

Abstract
Interferon regulatory factor 3 (IRF3) regulates early type I IFNs and other genes involved in innate immunity. We have previously shown that cells undergoing an endoplasmic reticulum (ER) stress response called the “Unfolded Protein Response” (UPR) produce synergistically augmented IFN-β when stimulated with pattern recognition receptor agonists such as LPS Concomitant ER stress and LPS stimulation resulted in greater recruitment of the IRF3 transcription factor to ifnb1 gene regulatory elements. In this study, we utilized murine cells to demonstrate that both oxygen-glucose deprivation and pharmacologic UPR inducers trigger phosphorylation and nuclear translocation of IRF3, even in the absence of exogenous LPS. Different ER stressors utilized distinct mechanisms to activate IRF3: IRF3 phosphorylation due to calcium-mobilizing ER stress (thapsigargin treatment, oxygen-glucose deprivation) critically depended upon Stimulator of interferon gene (STING), an ER-resident nucleic acid-responsive molecule. However, calcium mobilization alone by ionomycin was insufficient for IRF3 phosphorylation. In contrast, other forms of ER stress (e.g., tunicamycin treatment) promote IRF3 phosphorylation independently of STING and Tank binding kinase 1 (TBK1). Rather, IRF3 activation by tunicamycin and 2-deoxyglucose was inhibited by AEBSF, a serine protease inhibitor that blocks ATF6 processing. Interfering with ER stress-induced IRF3 activation abrogated IFN-β synergy. Together, these data suggest ER stress primes cells to respond to innate immune stimuli by activating the IRF3 transcription factor. Our results also suggest certain types of ER stress accomplish IRF3 phosphorylation by co-opting existing innate immune pathogen response pathways. These data have implications for diseases involving ER stress and type I IFN.
Introduction
Type I IFNs (IFN-α/β) play diverse roles in adaptive and innate immunity; Type I IFNs activate macrophages and NK cells, promote T cells survival and dendritic cell maturation, and increase the production of Th1-polarizing cytokines(1). Innate immune cells such as macrophages and dendritic cells produce large amounts of type I IFN following the ligation of diverse pattern recognition receptors (PRRs). PRRs recognize conserved molecular structural motifs on pathogens, as well as endogenous products released by tissue damage(2). The PRRs that mediate IFN-β induction in macrophages include the LPS receptor TLR4, the endosomal dsRNA sensor TLR3, and the cytoplasmic dsRNA responsive retinoic acid-inducible gene-I (RIG-I) family helicases(3). Interestingly, a recently identified molecule STimulator of INterferon Gene (STING, also known as MPYS/MITA/TMEM173/ERIS), located in the ER membrane, appears to play a critical role in the induction of IFN-β by cytoplasmic dsDNA and RNA, though STING does not directly bind nucleic acids(4-7).
Signaling by these various pathogen sensors converges at the activation of the Tank-binding kinase 1 (TBK1) family of kinases(8). TBK1 is a serine/threonine kinase that phosphorylates the transcription factor interferon regulatory factor 3 (IRF3)(9). IRF3 is constitutively expressed and resides in the cytoplasm in latent form. Upon phosphorylation, IRF3 dimerizes and translocates from the cytoplasm into the nucleus(10). At the ifnb1 locus, IRF3 cooperatively binds with other transcription factors including NF-κB, AP-1, and IRF7 to form a multi-molecular “enhanceosome” that promotes ifnb1 transcription(11). IRF3 is absolutely required for the induction of IFN-β and certain IFN-α species early during viral infections, and by LPS(12-14). IRF3-regulated early type I IFN production primes cells for greater IFN responses during viral infections by inducing IRF7(15). IRF3 also regulates other inflammatory mediators such as the chemokines CXCL10 and RANTES(16-18). In a murine model of hepatic ischemia-reperfusion injury, damage is significantly decreased in both type I IFN receptor and in IRF3-deficient animals(19, 20). In addition to its transcriptional role, IRF3 promotes apoptosis in virus-infected cells through association with Bax(21).
Even as innate immune cells are poised to counter external threats, conserved stress responses respond to intracellular derangements. We, and others have shown that type I IFN responses to PRR ligands are dramatically enhanced by an intracellular stress response originating in the ER called the “Unfolded Protein Response” (UPR)(22-25). The UPR represents a final common pathway in the response to a broad variety of stresses perturbing ER function, including oxygen and nutrient deprivation, calcium dysregulation, misfolded proteins and N-linked glycosylation inhibition(26). The three major signaling cascades of the UPR stem from activation of ER-resident molecules: protein kinase receptor-like ER kinase (PERK,) the proto-transcription factor ATF6, and inositol-requiring enzyme (IRE-1). IRE-1 is both a kinase and endonuclease that cleaves 26bp from the X-box binding protein (XBP1) transcription factor mRNA. This atypical splicing eliminates a premature stop codon, and thus enables translation of full length active XBP1(26). XBP1 is essential for synergistic type I IFN responses to PRR agonists(22). We have shown that XBP1 binds an enhancer element 6kb downstream of the ifnb1 gene(23). Interestingly, during concomitant UPR and LPS stimulation, IRF3 recruitment increased at both ifnb1 promoter and enhancer(23). IRF3 binding was only observed in the presence of LPS(23). Although XBP1, a member of the CREB family of transcription factors is known to heterodimerize with other transcription factors, IRF3 is not a known binding partner(27, 28).
Investigations of gene expression in macrophages from rats overexpressing the misfolding protein HLA-B27 reveal both UPR and IFN signature, including several IRF3-regulated genes (IFN-β, OAS and IP-10)(29). However the relationship between ER stress and IRF3 activation was unclear. A previous study had demonstrated activation of IRF3, evident by phosphorylation, nuclear translocation and CREB-binding protein (CBP) association in response to DNA-damaging agents such as doxorubicin in Hela cells(30). However a later study employing a wider range of stessors failed to validate these findings in HEK293 cells(31). In a recent study by Hu et al., the UPR inducer thapsigargin augmented Poly I:C dependent IRF3-phosphorylation, supporting the involvement of UPR in IRF3 regulation(25). This study also supported a role for XBP1 in promoting anti-viral activity.
In this study we sought to determine whether ER stress exerts a direct effect on IRF3 activation. We found that multiple pharmacologic ER stress inducers and oxygen-glucose deprivation triggered activation of IRF3, as indicated by IRF3 phosphorylation and nuclear translocation. Mechanism of IRF3 activation varied by type of ER stressor: Some forms of ER stress, in particular those that also involve calcium mobilization, require TBK1 and STING for phosphorylation and nuclear translocation of IRF3. Indeed, STING was essential for synergistic IRF3 phosphorylation and optimal IFN-β induction by concurrent thapsigargin and LPS. These data suggest intracellular stress responses may utilize innate immune pathogen sensing pathways to augment IRF3-regulated cytokine and chemokine production. In contrast, other ER stress inducers activated IRF3 by a pathway inhibited by 4-(2-Aminoethyl)-Benzenesulfonyl Fluoride Hydrochloride (AEBSF), a serine protease inhibitor that prevents UPR-dependent processing of ATF6. Thus, at least 2 distinct pathways link ER stress to IRF3 activation.
Materials and Methods
Cells, reagents and stimulations
The RAW264.7 macrophage cell line (ATCC) was maintained in DMEM/high glucose (Mediatech) with 10%FBS (Hyclone), and 1X antibiotic-antimycotic solution (Mediatech). Murine bone marrow macrophages were isolated from C57Bl/6 femurs with Histopaque 1083 (Sigma) and plated 3 days in non-tissue culture petri dishes in DMEM (as above) supplemented with 5% M-CSF-containing conditioned supernatant from CMG-14-12 cells(23, 32). Adherent cells were detached by 10mM EDTA and re-plated in tissue culture dishes with CMG-14-12 supernatant 3 more days. IRF3-/- mice were previously described(13). WT and STING-/- MEF were maintained in DMEM supplemented with 10% FBS (not heat inactivated, Invitrogen), and 1X antibiotic-antimycotic solution(4). XBP1-/- MEFs were kindly provided by Laurie Glimcher (Harvard University, Boston, MA). For RNAi studies, RAW 264.7 cells were transiently transfected with STING or control RNAi (Dharmacon) using AMAXA (Lonza).
To induce ER stress, cells were treated with 10μg/mL tunicamycin (Tm), 20mM 2-deoxyglucose (2DG), 10μM A23187, or 1μM thapsigargin (Tg, all from Sigma). Oxygen–glucose deprivation (OGD): cells were washed 3 times with a glucose-free isotonic salt solution (OGD buffer pH 7.4: 20mM NaHCO3, 120mM NaCl, 5.36mM KCl, 0.33 Na2HPO4mM, 0.44mM KH2PO4, 1.27mM CaCl2, and 0.81mM MgSO4), then incubated in OGD buffer in hypoxic condition containing 94% N2, 5% CO2 and 1%O2(33). Following OGD, cells were provided with growth media with or without LPS and transferred to a standard 5% CO2 incubator. For LPS stimulations, RAW 264.7 macrophage cells were treated with 100ng/mL and primary bone marrow macrophages and MEFs treated with 10ng/mL 3h unless otherwise indicated. Cytokine and chemokine protein assessment: IFN-α and IFN-β ELISA kits were from PBL Interferon Source, and eBioscience (RANTES). TBK1/IKKε inhibition: cells were pre-treated with 2 μM MRT67307 (generous gift from Dr. Philip Cohen, University of Dundee, Scotland) (34). The serine protease inhibitor AEBSF (4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride) was from Sigma.
Antibodies
β-actin, mouse mAb, NF-κB rabbit polyclonal Ab, and USF2 rabbit polyclonal Ab were from Santa Cruz; HA-Tag mouse mAb, IRF3 rabbit mAb (detects 1 IRF3 band) and phospho-IRF3 (pS396) rabbit mAb from Cell Signaling; Phospho-IRF3 (pS386) rabbit mAb from Epitomics; IRF3 rabbit polyclonal (detects 2 IRF3 forms), STING rabbit polyclonal Ab, and ATF6 mAb from Prosci; TBK1 rabbit mAb from Abcam; normal rabbit IgG from Millipore. Secondary antibodies: goat anti-mouse-IgG-Alexa Fluor 594 and anti-rabbit IgG-Alexa Fluor 488 (Invitrogen).
Immunofluorescence microscopy
Cells were plated on cover slips in 60 mm dishes with 24hr prior to treatment. For STING-TBK1 co-localization, MEFs were transiently transfected with MPYS-HA (generous gift from John Cambier, University of Colorado, Denver) using Fugene HD (Roche) 24h prior to stimulation(5). After treatment, cells were washed (3×5 min PBS), and then fixed in 4% paraformaldehyde 30 min at room temperature. Cells were then washed with PBS, Tris A buffer (0.1M pH 7.6 Tris, 0.1% Triton X-100) and Tris B buffer (0.1M pH 7.6 Tris, 0.1% Triton X-100, 0.2% bovine serum albumin) 3×5 min each, and incubated with 10% goat serum in Tris B buffer 1h. Primary antibodies were added in Tris B buffer and cells incubated at 4°C overnight. After washing the cells with Tris A 3×5 min, secondary fluorescence-conjugated antibody was added and samples incubated 1 h at room temperature. Cells were washed with PBS 3×5 min, and the coverslips mounted on slides with ProLong® Gold antifade reagent with DAPI nuclear stain (Invitrogen). For negative controls, the same concentration of primary mouse IgG (Sigma) or rabbit IgG (Sigma) was added. Images were acquired on Nikon Eclipse 50i fluorescence microscope. Bars in images are 50μM.
Quantitative PCR
Cells were lysed with Trizol (Invitrogen) and processed according to manufacturer's instructions. Briefly, RNA was extracted with chloroform and precipitated with isopropanol. Total RNA was purified with and treated with DNase I (Invitrogen) prior to reverse transcription using random primers (Promega). Gene expression level in cDNA was quantitated by SYBR Green (Bio-Rad) fluorescence, using an I-cycler or My-IQ (Bio-Rad). Relative mRNA expression was normalized to 18S rRNA housekeeping gene. Primers were designed using Beacon design software (Premier Biosoft): 18S rRNA: Forward (F) GGA CAC GGA CAG GAT TGA CAG and Reverse (R) ATC GCT CCA CCA ACT AAG AAC G; IFN-β: F-ACT AGA GGA AAA GCA AGA GGA AAG -3’ and R-CCA CCA TCC AGG CGT AGC; IFN-α4: F-CAC AAT GGC TAG GCT CTG and R-TGT TGG TTA TCC ACC TTC TC; IL-6: F-CTT CCA TCC AGT TGC CTT C and R-ATT TCC ACG ATT TCC CAG AG; IL1β: F-CTC GCA GCA GCA CAT CAA C and R-ACG GGA AAG ACACAG GTA GC; STING: F-TGC CGC CTC ATT GTC TAC and R-GCT GAT CCA TAC CAC TGA TG; BiP: F-AGG ATG CGG ACA TTG AAG AC and R-AGG TGA AGA TTC CAA TTA CAT TCG; spliced XBP1: F-GAG TCC GCA GCA GGT G and R-GTG TCA GAG TCC ATG GGA; ifit2: F-GAT TCT GTC TAC CAC CAT and R-CAA GCA TCA ATC CAA GTT ; RANTES: F-GAA TAC ATC AAC TAT TTG GAG AT and R-TAG AGC AAG CAA TGA CAG.
Western blotting and co-immunoprecipitations
Cells were lysed with RIPA buffer containing protease and phosphatase inhibitor cocktails (Sigma) and whole cell lysates, or cytoplasmic and nuclear fractions (Nuclear extraction kit, Sigma) resolved by 10% SDS-PAGE. Samples were transferred to PVDF membrance (Amersham), immunoblotted with primary antibody, followed by horseradish peroxidase conjugated secondary antibody (Bio-Rad). Proteins were visualized by ECL (Amersham) and film exposure.
MEF lysates (150mM NaCl, 1% Triton X-100, 1mM EDTA, 50mM Tris pH7.5, DTT, Protease inhibitor cocktail) were incubated 1h with anti-STING at 4°C, and then 1%BSA/PBS suspended protein A/G agarose (Santa cruz). Samples were rotated at 4°C over night. Beads were washed 2X with PBS, added to loading buffer, and then samples were denatured and resolved by SDS-PAGE.
Statistics
Statistical significance between different groups of data was determined by 2-tailed Student's T-test. In all figures, error bars represent standard errors of the mean from combined experiments or standard deviation within a representative experiment.
Results
Oxygen-glucose deprivation synergistically augments LPS-induced IFN-β and activates IRF3
In previous studies, we determined that multiple pharmacologic inducers of the UPR, or an overexpressed misfolding MHC allele HLA-B27, greatly augment LPS-dependent IFN-β mRNA and protein production(22, 23). This robust synergy was observed in primary macrophages, macrophage cell lines, murine embryonic fibroblasts (MEFs), and LPS-receptor-transfected HEK293 cells(22, 23). However, it was unclear if more “physiologic” stress, such as the oxygen and nutrient deprivation that might occur with ischemia, would enhance IFN-β production. MEFs were subjected to oxygen and glucose deprivation (OGD) for various times, and then stimulated with LPS for 3 more hours (Figure 1A). OGD alone did not induce IFN-β mRNA; however OGD dramatically augmented LPS-induced IFN-β, with maximal enhancement of a log-fold occurring after 1h of OGD pretreatment. Thus in vitro “ischemia” augmented IFN-β mRNA levels to a similar degree as the pharmacologic UPR inducers. In vitro OGD has been shown to induce early UPR events within 15 minutes(35). We confirmed BiP induction and increase in spliced XBP1 mRNA during the culture time frame (Figure 1B). OGD also induced IFN-β and BiP in RAW 264.7 macrophages (data not shown).
Figure 1.
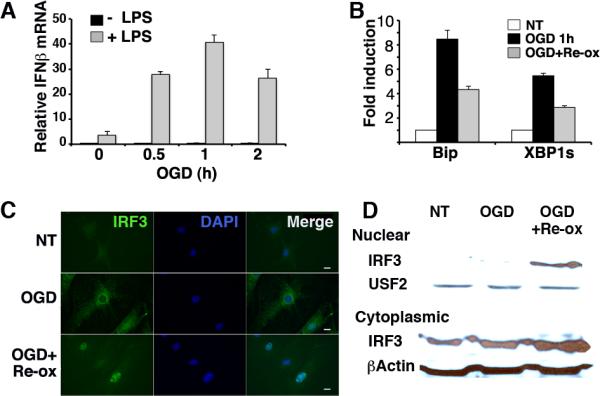
Oxygen glucose deprivation (OGD) synergistically augments LPS-induced IFN-β and causes IRF3 nuclear translocation. A) MEFs were subjected to 0, 0.5, 1, or 2 h OGD, followed by media with, or without 10ng/mL LPS for 3h in standard culture conditions. IFN-β expression was determined by quantitative PCR (qPCR) with normalization to 18S rRNA (Relative mRNA). Bars represent combined results from 3 independent experiments with error bars showing SEM. p<0.0005 comparing OGD pretreated conditions and no OGD. Comparable results have been obtained in 5 independent experiments. B) MEFs were subjected to 1h OGD followed by standard culture conditions as above. Expression of BiP and spliced XBP1 was detected by qPCR and normalized to untreated controls for fold induction (NT=1). Results are representative of 2 individual experiments and error bars denote S.D. P<0.0004 comparing OGD treated and NT cells. C) MEFs were subjected to OGD for 1h followed by 2h re-oxygenation (Re-ox) in standard culture as indicated. Fixed cells were incubated with anti-IRF3 followed by anti-rabbit IgG Alexa Fluor 488, and visualized by immunofluorescence microscopy. Results are representative of 3 independent experiments. D) Nuclear and cytoplasmic lysates were resolved by SDS PAGE and immunoblotted with anti-IRF3, β-actin, or the nuclear protein anti-USF2. Results are representative of 2 separate experiments.
In dissecting the mechanism behind UPR-TLR4 synergistic IFN-β production, we observed increased IRF3 recruitment to both the ifnb1 promoter and to a newly described enhancer in the context of ER stress(23). Although ER stress alone was not sufficient for recruitment of IRF3 to DNA, these findings raised the possibility that ER stress might contribute to the activation of IRF3. In unstimulated cells, IRF3 resides in the cytoplasm as monomers. Upon stimulation through pattern recognition receptors, serine-threonine kinases such as TBK1 and IKKe/IKKi phosphorylate IRF3 at multiple sites, resulting in dimerization of IRF3 and nuclear translocation(10). We tested whether OGD would affect IRF3 localization in MEFs in the absence of exogenous LPS (Figure 1C, D). After 1h of OGD, IRF3 resided in the cytoplasm. However, after 2h of re-oxygenation, IRF3 appeared predominantly nuclear, co-localizing with DAPI by immunofluorescence microscopy. Nuclear translocation was also evident as assessed by western blot (Figure 1D).
ER stress results in the phosphorylation and nuclear translocation of IRF3
Oxygen-glucose deprivation induces various stress responses including the UPR(35, 36). To determine if pharmacologic UPR induction stimulates the nuclear translocation of IRF3, MEFs were treated with thapsigargin, a SERCA pump inhibitor frequently utilized to study the UPR in vitro.(37) By 2h of thapsigargin treatment, IRF3 levels increased in nuclear extracts as detected by western blot (Figure 2A). Differentially migrating forms of IRF3 have been reported in unstimulated cells (referred to as “Forms I and II”) and stimulated cells (Forms III and IV); these forms may represent variably phosphorylated or alternatively spliced forms of IRF3 detected by polyclonal antibody preparations(31, 38, 39). Similar results were observed in RAW 264.7 cells (data not shown). The kinetics observed by immunofluorescence microscopy correlated well with these findings (Figure 2B). Previous studies had identified a critical role for the UPR regulated transcription factor XBP1 in synergistic IFN induction(22, 23, 25). To determine if XBP1 was required for IRF3 activation, nuclear translocation was examined in XBP1-/- MEFs (Figure 2C). Thapsigargin-induced nuclear translocation of IRF3 was intact, suggesting that the UPR activates IRF3 independently of the XBP1 pathway. Similar results were obtained with other ER stressors (data not shown).
Figure 2.

ER stress induces IRF3 nuclear translocation and phosphorylation. A) MEFs were treated with 1 μM Tg for the times indicated. Nuclear lysates were resolved by SDS page and immunoblotted with anti-IRF3 or β-actin control. Results are representative of 4 independent experiments. B) MEFs were treated with 1 μM Tg for 1 or 2h, fixed, incubated with anti-IRF3 followed by anti-rabbit IgG Alexa Fluor 488, and visualized by immunofluorescence microscopy. Results are representative of 3 independent experiments. C) XBP1-/- MEFs were treated with Tg for 0, 60 or 90 min and western blots incubated with anti-IRF3 or anti USF2. Results are representative of 3 independent experiments. D) MEFs were left untreated or subjected to OGD for 1h, 10μg/mL tunicamycin (Tm) 6h, 20mM 2-deoxyglucose (2DG) 6h, 1 μM Tg for 2h, or LPS 10ng/mL for 2h. Fixed cells were incubated with anti-pS386 IRF3 followed by anti-rabbit IgG Alexa Fluor 488, and visualized by immunofluorescence microscopy. Results are representative of 4 independent experiments. E) Wild type (WT) or IRF3-/- primary bone marrow derived macrophages were treated with 1μM Tg for 2h prior to fixation and staining for p-IRF3 (Ser 386). Results are representative of 2 independent experiments.
Nuclear translocation of IRF3 theoretically implies preceding phosphorylation. To directly test if the UPR elicits IRF3 phosphorylation, MEFs were stimulated with multiple commonly utilized pharmacologic UPR inducers, including thapsigargin, tunicamycin (N-linked glycosylation inhibitor), 2-deoxyglucose (glucose deprivation simulator), and OGD, with LPS as a positive control (Figure 2C). Treatment times were based upon mechanism of action, time required to induce XBP1 splicing (data not shown), and required pre-treatment times for synergistic IFN-β induction(23). All UPR agents induced detectable IRF3 phosphorylation at S386, comparable with LPS treatment (Figure 2D). IRF3 phosphorylation was not detected in IRF3-/- primary macrophages, confirming immunofluorescence specificity (Figure 2E). Synergistic IRF3 phosphorylation with dual thapsigargin and LPS treatment was not evident by immunofluorescence (data not shown) in comparison to western blot (see below). The pattern of phospho-IRF3 localization differed between LPS and UPR inducers; LPS treatment resulted in qualitatively higher order clustering (dots arranged in small circular clusters, compare OGD vs. LPS).
ER stress does not augment all IRF3 regulated genes
Our previous studies examining UPR-TLR synergy had focused on IFN-β production(23). However, the activation of IRF3 by ER stress raised the possibility that ER stress might augment the production of other known IRF3-regulated genes. Martinon et al. reported log-fold synergistic induction of the IRF3 regulated gene ISG15 in tunicamycin+LPS stimulated cells(24). IFN-α4 transcription is an early type I IFN, whose expression is highly dependent upon IRF3(15, 40). In addition to increased IFN-β production, primary bone marrow-derived mouse macrophages treated concurrently with thapsigargin and LPS produced increased levels of IFN-α4 (Figure 3). Thus the effect of ER stress on IRF3 activation has consequences for other anti-viral and inflammatory mediators besides IFN-β. As previously described, synergistic IFN-β mRNA induction correlated well with protein production(23). IFN-α was produced at such low level; only thapsigargin+LPS stimulated cytokine was present above the limit of detection (data not shown). However, synergistic induction of IRF3 regulated genes was not universal: neither Ifit2/ISG54 nor RANTES (as noted by others) were significantly augmented by UPR induction(24).
Figure 3.

ER stress augments LPS induction of some, but not all IRF3 regulated genes. A) Bone marrow derived macrophages were treated with 1μM Tg, 10ng/ml LPS or 1μM Tg+ 10ng/ml LPS for 3 hours. Relative expression of IFN-β, IFN-α4, RANTES, and ifit2 was determined by qPCR with normalization to 18S rRNA. Bars depict combined means of 6 (IFN-β, *p=0.001), 3 (IFN-α4, *p<0.006), and 4 (RANTES and ifit2) independent experiments with error bars showing the SEM. To combine IFN-α4, RANTES and ifit2 experiments, relative expression was normalized (fold induction vs. LPS=1). B) Macrophages were treated as above for 8h and supernatants assessed for cytokine and chemokine protein by ELISA. Bars represent standard error of triplicate determinations. *P<0.00002 (IFN-β).
Activation of IRF3 by thapsigargin requires STING and TBK1
To begin elucidating the mechanism of UPR-induced IRF3 phosphorylation, we sought to determine the relevant IRF3 kinase. TBK1 is an upstream serine/threonine kinase that serves as the final point of convergence for multiple innate immune sensing pathways culminating in the phosphorylation of IRF3(8, 9, 16). Within the past few years, STING has been identified as an ER resident transmembrane protein that plays a critical role in the induction of IRF3 and thus IFN-β by cytoplasmic nucleic acids(4). The direct nucleic acid-binding molecule(s) upstream of STING have not yet been well defined, but may include the helicase DDX41(41). Upon activation by nucleic acids, STING associates with, and phosphorylates IRF3 via TBK1(4, 6).
The ER resident location of STING raised the possibility that ER stress induced activation of IRF3 might proceed through this pathway. However, it was not clear if ER stress-induced IRF3 phosphorylation required TBK1 or STING, or induced TBK1-STING association. To determine the requirement for TBK1, MEFs were pre-treated with MRT67307, a TBK1/IKKε family kinase inhibitor, prior to thapsigargin stimulation(34). MRT67307 abrogated thapsigargin-dependent IRF3 phosphorylation (Figure 4A). To determine if ER stress induced association between TBK1 and STING, co-immunoprecipitation was performed. TBK1 and STING associated even before stimulation, and the association continues with thapsigargin treatment (Figure 4B). Similar results were obtained in primary macrophages (data not shown). However, by immunofluorescence microscopy, a striking ER stress-induced relocalization of STING and TBK1 was evident, with association into larger order clusters around the nucleus (Figure 4C). Together these data suggested that thapsigargin-induced IRF3 phosphorylation requires TBK1 and that thapsigargin mobilizes STING and TBK1. However, it was not clear if STING was required for thapsigargin-induced IRF3 phosphorylation via TBK1.
Figure 4.

Thapsigargin induces co-localization of TBK1 and STING and requires STING and TBK1 family kinases for IRF3 phosphorylation. A) MEFs were pre-treated with 2μM MRT67307 for 30 min prior to 1μM Tg for 2h. Fixed cells were incubated with anti-pIRF3 (pS386) followed by Alexafluor488 coupled secondary antibody and immunofluorescence detected by microscopy. Results are representative of 4 independent experiments. B) MEFs were untreated or treated with 1μM Tg for 2h. Samples were immunoprecipitated with control IgG or anti-STING. Whole cell lysate (input) or immunoprecipitations were resolved by SDS-PAGE and immunoblotted with anti-TBK1 or anti-STING. Results are representative of 4 independent experiments. C) MEFs were transfected with STING/MPYS-HA 24h prior to stimulation with 1μM TPG for 2h. Fixed cells were incubated with rabbit anti-TBK1 and mouse anti-HA followed by anti-rabbit Alexa Fluor 488 (green) or anti-mouse Alexa Fluor 594 (red) secondary antibodies. Results are representative of 3 independent experiments. D) WT or STING-/- MEFs were treated with 1 μM Tg for 2h as indicated. Cells were then fixed, incubated with anti-pS386 IRF3 followed by anti-rabbit IgG Alexa Fluor 488, and visualized by immunofluorescence microscopy. Results are representative of 4 independent experiments. E) WT and STING-/- MEFs were stimulated with 1μM Tg, 10ng/mL LPS, or 1μM Tg+10ng/ml LPS for 2 hrs. Nuclear lysates were resolved by SDS-PAGE and immunoblotted with antibodies specific for pIRF3 (S396), total IRF3, NF-κB, or USF2. Results are representative of 3 independent experiments.
To determine if STING was specifically required for ER stress-induced IRF3 phosphorylation, STING-/- MEFs were stimulated with thapsigargin and assessed by immunofluorescence for IRF3 phosphorylation(4). Neither IRF3 phosphorylation nor nuclear translocation were evident in STING-/- MEFs (Figure 4D and IRF3 immunofluorescence not shown). By immunoblot, in wild type MEFs, p-IRF3 (S396) is detectable in LPS-stimulated nuclear lysates, although we did not detect thapsigargin-induced p-IRF3 nuclear phosphorylation. In the WT cells, thapsigargin and LPS co-treatment increased levels of p-IRF3 over that observed with LPS alone (Figure 4E). These results are consistent with those observed by Hu et al, where thapsigargin augmented poly I:C-dependent IIRF3 phosphorylation but did not appear to induce IRF3 on its own(25). However, in the STING-/- MEFs, no p-IRF3 was observed by immunoblot. The lack of p-IRF3 was not because of IRF3 deficiency or generally defective LPS signaling in the MEFs, as LPS-induced NF-κB nuclear translocation was intact in STING-/- MEFs. Together these results support the idea that thapsigargin utilizes STING and downstream TBK1 to phosphorylate IRF3, and that synergistic p-IRF3 induction by LPS and thapsigargin requires STING.
Optimal synergistic induction of IFN-β by thapsigargin and LPS requires STING
STING appeared to be required for phosphorylation of IRF3 during ER stress and synergistically induced p-IRF3 during thapsigargin and LPS stimulation. However the consequences for an IRF3 regulated gene, such as IFN-β, were not clear. Thus, synergistic IFN-β induction was evaluated in RNAi transfected RAW 264.7 macrophages and STING-/- bone marrow- derived macrophages. Even moderate knockdown of STING with RNAi (Figures 5A and 5B) decreased IFN-β induction by combined thapsigargin and LPS stimulation. There was no significant effect of STING knockdown on LPS-induced IFN-β alone or the induction of two IRF3-independent cytokines, IL-1β and IL-6, by any of the stimuli. The effect of STING deficiency was more evident in the primary STING-/- bone marrow macrophages in comparison with the RNAi knockdown (Figure 5C). Again, IL-6 induction was intact. Synergy was not completely abrogated in the STING-/- macrophages or in STING-/- MEFs (data not shown). However, the magnitude of synergistic IFN-β expression was decreased by about a log-fold in primary macrophages.
Figure 5.

STING is required for optimal synergistic induction of IFN-β by thapsigargin and LPS. A) RAW264.7 macrophages were transfected with 100-200nM Control or STING specific RNAi. After 24h, cells were treated with 1h 1μM Tg followed by 100ng/mL LPS for 3h. Relative expression of STING, IFN-β, IL-1β and IL-6 was determined by q-PCR with normalization to 18S rRNA. Results were combined from 3 (STING and IL-1β) or 4 (IFN-β and IL-6) independent experiments with error bars representing the SEM. *P≤ 0.032, **p=0.011. B) RAW264.7 macrophages were transfected with control or STING specific RNAi and lysed at 24, 48 or 72h. Lysates were resolved by SDS-PAGE and immunoblotted with anti-STING. Western blot was performed twice. C) WT or STING-/- bone marrow derived macrophages were treated with 1μM Tg, 10ng/mL LPS or TPG+LPS for 3h. Cells were processed as in (A) by qPCR to assess IFN-β and IL-6 mRNA expression. Results were combined from 3(WT) and 5(STING-/-) independent experiments, with error bars representing the SEM. **p=6.1e-6.
Not all ER stressors require TBK1 and STING for IRF3 phosphorylation; ER stressors mobilizing calcium depend upon STING for IRF3 phosphorylation
Although all of the frequently utilized pharmacologic ER stress-inducing agents activate the three primary signaling pathways encompassed by the UPR and have final common endpoints, their mechanisms of action vary. Ultimately, the quality and magnitude of all the various UPR stress pathways induced by these agents are likely to differ as well. To determine if the dependence on STING was a general property of UPR induction, STING+/+ and -/- MEFs were stimulated with tunicamycin, an agent that induces the UPR independently of calcium (Figure 6A). There were no evident differences in tunicamycin-induced IRF3 phosphorylation in the presence or absence of STING. Likewise, 2-deoxyglucose-induced IRF3 phosphorylation occurred in the absence of STING. Consistent with these findings, tunicamycin-induced IRF3 phosphorylation also did not require TBK1 family kinases, since MRT67307 did not prevent tunicamycin induced IRF3 phosphorylation (Figure 6A). Thus, although thapsigargin and tunicamycin treatment may lead to similar common endpoints, the ER stress responses are qualitatively different. These data suggest that different UPR inducers activate IRF3 through a variety of signaling pathways.
Figure 6.

STING-dependent IRF3 phosphorylation requires both calcium mobilization and ER stress. A) WT or STING-/- MEFs were pre-treated with 2μM MRT67307 for 30 min then 10μg/mL Tm for 6h as indicated. Cells were incubated with anti-pIRF3 (S386) followed by Alexa Fluor 488 secondary antibody and visualized by immunofluorescence microscopy. Results are representative of 4 independent experiments. B) WT or STING-/- MEFs were untreated (NT), treated with 1μM ionomycin or 2μM A23187 as indicated. Fixed cells were incubated with anti-pIRF3 followed by Alexa Fluor 488 secondary antibody and visualized by immunofluorescence microscopy. Results are representative of 3 independent experiments. C) Wild type or STING-/- MEFs were subjected to OGD (as in Figure 1) for 1h followed by 2 hours re-oxygenation. Cells were fixed (1C) or lysed (1D) and IRF3 nuclear translocation detected by immunofluorescence or western blot, respectively.
One profound difference between thapsigargin and tunicamycin is that thapsigargin induces the UPR by inhibiting the SERCA pump, severely depleting ER calcium stores(37). Thus it was possible that mobilization of STING reflected altered calcium metabolism and not the UPR at all. To test this hypothesis, cells were stimulated with ionomycin, a calcium ionophore that mobilizes calcium by influx, but does not trigger the UPR at 1μM (data not shown and (42, 43)). Even at higher doses, ionomycin is much less effective at upregulating the UPR gene BiP as compared to another ionophore A23187(44). In contrast to thapsigargin, ionomycin did not induce significant IRF3 phosphorylation (Figure 6B). However, A23187, an agent widely utilized to induce the UPR, did induce IRF3 phosphorylation(45). These results indicate that collapsing the calcium gradient is not sufficient for IRF3 phosphorylation, and that some component of the ER stress response, or ER calcium depletion, is required as well. Furthermore, A23187 also required STING for IRF3 phosphorylation. Thus thapsigargin is not the only UPR inducer dependent upon STING for IRF3 activation. It was not clear if the STING-TBK1 pathway would be involved in a more “physiologic” setting. During ischemia-reperfusion injury, ATP depletion disrupts sequestration of calcium in the ER, resulting in excess cytosolic calcium, mitochondrial calcium uptake, and ER dysfunction(46, 47). Intracellular disruptions in calcium handling are reproduced by in vitro OGD(48). As seen in Figure 6C, OGD-dependent nuclear IRF3 translocation was impaired in STING-/- cells. Together these data suggest that ER stressors that dysregulate calcium metabolism phosphorylate IRF3 through the STING-TBK1 pathway.
A site 1 protease inhibitor of ATF6 processing prevents tunicamycin-induced activation of IRF3
We sought to determine which of the 3 major UPR pathways, stemming from IRE1/XBP1, PERK and ATF6 might be playing a role in IRF3 activation in response to ER stressors. As mentioned above (Figure 2), XBP1 is not absolutely required for nuclear translocation. PERK was not obligatory for synergistic IFN-β mRNA induction by either thapsigargin or tunicamycin(22). In response to ER stress, ATF6 transits to the Golgi, where it is cleaved to an active transcription factor by site 1 and site 2 specific proteases. AEBSF, a serine protease inhibitor has been shown to block this processing event by inhibiting the site 1 protease(49). We confirmed this activity (Figure 7). AEBSF prevented tunicamycin and 2-deoxyglucose, but not thapsigargin-dependent IRF3 phosphorylation, as visualized by immunofluorescence (Figure 7A). By western blot, the presence of AEBSF prevented synergistic p-IRF3 in dually treated LPS+Tm, reducing p-IRF3 to the level of LPS alone (Figure 7B); however AEBSF did not prevent LPS+Tg enhanced p-IRF3. Consistent with these results, AEBSF significantly diminished tunicamycin, but not thapsigargin dependent IFN-β mRNA synergy (Figure 7C). The lack of effect on thapsigargin-induced synergy argues against non-specific toxicity. Together these results indicate that AEBSF sensitivity distinguishes between IRF3 activation by thapsigargin and tunicamycin/2-deoxyglucose and is consistent with a possible role for ATF6 in the latter. Furthermore ER stressors activate IRF3, leading to synergistic IFN-β mRNA induction, by at least 2 distinguishable pathways.
Figure 7.

AEBSF, a site 1 protease inhibitor, blocks tunicamycin and 2-deoxyglucose induced IRF3 phosphorylation and tunicamycin-dependent synergistic IFN-β induction. A) RAW cells were pre-treated with 300 μM AEBSF for 1h, then stimulated with 1μM Tg for 2h, 20mM 2DG for 5h, or 10 μg/mL Tm for 5h. Cells were fixed and then stained with anti-pIRF3 (S386) plus secondary anti-rabbit Alexa Fluor488. Results are representative of 2 independent experiments. B) RAW cells were pretreated with AEBSF as in (A) and then untreated (NT), stimulated with Tg 1h, or Tm 5h, followed by an additional 3h media or LPS as indicated. Whole cell lysates were resolved by SDS PAGE and immunoblotted with anti-pIRF3 (S396), IRF-3 or actin. Results are representative of 2 independent experiments. C) RAW cells were pretreated with AEBSF as in (A), then stimulated with 1h Tg or 5h Tm followed by an additional 3h LPS as indicated. Relative IFN-β mRNA was quantified by qPCR with normalization to 18S rRNA. Results were combined from 4-5 independent experiments and error bars represent the standard error of the mean. * P<0.007. D) Cells were stimulated as in (B). Whole cell lysates were resolved by SDS PAGE and immunoblotted for precursor (P) and mature (M) cleaved forms of ATF6. Results are representative of 3 independent experiments.
Discussion
This study describes the novel observation that ER stress, even in absence of pattern recognition receptor stimulation, activates IRF3. Different forms of ER stress accomplish this through at least 2 distinct pathways, requiring either TBK1/STING or AEBSF-sensitive signaling. The synergistic induction of several IRF3-regulated inflammatory mediators by concurrent UPR and LPS stimulation suggest the activation of IRF3 by ER stress may have a wider impact in innate immunity, beyond augmenting IFN-β production. Even though ER stress alone is not sufficient to trigger the induction of an IRF3-regulated gene, activation of IRF3 by ER stress is necessary for the dramatic IFN-β synergism observed with LPS. In evidence of this requirement, interfering with thapsigargin-dependent IRF3 phosphorylation through the modulation of STING severely impacts the magnitude of thapsigargin-induced synergy. Similarly, disrupting tunicamycin-dependent IRF3 phopshorylation with AEBSF significantly diminished synergistic IFN-β expression. Residual IFN-β induction in the LPS+Tg treated STING-/- MEFs may relate to IRF3 phosphorylation below our limits of detection, or compensation by another IRF3 serine (e.g. Ser 339) for which we did not assay(50).
This study supports the novel concept that intracellular stress responses may coopt innate immune signaling pathways previously thought to be dedicated to pathogen sensing. The proximity of STING to the mitochondria-associated membrane (MAMs) a site of inter-organelle calcium transport and regulation may suggest why UPR inducers that affect calcium also mobilize STING(4, 51). Our findings suggest that at least in MEFs and macrophages, STING and TBK1 associate even prior to stimulation. Upon ER stress induction, STING and TBK1 dramatically reorganize into larger macroscopic collections. This mobilization may be a result of reorganization of the ER membranes themselves (containing STING), translocation to another organelle (e.g. Golgi), or association with other unidentified molecules in a multi-molecular complex. The augmentation of type I IFN responses by ER stress/calcium dysregulation and STING may be of particular relevance to viral infections, such as Hepatitis C, that induce ER stress and cause calcium leak(52). Our results obtained with in vitro OGD (Figure 6) have direct implications for in vivo ischemia-reperfusion injury: they suggest the dysregulation of calcium, ER stress and type I IFN-dependent inflammatory injury may be critically interrelated (20, 53-57).
The UPR has been shown to activate both NF-κB and AP-1 family member transcription factors(58). Thus it is unclear why UPR-induced IRF3 phosphorylation is not sufficient to induce IFN-β expression in vitro. Even though the UPR induces nuclear translocation of IRF3, translocation does not automatically confer transcriptional activity: Dissociation between translocation and transcriptional activity has been noted in multiple models of viral IRF3 inhibition (59-62). IRF3 has 2 activation clusters comprising 7 potentially phosphorylated serines and threonines (S385, S386, and S396, S398, S402, S405, T404). Some controversy remains regarding serine phosphorylation requirements for IRF3 activity: Phosphorylation of S396 has been proposed as an essential minimal acceptor site for responses to Sendai virus, and may be critical for homodimerization (50, 63). More recently S396 has been shown to promote higher order oligomerization(64). However, others have identified S386 as the critical site for homodimerization and nuclear translocation(65, 66). Unfortunately, it is not yet clear exactly which sites on IRF3 correspond with optimal transcriptional activity in response to LPS and other specific pathogens. Ultimately, both may act cooperatively to bind CBP/p300 with higher affinity (64). Thus one possibility is that even though ER stress induces S386 phosphorylation, ER stress in isolation does not induce strong enough phosphorylation at S396, as suggested by western blot (e.g. Figure 4E); the UPR may only induce partial phosphorylation of IRF3 and LPS remains necessary for additional phosphorylation at other serines/threonines. Alternatively, as suggested by our western blot data, UPR induced phosphorylation at S386 may facilitate or enhance LPS dependent S396 phosphorylation. The requirement for multiple-site IRF3 phosphorylation to promote oligomerization may explain the qualitative differences in immunofluorescence between LPS and ER stressors such as OGD (Figure 2)(64). Our data would suggest that ultimate phosphorylation at S396 correlates best with IRF3 DNA binding by chromatin immunoprecipitation and transcriptional activation of IFN-β(23).
Apart from suboptimal IRF3 activation, there are other possible explanations: IRF3 alone is not sufficient for IFN gene transcription; the enhanceosome also contains NF-kB and AP-1 transcription factors. Transcriptional activation following enhanceosome formation requires binding of multiple elements including critical scaffolding molecules (HMGA1) and histone acetyltransferases (e.g. CBP/p300)(11). LPS stimulation may be required to recruit these other molecules. Another possibility is that a stronger NF-κB signal may be required than that generated during ER stress alone. Finally, there could be a cell type issue, since our studies are conducted in macrophages and MEFs. When mice are treated in vivo with tunicamycin alone, we observed detectable serum IFN-β (preliminary data not shown), suggesting that an unidentified cell type is capable of producing IFN during a UPR.
In this study and others, ER stress has been noted to augment transcription of select IRF3-regulated genes (e.g. IFN-β but not RANTES)(24). IRF3 binds similar DNA sequences within gene promoters designated as interferon stimulated response elements (ISRE) or positive regulatory domains (PRD I and III in the IFN-β promoter)(67). The selectivity in synergism may relate to promoter complexity and requirement for multiple transcription factors, as mentioned above. Constitutively activated IRF3 (an aspartate containing phosphomimetic) is sufficient to activate only a small subset of ISRE containing genes, including ifit2/ISG54, ISG56, ISG60, CIG5 and PMA inducible protein 1(68). However, we did not detected robust activation of ISG54 by thapsigargin alone. This failure may reflect suboptimal IRF3 activation at specific serines. Alternatively, given the independence of XBP1 and IRF3 translocation (Figure 2), and the discovery of XBP1 binding sites in cytokine promoters and enhancers, significant synergy might require DNA binding sites for both IRF3 and UPR-dependent transcription factors(22-24). The experience with IFN-β would favor this “multi-hit” hypothesis.
It is not clear which aspects of the UPR are necessary for IRF3 phosphorylation and nuclear translocation. The answer may differ depending upon type of ER stress. Our studies would suggest that XBP1 is not required for ER stress-induced IRF3 nuclear translocation. PERK is not necessary for synergistic IFN induction ((22) and data not shown). AEBSF, a protease inhibitor that prevents ATF6 processing, blocked tunicamycin but not thapsigargin-dependent IRF3 phosphorylation and synergy (Figure 7)(22). Thapsigargin may utilize an IRE1 kinase mediated pathway to activate IRF3. Alternatively, thapsigargin and A23187 could mobilize a non-classical UPR ER stress pathway related to calcium flux that has not been described. Another possibility is that IRF3 activation resulting from profound ER calcium depletion, and the UPR are independent outcomes of treatment with these stressors.
Our results are consistent with the hypothesis that tunicamycin and 2-deoxyglucose-induced IRF3 phosphorylation proceed through ATF6 or a related protein. ATF6 belongs to the OASIS family of transcription factors that is processed by the site 1 proteases. However, protein distribution of these other family members is much more restricted than ATF6(69). AEBSF also inhibits reactive oxygen species generation (ROS) by NADPH(70). Thapsigargin, tunicamycin and ER stress related to cholesterol loading have all been found to induce oxidative stress via NADPH(71-73). ROS potentiate IRF3 activation(74). However, NADPH oxidase inhibition by AEBSF would not explain the divergent effects on thapsigargin and tunimcamycin, particularly given the involvement of calcium in ER stress mediated NADPH oxidase activation(71). The specific AEBSF sensitive signaling event remains to be confirmed.
Another outstanding question is the identity of the kinase activated by non-thapsigargin UPR inducers that is responsible for phosphorylating IRF3. In this study, tunicamycin led to IRF3 phosphorylation in the presence of a TBK1/IKKε inhibitor. The kinase cascade involving NIK and IKK-α has been reported to phosphorylate IRF3 independently of TBK1(75). The MAP-kinase cascade initiated by IRE-1 that includes p38 may also play a role(76, 77). Dissecting the exact mechanism of IRF3 activation by all ER stressors, at the level of the UPR and subsequent kinase cascades, is outside the scope of this study but will be interesting to tease apart in the future.
Ultimately, our findings raise the possibility that different forms of ER stress/UPR inducers may utilize innate immune sensing pathways including STING. Thus, ER stress is poised to significantly augment type I IFN and innate immune responses in the setting of pathogen challenge, or endogenous damage. These findings have implications for conditions involving type I IFN and ER stress such as viral infections, certain bacterial infections, ischemia-reperfusion injury, and potentially rheumatic inflammatory disease. The recruitment of innate immune sensing molecules such as STING represents a newly described interface between intracellular stress and innate immunity.
Acknowledgments
Grant support: K08 AI081045
Other funding: University of Wisconsin-Madison Graduate School, School of Medicine and Public Health, and Department of Pediatrics.
References
- 1.Theofilopoulos AN, Baccala R, Beutler B, Kono DH. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu Rev Immunol. 2005;23:307–336. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- 2.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 3.Kawai T, Akira S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity. 2011;34:637–650. doi: 10.1016/j.immuni.2011.05.006. [DOI] [PubMed] [Google Scholar]
- 4.Ishikawa H, Ma Z, Barber GN. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature. 2009;461:788–792. doi: 10.1038/nature08476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jin L, Waterman PM, Jonscher KR, Short CM, Reisdorph NA, Cambier JC. MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Molecular and cellular biology. 2008;28:5014–5026. doi: 10.1128/MCB.00640-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, Lei C, He X, Zhang L, Tien P, Shu HB. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29:538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]
- 7.Sun W, Li Y, Chen L, Chen H, You F, Zhou X, Zhou Y, Zhai Z, Chen D, Jiang Z. ERIS, an endoplasmic reticulum IFN stimulator, activates innate immune signaling through dimerization. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:8653–8658. doi: 10.1073/pnas.0900850106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hiscott J. Convergence of the NF-kappaB and IRF pathways in the regulation of the innate antiviral response. Cytokine Growth Factor Rev. 2007;18:483–490. doi: 10.1016/j.cytogfr.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 9.Fitzgerald KA, McWhirter SM, Faia KL, Rowe DC, Latz E, Golenbock DT, Coyle AJ, Liao SM, Maniatis T. IKKepsilon and TBK1 are essential components of the IRF3 signaling pathway. Nature immunology. 2003;4:491–496. doi: 10.1038/ni921. [DOI] [PubMed] [Google Scholar]
- 10.Hiscott J. Triggering the innate antiviral response through IRF-3 activation. The Journal of biological chemistry. 2007;282:15325–15329. doi: 10.1074/jbc.R700002200. [DOI] [PubMed] [Google Scholar]
- 11.Ford E, Thanos D. The transcriptional code of human IFN-beta gene expression. Biochim Biophys Acta. 2010;1799:328–336. doi: 10.1016/j.bbagrm.2010.01.010. [DOI] [PubMed] [Google Scholar]
- 12.Sakaguchi S, Negishi H, Asagiri M, Nakajima C, Mizutani T, Takaoka A, Honda K, Taniguchi T. Essential role of IRF-3 in lipopolysaccharide-induced interferon-beta gene expression and endotoxin shock. Biochem Biophys Res Commun. 2003;306:860–866. doi: 10.1016/s0006-291x(03)01049-0. [DOI] [PubMed] [Google Scholar]
- 13.Sato M, Suemori H, Hata N, Asagiri M, Ogasawara K, Nakao K, Nakaya T, Katsuki M, Noguchi S, Tanaka N, Taniguchi T. Distinct and essential roles of transcription factors IRF-3 and IRF-7 in response to viruses for IFN-alpha/beta gene induction. Immunity. 2000;13:539–548. doi: 10.1016/s1074-7613(00)00053-4. [DOI] [PubMed] [Google Scholar]
- 14.Doyle S, Vaidya S, O'Connell R, Dadgostar H, Dempsey P, Wu T, Rao G, Sun R, Haberland M, Modlin R, Cheng G. IRF3 mediates a TLR3/TLR4-specific antiviral gene program. Immunity. 2002;17:251–263. doi: 10.1016/s1074-7613(02)00390-4. [DOI] [PubMed] [Google Scholar]
- 15.Marie I, Durbin JE, Levy DE. Differential viral induction of distinct interferon-alpha genes by positive feedback through interferon regulatory factor-7. The EMBO journal. 1998;17:6660–6669. doi: 10.1093/emboj/17.22.6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McWhirter SM, Fitzgerald KA, Rosains J, Rowe DC, Golenbock DT, Maniatis T. IFN-regulatory factor 3-dependent gene expression is defective in Tbk1-deficient mouse embryonic fibroblasts. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:233–238. doi: 10.1073/pnas.2237236100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lin R, Genin P, Mamane Y, Hiscott J. Selective DNA binding and association with the CREB binding protein coactivator contribute to differential activation of alpha/beta interferon genes by interferon regulatory factors 3 and 7. Molecular and cellular biology. 2000;20:6342–6353. doi: 10.1128/mcb.20.17.6342-6353.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lu X, Masic A, Liu Q, Zhou Y. Regulation of influenza A virus induced CXCL-10 gene expression requires PI3K/Akt pathway and IRF3 transcription factor. Mol Immunol. 2011;48:1417–1423. doi: 10.1016/j.molimm.2011.03.017. [DOI] [PubMed] [Google Scholar]
- 19.Zhai Y, Shen XD, O'Connell R, Gao F, Lassman C, Busuttil RW, Cheng G, Kupiec-Weglinski JW. Cutting edge: TLR4 activation mediates liver ischemia/reperfusion inflammatory response via IFN regulatory factor 3-dependent MyD88-independent pathway. J Immunol. 2004;173:7115–7119. doi: 10.4049/jimmunol.173.12.7115. [DOI] [PubMed] [Google Scholar]
- 20.Zhai Y, Qiao B, Gao F, Shen X, Vardanian A, Busuttil RW, Kupiec-Weglinski JW. Type I, but not type II, interferon is critical in liver injury induced after ischemia and reperfusion. Hepatology. 2008;47:199–206. doi: 10.1002/hep.21970. [DOI] [PubMed] [Google Scholar]
- 21.Chattopadhyay S, Marques JT, Yamashita M, Peters KL, Smith K, Desai A, Williams BR, Sen GC. Viral apoptosis is induced by IRF-3-mediated activation of Bax. The EMBO journal. 2010;29:1762–1773. doi: 10.1038/emboj.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith JA, Turner MJ, DeLay ML, Klenk EI, Sowders DP, Colbert RA. Endoplasmic reticulum stress and the unfolded protein response are linked to synergistic IFN-beta induction via X-box binding protein 1. Eur J Immunol. 2008;38:1194–1203. doi: 10.1002/eji.200737882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zeng L, Liu YP, Sha H, Chen H, Qi L, Smith JA. XBP-1 couples endoplasmic reticulum stress to augmented IFN-beta induction via a cis-acting enhancer in macrophages. J Immunol. 2010;185:2324–2330. doi: 10.4049/jimmunol.0903052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinon F, Chen X, Lee AH, Glimcher LH. TLR activation of the transcription factor XBP1 regulates innate immune responses in macrophages. Nat Immunol. 2010;11:411–418. doi: 10.1038/ni.1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hu F, Yu X, Wang H, Zuo D, Guo C, Yi H, Tirosh B, Subjeck JR, Qiu X, Wang XY. ER stress and its regulator X-box-binding protein-1 enhance polyIC-induced innate immune response in dendritic cells. Eur J Immunol. 2011;41:1086–1097. doi: 10.1002/eji.201040831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schroder M, Kaufman RJ. The mammalian unfolded protein response. Annu Rev Biochem. 2005;74:739–789. doi: 10.1146/annurev.biochem.73.011303.074134. [DOI] [PubMed] [Google Scholar]
- 27.Ono SJ, Liou HC, Davidon R, Strominger JL, Glimcher LH. Human X-box-binding protein 1 is required for the transcription of a subset of human class II major histocompatibility genes and forms a heterodimer with c-fos. Proc Natl Acad Sci U S A. 1991;88:4309–4312. doi: 10.1073/pnas.88.10.4309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Acharya A, Rishi V, Moll J, Vinson C. Experimental identification of homodimerizing B-ZIP families in Homo sapiens. J Struct Biol. 2006;155:130–139. doi: 10.1016/j.jsb.2006.02.018. [DOI] [PubMed] [Google Scholar]
- 29.Turner MJ, Sowders DP, DeLay ML, Mohapatra R, Bai S, Smith JA, Brandewie JR, Taurog JD, Colbert RA. HLA-B27 misfolding in transgenic rats is associated with activation of the unfolded protein response. Journal of immunology. 2005;175:2438–2448. doi: 10.4049/jimmunol.175.4.2438. [DOI] [PubMed] [Google Scholar]
- 30.Kim T, Kim TY, Song YH, Min IM, Yim J, Kim TK. Activation of interferon regulatory factor 3 in response to DNA-damaging agents. The Journal of biological chemistry. 1999;274:30686–30689. doi: 10.1074/jbc.274.43.30686. [DOI] [PubMed] [Google Scholar]
- 31.Servant MJ, ten Oever B, LePage C, Conti L, Gessani S, Julkunen I, Lin R, Hiscott J. Identification of distinct signaling pathways leading to the phosphorylation of interferon regulatory factor 3. The Journal of biological chemistry. 2001;276:355–363. doi: 10.1074/jbc.M007790200. [DOI] [PubMed] [Google Scholar]
- 32.Takeshita S, Kaji K, Kudo A. Identification and characterization of the new osteoclast progenitor with macrophage phenotypes being able to differentiate into mature osteoclasts. J Bone Miner Res. 2000;15:1477–1488. doi: 10.1359/jbmr.2000.15.8.1477. [DOI] [PubMed] [Google Scholar]
- 33.Lenart B, Kintner DB, Shull GE, Sun D. Na-K-Cl cotransporter-mediated intracellular Na+ accumulation affects Ca2+ signaling in astrocytes in an in vitro ischemic model. J Neurosci. 2004;24:9585–9597. doi: 10.1523/JNEUROSCI.2569-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clark K, Peggie M, Plater L, Sorcek RJ, Young ER, Madwed JB, Hough J, McIver EG, Cohen P. Novel cross-talk within the IKK family controls innate immunity. The Biochemical journal. 2011;434:93–104. doi: 10.1042/BJ20101701. [DOI] [PubMed] [Google Scholar]
- 35.Badiola N, Penas C, Minano-Molina A, Barneda-Zahonero B, Fado R, Sanchez-Opazo G, Comella JX, Sabria J, Zhu C, Blomgren K, Casas C, Rodriguez-Alvarez J. Induction of ER stress in response to oxygen-glucose deprivation of cortical cultures involves the activation of the PERK and IRE-1 pathways and of caspase-12. Cell Death Dis. 2011;2:e149. doi: 10.1038/cddis.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Almeida A, Delgado-Esteban M, Bolanos JP, Medina JM. Oxygen and glucose deprivation induces mitochondrial dysfunction and oxidative stress in neurones but not in astrocytes in primary culture. J Neurochem. 2002;81:207–217. doi: 10.1046/j.1471-4159.2002.00827.x. [DOI] [PubMed] [Google Scholar]
- 37.Denmeade SR, Isaacs JT. The SERCA pump as a therapeutic target: making a “smart bomb” for prostate cancer. Cancer Biol Ther. 2005;4:14–22. doi: 10.4161/cbt.4.1.1505. [DOI] [PubMed] [Google Scholar]
- 38.Servant MJ, Grandvaux N, Hiscott J. Multiple signaling pathways leading to the activation of interferon regulatory factor 3. Biochem Pharmacol. 2002;64:985–992. doi: 10.1016/s0006-2952(02)01165-6. [DOI] [PubMed] [Google Scholar]
- 39.Li Y, Hu X, Song Y, Lu Z, Ning T, Cai H, Ke Y. Identification of novel alternative splicing variants of interferon regulatory factor 3. Biochim Biophys Acta. 2011;1809:166–175. doi: 10.1016/j.bbagrm.2011.01.006. [DOI] [PubMed] [Google Scholar]
- 40.Civas A, Island ML, Genin P, Morin P, Navarro S. Regulation of virus-induced interferon-A genes. Biochimie. 2002;84:643–654. doi: 10.1016/s0300-9084(02)01431-1. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Z, Yuan B, Bao M, Lu N, Kim T, Liu YJ. The helicase DDX41 senses intracellular DNA mediated by the adaptor STING in dendritic cells. Nature immunology. 2011;12:959–965. doi: 10.1038/ni.2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fu M, Li L, Albrecht T, Johnson JD, Kojic LD, Nabi IR. Autocrine motility factor/phosphoglucose isomerase regulates ER stress and cell death through control of ER calcium release. Cell death and differentiation. 2011;18:1057–1070. doi: 10.1038/cdd.2010.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pino SC, O'Sullivan-Murphy B, Lidstone EA, Thornley TB, Jurczyk A, Urano F, Greiner DL, Mordes JP, Rossini AA, Bortell R. Protein kinase C signaling during T cell activation induces the endoplasmic reticulum stress response. Cell Stress Chaperones. 2008;13:421–434. doi: 10.1007/s12192-008-0038-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Werno C, Zhou J, Brune B. A23187, ionomycin and thapsigargin upregulate mRNA of HIF-1alpha via endoplasmic reticulum stress rather than a rise in intracellular calcium. J Cell Physiol. 2008;215:708–714. doi: 10.1002/jcp.21351. [DOI] [PubMed] [Google Scholar]
- 45.Li WW, Alexandre S, Cao X, Lee AS. Transactivation of the grp78 promoter by Ca2+ depletion. A comparative analysis with A23187 and the endoplasmic reticulum Ca(2+)-ATPase inhibitor thapsigargin. The Journal of biological chemistry. 1993;268:12003–12009. [PubMed] [Google Scholar]
- 46.Paschen W. Dependence of vital cell function on endoplasmic reticulum calcium levels: implications for the mechanisms underlying neuronal cell injury in different pathological states. Cell Calcium. 2001;29:1–11. doi: 10.1054/ceca.2000.0162. [DOI] [PubMed] [Google Scholar]
- 47.Zucchi R, Ronca F, Ronca-Testoni S. Modulation of sarcoplasmic reticulum function: a new strategy in cardioprotection? Pharmacol Ther. 2001;89:47–65. doi: 10.1016/s0163-7258(00)00103-0. [DOI] [PubMed] [Google Scholar]
- 48.Pisani A, Bonsi P, Centonze D, Giacomini P, Calabresi P. Involvement of intracellular calcium stores during oxygen/glucose deprivation in striatal large aspiny interneurons. J Cereb Blood Flow Metab. 2000;20:839–846. doi: 10.1097/00004647-200005000-00011. [DOI] [PubMed] [Google Scholar]
- 49.Okada T, Haze K, Nadanaka S, Yoshida H, Seidah NG, Hirano Y, Sato R, Negishi M, Mori K. A serine protease inhibitor prevents endoplasmic reticulum stress-induced cleavage but not transport of the membrane-bound transcription factor ATF6. The Journal of biological chemistry. 2003;278:31024–31032. doi: 10.1074/jbc.M300923200. [DOI] [PubMed] [Google Scholar]
- 50.Clement JF, Bibeau-Poirier A, Gravel SP, Grandvaux N, Bonneil E, Thibault P, Meloche S, Servant MJ. Phosphorylation of IRF-3 on Ser 339 generates a hyperactive form of IRF-3 through regulation of dimerization and CBP association. Journal of virology. 2008;82:3984–3996. doi: 10.1128/JVI.02526-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hajnoczky G, Csordas G, Yi M. Old players in a new role: mitochondria-associated membranes, VDAC, and ryanodine receptors as contributors to calcium signal propagation from endoplasmic reticulum to the mitochondria. Cell Calcium. 2002;32:363–377. doi: 10.1016/s0143416002001872. [DOI] [PubMed] [Google Scholar]
- 52.Robinson LC, Marchant JS. Enhanced Ca2+ leak from ER Ca2+ stores induced by hepatitis C NS5A protein. Biochemical and biophysical research communications. 2008;368:593–599. doi: 10.1016/j.bbrc.2008.01.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Janicki PK, Wise PE, Belous AE, Pinson CW. Interspecies differences in hepatic Ca(2+)-ATPase activity and the effect of cold preservation on porcine liver Ca(2+)-ATPase function. Liver Transpl. 2001;7:132–139. doi: 10.1053/jlts.2001.21459. [DOI] [PubMed] [Google Scholar]
- 54.Emadali A, Nguyen DT, Rochon C, Tzimas GN, Metrakos PP, Chevet E. Distinct endoplasmic reticulum stress responses are triggered during human liver transplantation. J Pathol. 2005;207:111–118. doi: 10.1002/path.1798. [DOI] [PubMed] [Google Scholar]
- 55.DeGracia DJ, Montie HL. Cerebral ischemia and the unfolded protein response. J Neurochem. 2004;91:1–8. doi: 10.1111/j.1471-4159.2004.02703.x. [DOI] [PubMed] [Google Scholar]
- 56.Azfer A, Niu J, Rogers LM, Adamski FM, Kolattukudy PE. Activation of endoplasmic reticulum stress response during the development of ischemic heart disease. Am J Physiol Heart Circ Physiol. 2006;291:H1411–1420. doi: 10.1152/ajpheart.01378.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chang WJ, Chehab M, Kink S, Toledo-Pereyra LH. Intracellular calcium signaling pathways during liver ischemia and reperfusion. J Invest Surg. 2010;23:228–238. doi: 10.3109/08941939.2010.496036. [DOI] [PubMed] [Google Scholar]
- 58.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Spiegel M, Pichlmair A, Martinez-Sobrido L, Cros J, Garcia-Sastre A, Haller O, Weber F. Inhibition of Beta interferon induction by severe acute respiratory syndrome coronavirus suggests a two-step model for activation of interferon regulatory factor 3. Journal of virology. 2005;79:2079–2086. doi: 10.1128/JVI.79.4.2079-2086.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ren J, Liu T, Pang L, Li K, Garofalo RP, Casola A, Bao X. A novel mechanism for the inhibition of interferon regulatory factor-3-dependent gene expression by human respiratory syncytial virus NS1 protein. J Gen Virol. 2011;92:2153–2159. doi: 10.1099/vir.0.032987-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rose KM, Elliott R, Martinez-Sobrido L, Garcia-Sastre A, Weiss SR. Murine coronavirus delays expression of a subset of interferon-stimulated genes. Journal of virology. 2010;84:5656–5669. doi: 10.1128/JVI.00211-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wang JT, Doong SL, Teng SC, Lee CP, Tsai CH, Chen MR. Epstein-Barr virus BGLF4 kinase suppresses the interferon regulatory factor 3 signaling pathway. Journal of virology. 2009;83:1856–1869. doi: 10.1128/JVI.01099-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Servant MJ, Grandvaux N, tenOever BR, Duguay D, Lin R, Hiscott J. Identification of the minimal phosphoacceptor site required for in vivo activation of interferon regulatory factor 3 in response to virus and double-stranded RNA. The Journal of biological chemistry. 2003;278:9441–9447. doi: 10.1074/jbc.M209851200. [DOI] [PubMed] [Google Scholar]
- 64.Chen W, Srinath H, Lam SS, Schiffer CA, Royer WE, Jr., Lin K. Contribution of Ser386 and Ser396 to activation of interferon regulatory factor 3. J Mol Biol. 2008;379:251–260. doi: 10.1016/j.jmb.2008.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mori M, Yoneyama M, Ito T, Takahashi K, Inagaki F, Fujita T. Identification of Ser-386 of interferon regulatory factor 3 as critical target for inducible phosphorylation that determines activation. The Journal of biological chemistry. 2004;279:9698–9702. doi: 10.1074/jbc.M310616200. [DOI] [PubMed] [Google Scholar]
- 66.Takahasi K, Horiuchi M, Fujii K, Nakamura S, Noda NN, Yoneyama M, Fujita T, Inagaki F. Ser386 phosphorylation of transcription factor IRF-3 induces dimerization and association with CBP/p300 without overall conformational change. Genes Cells. 2010;15:901–910. doi: 10.1111/j.1365-2443.2010.01427.x. [DOI] [PubMed] [Google Scholar]
- 67.Hiscott J, Pitha P, Genin P, Nguyen H, Heylbroeck C, Mamane Y, Algarte M, Lin R. Triggering the interferon response: the role of IRF-3 transcription factor. J Interferon Cytokine Res. 1999;19:1–13. doi: 10.1089/107999099314360. [DOI] [PubMed] [Google Scholar]
- 68.Grandvaux N, Servant MJ, tenOever B, Sen GC, Balachandran S, Barber GN, Lin R, Hiscott J. Transcriptional profiling of interferon regulatory factor 3 target genes: direct involvement in the regulation of interferon-stimulated genes. Journal of virology. 2002;76:5532–5539. doi: 10.1128/JVI.76.11.5532-5539.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Asada R, Kanemoto S, Kondo S, Saito A, Imaizumi K. The signalling from endoplasmic reticulum-resident bZIP transcription factors involved in diverse cellular physiology. J Biochem. 2011;149:507–518. doi: 10.1093/jb/mvr041. [DOI] [PubMed] [Google Scholar]
- 70.Wind S, Beuerlein K, Eucker T, Muller H, Scheurer P, Armitage ME, Ho H, Schmidt HH, Wingler K. Comparative pharmacology of chemically distinct NADPH oxidase inhibitors. Br J Pharmacol. 2010;161:885–898. doi: 10.1111/j.1476-5381.2010.00920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li G, Scull C, Ozcan L, Tabas I. NADPH oxidase links endoplasmic reticulum stress, oxidative stress, and PKR activation to induce apoptosis. J Cell Biol. 2010;191:1113–1125. doi: 10.1083/jcb.201006121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Liao J, Sun A, Xie Y, Isse T, Kawamoto T, Zou Y, Ge J. Aldehyde Dehydrogenase-2 Deficiency Aggravates Cardiac Dysfunction Elicited by Endoplasmic Reticulum Stress Induction. Mol Med. 2012 doi: 10.2119/molmed.2011.00466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang Y, Ren J. Thapsigargin triggers cardiac contractile dysfunction via NADPH oxidase-mediated mitochondrial dysfunction: Role of Akt dephosphorylation. Free Radic Biol Med. 2011;51:2172–2184. doi: 10.1016/j.freeradbiomed.2011.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 74.Soucy-Faulkner A, Mukawera E, Fink K, Martel A, Jouan L, Nzengue Y, Lamarre D, Vande Velde C, Grandvaux N. Requirement of NOX2 and reactive oxygen species for efficient RIG-I-mediated antiviral response through regulation of MAVS expression. PLoS Pathog. 2010;6:e1000930. doi: 10.1371/journal.ppat.1000930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang RP, Zhang M, Li Y, Diao FC, Chen D, Zhai Z, Shu HB. Differential regulation of IKK alpha-mediated activation of IRF3/7 by NIK. Mol Immunol. 2008;45:1926–1934. doi: 10.1016/j.molimm.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 76.Chiang E, Dang O, Anderson K, Matsuzawa A, Ichijo H, David M. Cutting edge: apoptosis-regulating signal kinase 1 is required for reactive oxygen species-mediated activation of IFN regulatory factor 3 by lipopolysaccharide. Journal of immunology. 2006;176:5720–5724. doi: 10.4049/jimmunol.176.10.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hetz C, Glimcher LH. Fine-tuning of the unfolded protein response: Assembling the IRE1alpha interactome. Molecular cell. 2009;35:551–561. doi: 10.1016/j.molcel.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]