

Abstract
Excess lipid availability causes insulin resistance. We examined the effect of acute exercise on lipid-induced insulin resistance and TBC1 domain family member 1/4 (TBCD1/4)-related signaling in skeletal muscle. In eight healthy young male subjects, 1 h of one-legged knee-extensor exercise was followed by 7 h of saline or intralipid infusion. During the last 2 h, a hyperinsulinemic-euglycemic clamp was performed. Femoral catheterization and analysis of biopsy specimens enabled measurements of leg substrate balance and muscle signaling. Each subject underwent two experimental trials, differing only by saline or intralipid infusion. Glucose infusion rate and leg glucose uptake was decreased by intralipid. Insulin-stimulated glucose uptake was higher in the prior exercised leg in the saline and the lipid trials. In the lipid trial, prior exercise normalized insulin-stimulated glucose uptake to the level observed in the resting control leg in the saline trial. Insulin increased phosphorylation of TBC1D1/4. Whereas prior exercise enhanced TBC1D4 phosphorylation on all investigated sites compared with the rested leg, intralipid impaired TBC1D4 S341 phosphorylation compared with the control trial. Intralipid enhanced pyruvate dehydrogenase (PDH) phosphorylation and lactate release. Prior exercise led to higher PDH phosphorylation and activation of glycogen synthase compared with resting control. In conclusion, lipid-induced insulin resistance in skeletal muscle was associated with impaired TBC1D4 S341 and elevated PDH phosphorylation. The prophylactic effect of exercise on lipid-induced insulin resistance may involve augmented TBC1D4 signaling and glycogen synthase activation.
Studies in human and rodent models have revealed deleterious effects of excess lipid availability on peripheral insulin sensitivity (1,2). Intracellular increases in fatty acid metabolites, such as diacylglycerol (DAG) and ceramide, may play critical roles in mediating lipid-induced insulin resistance by inducing serine phosphorylation of insulin-receptor substrate 1 (IRS-1) (3–6) and consequently inhibiting downstream signaling to GLUT4 translocation. However, recent reports challenge such causality. These studies revealed unaltered signal transduction at the level of IRS-1, IRS-1–associated phosphatidylinositol-3-kinase (PI3K) activity, Akt, and TBC1 domain family member 4 (TBC1D4) phosphorylation (phospho-Akt-substrate [PAS] an unspecific antibody recognizing phosphorylated Akt substrate motifs), after 2–7 h of lipid infusion (7–11). When DAG and/or ceramide levels were reported, no changes in skeletal muscle DAG or ceramide levels were found after lipid infusion (7,11).
We recently showed that lactate release in human skeletal muscle is augmented along with reduced respiratory exchange ratio (RER) values during lipid infusion (11). This could indicate suppressed activity of the pyruvate dehydrogenase (PDH) complex, which in turn could lead to a reduction in glucose uptake according to the Randle cycle (12). Here, we wished to investigate whether this increase in leg lactate release and reduced RER values were accompanied by altered regulation of PDH, measured by site-specific phosphorylation.
Exercise increases peripheral insulin sensitivity (13–15). After an acute bout of exercise, the ability for insulin to stimulate glucose uptake in skeletal muscle is increased several hours into recovery (14,16). This effect can be ascribed to adaptations in the exercised muscle rather than changes in systemic factors (13,17,18) and is observed in both healthy and insulin-resistant states (e.g., obesity) (19) and type 2 diabetes (20). A recent study has shown that a single bout of exercise can prevent subsequent lipid-induced impairments in whole-body glucose tolerance assessed by an intravenous glucose tolerance test (IVGTT) (2). It was hypothesized that repartitioning fatty acids toward intramuscular triacylglycerol (IMTG) synthesis and storage rather than DAG or ceramide might be a primary mediator of the beneficial effects of exercise on lipid-induced impairments in glucose tolerance (2). Enhanced insulin sensitivity after a bout of exercise is associated with increased GLUT4 recruitment to the plasma membrane (21) and not with altered protein synthesis (e.g., GLUT4 protein) (22), but has not been associated with altered signal transduction through the insulin receptor, IRS-1, PI3K, or Akt (13,22,23). Recently, the hypothesis was put forward (24) that the guanosine triphosphatase (GTPase) activating proteins TBC1 domain family member 1 (TBC1D1) and 4 (TBC1D4) might serve as points of convergence for insulin dependent and independent signaling pathways to GLUT4 translocation. In agreement with this hypothesis, PAS phosphorylation of TBC1D4 is elevated along with insulin-stimulated glucose uptake for up to 27 h after exercise in skeletal muscle of rats (25), and we recently showed that phosphorylation of TBC1D4 on specific residues was elevated 4 h after a single bout of exercise in human skeletal muscle (26).
TBC1D4/D1 are multikinase substrates proposed to be involved in contraction- and insulin-stimulated glucose uptake in mice (27,28), and exercise and insulin both substantially increase TBC1D4/D1 phosphorylation in human skeletal muscle (29,30). TBC1D4/D1 contain several phosphorylation sites distinctly phosphorylated by various kinases, including Akt and 5′AMP-activated protein kinase (AMPK) (28,31–33). Phosphorylation of TBC1D4/D1 and subsequent 14-3-3 binding is proposed to lead to inactivation of the GTPase-activating proteins, decreasing their inhibitory function on the GLUT4 translocation process and thus, potentially, increasing the GLUT4 capacity of the surface membrane.
In the current study we tested the hypothesis that prior exercise prevents subsequent lipid-induced insulin resistance in human skeletal muscle through regulation of the signaling molecules TBC1D4/TBC1D1.
RESEARCH DESIGN AND METHODS
Subjects.
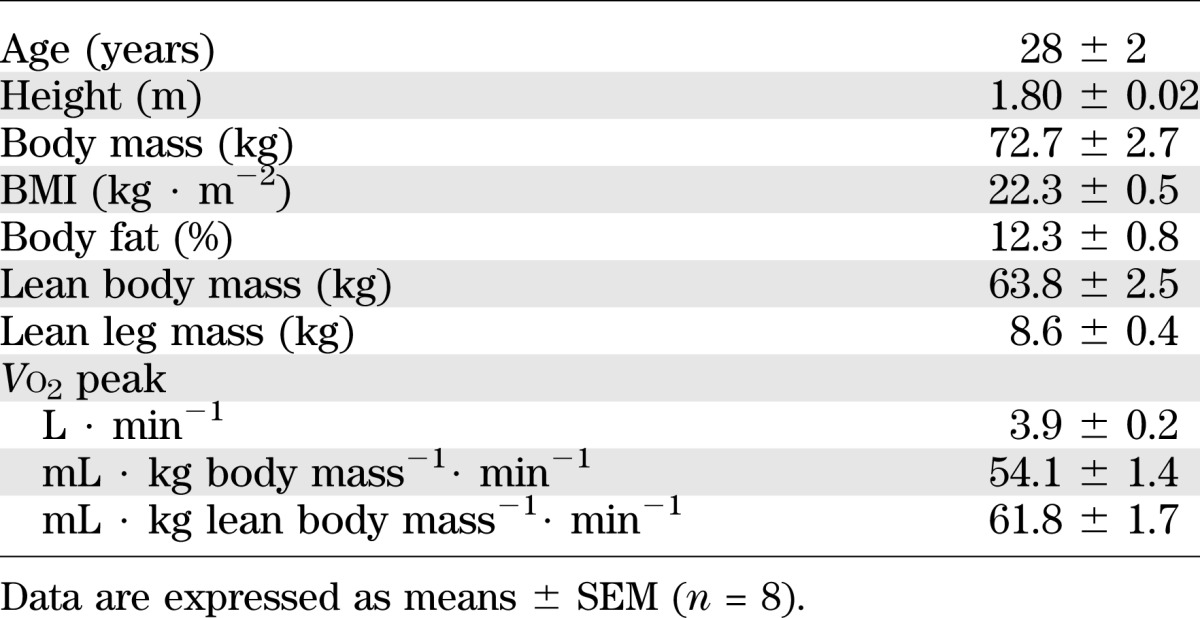
Eight healthy, moderately fit men (Table 1) were included in the study and gave their written informed consent. The study was approved by the local ethics committee in the municipality of Copenhagen (KF 01 26 1127) and performed in accordance with the Helsinki Declaration II.
TABLE 1.
Subject characteristics
Experimental design.
All subjects underwent two experimental trials only differing by infusion of intralipid and heparin or saline for 7 hours in a randomized order. The trials were separated by at least 2 weeks. After 8 days of a standardized diet (2.5 MJ; 60E% carbohydrate [CHO], 25E% fat, and 15E% protein) that was provided in weighed portions to the subjects, the subject arrived at the laboratory in the morning after having consumed a meal corresponding to 20% of daily energy intake (60E% CHO, 25E% fat, and 15E% protein) 3 h earlier. The meal was added to avoid too high plasma fatty acid concentrations in the few hours after exercise in the saline trial as a consequence of a long fast. Previous studies have shown that when a pre-exercise meal of 3 MJ is consumed, plasma glucose and insulin concentrations returned to baseline values between 4 and 6 h after ingestion (34). Because the clamp was initiated 8 h after the meal in the current study, we assume that the meal would have no or at least a very minor impact on the metabolic responses. Nevertheless, the protocol was identical for both of the following trials.
Upon arrival, the subjects performed 60 min of dynamic one-legged knee extensor exercise at 80% peak workload of the knee extensors. During the 60 min of exercise two 5-min intervals at 100% peak workload were performed (at the 20–25 and 40–45 min time points) to ensure activation of the majority of the vastus lateralis muscle fibers. At 20 min after termination of exercise, a 7-h infusion of intralipid (20% soybean oil emulsion, Fresenius Kabi, Uppsala, Sweden; 1.15 mL · kg−1· h−1) + heparin (0.2 units · kg−1· min−1) or saline was initiated through a catheter inserted in an antecubital vein. Teflon catheters were inserted into the femoral artery and both femoral veins and a thermistor (EDSLAB T.D. Model 94-030-2.F, Baxter Healthcare Corporation, CA) was inserted through the femoral vein catheter for blood flow determination. After 3 h of infusion of intralipid or saline, a bolus injection of [6,6-2H] glucose was administered (3.203 mg · kg−1) within 1 min, followed by a constant infusion (0.055 mg · kg−1· min−1) for the remaining experimental period (4 h). After 5 h of intralipid or saline infusion, subjects underwent a 120-min hyperinsulinemic-euglycemic clamp (1.42 mU · kg−1· min−1 insulin) initiated with a bolus injection of insulin (9.0 mU · kg−1; Actrapid, Novo Nordisk, Denmark). Femoral venous blood flow was determined, and blood was collected simultaneously from the femoral arteries and veins at −60, 0, 15, 30, 45, 60, 80 100, and 120 min of the clamp. Expired air was collected in Douglas bags before the clamp and at the end of the clamp. The subjects did not consume any food throughout the experimental day.
Muscle biopsies.
Biopsy specimens were obtained from both vastus lateralis muscles 30 min before the clamp and at 30 and 120 min into the clamp. Muscle samples were snap frozen in liquid nitrogen and stored at −80°C for later processing. Biopsy specimens from both legs were obtained within <2 min.
Insulin sensitivity.
Insulin sensitivity at whole-body level was expressed as the average glucose infusion rate during the last 40 min of the clamp normalized to the steady-state plasma insulin concentration in the same period. Glucose uptake across the thigh was calculated as the arterial – venous glucose difference multiplied by blood flow (Fick’s Principle), and insulin-stimulated glucose uptake across the leg was expressed as a mean of the last 40 min of the clamp.
Methods for blood chemistry, RER, body composition, stable isotopes, muscle tissue processing, glycogen, IMTG content, SDS PAGE and Western blotting, antibodies, 14-3-3 overlay and glycogen synthase (GS) activity, and calculation of hepatic glucose production are all presented in the Supplementary Data.
Statistics.
Data are expressed as means ± SEM. Statistical analyses were performed in SPSS 17.0 software (SPSS, Chicago, IL) and Sigma Plot 11.0 software (Systat Software Inc, Erkrath, Germany) using two- or three-way ANOVA with repeated measures (RM). Three-way ANOVA RM was used for comparison of exercise, insulin, and intralipid and two-way ANOVA RM for comparison of insulin and intralipid or exercise and insulin. The Tukey post hoc test was performed when ANOVA revealed significant interaction. Finally, paired Student t test was used for data shown in Fig. 1A. Correlation analyses were performed in Sigma Plot 11.0 using Pearson product moment correlation. Correlation of TBC1D4 phosphorylation with leg glucose uptake was calculated during insulin stimulation and included values from all four interventions (saline/lipid and rest/exercise, n = 28).
FIG. 1.

A: Glucose infusion rate/arterial insulin concentration the last 40 min of the clamp. B: Insulin-stimulated leg glucose uptake during the last 40 min of the clamp (LLM, lean leg mass). Insulin-stimulated leg glucose uptake in the rested and previously exercised (Ex’d) leg during the 120 min of the clamp in the saline trial (C) and intralipid trial (D). Data are expressed as means ± SEM (n = 8). #P < 0.05, ##P < 0.01 vs. saline trial; ††P < 0.01 vs. rested leg; *P < 0.05, **P < 0.01, ***P < 0.001 vs. basal (0 min) in respective trial.
RESULTS
Subject characteristics.
Subject characteristics are presented in Table 1.
Insulin sensitivity.
Preclamp plasma insulin levels were not different between trials, and insulin infusion resulted in arterial insulin levels ∼100 μU · mL−1 in both trials (Table 2).
TABLE 2.
RER, femoral venous blood flow, arterial blood glucose concentration, plasma substrate and hormone concentration, lactate release, and skeletal muscle glycogen and IMTG content

Insulin-stimulated whole-body glucose infusion rate (GIR) to maintain arterial blood glucose concentration at 5.2 ± 0.1 mmol · L−1 (Table 2) leveled at 42.9 ± 0.9 μmol · min−1· kg−1 body mass (BM) during the last 40 min of the clamp in the saline trial. GIR/insulin was ∼32% (P < 0.001) lower in the intralipid trial compared with the saline trial (Fig. 1A).
Insulin-stimulated leg glucose uptake increased during the clamp regardless of the intervention (P < 0.001; Fig. 1C and D), but was ∼30% (P < 0.01) lower in the rested and previously exercised leg in the intralipid trial compared with the saline trial (Fig. 1B). Insulin-stimulated leg glucose uptake was higher in the previously exercised leg compared with the rested leg in both trials (P < 0.05).
Hepatic glucose production (HGP).
HGP was significantly suppressed during the last 40 min of the clamp in both trials (P < 0.001). Although HGP was fully suppressed in the saline trial, there was still a small but borderline significantly higher HGP in the lipid trial compared with the saline trial (P = 0.051; power = 0.444; Table 2).
RER.
RER was 0.79 ± 0.02 preclamp in the saline trial and increased to 0.85 ± 0.02 (P < 0.01) at the end of the clamp (Table 2). Insulin did not affect RER in the intralipid trial. Thus, RER was significantly lower at the end of the clamp in the intralipid trial compared with the saline trial (P < 0.01; Table 2).
Arterial plasma long-chain fatty acids (LCFAs).
Arterial plasma LCFA levels were approximately fivefold higher in the intralipid trial compared with the saline trial (P < 0.001). Insulin suppressed plasma LCFA levels in both trials by approximately the same absolute magnitude. Thus, plasma LCFA levels were almost completely suppressed in the saline trial, whereas in the intralipid trial, plasma LCFA levels remained markedly elevated (1,894 ± 300 μmol · L−1) above levels observed in the saline trial (27.7 ± 7.50 μmol · L−1; P < 0.001; Table 2).
Blood flow and plasma parameters.
Venous blood flow increased during the clamp (P < 0.05) in both trials and tended (∼17%, P = 0.058) toward being higher in the previously exercised leg compared with the rested leg (Table 2). Leg blood flow was not affected by intralipid infusion. Arterial plasma concentrations of epinephrine and norepinephrine were not affected by insulin, previous exercise, or intralipid infusion (Table 2). Leg lactate release was higher in the intralipid compared with the saline trial, irrespective of the clamp (P < 0.01), but was not different between the rested and exercised leg. Leg lactate release did not change in response to insulin infusion (Table 2). The increased lactate release in the intralipid trial did not result in detectable increases in arterial lactate concentrations (Table 2).
Muscle glycogen and IMTG content.
Muscle glycogen content was lower in the previously exercised leg compared with the rested leg before the clamp (P < 0.05; Table 2). Muscle glycogen content increased marginally during the clamp only in the previously exercised leg (P < 0.05), and thus, the levels reached at the end of the clamp were still lower compared with the rested leg (P < 0.05; Table 2). Intralipid infusion did not affect IMTG levels in the vastus lateralis muscle, and IMTG levels were not different between the rested and previously exercised leg before or at the end of the clamp (Table 2).
Insulin signaling in skeletal muscle.
Akt regulation was evaluated by phosphorylation on two regulatory sites, S473 and T308. Insulin markedly increased S473 and T308 phosphorylation in all situations, but there was no effect of prior exercise or intralipid infusion on either site (Fig. 2A and C). In addition, Akt protein expression was not affected by any of the interventions (Fig. 2D).
FIG. 2.

Akt T308 phosphorylation (A), Akt S473 phosphorylation (B), and AMPK T172 phosphorylation/AMPK-α2 expression (C) in rested or exercised (Ex’d) vastus lateralis muscle in the saline and lipid trials. D: Representative Western blots of Akt protein expression and site-specific phosphorylation and AMPK-α2 protein expression and phosphorylation. Blots show the quality and signal obtained with the corresponding antibody and represent one subject; thus, they do not necessarily represent an exact mean of all subjects. Data are expressed as means ± SEM (n = 8). ***P < 0.001 vs. basal (0 min.) in respective trial. AU, arbitrary units.
Insulin increased TBC1D1 phosphorylation at T596 in all situations (P < 0.01), whereas S237 phosphorylation was not affected (Fig. 3A and B). Neither prior exercise nor intralipid affected basal and insulin-induced phosphorylation of these two sites. TBC1D1 protein expression was not affected by the interventions (Fig. 3C).
FIG. 3.

Site-specific phosphorylation in rested or exercised (Ex’d) vastus lateralis muscle in the saline and lipid trials. A: TBC1D1 pS237 phosphorylation/TBC1D1 protein expression. B: TBC1D1 pT596 phosphorylation/TBC1D1 protein expression. C: Representative Western blots of TBC1D1 protein expression and phosphorylation show the quality and signal obtained with the corresponding antibody and represent one subject; thus, they do not necessarily represent an exact mean of all subjects. Data are expressed as means ± SEM (n = 8). **P < 0.01 vs. basal (0 min) in respective trial. AU, arbitrary units.
Insulin increased phosphorylation of all TBC1D4 phosphorylation sites investigated in all situations (all P < 0.001; Fig. 4B–G). In both the saline and intralipid trials, the basal and insulin-induced phosphorylation at all sites was significantly higher in the previously exercised muscle compared with the rested muscle. Lipid infusion significantly impaired phosphorylation of S341 by ∼20% compared with the saline trial (P < 0.05; Fig. 4C) under both basal and during insulin-stimulated conditions. TBC1D4 S588 phosphorylation showed a similar pattern, but this did not reach statistical significance (Fig. 4D). Although prior exercise increased phosphorylation at all sites, the impairing effect of intralipid on TBC1D4 S341 phosphorylation was not fully rescued to the level seen in the saline trial. At present we are not able to evaluate TBC1D4 GAP activity in skeletal muscle biopsy specimens. We tested whether the lower TBC1D4 S341 phosphorylation had any consequence for the ability of TBC1D4 to bind to 14-3-3 proteins. Although 14-3-3 binding to TBC1D4 increased during the clamp, no differences were observed between the saline and intralipid trial or between the rested and exercised leg (Fig. 4A). Nevertheless, correlation analyses reveal a relatively weak, but statistically significant, positive correlation between TBC1D4 S341 phosphorylation and leg glucose uptake during insulin stimulation (R2 = 0.140, P < 0.005; n = 28 data not shown). TBC1D4 protein expression was not affected by the interventions (Fig. 4H).
FIG. 4.

14-3-3 binding to TBC1D4 and site-specific phosphorylation in rested or exercised (Ex’d) vastus lateralis muscle in the saline and lipid trials. A: 14-3-3 binding to TBC1D4/TBC1D4 protein expression. B: TBC1D4 S318 phosphorylation/TBC1D4 protein expression. C: TBC1D4 S341 phosphorylation/TBC1D4 protein expression. D: TBC1D4 S588 phosphorylation/TBC1D4 protein expression. E: TBC1D4 T642 phosphorylation/TBC1D4 protein expression. F: TBC1D4 S704 phosphorylation/TBC1D4 protein expression. G: TBC1D4 S751 phosphorylation/TBC1D4 protein expression. H: Representative Western blots of TBC1D4 phosphorylation and 14-3-3 binding to TBC1D4 show the quality and signal obtained with the corresponding antibody and represent one subject; thus, they do not necessarily represent an exact mean of all subjects. Data are expressed as means ± SEM (n = 8). #P < 0.05 vs. saline trial; *P < 0.05, **P < 0.01, ***P < 0.001 vs. basal (0 min.) in respective trial; †P < 0.05, ††P < 0.01, †††P < 0.001 vs. rested leg. AU, arbitrary units.
AMPK is a proposed upstream kinase for TBC1D1 and TBC1D4, and AMPK function was evaluated by measuring phosphorylation of T172 in the activating loop of the α2 subunit. AMPK phosphorylation was not affected by either of the interventions (prior exercise, intralipid or insulin; Fig. 2E).
Regulation of PDH.
Regulation of PDH was evaluated by phospho-specific antibodies toward two regulatory (deactivating) phosphorylation sites of PDH (site 1 and 2). Phosphorylation of PDH on site 1 was not affected by any of the interventions (Fig. 5A). However, phosphorylation of PDH site 2 was elevated in the previously exercised muscle compared with the rested muscle in all situations (P < 0.05) and showed a trend (P = 0.08; power = 0.403) toward higher phosphorylation in the lipid trial compared with the saline trial (Fig. 5B). Protein expression of PDH was not affected by any of the interventions (Fig. 5C).
FIG. 5.

PDH site 1 and site 2 phosphorylation in rested or exercised (Ex’d) vastus lateralis muscle in the saline and lipid trials. A: PDH site 1 phosphorylation/PDH protein expression. B: PDH site 2 phosphorylation/PDH protein expression. C: Representative Western blots of PDH protein expression and site-specific phosphorylation protein expression and phosphorylation show the quality and signal obtained with the corresponding antibody and represent one subject; thus, they do not necessarily represent an exact mean of all subjects. Data are expressed as means ± SEM (n = 8). (#)P = 0.08 vs. saline trial; †P < 0.05 vs. rested leg. AU, arbitrary units.
GS activity.
Insulin increased muscle GS activity (% I-form and % fractional velocity [FV]) in all situations (P < 0.001), and lipid infusion did not affect this increase (Fig. 6A and B). Basal and insulin-stimulated GS activity (% I-form and % FV) were higher in the previously exercised muscle compared with the rested muscle (P < 0.001; Fig. 6A and B).
FIG. 6.

GS activity in rested or exercised (Ex’d) vastus lateralis muscle in the saline and lipid trials. A: GS activity (% I-form). B: GS activity (% FV). Data are expressed as means ± SEM (n = 8). ***P < 0.001 vs. basal (0 min.) in respective trial; †††P < 0.001 vs. rested leg. AU, arbitrary units.
DISCUSSION
Acute intralipid infusion dramatically impaired whole-body and leg insulin sensitivity. The latter effect was partly prevented by prior exercise. Although we observed substantial lipid-induced reduction in skeletal muscle insulin sensitivity (Fig. 1B), phosphorylation of Akt by insulin was not affected (Fig. 2A and B). This confirms our recent report where insulin-stimulated IRS-1–associated PI3K activity and Akt phosphorylation were also not impaired by 7 h of intralipid infusion despite pronounced insulin resistance (11). The unaltered proximal insulin signaling was also supported by the unaltered insulin-induced activation of GS. Hence, our data add to the accumulating body of evidence (7–9,11) challenging the view (3–6) linking acute lipid-induced insulin resistance to defects in proximal insulin signaling.
We recently reported that insulin-induced TBC1D4 phosphorylation detected with the PAS antibody was intact after lipid infusion in human skeletal muscle (11). Also, a study on lipid-induced insulin resistance in rat skeletal muscle showed unaltered TBC1D4 phosphorylation at T642 (7), the predominant site on TBC1D4 recognized by the PAS antibody (33). In line with these reports, we observed intact phosphorylation of TBC1D4 T642 along with intact phosphorylation of other Akt-regulated phosphorylation sites S318, S704, and S751 (Fig. 4B and E–G). Although the ability for insulin to induce TBC1D4 S341 phosphorylation was normal (Δ values), the general level of phosphorylation was impaired.
Previous reports have demonstrated that 14-3-3 binding to TBC1D4 by itself was sufficient to increase GLUT4 translocation (35) suggesting that 14-3-3 binding may modify TBC1D4 GAP activity. Hence, because insulin-induced 14-3-3 binding to TBC1D4 was not altered in the intralipid trial, this could indicate that impaired S341 phosphorylation did not result in impaired GAP activity. Knowledge of the regulation of TBC1D4 GAP activity by phosphorylation and 14-3-3 binding and the contribution of each individual phosphorylation site is still very limited, and hence, whether 14-3-3 binding can be used as a surrogate measure of GAP activity is not known. Nevertheless, it seems well established that TBC1D4 phosphorylation is involved in the regulation of insulin-stimulated GLUT4 translocation in adipocytes (32) and in insulin-stimulated glucose uptake in mouse skeletal muscle (27). However, no conclusive data exist on the role TBC1D4 S341 in insulin-stimulated glucose uptake because the studies examining this matter (27,32) primarily have used a TBC1D4 4P mutant in which four predicted Akt phosphorylation sites (S318, S588, T642, and S751) have been mutated to alanine. The TBC1D4 4P mutant does not include S341. In fact, over-expressing a TBC1D4 S341 to an alanine mutant, along with the 4P mutant in 3T3-L1 adipocytes, does not exacerbate the effects of overexpressing the 4P mutant (32). However, the TBC1D4 S341 mutant in that study was only expressed simultaneously with the 4P mutant. Thus, it cannot be ruled out that overexpressing mutated TBC1D4 S341 alone could negatively affect glucose uptake. Nonetheless, given the involvement of TBC1D4 in insulin-stimulated glucose uptake and that all measured TBC1D4 phosphorylation sites were potently phosphorylated by insulin (Fig. 4B–G), it seems plausible to propose that impaired TBC1D4 S341 phosphorylation could partially inhibit the regulatory function of the protein. The positive correlation between leg glucose uptake and TBC1D4 S341 phosphorylation during insulin stimulation in the current study provides further support for this notion.
The beneficial effect of prior exercise on insulin sensitivity is characterized by an increased insulin-induced abundance of GLUT4 at the plasma membrane (21) and by increased activation of GS (22). In line with the latter, we also observed increased GS activity in the previously exercised leg in the current study (Fig. 6A and B). Corresponding with the similar GS activity in the saline versus the lipid trial, we did not observe any differences in postexercise glycogen resynthesis between these two trials (Table 2). This was despite the rather large relative differences in leg glucose uptake in the rested and exercised leg between the two trials (Fig. 1). We note here that the numeric differences in glucose uptake between the two trials are relatively small compared with the levels of glycogen in the muscle. Taking the accuracy of the glycogen measurements into account, this could explain why we do not observe any differences in glycogen resynthesis between the trials.
Recently, and as confirmed by the current study, we showed that the signaling nexus TBC1D4 could play a potential role in the enhanced insulin action observed after a single bout of exercise because the general level of phosphorylation of all sites investigated was elevated by prior exercise (26). In fact, a novel observation was the very potent elevation of S704 phosphorylation. Although phosphorylation was elevated on all sites in the previously exercised leg, 14-3-3 binding to TBC1D4 did not follow this pattern, confirming our previous report (26). This underlines the imperative need for a better understanding of TBC1D4 GAP activity regulation (Fig. 4A).
We anticipated that the closely related paralog TBC1D1 would show a similar pattern of regulation as TBC1D4 because these proteins share several similar structural features. Both proteins contain two clusters of phosphorylation sites, each containing a potential 14-3-3 binding site (Ser237 and Thr596 on TBC1D1 and Ser341 and Thr642 on TBC1D4) (35,36), they show specificity toward the same Rabs (37), and are potentially regulated by the same kinases. Although it is well established that supraphysiologic insulin concentrations regulate TBC1D1 phosphorylation in cell systems and in rodent skeletal muscle (37,38), we show, for the first time, that near-physiologic insulin concentrations regulate TBC1D1 T596 phosphorylation in human skeletal muscle in vivo. TBC1D1 S237 phosphorylation increases acutely during exercise in human skeletal muscle (30,39). Although a different exercise protocol was applied (the present protocol was relatively more intense), we anticipate a similar acute phosphorylation of TBC1D1 during the exercise in the current study. Interestingly, such phosphorylations were normalized 5 h after exercise. Furthermore and in line with the intact proximal insulin signaling, lipid infusion had no effect on insulin-induced TBC1D1 phosphorylation.
Thus, our observations suggest that these two GTPase-activating proteins are differentially regulated by prior exercise and lipid infusion. Under the conditions investigated, TBC1D4 was affected by prior exercise and lipid infusion, whereas TBC1D1 was not affected. The current study does not bring evidence to clarify the basis for this difference.
TBC1D4 is a multikinase substrate phosphorylated by various kinases. In cell-free assays, these include serum- and glucocorticoid-induced protein kinase 1 (SGK1), p90 ribosomal S6 kinase 1 (RSK1), Akt, and AMPK (33), and in Chinese hamster ovary cells, overexpressing insulin receptor, conventional/novel protein kinase C isoforms have been proposed as upstream regulators of TBC1D4 (40). In skeletal muscle, Akt and AMPK have been confirmed as upstream regulators of TBC1D4 phosphorylation (41,42). AMPK is an important energy sensor in skeletal muscle and can induce uptake of glucose when activated (43). Moreover, decreased activity and/or protein expression of AMPK has been observed in states of insulin resistance in rodents (44). Although most studies in type 2 diabetic patients do not observe impaired AMPK activity and/or expression (45,46), the observations in rodents, along with the impaired phosphorylation of TBC1D4 S341, which is phosphorylated upon AMPK activation with AICAR in human embryonic kidney 293 cells, led us to examine AMPK phosphorylation in the current study (Fig. 2C). Confirming our previous report (11), AMPK T172 phosphorylation was completely unaffected by lipid infusion (Fig. 2), and thus, the activity of other kinases and/or phosphatases could be regulating TBC1D4 phosphorylation in vivo as well.
We show that intralipid infusion increased leg lactate release, confirming our recent finding (11) and leading us to investigate the regulation of PDH. PDH phosphorylation has been shown to inactivate the enzyme (47), and our observations thus suggest that PDH activity was decreased in the intralipid trial in line with the Randle cycle hypothesis (12). In support of our observation, intralipid was previously shown to inhibit the insulin-mediated decrease in PDK4 mRNA in humans (8) and rats (7,48). However, we recently reported unaltered PDH activity in human skeletal muscle, despite lipid-induced increases in PDK4 mRNA expression and PDH-E1α phosphorylation (49). Whether this discrepant finding relates to the sensitivity of the activity assay is uncertain. However, the lower RER values at the end of the clamp in the intralipid trial compared with the saline trial in the current study, along with higher leg lactate release in the intralipid trial, indicates a lower degree of carbohydrate oxidation consistent with lower conversion of pyruvate to acetyl-CoA. Whether this reduced glucose oxidation could result in decreased glucose uptake is still uncertain because we did not observe any accumulation of glucose-6-phosphate or free glucose in response to intralipid infusion in our recent study (11). However, due to inherent difficulties in measuring these variables, one cannot put too much weight on these negative results. We, therefore, cannot rule out that the Randle cycle hypothesis is indeed applicable in this context.
In conclusion, the current study proposes two molecular mechanisms—site-specific TBC1D4 and PDH phosphorylation—that both potentially account for part of the impairing effect of intralipid on skeletal muscle glucose uptake. We furthermore show that a previous bout of exercise partly protects from subsequent lipid-induced insulin resistance in humans at the level of skeletal muscle. This may potentially occur through enhanced insulin action on specific TBC1D4 phosphorylation sites. Finally, we show that insulin increased phosphorylation of the signaling nexus TBC1D1 in humans independently of lipid infusion and prior exercise.
ACKNOWLEDGMENTS
This study was supported by grants from the Danish Medical Research Council (J.F.P.W.); The Lundbeck Research Foundation (J.F.P.W.); the Novo Nordisk Research Foundation (J.F.P.W.); Danish Ministry of Food, Agriculture and Fisheries (B.K.); and an integrated project funded by the European Union (LSHMCT-2004-005272) (E.A.R., B.K., J.F.P.W.). This work was carried out as part of the research program of the UNIK: Food, Fitness and Pharma for Health and Disease (see www.foodfitnesspharma.ku.dk) (E.A.R., B.K., J.F.P.W.). The UNIK project is supported by the Danish Ministry of Science, Technology and Innovation.
No potential conflicts of interest relevant to this article were reported.
C.P. performed the biochemical analyses, wrote the manuscript, and contributed to the final version of the manuscript. N.B., L.D.H., and K.A.S. designed the study, carried out the experiments, performed the biochemical analyses, and contributed to the final version of the manuscript. J.B.B. performed the biochemical analyses and contributed to the final version of the manuscript. L.J.G. contributed reagents, reviewed and edited the manuscript, and contributed to the final version of the manuscript. B.K., E.A.R., and J.F.P.W. designed the study, carried out the experiments, wrote the manuscript, and contributed to the final version of the manuscript. J.F.P.W. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors acknowledge the skilled technical assistance of Irene Bech Nielsen and Betina Bolmgren, Molecular Physiology Group, University of Copenhagen, Denmark. The authors thank Associate Professor Henriette Pilegaard, Department of Molecular Biology & Center of Inflammation and Metabolism, University of Copenhagen, Copenhagen, Denmark, for fruitful discussions on the experimental protocol and on the data obtained. The authors also appreciate the kind donation of necessary antibodies (α2AMPK, pAMPK, PDH total, and pPDH) by Prof. D. Grahame Hardie, Division of Molecular Physiology, College of Life Sciences, University of Dundee, Dundee, Scotland, U.K.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db11-1572/-/DC1.
REFERENCES
- 1.Boden G, Lebed B, Schatz M, Homko C, Lemieux S. Effects of acute changes of plasma free fatty acids on intramyocellular fat content and insulin resistance in healthy subjects. Diabetes 2001;50:1612–1617 [DOI] [PubMed] [Google Scholar]
- 2.Schenk S, Horowitz JF. Acute exercise increases triglyceride synthesis in skeletal muscle and prevents fatty acid-induced insulin resistance. J Clin Invest 2007;117:1690–1698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Itani SI, Ruderman NB, Schmieder F, Boden G. Lipid-induced insulin resistance in human muscle is associated with changes in diacylglycerol, protein kinase C, and IkappaB-alpha. Diabetes 2002;51:2005–2011 [DOI] [PubMed] [Google Scholar]
- 4.Chavez JA, Summers SA. Characterizing the effects of saturated fatty acids on insulin signaling and ceramide and diacylglycerol accumulation in 3T3-L1 adipocytes and C2C12 myotubes. Arch Biochem Biophys 2003;419:101–109 [DOI] [PubMed] [Google Scholar]
- 5.Itani SI, Zhou Q, Pories WJ, MacDonald KG, Dohm GL. Involvement of protein kinase C in human skeletal muscle insulin resistance and obesity. Diabetes 2000;49:1353–1358 [DOI] [PubMed] [Google Scholar]
- 6.Dresner A, Laurent D, Marcucci M, et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J Clin Invest 1999;103:253–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hoy AJ, Brandon AE, Turner N, et al. Lipid and insulin infusion-induced skeletal muscle insulin resistance is likely due to metabolic feedback and not changes in IRS-1, Akt, or AS160 phosphorylation. Am J Physiol Endocrinol Metab 2009;297:E67–E75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsintzas K, Chokkalingam K, Jewell K, Norton L, Macdonald IA, Constantin-Teodosiu D. Elevated free fatty acids attenuate the insulin-induced suppression of PDK4 gene expression in human skeletal muscle: potential role of intramuscular long-chain acyl-coenzyme A. J Clin Endocrinol Metab 2007;92:3967–3972 [DOI] [PubMed] [Google Scholar]
- 9.Storgaard H, Jensen CB, Björnholm M, et al. Dissociation between fat-induced in vivo insulin resistance and proximal insulin signaling in skeletal muscle in men at risk for type 2 diabetes. J Clin Endocrinol Metab 2004;89:1301–1311 [DOI] [PubMed] [Google Scholar]
- 10.Gormsen LC, Jessen N, Gjedsted J, et al. Dose-response effects of free fatty acids on glucose and lipid metabolism during somatostatin blockade of growth hormone and insulin in humans. J Clin Endocrinol Metab 2007;92:1834–1842 [DOI] [PubMed] [Google Scholar]
- 11.Høeg LD, Sjøberg KA, Jeppesen J, et al. Lipid-induced insulin resistance affects women less than men and is not accompanied by inflammation or impaired proximal insulin signaling. Diabetes 2011;60:64–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963;1:785–789 [DOI] [PubMed] [Google Scholar]
- 13.Wojtaszewski JF, Hansen BF, Kiens B, Richter EA. Insulin signaling in human skeletal muscle: time course and effect of exercise. Diabetes 1997;46:1775–1781 [DOI] [PubMed] [Google Scholar]
- 14.Richter EA, Garetto LP, Goodman MN, Ruderman NB. Muscle glucose metabolism following exercise in the rat: increased sensitivity to insulin. J Clin Invest 1982;69:785–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mikines KJ, Sonne B, Farrell PA, Tronier B, Galbo H. Effect of physical exercise on sensitivity and responsiveness to insulin in humans. Am J Physiol 1988;254:E248–E259 [DOI] [PubMed] [Google Scholar]
- 16.Frøsig C, Roepstorff C, Brandt N, et al. Reduced malonyl-CoA content in recovery from exercise correlates with improved insulin-stimulated glucose uptake in human skeletal muscle. Am J Physiol Endocrinol Metab 2009;296:E787–E795 [DOI] [PubMed] [Google Scholar]
- 17.Richter EA, Garetto LP, Goodman MN, Ruderman NB. Enhanced muscle glucose metabolism after exercise: modulation by local factors. Am J Physiol 1984;246:E476–E482 [DOI] [PubMed] [Google Scholar]
- 18.Richter EA, Mikines KJ, Galbo H, Kiens B. Effect of exercise on insulin action in human skeletal muscle. J Appl Physiol 1989;66:876–885 [DOI] [PubMed] [Google Scholar]
- 19.Devlin JT, Horton ES. Effects of prior high-intensity exercise on glucose metabolism in normal and insulin-resistant men. Diabetes 1985;34:973–979 [DOI] [PubMed] [Google Scholar]
- 20.Devlin JT, Hirshman M, Horton ED, Horton ES. Enhanced peripheral and splanchnic insulin sensitivity in NIDDM men after single bout of exercise. Diabetes 1987;36:434–439 [DOI] [PubMed] [Google Scholar]
- 21.Hansen PA, Nolte LA, Chen MM, Holloszy JO. Increased GLUT-4 translocation mediates enhanced insulin sensitivity of muscle glucose transport after exercise. J Appl Physiol 1998;85:1218–1222 [DOI] [PubMed] [Google Scholar]
- 22.Wojtaszewski JF, Hansen BF, Gade J, et al. Insulin signaling and insulin sensitivity after exercise in human skeletal muscle. Diabetes 2000;49:325–331 [DOI] [PubMed] [Google Scholar]
- 23.Frøsig C, Sajan MP, Maarbjerg SJ, et al. Exercise improves phosphatidylinositol-3,4,5-trisphosphate responsiveness of atypical protein kinase C and interacts with insulin signalling to peptide elongation in human skeletal muscle. J Physiol 2007;582:1289–1301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cartee GD, Wojtaszewski JF. Role of Akt substrate of 160 kDa in insulin-stimulated and contraction-stimulated glucose transport. Appl Physiol Nutr Metab 2007;32:557–566 [DOI] [PubMed] [Google Scholar]
- 25.Funai K, Schweitzer GG, Sharma N, Kanzaki M, Cartee GD. Increased AS160 phosphorylation, but not TBC1D1 phosphorylation, with increased postexercise insulin sensitivity in rat skeletal muscle. Am J Physiol Endocrinol Metab 2009;297:E242–E251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Treebak JT, Frøsig C, Pehmøller C, et al. Potential role of TBC1D4 in enhanced post-exercise insulin action in human skeletal muscle. Diabetologia 2009;52:891–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kramer HF, Witczak CA, Taylor EB, Fujii N, Hirshman MF, Goodyear LJ. AS160 regulates insulin- and contraction-stimulated glucose uptake in mouse skeletal muscle. J Biol Chem 2006;281:31478–31485 [DOI] [PubMed] [Google Scholar]
- 28.Treebak JT, Taylor EB, Witczak CA, et al. Identification of a novel phosphorylation site on TBC1D4 regulated by AMP-activated protein kinase in skeletal muscle. Am J Physiol Cell Physiol 2010;298:C377–C385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Howlett K, Mathews A, Garnham A, Sakamoto K. The effect of exercise and insulin on AS160 phosphorylation and 14-3-3 binding capacity in human skeletal muscle. Am J Physiol Endocrinol Metab 2008;294:E401–E407 [DOI] [PubMed] [Google Scholar]
- 30.Frøsig C, Pehmøller C, Birk JB, Richter EA, Wojtaszewski JF. Exercise-induced TBC1D1 Ser237 phosphorylation and 14-3-3 protein binding capacity in human skeletal muscle. J Physiol 2010;588:4539–4548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mîinea CP, Sano H, Kane S, et al. AS160, the Akt substrate regulating GLUT4 translocation, has a functional Rab GTPase-activating protein domain. Biochem J 2005;391:87–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sano H, Kane S, Sano E, et al. Insulin-stimulated phosphorylation of a Rab GTPase-activating protein regulates GLUT4 translocation. J Biol Chem 2003;278:14599–14602 [DOI] [PubMed] [Google Scholar]
- 33.Geraghty KM, Chen S, Harthill JE, et al. Regulation of multisite phosphorylation and 14-3-3 binding of AS160 in response to IGF-1, EGF, PMA and AICAR. Biochem J 2007;407:231–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Montain SJ, Hopper MK, Coggan AR, Coyle EF. Exercise metabolism at different time intervals after a meal. J Appl Physiol 1991;70:882–888 [DOI] [PubMed] [Google Scholar]
- 35.Ramm G, Larance M, Guilhaus M, James DE. A role for 14-3-3 in insulin-stimulated GLUT4 translocation through its interaction with the RabGAP AS160. J Biol Chem 2006;281:29174–29180 [DOI] [PubMed] [Google Scholar]
- 36.Chen S, Murphy J, Toth R, Campbell DG, Morrice NA, Mackintosh C. Complementary regulation of TBC1D1 and AS160 by growth factors, insulin and AMPK activators. Biochem J 2008;409:449–459 [DOI] [PubMed] [Google Scholar]
- 37.Roach WG, Chavez JA, Mîinea CP, Lienhard GE. Substrate specificity and effect on GLUT4 translocation of the Rab GTPase-activating protein Tbc1d1. Biochem J 2007;403:353–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pehmøller C, Treebak JT, Birk JB, et al. Genetic disruption of AMPK signaling abolishes both contraction- and insulin-stimulated TBC1D1 phosphorylation and 14-3-3 binding in mouse skeletal muscle. Am J Physiol Endocrinol Metab 2009;297:E665–E675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jessen N, An D, Lihn AS, et al. Exercise increases TBC1D1 phosphorylation in human skeletal muscle. Am J Physiol Endocrinol Metab 2011;301:E164–E171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thong FS, Bilan PJ, Klip A. The Rab GTPase-activating protein AS160 integrates Akt, protein kinase C, and AMP-activated protein kinase signals regulating GLUT4 traffic. Diabetes 2007;56:414–423 [DOI] [PubMed] [Google Scholar]
- 41.Kramer HF, Witczak CA, Fujii N, et al. Distinct signals regulate AS160 phosphorylation in response to insulin, AICAR, and contraction in mouse skeletal muscle. Diabetes 2006;55:2067–2076 [DOI] [PubMed] [Google Scholar]
- 42.Treebak JT, Glund S, Deshmukh A, et al. AMPK-mediated AS160 phosphorylation in skeletal muscle is dependent on AMPK catalytic and regulatory subunits. Diabetes 2006;55:2051–2058 [DOI] [PubMed] [Google Scholar]
- 43.Jørgensen SB, Viollet B, Andreelli F, et al. Knockout of the alpha2 but not alpha1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. J Biol Chem 2004;279:1070–1079 [DOI] [PubMed] [Google Scholar]
- 44.Richter EA, Ruderman NB. AMPK and the biochemistry of exercise: implications for human health and disease. Biochem J 2009;418:261–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vind BF, Pehmøller C, Treebak JT, et al. Impaired insulin-induced site-specific phosphorylation of TBC1 domain family, member 4 (TBC1D4) in skeletal muscle of type 2 diabetes patients is restored by endurance exercise-training. Diabetologia 2011;54:157–167 [DOI] [PubMed] [Google Scholar]
- 46.Højlund K, Mustard KJ, Staehr P, et al. AMPK activity and isoform protein expression are similar in muscle of obese subjects with and without type 2 diabetes. Am J Physiol Endocrinol Metab 2004;286:E239–E244 [DOI] [PubMed] [Google Scholar]
- 47.Sale GJ, Randle PJ. Analysis of site occupancies in [32P]phosphorylated pyruvate dehydrogenase complexes by aspartyl-prolyl cleavage of tryptic phosphopeptides. Eur J Biochem 1981;120:535–540 [DOI] [PubMed] [Google Scholar]
- 48.Kim YI, Lee FN, Choi WS, Lee S, Youn JH. Insulin regulation of skeletal muscle PDK4 mRNA expression is impaired in acute insulin-resistant states. Diabetes 2006;55:2311–2317 [DOI] [PubMed] [Google Scholar]
- 49.Pilegaard H, Birk JB, Sacchetti M, et al. PDH-E1alpha dephosphorylation and activation in human skeletal muscle during exercise: effect of intralipid infusion. Diabetes 2006;55:3020–3027 [DOI] [PubMed] [Google Scholar]