were metabolically labeled with [32P] orthophosphate and treated with 10ug/mL BIM-I or control reagent BIM-V at 37°C for 60 minutes prior to receptor cross-linking for 5 min at 25°C. Immunoprecipitated CD16A was detected by autoradiogram and parallel gels blotted with rabbit anti-CD16A-CY. Data are representative of three experiments. (E) [RSSTR] or [RAAAR] CD16A-stably transfected RBL-2H3 cells were metabolically labeled with [32P] orthophosphate and receptor was cross-linked for 5 min at 25°C. Immunoprecipitated CD16A was analyzed by autoradiography, western blotting, and densitometry (upper panel). The density of [32P] CD16A was plotted for each treatment condition (lower panel). Data are means of three experiments.
In NK3.3 and P388D1 cells stably-expressing human CD16A, CD16A phosphorylation was enhanced by PMA treatment and inhibited by selective PKC inhibitor (Fig. S2B and C). CD16A phosphorylation was induced by CD16A cross-linking in P388D1 cells stably-expressing human CD16A. Selective PKC inhibitors BIM-I and Go6976 inhibited the phosphorylation (Fig. 1D and S2A), indicating that CD16A is phosphorylated by PKC. Phosphorylation of CD16A in [RSSTR], but not [RAAAR] CD16A RBL-2H3 cells indicates that the [RSSTR] motif is responsible for the phosphorylation (Fig. 1E).
The PKC motif in CD16A-CY differentially regulates induction of pro-inflammatory cytokines and degranulation
Activation of monocytes/macrophages by CD16A results in the synthesis and secretion of TNFα, IL-6 and IL-1β (30, 31). The levels of secretion of IL-6, IL-1β, and TNFα in [RSSTR] CD16A expressing P388D1 cells following receptor cross-linking were markedly higher than that in [RAAAR] cells expressing equivalent levels of CD16A (Fig. 2A and B). Comparable levels of γ-chain were associated with CD16A, consistent with the requirement of γ-chain for CD16A surface expression (Fig. 2C). Tail less (TL) CD16A expressing cells gave similar results as [RAAAR] cells (Fig. S3). LPS induced cytokine production was at similar levels in these cell lines (Fig. S3 and data not shown). The levels of secretion of IL-4 in [RAAAR] CD16A expressing RBL-2H3 cells following receptor cross-linking were significantly reduced in comparison with that in [RSSTR] cells expressing equivalent levels of CD16A (Fig. 2D). CD16A cell surface expression was comparable (Fig. 2E) and γ-chain association with CD16A was equivalent (Fig. 2F) in the transfectants. In contrast, the [RAAAR] transfected cells showed enhanced receptor-mediated degranulation compared to [RSSTR] CD16A transfected RBL-2H3 cells (Fig. 3A). Both [RSSTR] and [RAAAR] CD16A expressing cells underwent comparable levels of degranulation when activated through endogenous FcεR, indicating equivalent capacities for degranulation (Fig. 3B). As a complementary approach, RBL-2H3 cells stably expressing [RSSTR] or [RAAAR] CD16A were transiently transfected with wild type PKCθ or constitutively active PKCθ A148E constructs. Both the wild type PKCθ and the constitutively active PKCθ resulted in a significant decrease (40.2% and 39.5% reduction at 30 min, p = 0.0004 and 0.0036, respectively) in receptor induced degranulation in cells expressing [RSSTR] CD16A when compared to the vector control (Fig. 3C). However, the wild type and the constitutively active PKC constructs reduced receptor induced degranulation much less in cells expressing the [RAAAR] receptor (9.4% and 15.2% at 30 min, respectively) (Fig. 3D) Taken together, these results provide strong evidence for the role of PKC-mediated phosphorylation of CD16A α-chain in determining the balance of cell programs initiated by receptor engagement.
FIGURE 2.
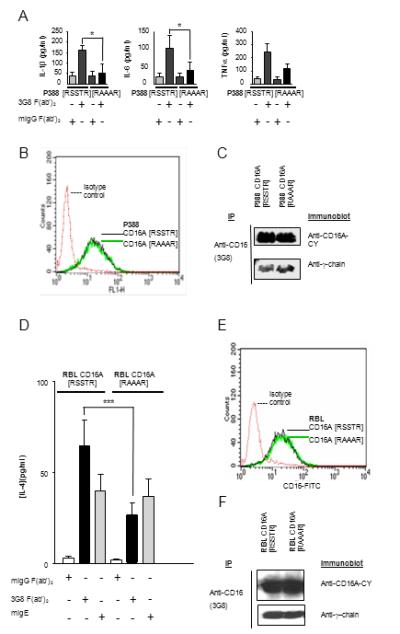
Receptor-mediated production of pro-inflammatory cytokines is impaired in transfectants expressing non-phosphorylatable CD16A. (A) P388D1 stable transfectants expressing CD16A [RSSTR] or [RAAAR] were stimulated in a receptor specific manner as described previously (13) . Cytokine proteins in supernatants were measured by ELISA. Data are the mean ±SD from six experiments. *p < 0.5 by Student paired t-test. (B) Expression of human CD16A [RSSTR] and [RAAAR] on the surface of P388D1 stable transfectants measured by FITC-3G8 staining. (C) Association of γ-chain with CD16A [RSSTR] and [RAAAR] in P388D1 stable transfectants by co-immunoprecipitation. CD16A was immunoprecipitated from cell lysates with mAb 3G8 and immunoblotted sequentially with polyclonal anti-CD16A-CY and polyclonal anti-γ-chain. (D) RBL-2H3 stable transfectants expressing CD16A [RSSTR] or [RAAAR] were stimulated in a receptor specific manner as described previously (13) . IL-4 protein in supernatants was measured by ELISA. Data are the mean ±SD from six experiments. ***p < 0.001 by Student paired t-test. (E) Expression of human CD16A [RSSTR] and [RAAAR] on the surface of RBL-2H3 stable transfectants measured by FITC-3G8 staining. (F) Association of γ-chain with CD16A [RSSTR] and [RAAAR] in RBL-2H3 stable transfectants by co-immunoprecipitation. CD16A was immunoprecipitated from cell lysates with mAb 3G8 and immunoblotted sequentially with polyclonal anti-CD16A-CY and polyclonal anti-γ-chain.
FIGURE 3.

Receptor-mediated degranulation is augmented in RBL-2H3cells expressing non-phosphorylatable CD16A. (A) Degranulation in RBL-2H3 stable transfectants expressing CD16A [RSSTR] or [RAAAR] was induced by receptor cross-linking and measured as hexosaminidase activity. Data are mean ±SD from three experiments. (B) Degranulation induced by cross-linking FcεR with mouse IgE and goat anti-mouse cross-linking. Results are representative of three experiments with p>0.05 for receptor-specific degranulation in CD16A [RSSTR] versus [RAAAR] RBL-2H3 stable transfectants. (C and D) PKCθ wild type (WT), PKCθ A148E (constitutively active form) or control vector DNA were introduced into CD16A [RSSTR] (C) and [RAAAR] (D) RBL-2H3 stable transfectants. After CD16A cross-linking, degranulation was measured as described. Data are presented as mean ±SD of three experiments. *p < 0.05 by Student paired t-test.
The PKC motif in CD16A-CY regulates γ chain-mediated signaling
The CD16A-CY regulates receptor mediated early signaling including calcium mobilization and tyrosine phosphorylation of Syk (15 and data not shown). We investigated the role of CD16A-CY [RSSTR] motif in receptor-mediated early signaling in cell lines expressing functionally competent endogenous γ-chain (13, 14, 29). Receptor cross-linking induced a rise in intracellular free calcium [Ca2+]i of 241 nM in RBL-2H3 cells expressing [RSSTR] CD16A, but only 127 nM of calcium, on average, in RBL-2H3 cells expressing [RAAAR] CD16A at equivalent levels (Fig. 4A). Endogenous FcεR engagement induced comparable calcium fluxes in both cell lines. Receptor-induced tyrosine phosphorylation of total cellular γ-chain and co-precipitated Syk was reduced markedly in P388D1 cells expressing [RAAAR] CD16A in comparison with [RSSTR] CD16A (Fig. 4B). Furthermore, although association of γ-chain with the CD16A was equivalent in [RSSTR] and [RAAAR] cells, receptor-induced tyrosine phosphorylation of receptor-associated γ-chain and γ-chain-associated Syk was significantly reduced in P388D1 cells expressing [RAAAR] CD16A in comparison with [RSSTR] CD16A (data not shown). Thus, key components of γ-chain mediated signaling are regulated by the PKC motif in CD16A.
FIGURE 4.
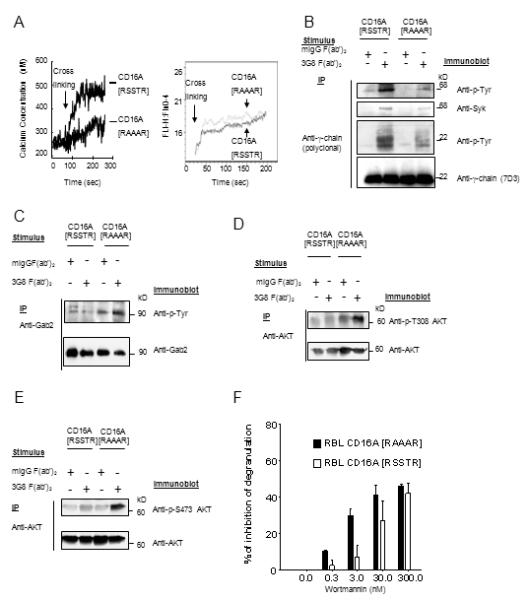
Early signaling following CD16A cross-linking was altered in cells expressing non-phosphorylatable CD16A. (A) CD16A receptor cross-linking triggered calcium mobilization in RBL cells expressing human CD16A [RSSTR] or [RAAAR] (left panel). Data are representative of three experiments. IgE receptor cross-linking triggered calcium mobilization in RBL cells expressing human CD16A [RSSTR] or [RAAAR] (right panel). Data represent one of five experiments giving similar results. (B) Tyrosine phosphorylation of γ-chain and γ-chain-associated Syk in P388D1 stable transfectants expressing CD16A [RSSTR] or [RAAAR]. After receptor cross-linking for 5 min at 25°C, cell lysates were immunoprecipitated with polyclonal anti-γ-chain and separated by SDS-PAGE under non-reducing conditions. Tyrosine phosphorylated γ-chain and Syk were analyzed by sequentially blotting with anti-phosphotyrosine (4G10), anti-Syk and anti-γ-chain. Data are representative of three experiments. (C) After CD16A cross-linking in RBL-2H3 stable transfectants for 5 min, Gab2 in cell lysates was immunoprecitated and probed sequentially with anti-p-Tyr (4G10) and polyclonal anti-Gab2. (D and E), Cells were stimulated as above, AKT was immunoprecipitated and probed with anti-phospho-AKT Thr308 (D), anti-phospho-AKT Ser473 (E), and anti-AKT. (F) RBL-2H3 stable transfectants expressing [RSSTR] or [RAAAR] CD16A were pre-incubated for 15 min at 37°C with the indicated concentrations of wortmannin or DMSO vehicle (35). The cells were washed, and CD16A was cross-linked for 30 min. Degranulation was measured as hexosaminidase activity. Data are summarized from three experiments.
The PKC motif in CD16A-CY regulates degranulation via a Gab2/PI3K-dependent pathway
FcR induced degranulation is likely mediated by both calcium dependent and independent pathways (32). The adaptor protein Gab2 is critical for activation of the PI3K/Akt pathway (33) and the Fyn/Gab2/RhoA-dependent, calcium-independent pathway in mast cell degranulation (32). Engagement of CD16A on NK cells initiates a cascade of signaling events including PI3K activation (34), which is required for CD16A mediated granule exocytosis and ADCC in NK cells (35). The enhanced degranulation in the [RAAAR] CD16A expressing RBL-2H3 cells is unlikely caused by the calcium-dependent pathway since there is impaired calcium signaling in these cells. We therefore investigated the effect of the PKC motif on the calcium-independent Gab2/PI3K/AKT pathway. After CD16A cross-linking, tyrosine phosphorylation of Gab2 was induced in the [RAAAR] but not the [RSSTR] CD16A expressing RBL-2H3 and P388D1 cells (Fig. 4C and data not shown). Similarly, levels of phosphorylation of serine473 and threonine308 of AKT, an indication of maximum activation of the enzyme, markedly increased in the [RAAAR] CD16A cells following receptor cross-linking compared to that in [RSSTR] CD16A cells (Fig. 4D and E). Treatment with wortmannin, a highly selective inhibitor for PI3K with an in vitro IC50 of 2-4 nM for PI3K (36), at levels of 0.3-3 nM markedly reduced receptor-mediated degranulation in the [RAAAR] CD16A RBL-2H3 cells (10.4% and 29.9% at 0.3 and 3 nM, respectively), but only reduced degranulation modestly in the [RSSTR] CD16A cells (2.1% and 7.3% at 0.3 and 3 nM, respectively) (Fig. 4F). The percentage of β-hexosaminidase release without inhibitor was 17.9% in [RSSTR] CD16A cells and 51.3% in [RAAAR] CD16A RBL-2H3 cells. These results indicate that the enhanced degranulation in [RAAAR] CD16A RBL-2H3 cells is likely mediated by a wortmannin-sensitive, PI3K-dependent pathway.
S100A4 interacts with CD16A-CY
To identify protein partner(s) which might interact with the CD16A cytoplasmic domain and contribute to the modulation of CY function, we performed yeast two hybrid studies using a pEG202-CD16A-CY bait vector and the human PBL cDNA library and identified S100A4 as a specific CD16A-CY-interacting protein (Fig. 5A). To map the region of CD16A-CY that is essential for the interaction with S100A4, a series of CD16A mutant variants were constructed (Fig. 5B and data not shown). These variants were tested for interaction of CD16A-CY with S100A4 using the yeast two-hybrid system. Deletion of the six most C-terminal residues of CD16A abrogated the optimal interaction with S100A4, implicating these to be part of the S100A4 binding site of CD16A (Fig. 5C). To verify interaction between CD16A-CY and S100A4, we used GST fusion proteins and in vitro pull-down assays to demonstrate that that GST-S100A4, but not GST or GST-CD64-CY, was pulled-down by GST-CD16A-CY (Fig. 6A). Similarly, GST-CD16A-CY, but not GST-CD64-CY, was pulled-down by GST-S100A4 or purified S100A4 (data not shown). Furthermore, GST-CD16A-CY, GST-CD64-CY, GST-CD89-CY or GST was incubated with U937 cell lysates containing endogenous S100A4, S100A4 was only pulled down with GST-CD16A-CY in the presence of Ca2+ (Fig. 6B). These data demonstrated that the interaction between CD16A-CY and S100A4 is direct, Ca2+-dependent, and specific. To confirm that these two proteins interact in cells, CD16A was immunoprecipitated from NK3.3 cell lysates and probed for CD16A and S100A4, which co-precipitated with CD16A in these cells in a Ca2+-dependent manner (Fig. 6C). CD16A or CD64, immunoprecipitated from 293 cells transiently expressing CD16A or CD64 and S100A4, showed co-immunoprecipitation of Sl00A4 with CD16A but not with CD64 in a Ca2+-dependent manner (Fig. 6D). Furthermore, S100A4 was co-immunoprecipitated with CD16A but not CD64 in lysates from primary mononuclear cells in the presence of calcium (Fig. 6E), indicating specific interaction of the two proteins at physiological expression levels. To study the subcellular localization of both proteins in freshly prepared primary mononuclear cells, cells were stained for S100A4 (green) and CD16 (red) and examined by scanning laser microscopy. About 20-30% of the cells stained positive for CD16A, presumably NK cells, monocytes, and macrophages. S100A4 was also present in these cells, with a cytoplasmic distribution and a few patches of co-localization with CD16 (Fig. 6F). When CD16 was cross-linked, enhanced co-localization of CD16A and S100A4 was evident with some co-migration to the cell’s interior. Thus, modest co-localization of S100A4 with CD16A is enhanced following receptor cross-linking. In cells pre-treated with BAPTA-AM, however, S100A4 no longer co-localized with CD16A following receptor cross-linking, indicating the co-localization is Ca2+-dependent. CD64 and S100A4 were not co-localized in receptor cross-linked cells.
FIGURE 5.
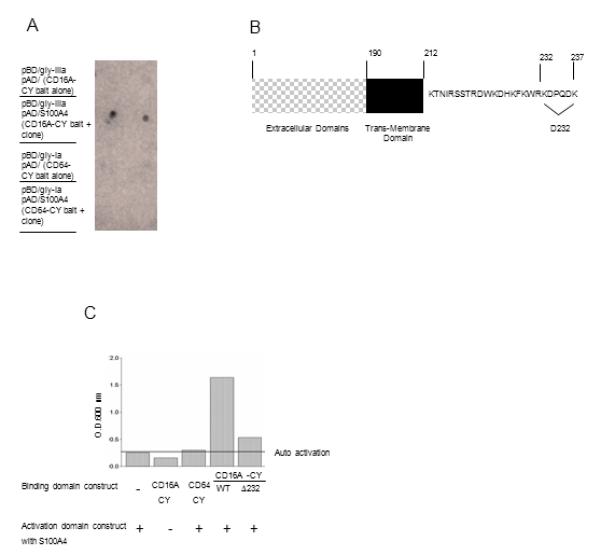
CD16A-CY specifically interacts with S100A4 protein in the yeast LexA two-hybrid analysis. Initial screen of 52×106 yeast cells carrying the CD16A-CY bait vector identified 399 clones that were able to survive on minimal media. A secondary screen using a LacZ reporter containing the Gal4 minimal promoter identified 150 LacZ positive clones. Sequencing identified that 84% of the lacZ positive clones were S100A4. (A) The colonies for each combination of cDNA were transferred to leucine-, tryptophan-, and histidine-free plates and tested for β-galactosidase activity (indicated by blue staining within 24 hours). CD16A-CY bait, but not CD64-CY bait mediated LacZ expression (n=3). (B) Schematic representation of mutations in the cytoplasmic domain of CD16A. (C) Deletion of the C terminal six amino acids greatly reduced the interaction of CD16A with S100A4 (n=3).
FIGURE 6.
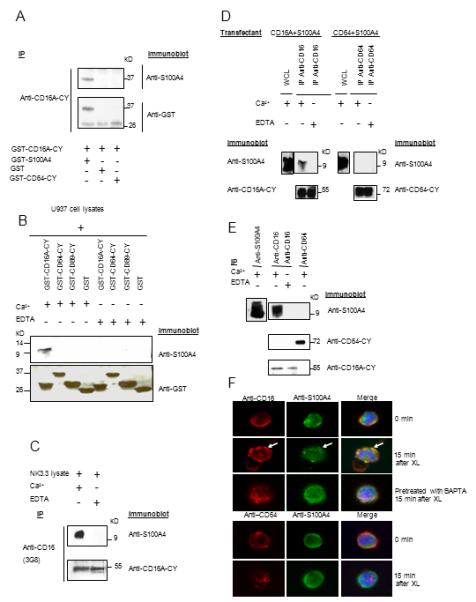
The interaction of CD16A-CY with S100A4 is direct, specific and calcium-dependent. (A) GST-CD16A-CY in binding buffer containing 1 mM CaCl2 or 10 mM EDTA, was incubated with GST-S100A4, GST-CD64-CY, or GST. GST-CD16A-CY was precipitated with polyclonal anti-CD16A-CY and sequentially blotted with polyclonal anti-S100A4 and anti-GST (n=3). (B) U937 cell lysates were incubated with GST-CD16A-CY, GST-CD64-CY, GST-CD89-CY, or GST on glutahione-Sepharose 4B beads in the presence of 1mM Ca2+ or 10 mM EDTA overnight at 4°C. The beads were washed and the proteins were resolved and immunoblotted sequentially with anti-S100A4 and anti-GST (n=3). (C) Interaction between CD16A and S100A4 in NK3.3 cell line in the presence of 1mM Ca2+ or 10mM EDTA. Cell lysates were immunoprecipitated with 3G8 and analyzed by sequential immunoblotting with anti-S100A4 and anti-CD16-CY (n=3). (D) 293 cells transiently expressing S100A4 and CD16A or CD64 were lysed with NP40 lysis buffer with or without Ca2+ 48 hr after transfection. CD16A and CD64 immunoprecipitates were sequentially blotted with anti-S100A4, anti-CD16-CY, and anti-CD64. S100A4 expression in the cells was examined by immunoblotting with anti-S100A4 in the cell lysate (upper panels) (n=3). (E) Freshly prepared mononuclear cells were lysed with NP40 lysis buffer with 1mM Ca2+ or 10 mM EDTA. CD16A or CD64 immunoprecipitates were blotted with anti-S100A4, anti-CD16-CY, and anti-CD64-CY sequentially. S100A4 expression in the cells was examined by immunoblotting with anti-S100A4 in the cell lysate (upper left panel) (n=3). (F) Mononuclear cells from donors were incubated with Alexa Fluor 594 conjugated 3G8 F(ab’)2 (10 ug/ml) for 40 min at 4°C to label surface CD16 (upper 3 panels). Cells were then washed with PBS and cross linked at 37°C for 15 min. Cells were fixed with 4% paraformaldehyde in PBS for 20 min at room temperature and permeabilized with 0.2 % Triton in PBS for 2 min at room temperature. Cells were incubated with Alexa Fluor 488 conjugated anti-S100A4 for 1 hr at room temperature. Alternatively, the cells were pre-treated with 10 mM BAPTA-AM for 30 min at 37°C before cross-linking and staining. Mononuclear cells were incubated with Alexa Fluor 594 conjugated anti-CD64 for 40 min at 4°C to label surface CD64, cross-linked and analyzed as above. Data are representative of 6 experiments.
S100A4 inhibits phosphorylation of CD16A by PKC and affects receptor mediated degranulation
S100A4 inhibits phosphorylation of its target proteins by PKC and/or CK2 (20, 22, 23, 37). IVK assays with GST-CD16A-CY and PKC in the presence or absence of S100A4 demonstrated that GST-S100A4, but not GST, inhibited phosphorylation of GST-CD16A-CY by PKC (Fig. 7A, Fig. S4A). Since GST-S100A4 inhibited phosphorylation of GST-CD16A-CY and p53 but did not affect the phosphorylation of MBP by PKC under the same conditions, we exclude the possibility that GST-S100A4 acts as a competitive substrate or results in substrate inhibition of the kinase (Fig. S4B and C). Notably, the level of inhibition of CD16A phosphorylation by PKC in the presence of S100A4 was similar to that reported for other target proteins of S100A4 including p53 and liprin β-1 (20-23). To assess the effect of S100A4 binding on CD16A phosphorylation in vivo, phosphorylation of CD16A was measured in S100A4 siRNA or control RNA treated RBL-2H3 cells. S100A4 was reduced to an undetectable level in S100A4 siRNA treated cells (data not shown). The ratio of 32P-CD16A to CD16A protein in anti-CD16 immunoprecipitates increased 40.5% in cells treated with control RNA, whereas the ratio increased 78.6% in S100A4 siRNA treated cells after receptor cross-linking (Fig. 7B), indicating interference of S100A4 with induced phosphorylation of CD16A. Thus, S100A4 may regulate CD16A specific functions by modulating its phosphorylation by PKC. Correspondingly, degranulation measured at 10 min after CD16A cross-linking was reduced in cells treated with S100A4 siRNA (Fig. 7C).
FIGURE 7.

S100A4 inhibits phosphorylation of CD16A by PKC and attenuate receptor-mediated degranulation. (A) GST-CD16A (1uM) was phosphorylated in the absence and presence of various concentrations of GST-S100A4 by active PKC for 30 min at 30°C (23) (upper panel). A parallel set of samples without 32P ATP was stained with Coomassie to control protein loading (lower panel). (B) RBL-2H3 [RSSTR] or [RAAAR] CD16A stable transfectants were transfected with S100A4 siRNA or nonsilencing control RNA (45). After 24 hours, phosphorylation of CD16A in 32P metabolically-labeled RBL-2H3 cells was measured as described above. Data are representative of three experiments. (C) RBL-2H3 stable transfectants expressing CD16A were transfected with S100A4 siRNA or control RNA. After 24 hours, degranulation was measured at 10 min after CD16A crosslinking at the indicated concentrations. Data represent the means +/- SD for six experiments, **p <0.01, ***p <0.001 by Student paired t-test.
Discussion
The [RSSTR] sequence in the cytoplasmic domain of CD16A is phosphorylated by PKC in in vitro kinase assays, in human cell lines and in primary human cells. The phosphorylation is specific to this motif, and the phosphorylated and non-phosphorylatable states differentially regulate receptor initiated signaling and downstream cell programs. Phosphorylated CD16A favors more robust tyrosine phosphorylation of FcRγ-chain with greater flux in [Ca2+]i and production of IL-1β, IL-6, TNF-α and IL-4, a provocative observation given the hyperactivation of PKCθ in a subset of lupus patients (38). In contrast, the [RAAAR] CD16A leads to greater activation of Gab2, AKT and greater dependence of degranulation on the PI3K pathway. Furthermore, the calcium-sensitive S100A4, known to inhibit the phosphorylation of its target proteins by PKC and CK2, interacts specifically with CD16A, among FcRs, inhibits phosphorylation of CD16A-CY by PKC in vitro, and modulates in vivo CD16A phosphorylation. Taken together, these data suggest that S100A4 may serve as a modulator of CD16A [RSSTR] phosphorylation. Its calcium dependent binding of CD16A may diminish further phosphorylation, providing both a negative feedback loop for cytokine production and a mechanism favoring degranulation by receptors newly recruited to structures such as the immunological synapse in conjugate formation.
Among the FcRγ-chain associated Fc receptors, total truncation mutants suggest a contribution of the α-chain cytoplasmic domain to receptor signaling and cell programs (29, 39). We hypothesized that ser/thr phosphorylation events might provide a window on the mechanism of α-chain contributions, and in silico analysis identified the PKC phosphorylation consensus motif [RSSTR] within the CD16A cytoplasmic domain, while FcγRI (CD64) and FcαR (CD89) have CK2 and CK1 sites respectively. The predicted specificity for kinases has been supported experimentally, suggesting important mechanisms for differential regulation. Further evidence for receptor specific mechanisms for regulation is provided by the specific interaction of S100A4 with the cytoplasmic domain of CD16A, but not with those of CD64 or CD89. Not only do CD16A and S100A4 co-immunoprecipitate in a Ca2+ dependent manner in human cell lines and primary mononuclear cells, but they also co-localize in primary mononuclear cells.
S100A4 inhibits phosphorylation of its binding partner proteins by PKC and/or CK2 (20, 22, 23, 37). Consistent with these observations, we show that S100A4 inhibits phosphorylation of CD16A-CY by PKC in vitro. Furthermore, in cells treated with S100A4 siRNA, phospho-CD16A induced by receptor cross-linking was increased, indicating interference of S100A4 with induced phosphorylation of CD16A.
Non-phosphorylatable CD16A enhanced degranulation, implying that phosphorylation of the PKC motif down regulates receptor-mediated degranulation. Indeed, over expression of a constitutively active PKCθ construct decreased receptor-mediated degranulation in cells expressing WT, but only minimally in cells with the non-phosphorylatable CY receptor, clearly supporting the notion that phosphorylation of the [RSSTR] motif is responsible for this effect. While PKCs are important mediators in initiation of many effector functions of Fcγ receptors including NK killing and phagocytosis (40, 41), their effects are no doubt multiple. For example, PKCδ-deficient mast cells exhibited a significantly higher level of degranulation, suggesting that PKCδ is a negative regulator of antigen-induced mast cell degranulation (42) . In addition to the well-known calcium-dependent pathway, degranulation in mast cells requires a calcium-independent, Fyn/Gab2/RhoA-dependent pathway, which plays a critical role in the microtubule-dependent translocation of granules to the plasma membrane (32). Gab2 is critical for PI3K recruitment and activation of the PI3K/AKT pathway (33) and Rho GTPases, involved in microtubule organization and granule translocation, and is a downstream effector of PI3K (reviewed by (43)). Engagement of CD16A on NK cells initiates PI3K activation (34) which is required for CD16 mediated granule exocytosis and ADCC by NK cells (35). Hyper-responsive CD16A-initiated degranulation in RBL-2H3 cells with non-phosphorylableCD16A is unlikely due to the effects on the calcium-dependent pathway, which is down-regulated in cells with non-phosphorylatable receptor. In contrast, the Gab2/PI3K/AKT pathway is up-regulated in cells with non-phosphorylableCD16A and degranulation is more sensitive to wortmannin inhibition in these cells. Thus, phosphorylation of CD16A regulates two distinct functions mediated by the receptor via at least two distinct pathways: a calcium-dependent pathway is concurrently up-regulated, leading to augmented production of pro-inflammatory cytokines; and a calcium-independent, PI3K-dependent pathway is down-regulated, leading to attenuated degranulation. That activation of PI3K after CD16 cross-linking in human primary monocytes limited the expression of TNFα, IL-1β and IL-6(44) supports the notion that PI3K activation plays distinct roles in CD16A-mediated degranulation and production of pro-inflammatory cytokines.
Our proposed model of PKC-mediated, phosphorylation-dependent balancing of pro-inflammatory cytokine production and degranulation, with a feedback loop involving the calcium-activated binding of S100A4, focuses attention on a critical role of the ligand-binding α-chain of CD16A and other Fc common γ-chain associated receptors. The calcium activated binding of S100A4 to CD16A may serve as such an inhibitory modulator on the Syk/cytokine pathways during sustained signaling and favor a shift to degranulation upon conjugate formation between CD16A+ effector cells and target cells. Such a molecular switch points to new therapeutic targets. Further, the subtle modulation of the contributions of the ligand-binding α-chain through genetic variation in the cytoplasmic region, -- a theme clearly established within the extracellular domain, -- may provide additional insights into receptor function and into risk for immune-mediated diseases.
Supplementary Material
FIGURE 8.
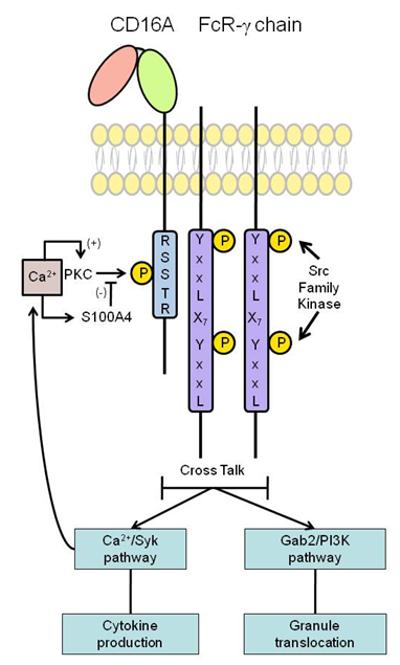
Roles of CD16A CY phosphorylation in receptor-mediated functions. Upon engagement, phosphorylation of CD16A regulates two distinct functions mediated by the receptor via at least two distinct pathways: a calcium-dependent pathway is concurrently up-regulated, leading to augmented production of pro-inflammatory cytokines; and a calcium-independent, PI3K-dependent pathway is down-regulated, leading to attenuated degranulation. Furthermore, calcium dependent binding of S100A4 to CD16A diminishes further phosphorylation, providing both a negative feedback loop, potentially restricting cytokine production and a mechanism favoring degranulation.
Acknowledgements
The authors thank Dr. Xiaoli Yang for performing confocal microscopy experiments; Dr. Tong Zhou for making monoclonal anti-S100A4; Dr. Jianming Wu for making CD16A-TL construct; Dr. Koichi Fujii for providing PKCθ constructs; Dr. Steven Pittler for quantitative analysis of PKC binding; Dr. Kelly Andringa for graphic assistance.
This work was supported by grants R01-AR33062, P01-AR49084, P60-AR49095 (Methodology Core) and P30-AR48311 (Flow Cytometry Core) from the National Institutes of Health.
The abbreviations used are
- FcR
Fc receptor
- PKC
protein kinase C
- ITAM
immunoreceptor tyrosine-based activating motif
- GST
glutathione S-transferase
- RT-PCR
reverse transcript PCR
- CY
cytoplasmic domain
- MNC
mononuclear cells
Footnotes
Disclosures The authors have no financial conflicts of interests.
References
- 1.Anderson CL, Shen L, Eicher DM, Wewers MD, Gill JK. Phagocytosis mediated by three distinct Fc gamma receptor classes on human leukocytes. J Exp Med. 1990;171:1333–1345. doi: 10.1084/jem.171.4.1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Graziano RF, Fanger MW. Fc gamma RI and Fc gamma RII on monocytes and granulocytes are cytotoxic trigger molecules for tumor cells. J Immunol. 1987;139:3536–3541. [PubMed] [Google Scholar]
- 3.Hazenbos WL, Gessner JE, Hofhuis FM, Kuipers H, Meyer D, Heijnen IA, Schmidt RE, Sandor M, Capel PJ, Daeron M, van de Winkel JG, Verbeek JS. Impaired IgG-dependent anaphylaxis and Arthus reaction in Fc gamma RIII (CD16) deficient mice. Immunity. 1996;5:181–188. doi: 10.1016/s1074-7613(00)80494-x. [DOI] [PubMed] [Google Scholar]
- 4.Nimmerjahn F RJ. Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science. 2005;310(5753):1510–1512. doi: 10.1126/science.1118948. [DOI] [PubMed] [Google Scholar]
- 5.Wu J, Edberg JC, Redecha PB, Bansal V, Guyre PM, Coleman K, Salmon JE, Kimberly RP. A novel polymorphism of FcgammaRIIIa (CD16) alters receptor function and predisposes to autoimmune disease. J Clin Invest. 1997;100:1059–1070. doi: 10.1172/JCI119616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Edberg JC, Langefeld CD, Wu J, Moser KL, Kaufman KM, Kelly J, Bansal V, Brown WM, Salmon JE, Rich SS, Harley JB, Kimberly RP. Genetic linkage and association of Fcgamma receptor IIIA (CD16A) on chromosome 1q23 with human systemic lupus erythematosus. Arthritis Rheum. 2002;46:2132–2140. doi: 10.1002/art.10438. [DOI] [PubMed] [Google Scholar]
- 7.Lanier LL, Yu G, Phillips JH. Analysis of Fc gamma RIII (CD16) membrane expression and association with CD3 zeta and Fc epsilon RI-gamma by site-directed mutation. J Immunol. 1991;146:1571–1576. [PubMed] [Google Scholar]
- 8.Wirthmueller U, Kurosaki T, Murakami MS, Ravetch JV. Signal transduction by Fc gamma RIII (CD16) is mediated through the gamma chain. J Exp Med. 1992;175:1381–1390. doi: 10.1084/jem.175.5.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Indik ZK, Hunter S, Huang MM, Pan XQ, Chien P, Kelly C, Levinson AI, Kimberly RP, Schreiber AD. The high affinity Fc gamma receptor (CD64) induces phagocytosis in the absence of its cytoplasmic domain: the gamma subunit of Fc gamma RIIIA imparts phagocytic function to Fc gamma RI. Exp Hematol. 1994;22:599–606. [PubMed] [Google Scholar]
- 10.Lowry MB, Duchemin AM, Robinson JM, Anderson CL. Functional separation of pseudopod extension and particle internalization during Fc gamma receptor-mediated phagocytosis. J Exp Med. 1998;187:161–176. doi: 10.1084/jem.187.2.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ioan-Facsinay A, de Kimpe SJ, Hellwig SM, van Lent PL, Hofhuis FM, van Ojik HH, Sedlik C, da Silveira SA, Gerber J, de Jong YF, Roozendaal R, Aarden LA, van den Berg WB, Saito T, Mosser D, Amigorena S, Izui S, van Ommen GJ, van Vugt M, van de Winkel JG, Verbeek JS. FcgammaRI (CD64) contributes substantially to severity of arthritis, hypersensitivity responses, and protection from bacterial infection. Immunity. 2002;16:391–402. doi: 10.1016/s1074-7613(02)00294-7. [DOI] [PubMed] [Google Scholar]
- 12.Barnes N, Gavin AL, Tan PS, Mottram P, Koentgen F, Hogarth PM. FcgammaRI-deficient mice show multiple alterations to inflammatory and immune responses. Immunity. 2002;16:379–389. doi: 10.1016/s1074-7613(02)00287-x. [DOI] [PubMed] [Google Scholar]
- 13.Edberg JC, Yee AM, Rakshit DS, Chang DJ, Gokhale JA, Indik ZK, Schreiber AD, Kimberly RP. The cytoplasmic domain of human FcgammaRIa alters the functional properties of the FcgammaRI.gamma-chain receptor complex. J Biol Chem. 1999;274:30328–30333. doi: 10.1074/jbc.274.42.30328. [DOI] [PubMed] [Google Scholar]
- 14.Qin H, Edberg JC, Gibson AW, Page GP, Teng L, Kimberly RP. Differential gene expression modulated by the cytoplasmic domain of Fc gamma RIa (CD64) alpha-chain. J Immunol. 2004;173:6211–6219. doi: 10.4049/jimmunol.173.10.6211. [DOI] [PubMed] [Google Scholar]
- 15.Hou X, Dietrich J, Geisler NO. The cytoplasmic tail of FcgammaRIIIAalpha is involved in signaling by the low affinity receptor for immunoglobulin G. J Biol Chem. 1996;271:22815–22822. doi: 10.1074/jbc.271.37.22815. [DOI] [PubMed] [Google Scholar]
- 16.Barraclough R, Savin J, Dube SK, Rudland PS. Molecular cloning and sequence of the gene for p9Ka. A cultured myoepithelial cell protein with strong homology to S-100, a calcium-binding protein. J Mol Biol. 1987;198:13–20. doi: 10.1016/0022-2836(87)90453-0. [DOI] [PubMed] [Google Scholar]
- 17.Schafer BW, Heizmann CW. The S100 family of EF-hand calcium-binding proteins: functions and pathology. Trends Biochem Sci. 1996;21:134–140. doi: 10.1016/s0968-0004(96)80167-8. [DOI] [PubMed] [Google Scholar]
- 18.Kriajevska MV, Cardenas MN, Grigorian MS, Ambartsumian NS, Georgiev GP, Lukanidin EM. Non-muscle myosin heavy chain as a possible target for protein encoded by metastasis-related mts-1 gene. J Biol Chem. 1994;269:19679–19682. [PubMed] [Google Scholar]
- 19.Ford HL, Zain SB. Interaction of metastasis associated Mts1 protein with nonmuscle myosin. Oncogene. 1995;10:1597–1605. [PubMed] [Google Scholar]
- 20.Grigorian M, Andresen S, Tulchinsky E, Kriajevska M, Carlberg C, Kruse C, Cohn M, Ambartsumian N, Christensen A, Selivanova G, Lukanidin E. Tumor suppressor p53 protein is a new target for the metastasis-associated Mts1/S100A4 protein: functional consequences of their interaction. J Biol Chem. 2001;276:22699–22708. doi: 10.1074/jbc.M010231200. [DOI] [PubMed] [Google Scholar]
- 21.Takenaga K, Nakamura Y, Sakiyama S, Hasegawa Y, Sato K, Endo H. Binding of pEL98 protein, an S100-related calcium-binding protein, to nonmuscle tropomyosin. J Cell Biol. 1994;124:757–768. doi: 10.1083/jcb.124.5.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kriajevska M, Bronstein IB, Scott DJ, Tarabykina S, Fischer-Larsen M, Issinger O, Lukanidin E. Metastasis-associated protein Mts1 (S100A4) inhibits CK2-mediated phosphorylation and self-assembly of the heavy chain of nonmuscle myosin. Biochim Biophys Acta. 2000;1498:252–263. doi: 10.1016/s0167-4889(00)00100-2. [DOI] [PubMed] [Google Scholar]
- 23.Kriajevska M, Fischer-Larsen M, Moertz E, Vorm O, Tulchinsky E, Grigorian M, Ambartsumian N, Lukanidin E. Liprin beta 1, a member of the family of LAR transmembrane tyrosine phosphatase-interacting proteins, is a new target for the metastasis-associated protein S100A4 (Mts1) J Biol Chem. 2002;277:5229–5235. doi: 10.1074/jbc.M110976200. [DOI] [PubMed] [Google Scholar]
- 24.Li X, Wu J, Carter RH, Edberg JC, Su K, Cooper GS, Kimberly RP. A novel polymorphism in the Fcgamma receptor IIB (CD32B) transmembrane region alters receptor signaling. Arthritis Rheum. 2003;48:3242–3252. doi: 10.1002/art.11313. [DOI] [PubMed] [Google Scholar]
- 25.Hampe CS, Pecht I. Protein tyrosine phosphatase activity enhancement is induced upon Fc epsilon receptor activation of mast cells. FEBS Lett. 1994;346:194–198. doi: 10.1016/0014-5793(94)00471-4. [DOI] [PubMed] [Google Scholar]
- 26.Edberg JC, Lin CT, Lau D, Unkeless JC, Kimberly RP. The Ca2+ dependence of human Fc gamma receptor-initiated phagocytosis. J Biol Chem. 1995;270:22301–22307. doi: 10.1074/jbc.270.38.22301. [DOI] [PubMed] [Google Scholar]
- 27.Coligan J. Current protocols in immunology. Vol. 3. John Wiley & Sons, Inc.; 2002. Preperation and analysis of phosphorylated proteins; pp. 11.12.11–13. [DOI] [PubMed] [Google Scholar]
- 28.Wang G, Rudland PS, White MR, Barraclough R. Interaction in vivo and in vitro of the metastasis-inducing S100 protein, S100A4 (p9Ka) with S100A1. J Biol Chem. 2000;275:11141–11146. doi: 10.1074/jbc.275.15.11141. [DOI] [PubMed] [Google Scholar]
- 29.Edberg JC, Qin H, Gibson AW, Yee AM, Redecha PB, Indik ZK, Schreiber AD, Kimberly RP. The CY domain of the Fcgamma RIa alpha-chain (CD64) alters gamma-chain tyrosine-based signaling and phagocytosis. J Biol Chem. 2002;277:41287–41293. doi: 10.1074/jbc.M207835200. [DOI] [PubMed] [Google Scholar]
- 30.Abrahams VM, Cambridge G, Lydyard PM, Edwards JC. Induction of tumor necrosis factor alpha production by adhered human monocytes: a key role for Fcgamma receptor type IIIa in rheumatoid arthritis. Arthritis Rheum. 2000;43:608–616. doi: 10.1002/1529-0131(200003)43:3<608::AID-ANR18>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 31.Kramer PR, Kramer SF, Guan G. 17 beta-estradiol regulates cytokine release through modulation of CD16 expression in monocytes and monocyte-derived macrophages. Arthritis Rheum. 2004;50:1967–1975. doi: 10.1002/art.20309. [DOI] [PubMed] [Google Scholar]
- 32.Nishida K, Yamasaki S, Ito Y, Kabu K, Hattori K, Tezuka T, Nishizumi H, Kitamura D, Goitsuka R, Geha RS, Yamamoto T, Yagi T, Hirano T. Fc{epsilon}RI-mediated mast cell degranulation requires calcium-independent microtubule-dependent translocation of granules to the plasma membrane. J Cell Biol. 2005;170:115–126. doi: 10.1083/jcb.200501111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gu H, Maeda H, Moon JJ, Lord JD, Yoakim M, Nelson BH, Neel BG. New role for Shc in activation of the phosphatidylinositol 3-kinase/Akt pathway. Mol Cell Biol. 2000;20:7109–7120. doi: 10.1128/mcb.20.19.7109-7120.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kanakaraj P, Duckworth B, Azzoni L, Kamoun M, Cantley LC, Perussia B. Phosphatidylinositol-3 kinase activation induced upon Fc gamma RIIIA-ligand interaction. J Exp Med. 1994;179:551–558. doi: 10.1084/jem.179.2.551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bonnema JD, Karnitz LM, Schoon RA, Abraham RT, Leibson PJ. Fc receptor stimulation of phosphatidylinositol 3-kinase in natural killer cells is associated with protein kinase C-independent granule release and cell-mediated cytotoxicity. J Exp Med. 1994;180:1427–1435. doi: 10.1084/jem.180.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ferby IM, Waga I, Hoshino M, Kume K, Shimizu T. Wortmannin inhibits mitogen-activated protein kinase activation by platelet-activating factor through a mechanism independent of p85/p110-type phosphatidylinositol 3-kinase. J Biol Chem. 1996;271:11684–11688. doi: 10.1074/jbc.271.20.11684. [DOI] [PubMed] [Google Scholar]
- 37.Kriajevska M, Tarabykina S, Bronstein I, Maitland N, Lomonosov M, Hansen K, Georgiev G, Lukanidin E. Metastasis-associated Mts1 (S100A4) protein modulates protein kinase C phosphorylation of the heavy chain of nonmuscle myosin. J Biol Chem. 1998;273:9852–9856. doi: 10.1074/jbc.273.16.9852. [DOI] [PubMed] [Google Scholar]
- 38.Chuang HC, L. J, Chen DY, Yang CY, Chen YM, Li JP, Huang CY, Liu PE, Wang X, Tan TH. The kinase GLK controls autoimmunity and NF-κB signaling by activating the kinase PKC-θ in T cells. Nat Immunol. 2011;12:1113–1118. doi: 10.1038/ni.2121. [DOI] [PubMed] [Google Scholar]
- 39.Wu J, Ji C, Xie F, Langefeld CD, Qian K, Gibson AW, Edberg JC, Kimberly RP. FcalphaRI (CD89) alleles determine the proinflammatory potential of serum IgA. J Immunol. 2007;178:3973–3982. doi: 10.4049/jimmunol.178.6.3973. [DOI] [PubMed] [Google Scholar]
- 40.Zheleznyak A, Brown EJ. Immunoglobulin-mediated phagocytosis by human monocytes requires protein kinase C activation. Evidence for protein kinase C translocation to phagosomes. J Biol Chem. 1992;267:12042–12048. [PubMed] [Google Scholar]
- 41.Chow SC, Ng J, Nordstedt C, Fredholm BB, Jondal M. Phosphoinositide breakdown and evidence for protein kinase C involvement during human NK killing. Cell Immunol. 1988;114:96–103. doi: 10.1016/0008-8749(88)90257-2. [DOI] [PubMed] [Google Scholar]
- 42.Leitges M, Gimborn K, Elis W, Kalesnikoff J, Hughes MR, Krystal G, Huber M. Protein kinase C-delta is a negative regulator of antigen-induced mast cell degranulation. Mol Cell Biol. 2002;22:3970–3980. doi: 10.1128/MCB.22.12.3970-3980.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Procko E, McColl SR. Leukocytes on the move with phosphoinositide 3-kinase and its downstream effectors. Bioessays. 2005;27:153–163. doi: 10.1002/bies.20157. [DOI] [PubMed] [Google Scholar]
- 44.Kramer PR, Winger V, Reuben J. PI3K limits TNF-alpha production in CD16-activated monocytes. Eur J Immunol. 2009;39:561–570. doi: 10.1002/eji.200838801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saleem M, Kweon MH, Johnson JJ, Adhami VM, Elcheva I, Khan N, Hafeez B. Bin, Bhat KM, Sarfaraz S, Reagan-Shaw S, Spiegelman VS, Setaluri V, Mukhtar H. S100A4 accelerates tumorigenesis and invasion of human prostate cancer through the transcriptional regulation of matrix metalloproteinase 9. Proc Natl Acad Sci U S A. 2006;103:14825–14830. doi: 10.1073/pnas.0606747103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.