

Abstract
The success of model-based methods in phylogenetics has motivated much research aimed at generating new, biologically informative models. This new computer-intensive approach to phylogenetics demands validation studies and sound measures of performance. To date there has been little practical guidance available as to when and why the parameters in a particular model can be identified reliably. Here, we illustrate how Data Cloning (DC), a recently developed methodology to compute the maximum likelihood estimates along with their asymptotic variance, can be used to diagnose structural parameter nonidentifiability (NI) and distinguish it from other parameter estimability problems, including when parameters are structurally identifiable, but are not estimable in a given data set (INE), and when parameters are identifiable, and estimable, but only weakly so (WE). The application of the DC theorem uses well-known and widely used Bayesian computational techniques. With the DC approach, practitioners can use Bayesian phylogenetics software to diagnose nonidentifiability. Theoreticians and practitioners alike now have a powerful, yet simple tool to detect nonidentifiability while investigating complex modeling scenarios, where getting closed-form expressions in a probabilistic study is complicated. Furthermore, here we also show how DC can be used as a tool to examine and eliminate the influence of the priors, in particular if the process of prior elicitation is not straightforward. Finally, when applied to phylogenetic inference, DC can be used to study at least two important statistical questions: assessing identifiability of discrete parameters, like the tree topology, and developing efficient sampling methods for computationally expensive posterior densities.
Keywords: Bayesian estimation in Phylogenetics, Data Cloning, diagnostics, Maximum Likelihood, parameter estimability, Parameter Identifiability
In recent years, statistical phylogenetics has seen a profusion of model-based inference methods using either maximum likelihood or Bayesian methods Felsenstein (2004). This relatively recent emphasis on a computer-intensive approach to phylogenetics begs for compelling validation studies and examination of performance measures. The performance of statistical phylogenetic analyses is usually evaluated using either simulation studies (Abdo et al. 2005; Hillis et al. 1994; Huelsenbeck and Rannala 2004; Pickett and Randle 2005; Ripplinger and Sullivan 2008; Schwartz and Mueller 2010; Sullivan et al. 2005; Sullivan and Joyce 2005; Sullivan and Swofford 2001; Yang and Rannala 2005), studies using “known” phylogenies (Hillis et al. 1992; Naylor and Brown 1998), and mathematical analysis, which has revealed for instance that the parsimony and compatibility methods are inconsistent in some cases (Felsenstein 1978; Hendy and Penny 1989). Careful statistical analysis has also exposed the asymptotic properties of various methods of phylogenetic estimation (Chang 1996; Felsenstein 1983; Kim 2000; Matsen and Steel 2007). Finally, a probabilistic approach has been used to assess parameter identifiability for some of the models commonly used in maximum likelihood (ML) and Bayesian Markov chain Monte Carlo (MCMC) phylogenetic estimation (e.g., Allman et al. 2008; Allman and Rhodes 2006, 2008; Chai and Housworth 2011; Mossel and Vigoda 2005; Rogers 1997, 2001).
To a great extent, these validation approaches have been used as a justification for proposing new even more biologically relevant phylogenetic models. Still, the underlying processes of organismal evolution are certainly more complex than the models commonly used for phylogenetic estimation, such as the GTR+I+Γ model and its submodels. Indeed, the additional complexities associated with patterns of genomic evolution are likely to include population genetics factors (Cartwright et al. 2011; Maddison 1997), complex mutational patterns (Siepel and Haussler 2004), the potential for action of natural selection on genome-scale features (Akashi and Gojobori 2002), and other types of interactions between genomic properties and organismal phenotypes (e.g., Chojnowski and Braun 2008; Chojnowski et al. 2007; Jobson and Qiu 2011). The recognition of the complexity of patterns of genomic and organismal evolution has motivated the development of novel phylogenetic models that attempt to capture a large number of features enhancing biological realism (Drummond et al. 2006; Felsenstein 2004; Fisher 2008; Huelsenbeck et al. 2000; Thorne et al. 1998; Yang and Rannala 2006). Hierarchical models to reconstruct species trees from multilocus sequence data (e.g., Liu 2008) provide an excellent example of such an approach. However, the goal of a reliable understanding of phylogenetic processes through these complex models has often proved to be an elusive target, mostly due to the difficulties of implementing such models and because of the paucity of papers providing practical guidance as to when and why the parameters in a particular model can be identified reliably (Rannala 2002; Yang and Rannala 2006).
Nonestimability of parameters arises when the maximum value of the likelihood function given the data at hand occurs at more than one set of parameter values (Lele et al. 2010; Rannala 2002; Rothenberg 1971). In that case, the asymptotic properties of ML estimation cannot be used. Two different scenarios leading to a parameter being nonestimable must be distinguished. The first one occurs when the model has been written in such a way that two or more parameters are nonseparable. The nonseparable parameters are not estimable with any data set. In this case, the parameters are often referred to as nonidentifiable (NI). Rannala (2002) showed a simple exponential modeling case with this type of nonidentifiability. Importantly, the specification of further biological realism, like that brought about by the hidden components in a hierarchical stochastic process setting, often inadvertently results in the introduction of such NI parameters that even in the presence of an infinite amount of data cannot be teased apart (Lele et al. 2010; McCulloch and Searle 2001; Rannala 2002; Yang and Rannala 2006). The second scenario leading to nonestimability takes place when by pure happenstance, the sampled data contains absolutely no information about the parameter of interest, yet other data sets might. We term such cases as identifiable but nonestimable (INE). In phylogenetics, INE could materialize if there is no sequence variation within an alignment. Even if a parameter is estimable, the precision with which it can be estimated may vary. For instance, if the curvature of the likelihood is sharp, then the parameter will be estimated accurately (i.e., with smaller variance). On the other hand, if the curvature of the likelihood is almost nonexistent then the parameter estimates will have greater variance. We distinguish these last two cases as strongly estimable (SE) and weakly estimable (WE). Although WE and INE can be prevented by better sampling practices (like using more sequence data) or by bringing external information into the analysis (i.e., Huelsenbeck et al. 2008, but see Felsenstein 2004; Pickett and Randle 2005; Zwickl and Holder 2004 and Brandley et al. 2006), the first scenario (NI) is difficult to detect because it may be determined by complex interactions among the components of the model.
Nonidentifiability is not always regarded as an inferential problem. For instance, under subjective Bayesianism (Eberly and Carlin 2000; Gelfand and Sahu 1999; Robert 2007), when the data contain no information to estimate the parameter of interest, it is the prior distribution that conveys such information and thus provides a starting point for the Bayesian learning process (Lindley 1972, 2000; Rannala 2002) but see (Lele and Dennis 2009). Indeed, an often repeated justification for using the Bayesian solution in complex model-based problems is the ability to bring into the analysis external, a priori information concerning the parameters of interest (Huelsenbeck et al. 2002; Alfaro and Holder 2006; Huelsenbeck et al. 2008; Rannala 2002). Another case where nonidentifiability does not affect the statistical inference of the biological parameters of interest is when it occurs between parameters that can effectively be viewed as nuisance parameters.
Detecting nonidentifiability remains an important inferential problem when the prior elicitation process cannot easily be informed by any method, including expert opinion (Lele and Allen 2006). While analyzing Hidden Markov models and/or state-space models, it is not always clear how to go about the process of prior elicitation for a quantity that by definition cannot be observed (Hastie and Green 2012; Lele and Dennis 2009; Lele et al. 2010). For these complex hierarchical models, the only practical way to obtain a prior for Bayesian inference is the use of expert opinion, which may be difficult to justify under these circumstances. Finally, in the context of phylogenetics, nonidentifiability is particularly important when it implies that a section of the phylogeny of interest cannot be resolved even with infinite amounts of data.
In this article, we develop diagnostic tools for the early detection of nonidentifiability in phylogenetic analyses for continuous and discrete parameters. Such diagnostic tools use a recently developed computer-intensive method for ML estimation and inference called Data Cloning (DC) (Lele et al. 2007; Ponciano et al. 2009) that has been proved to work with continuous parameters (Lele et al. 2010). We show how this method can provide a very simple way to assess nonidentifiability for many phylogenetic models and tree topologies by embedding rooted binary trees in a metric space associated with the Billera, Holmes and Vogtmann (BHV) distance (Billera et al. 2001). Importantly, this diagnostic procedure can be easily implemented using existing Bayesian MCMC software for phylogenetic inference and used with empirical data matrices. We demonstrate that, despite the presence of a nearly flat likelihood, our diagnostic tool can distinguish cases of weak estimability from nonidentifiability. With this work, we hope to provide useful guidance for practitioners focused on empirical problems who wish to run a careful diagnosis of parameter identifiability in their data analyses.
Despite the simplicity of the DC methods, a phylogenetics practitioner can draw a remarkable set of conclusions using DC. In what follows, we illustrate the implementation of DC for phylogenetic analyses and discuss the breadth of implications of applying these results to currently relevant problems in phylogenetics.
Methods
DC: A Computationally-Intensive ML Method
Developed in the context of hierarchical models in ecology, DC is a general technique that uses MCMC algorithms to compute ML estimates along with their asymptotic variance estimates (Lele et al. 2007, 2010; Ponciano et al. 2009). DC was anticipated in the works of (Robert 1993), (Doucet et al. 2002), (Kuk 2003) and (Jacquier et al. 2007). With DC the ML estimates can be calculated whenever a Bayesian solution can be computed. Instead of relying on exact or numerical differentiation methods to maximize the likelihood, DC only relies on the computation of means and variances of posterior distributions.
An often repeated justification of the Bayesian approach is the fact that, as sample size increases, the Bayesian solution approaches the ML solution (Walker 1969). DC works by applying the Bayesian methodology to a data set constructed by duplicating (cloning) the original data set enough times so that the resulting posterior approaches the ML solution. Indeed, as the number of replicates, r, increases, the posterior distribution becomes nearly degenerate, and its mean vector converges to the ML estimates. Furthermore, for continuous parameters, its variance–covariance matrix converges to 1/r times the inverse of the observed Fisher's information matrix. Hence, the estimated variances can be used to obtain Wald-type confidence intervals (Lele et al., 2007, 2010). In what follows we provide a detailed explanation of DC. It is important to realize that cloning the data is not a remedy to alleviate the problems of having small sample sizes. It is just an algorithmic device to facilitate the computation of the ML estimates along with their variance. Should software already be available to calculate the likelihood and its second derivative, DC would not improve the analysis. Note, while (Lele et al. 2007, 2010; Ponciano et al. 2009) use k to denote the number of clones, we used r in order to keep the notation of (Rannala 2002) as well as the model parametrization which he extensively used.
Let Yt×n denote a sequence alignment, where n is the length of the sequence alignment and t is the number of taxa. Such observations are supposed to arise from a probabilistic, Markovian model of sequence evolution. The most efficient way to derive information about the evolutionary process from the sequence alignment data is to connect it with a proposed model of character evolution through the specification of a likelihood function. The likelihood function, denoted L(θ; Y), is a function of the vector of model parameters θ. The value of the likelihood at θ is the value of the joint probability density function for the random variables Yt×n=[Y1, Y2,…, Yn] evaluated at the data set at hand and at θ. Here, Yi, i=1, 2,…, n corresponds to the t×1 vector of character states for the t taxa at the i-th position in the sequence. The Markovian structure of the data allows the joint distribution of the observations, denoted by g(Y; θ), to be computed as an explicit function of the evolutionary parameters of interest, θ (Felsenstein 2004). Thus, the joint probability distribution is crucial as it provides the connection between the probabilistic description of the model of character evolution and the data set Y (but see (Sprott 2000), page 10 for a precise definition of the likelihood). The ML estimates of the vector of model parameters θ are those parameter values that make the observed data “most probable”, i.e., they maximize the likelihood function for the observations.
In a Bayesian framework, parameters are viewed as random variables and inference about the process and the parameters of interest is achieved via the posterior distribution
where C(Y)= ∫ L(θ; Y)π(θ)dθ, L(θ; Y)=g(Y; θ) and π(θ) corresponds to the joint prior distribution of the model parameters. Bayesian methods have been used with increasing frequency because computational methods allow the estimation of the posterior for many complex models where an explicit likelihood function is difficult to write and solve. However, the influence of the prior distribution in Bayesian inference has been the source of much debate (Brown et al. 2010; Felsenstein 2004; Yang and Rannala 2005).
A heuristic description of DC is as follows: suppose that in r independent experiments recording a sequence alignment for t taxa and n base pairs, just by pure coincidence, exactly the same set of observations Y is recorded every single time. Then, the likelihood function for these r independent experiments would be simply written as [L(θ; Y)]r and the posterior distribution of θ given the data Yt×n(1),…, Yt×n(r) would be written as
In the equation above, C(r, Y)= ∫[g(Y; θ)]r π(θ)dθ is the constant of integration and the posterior superscript (r) emphasizes the fact that this distribution is obtained by using the r identical realizations of the data. For large enough r and under certain regularity conditions (see Lele et al. 2010), the mean vector and the variance–covariance matrix of π(θ|Y)(r) converge respectively to the vector of ML estimates and to  times the inverse of the observed Fisher's information matrix. Thus, Wald's (Wald 1948) asymptotic variance of the ML estimate can be estimated by multiplying r times the variance of the r-th cloned posterior distribution, a quantity that in what follows we call the “DC variance estimate”. Proofs of these convergence results were published by Lele et al. (2010). These authors showed that, because the data set copies are not independent, the convergence results used by Walker (1969) do not apply. Rather than corresponding to the probabilistic convergence results used in (Walker, 1969), the convergences that validate the DC methodology are deterministic convergences of a sequence of functions.
times the inverse of the observed Fisher's information matrix. Thus, Wald's (Wald 1948) asymptotic variance of the ML estimate can be estimated by multiplying r times the variance of the r-th cloned posterior distribution, a quantity that in what follows we call the “DC variance estimate”. Proofs of these convergence results were published by Lele et al. (2010). These authors showed that, because the data set copies are not independent, the convergence results used by Walker (1969) do not apply. Rather than corresponding to the probabilistic convergence results used in (Walker, 1969), the convergences that validate the DC methodology are deterministic convergences of a sequence of functions.
(Robert 1993) described an alternative scheme to obtain π(θ|Y)(r) that he deemed the “prior feedback method”. The r-th posterior π(θ|Y)(r) can be thought of as resulting from the hypothetical experiment of computing the posterior distribution iteratively, starting with the original likelihood L(θ; Y) and prior distribution π(θ) and at each iteration using the preceding posterior distribution as the prior distribution. It is easy to show by induction that the two formulations are equivalent, and thus, as the number of iterations r tends to ∞, the ML convergence results proved by (Lele et al. 2010) applies. Importantly, the ML estimates and their covariances are invariant to the choice of the prior distribution.
In practice, the DC methodology poses no other difficulties. For a given value of r, DC is implemented by simply copying the original data set (see sample files in the online Appendix, available from http://sysbio.oxfordjournals.org) r times and feeding the cloned data to any Bayesian software for phylogenetic analysis. As the number of clones r increases, if the parameters are identifiable, the marginal variance of each parameter in the r-th posterior distribution should converge to 0 (see Lele et al. 2010) at a rate of 1/r. As a consequence, the largest eigenvalue of the variance–covariance matrix of the r-th posterior distribution should also converge to 0 at a rate of 1/r. If, however, the likelihood surface is flat, as the number of clones is increased, the effect of the prior in the posterior distribution decreases too, and the r-th posterior distribution is dominated by a flat likelihood function. In this case, as r grows large, the variances of such posterior distribution do not converge to 0. Therefore, a useful statistic to assess parameter identifiability is to perform DC with multiple values of r, and each time, compute the first eigenvalue of the resulting posterior distribution. If these eigenvalues are standardized by the first eigenvalue of the first posterior distribution's variance, and if the parameters are identifiable, then the plot of the first eigenvalue of each of these r-th posterior distributions variances as a function of r should decrease at a rate equal to 1/r. However, if the parameters are non-identifiable, then the r-th posterior distribution converges to a distribution truncated over the space of nonestimable parameter values (Lele et al. 2010). The largest eigenvalue of such a posterior distribution will not converge to 0. Thus, DC can be used as a precise tool to diagnose nonidentifiability.
Asymptotic likelihood based inference is available for a wide array of models, including hierarchical models and many problems that were heretofore only accessible through Bayesian inference. Indeed, the DC methodology was originally derived as a means to deal with any hierarchical model of the following form (Lele et al. 2007; Ponciano et al. 2009):
where Y is a vector of observations, and X is a vector of unobserved random quantities (often called latent variables or random effects) on which the observations depend. While modeling ecological or evolutionary dynamics, X often represents a stochastic process of biological relevance, specified using the parameters θ. For both, Bayesians and frequentists alike, X is a random vector. Bayesians also think of θ as random quantities. Frequentists, however, regard the parameters θ as constants, unless another hierarchy of stochasticity with biological meaning is added to the model (i.e., if a stochastic processes giving rise to these parameters is specified to get a better understanding of the biology). In the context of phylogenetics, for instance, X could denote a birth–death model of speciations and extinctions, θ the vector of rates parameters at which these processes occur, while Y would represent the random samples from this stochastic process, obtained by an adequate statistical sampling model with parameters ϕ. In those cases, the likelihood function is obtained by averaging the probability of the observations, specified by the statistical sampling model, over all the possible realizations of the process:
Note that the dimension of the integral is the same as the number of components in the vector X. Because the Bayesian approach completely circumvents the problem of high-dimensional integration by using MCMC algorithms to sample from the joint posterior
where π(θ, ϕ) is a joint prior of the model parameters, ML estimation for θ and ϕ is made possible using DC by sampling instead from the r-th joint posterior distribution
for r large enough. Provided enough data are available, further layers of hierarchy can be added to this basic model. Furthermore, model selection, hypothesis testing, and likelihood profiles can all be computed efficiently for these hierarchical models using “likelihood ratios for DC” (Ponciano et al. 2009). In the context of phylogenetics, using this modeling approach, one could assume that for each transition, nature selects at random from a set of models (Evans and Sullivan 2012; Huelsenbeck et al. 2004) and that the main parameter of interest is the tree topology. The model identity can therefore be specified as another layer of latent components and the tree topology estimation could be performed after averaging over all possible models. Finally, in other applications, it has been shown that nonparametric mixture distributions, change-point processes, and general stochastic process, models with added sampling error can all be analyzed using a likelihood framework eased by DC (Lele et al. 2010)
Instead of focusing on the reaches of DC as an estimation tool, here we would like to address the usefulness of this novel approach as a tool to distinguish between the different identifiability and estimability scenarios mentioned above. We show that in so doing, DC emerges as a practical diagnostic methodology, regardless of the inferential paradigm adopted.
Results
The results are divided in three sections. In the first section, we illustrate the implementation of DC under three different estimability scenarios: SE, NI, and WE. In all three cases cases, we present a closed analytical form of the cloned posterior distribution, its moments, and its limiting behavior as the number of clones increases. We use examples built on Rannala's (Rannala 2002) simple models used to illustrate nonidentifiability of parameters in Bayesian phylogenetics inference. In the second and third sections we explore the properties of DC as a diagnostic tool for the tree topology estimation problem and to assess the extent to which increasing the length of a sequence alignment improves the parameter estimation process. We illustrate INE in this later phylogenetic context.
Implementing DC: Three Simple Analytical Examples
Strong Estimable parameters.—Rannala (2002) demons-trated the issues associated with parameter identifiability using two simple Bayesian statistical models, one of which was over parametrized. In the first model, the parameter of interest is Strong-Estimable SE. The observations Y1, Y2,…, Yn are assumed to be iid exponentially distributed with parameter λ. Let ∑i=1nYi=Δ. Then, the likelihood of the observations is
Before delving into the Bayesian approach for this first model, note that by setting the derivative of L(λ) equal to 0 and solving for λ, the ML estimate of λ, 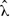 , is found to be
, is found to be 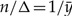 , i.e., the inverse of the sample mean. Because the set of values of λ that maximizes the likelihood contains only the point
, i.e., the inverse of the sample mean. Because the set of values of λ that maximizes the likelihood contains only the point 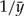 , here, the exponential rate λ is indeed an example of an identifiable parameter. Fisher's information ℐ(λ) and the asymptotic variance of the ML estimate
, here, the exponential rate λ is indeed an example of an identifiable parameter. Fisher's information ℐ(λ) and the asymptotic variance of the ML estimate 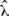 (Wald 1948) are
(Wald 1948) are
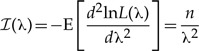 |
and
On the other hand, using the Bayesian approach, we could assume that the prior distribution for the model parameter λ is Gamma distributed with shape and scale parameters k and α, respectively, and pdf 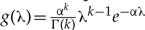 (Rannala 2002). Different priors can therefore be specified by changing the values of α and k. The posterior distribution of λ, π(λ|Y) is
(Rannala 2002). Different priors can therefore be specified by changing the values of α and k. The posterior distribution of λ, π(λ|Y) is
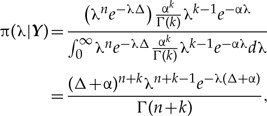 |
which is the pdf of a gamma distribution with parameters n+k and Δ+α. Note that the parameters of this gamma posterior distribution depend on the prior parameters α and k. Hence, specifying different prior distributions through changes in these parameters will result in changes in the posterior distribution. Recall now that in DC we write down the posterior distribution just as if by pure chance we had recorded r independent data sets that happen to be identical. That is, if we clone the data r times, the r-th posterior distribution is written as
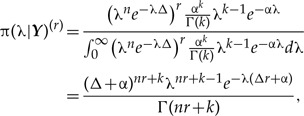 |
which is again a gamma distribution, but with parameters nr+k and Δr+α. Note that the mean and the variance of the r-th posterior distribution are now
and, just as it was mentioned above,
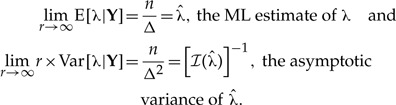 |
The above limits also imply that limr→∞ Var[λ|Y]=0, i.e., the variance of the r-th posterior distribution as r → ∞ converges to 0. Indeed, this is expected when a parameter is identifiable Lele et al. (2010). Finally, note that these convergence results are valid regardless of the specified prior distributions. Any change in the prior accomplished through changes in α and k does not affect the resulting limiting expressions for the mean and variance of the r-th posterior distribution. The above description is just a heuristic example. The formal proof of convergence of the mean and variance of the r-th posterior distribution to the ML estimate and to Wald's asymptotic variance of the ML estimate respectively, can be found in Lele et al. (2010). For now, it suffices to mention that such convergence occurs provided some regularity conditions are met (see Lele et al. 2010, for details).
NI parameters.—In Rannala's second example, the observations Y1, Y2,…, Yn are again exponentially distributed, but the exponential rate parameter λ is now assumed to be a sum of ℓ parameters, i.e., λ=λ1+λ2+···+λℓ. The likelihood of the observations is then written as
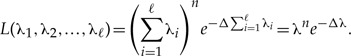 |
Note that when ∑i=1ℓ λi is replaced by λ, this likelihood is identical to the likelihood in the first example. Hence, there is a single value of λ that maximizes this likelihood function. However, because for every value of λ there is an infinite combination of values of λ1, λ2,…, λℓ such that λ = ∑i=1ℓ λi, there exists an infinite number of solutions to the problem of maximizing the likelihood function. Therefore, the parameters λi, i=1,…, ℓ are non-identifiable. The sum of these parameters, however, is identifiable and its ML estimate is identical to the ML estimate of λ in the first example, n/Δ.
While describing the Bayesian analysis for this model, Rannala (2002) assumed that the priors for the parameters λi were exponentially distributed with parameter α. Accordingly, the joint prior distribution is
It follows that the joint posterior distribution π(λ1, λ2,…, λℓ|Y) is
To keep the implementation of DC simple, in what follows we use ℓ=2, so that λ=λ1+λ2. Then, the joint posterior above simplifies to
When DC is implemented with r clones, the r-th joint posterior distribution is:
The simplicity of this joint posterior distribution allows us to write down a closed expression for the marginal posterior density for each of the two parameters, λ1 and λ2 (see Appendix 1). As a consequence, we can find the mean and variance for each marginal distribution, as well as the posterior covariance between these two parameters. Also, because the algebraic form of the joint posterior distribution for λ1 and λ2 is symmetric with respect to these two parameters, both marginal posterior distributions are identical. The expected value and the variance of the r-th marginal posterior for λi, i=1, 2 are (see Appendix 1):
| (1) |
| (2) |
Finally, the r-th posterior correlation between λ1 and λ2 is found to be (see Appendix 1)
| (3) |
Elementary calculations (Appendix 1) show that the posterior variances for both λ1 and λ2 [Equation (2)] do not converge to 0 as the number of clones tends to infinity. Instead, we have that the 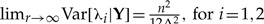 . Therefore, marginally, neither parameter is identifiable; however, the sum of the two parameters is identifiable. This result can be verified by looking at the distribution of a number (say 5000) of MCMC samples from the cloned posterior distribution of λ1, λ2 and λ1+λ2. As the number of clones goes from one to 200, the variance of the cloned posterior distribution of the sum goes to zero, whereas the variance of λ1 and λ2 does not (Figure 1). To see why the sum is identifiable whereas the individual components λ1 and λ2 are not in this case, first note that
. Therefore, marginally, neither parameter is identifiable; however, the sum of the two parameters is identifiable. This result can be verified by looking at the distribution of a number (say 5000) of MCMC samples from the cloned posterior distribution of λ1, λ2 and λ1+λ2. As the number of clones goes from one to 200, the variance of the cloned posterior distribution of the sum goes to zero, whereas the variance of λ1 and λ2 does not (Figure 1). To see why the sum is identifiable whereas the individual components λ1 and λ2 are not in this case, first note that 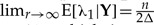 and hence limr→∞E[(λ1+λ2)|Y]=n/Δ, which is exactly the ML estimate of the sum of these parameters. Second, the variance of the sum of these parameters does converge to 0 as the number of clones increase. Indeed, noting that limr→∞ Corr(λ1, λ2|Y)=−1 we get that
and hence limr→∞E[(λ1+λ2)|Y]=n/Δ, which is exactly the ML estimate of the sum of these parameters. Second, the variance of the sum of these parameters does converge to 0 as the number of clones increase. Indeed, noting that limr→∞ Corr(λ1, λ2|Y)=−1 we get that
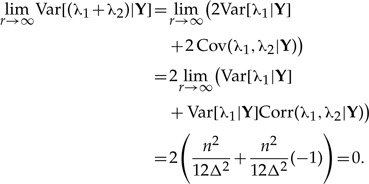 |
Figure 1.
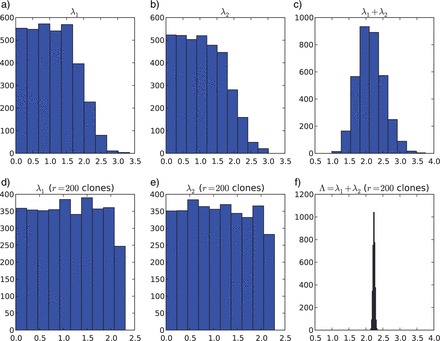
Posterior distribution for λ1, λ2 and λ1+λ2 in a standard Bayesian analysis (first row) and using DC, with the number of clones r=200 (second row). Note that as the number of clones increases (from no clones in a), b), c) to 200 in d), f), g)), the variance of the posterior distributions for λ1 and λ2 do not converge to 0, yet the variance of the posterior distribution for the sum does converge to 0 (bottom right pannel).
Rannala (2002), noting that the posterior correlation (without cloning) between λ1 and λ2 was equal to −n/(n+6), mentioned that, even as sample size increases, the correlation between the parameters does not decrease but rather converges to −1. He thus concluded that over parametrization can be diagnosed with the presence of a strong correlation in the posterior parameters despite having a very large sample size. Yet, he included a cautionary note mentioning that parameters may still be correlated and identifiable at the same time in some cases. Therefore, such correlation among parameters is not an unequivocal diagnostic tool. In contrast, DC does provide an unequivocal diagnostic of nonidentifiability, namely, the nonconvergence to 0 of the variance of the r-th posterior distribution of the parameter of interest. To check for such convergence, the first eigenvalue of the posterior distribution can be compared to the first (scaled) eigenvalue for the r-th posterior distribution, for various values of r (Lele et al. 2010).
WE parameters.—Here, we present an extension of Rannala's second model in which we add increasing amounts of information to tease apart the two parameters λ1 and λ2 from each other, and we implement DC with simulated samples from such scenarios. When the model parameters are identifiable but there is little data to estimate separately the parameters of interest, the likelihood surface will be nearly flat and thus result in WE parameters. This scenario poses practical difficulties if one wishes to ascertain whether the problem at hand cannot be resolved because of lack of data or because it is impossible to resolve, even with infinite amounts of data. Here, we show that weak identifiability can be clearly diagnosed and distinguished from the case where there is exact nonidentifiability.
As we have established above, in Rannala's second model, λ1 and λ2 are not identifiable. If, however, another sample of size m from an exponential distribution with parameter λ1 is observed and that information taken into account, then λ1 and λ2 could be easily estimated. As m decreases however, the extra information content about the parameter λ1 in such sample decreases and the curvature of the likelihood around the ML estimate of λ1 becomes more and more flat, and hence, λ1 becomes WE. Let Y11, Y12,…, Y1n be the observed samples from Rannala's statistical model, an exponential distribution with parameter λ1+λ2. Also, let Y21, Y22,…, Y2m be a set of samples from an exponential distribution with parameter λ1. Using the notation ∑i=1nY1i = Δn, ∑i=1mY2i = Δm and Δ=Δn+Δm, the joint likelihood of the two sets of observations is
and the ML estimates for the parameters are found to be
Using the Bayesian framework the joint posterior distribution of the parameters is (see Appendix 2)
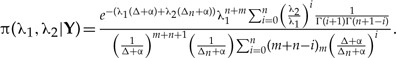 |
(4) |
Here again, we assumed an exponential prior distribution with parameter α for each of the parameters. Also, Y denotes a concatenated vector containing the two sets of observations and the notation (x)θ=x(x−1)(x−2)…(x−θ+1) represents the Pochhammer symbol, or falling factorial. Just as with the second model, a closed form expression for the r-th joint and marginal posterior distributions can be found, along with their corresponding mean and variances. In Figure 2, we explored via simulations and using the exact analytical formulae presented in Appendix 2 what happens to the mean and variance of the marginal r-th posterior distribution for λ1 in this third model, when the sample size of the second experiment that allows the separation between λ1 and λ2 decreases progressively to 0. When the extra sample size (m) is large, the empirical mean (blue line) of the MCMC samples from the r-th posterior distribution matches the ML estimate (horizontal black line) when the number of clones r is about 11. As a check, we also plotted the value of the analytical expression for the mean of the r-th posterior distribution in that model [black crosses in Figure 2, see equation also (A.8)]. Note that by the time m=2, it takes a much higher number of clones (about 140) for the mean of the r-th distribution to match the value of the ML estimate. Figure 2 also shows that as the extra sample size diminishes, the size of the asymptotic Wald confidence intervals increases. These confidence intervals were computed by multiplying by 1.96 the square root of the empirical variance–covariance matrix of the MCMC samples multiplied by r, i.e., 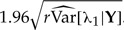 . Of particular interest is the comparison between the size of the confidence intervals when m=2 vs. the case when m=0, that is, when no extra information about the parameter λ1 is available. As the number of clones increases, but before the mean of the cloned posterior reaches the ML estimate, the size of the confidence intervals seems to increase in both cases. However, in the first case, the slope of the DC standard deviation curve does not change as the number of clones increase, whereas when m=0 the slope of such curve grows notably with an increasing cloning size. Thus, even if there is a very small amount of extra information about a parameter that allows it to be identifiable, like when m=2, as the number of clones increases, the changes in the size of the confidence intervals are linear. However, when the parameter is non-identifiable (m=0), an early indication that the DC asymptotic result will never be reached is the fact that the size of the confidence intervals grows at a highly nonlinear rate.
. Of particular interest is the comparison between the size of the confidence intervals when m=2 vs. the case when m=0, that is, when no extra information about the parameter λ1 is available. As the number of clones increases, but before the mean of the cloned posterior reaches the ML estimate, the size of the confidence intervals seems to increase in both cases. However, in the first case, the slope of the DC standard deviation curve does not change as the number of clones increase, whereas when m=0 the slope of such curve grows notably with an increasing cloning size. Thus, even if there is a very small amount of extra information about a parameter that allows it to be identifiable, like when m=2, as the number of clones increases, the changes in the size of the confidence intervals are linear. However, when the parameter is non-identifiable (m=0), an early indication that the DC asymptotic result will never be reached is the fact that the size of the confidence intervals grows at a highly nonlinear rate.
Figure 2.
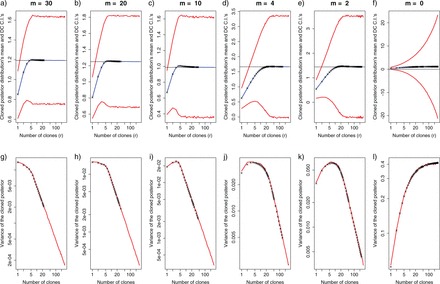
Empirical and analytical mean and variance of the r-th posterior distribution for the third analytical example with exponential random variables. The first row (panels a) to f)) shows in blue the empirical mean of π(λ1|Y)(r) calculated from MCMC samples of the joint r-th posterior distribution. In red, are the empirical Wald confidence intervals from those MCMC samples, also as a function of the number of clones and as the sample size m of the extra sample that allows identifying separately λ1, decreases to 0. The black crosses are the analytical means of the r-th marginal posterior distribution of λ1. The horizontal black lines denote the location of the analytical ML estimate. The second row [(panels g to l)] shows the changes in the empirical variance from the MCMC samples of the r-th posterior distribution π(r)(λ1|Y) as the number of clones increases, for the same sizes of the second sample. The black crosses denote the corresponding analytical variance of the r-th marginal posterior distribution of λ1
DC Inference for Tree Topology: The Analysis of a Chloroplast Data Set
One of the parameters of interest in phylogenetic analysis, the tree topology, is discrete in nature. Unfortunately, the asymptotic results for ML estimation in the discrete case are restricted (see for instance Lindsay and Roeder 1987 and subsequent work). Further, the regularity conditions under which DC has been proven seem not to be met (Lele et al., 2010, Appendix). Nevertheless, biological intuition indicates that better resolution of the tree topology can be brought about by increasing the amount of sequence data. But, how can we know if enough new data have been added to reliably resolve the phylogeny of interest? Answering this question amounts to assessing the changes in the quality of the tree topology estimate as the amount of sequence data included in the alignment is increased. Thus, in this section we investigate the properties of DC as a diagnostic tool for the tree estimation problem despite the inherent discreteness of topologies.
We used the 83 gene chloroplast genome data set from (Moore et al. 2010), which includes sequences from 86 seed plant taxa, to assess the properties of the tree topology estimate when different amounts of data were used for the analysis. We implemented DC using three single-gene (atpB, ndhF, and rbcL) subsets of this data as well as the entire 83 gene concatenated alignment.
Because the key DC diagnostic of estimability in continuous parameters is the decline of the variance of the posterior distribution toward 0 as the number of clones increases, we studied the changes in total variability in the posterior samples of trees for increasing number of clones to look for analogous behavior. To study the variation in topology space as the number of clones increased, we adopted the approach of (Chakerian and Holmes 2010). By embedding rooted binary trees in a metric space associated with the BHV distance Billera et al. (2001), these authors capitalized on statistical methods such as multi-dimensional scaling (MDS) to evaluate and study the properties of a posterior distribution of trees. A measure of the variance of the posterior distribution of trees can be taken to be the sum of squares of all the elements of the BHV distance matrix among the trees sampled. Therefore, we simply studied the changes in the total sum of squared distances in the posterior distribution of trees as the number of clones increased and compared the rate of these changes with the predicted rate of 1/r by the DC theory of (Lele et al. 2010) for continuous parameters.
Using the software MrBayes version 3.1.2 (Ronquist et al. 2005) in the default settings for the GTR +I+Γ model we ran an MCMC analysis for 10 million generations using 1,2,4,8,16, and 32 clones. Samples from each of these Markov Chains were taken every 1000 generations. The last 5000 samples of the chains were used for the analyses. In each case, besides the 5000 samples of the continuous parameters, we also stored the 5000 samples of the posterior tree topologies and branch lengths. We then randomly took a subsample of size 500 from the sampled posterior distribution of trees and computed the matrix of BHV pairwise distances between each of these 500 trees using the methodology and R software described in (Chakerian and Holmes 2010). Next, we used that matrix of distances to obtain the best representation in Euclidean space of the relative positions of the sample of trees using nonlinear MDS in R (Borcard et al. 2011).
The Euclidean representation of the sampled trees and the total number of different topologies in the sample, for each cloning size of the entire data set is shown in Figure 3. Also shown in Figure 3 is the progression of the total sum of squared BHV (TSS-BHV) distances as the number of clones r increases from 1 to 16. Each of these TSS-BHV distances was divided by the TSS-BHV distances when r=1 to obtain a relative measure of the total variance in the posterior sample of tree topologies. As it can be seen in Figure 3, the TSS-BHV distances decay very much like 1/r, which strongly suggests that DC as a tool for ML estimation for the tree topology works, and in particular, that the topology is indeed estimable in this case. Note also how the number of different topologies as well as the overall dispersion in the non-metric MDS (NMDS) axis of the posterior sample of the trees tends to decrease. Finally, we point that the sum of squares used here is not traditional. Usually, sums of squares are defined from distances to a central point, which would correspond to a central tree here. Both, the formal definition and proper calculation of a “central” tree for the BHV distance is still an active area of research (e.g., Nye 2011; Owen 2008; Owen and Provan 2011).
Figure 3.
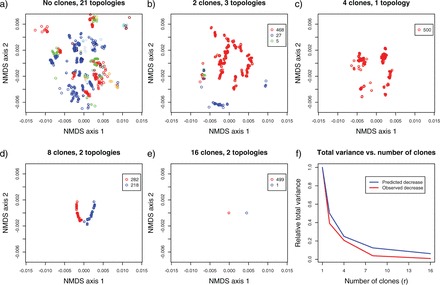
NMDS representation in Euclidean space of a sample of size 500 trees from the r-th posterior distribution in geodesic space of the tree topologies, as the number of clones increase [(panels a) to e)], for the complete chloroplast data set (86 genes). Each color represents a different topology, and the number of different topologies is specified above each plot. Panel f) depicts the reduction in the relative total variance as the number of clones increases (see text for details).
In Figure 4, we present the results from the analysis of the atpB single gene data set. In this case, the overall variance of the posterior distribution of topologies also decreases (see insert in Figure 4, panel f), but not at the expected rate of 1/r. This finding suggests that the number of clones at which DC begins to stabilize (e.g., Figure 2) and the variance begins to drop like 1/r has not been reached yet, but that the topology parameter is indeed identifiable. Just like in the third exponential model however, although the parameter of interest is still identifiable with a small sample size, it may take many more clones to get the ML estimate of the topology and we expect the quality of the inferences (e.g., precision of the estimate) to be poor. Different rates of decrease were found with the rbcL and ndhF data sets (see online Appendix with the program), which indicates the different amounts of information borne by the different genes.
Figure 4.
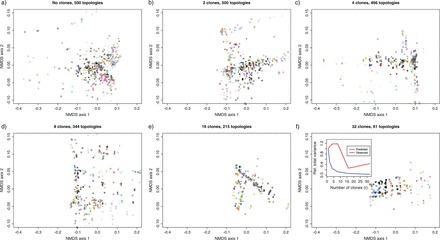
NMDS representation in Euclidean space of a sample of size 500 trees from the r-th posterior distribution in geodesic space of the tree topologies, as the number of clones increase [panels a) to f)], for a single gene (atpB) from the chloroplast data set. Each color represents a different topology, and the number of different topologies is specified above each plot. The insert in panel f) depicts the reduction in the relative total variance as the number of clones increases (see text for details).
Chance and Estimability in Phylogenetic Analyses
We have asserted above that chance events in the sampling of data may lead the analysis of some data sets to have parameters that are identifiable but not estimable while other data sets of the same size may contain parameters that are SE or WE. We have also indicated that estimability is not an all or nothing property. Some data sets may have a group of INE parameters but still have other parameters that are identifiable. Here, we demonstrate both these claims with the re-analysis of a real data set.
Increasing the length of the sequence alignment used for parameter estimation certainly is a means to obtain more precise estimates of the continuous evolutionary model parameters. The extent to which the added information improves the parameter estimation process can be directly measured using statistics such as the mean squared error (MSE), or by computing the amount by which the observed Fisher's information improves. Furthermore, classical asymptotic results for continuous ML parameter estimates are readily available, and, as a consequence, the DC methodology can unequivocally be used as an identifiability diagnostic tool. To illustrate such scenario, we analyzed data sets of varying sequence length constructed by subsampling at random data sets from increasing sequence lengths out of the well-known primate data set (Hayasaka et al. 1988).
The “primates data set” is an alignment of 898 bp including 12 taxa. In our analysis, we re-sampled the original data set with replacement to form nonparametric bootstrap replicates of 14, 28, 56, 112, 225, 449, and 898 bp in length. For each of these lengths, 250 random alignments were formed, thus obtaining 7×250=1750 data sets. For each data set, we performed DC using 2, 4, 16, and 32 clones with MrBayes by implementing a standard Bayesian analysis using the default settings for the GTR +I+Γ model. The MCMC was ran for 10 million generations, and samples were taken every 1000 generations. The last 5000 samples of the chain were used for the analyses.
For each combination of alignment length, random replicate number, and total number of clones used, 5000 samples of the multivariate posterior distribution of the (continuous) model parameters were saved, as well as their corresponding tree topologies and branch lengths. For each of these matrices of 5000 rows of the continuous parameters, the empirical variance–covariance matrix was computed as an estimate of the joint posterior variance–covariance matrix, from which the first eigenvalue was computed and saved. The boxplots for each set of 250 estimates of the first eigenvalue are plotted in Figure 5, for each combination of base pair length and number of clones used. Note how, as the number of clones and the sequence length increases, both the median and the variance of the distributions of the first eigenvalues decrease. The plot in the second upper panel highlights the fact that for the same sample size, different amounts of information may be borne by different data sets of the same size. Thus, for the same alignment length, some randomly sampled data sets end up containing much more information about the continuous model parameters of interest than others (see Figure legend for details). Note that, although we are calling the resampled data set “bootstrap” samples, we are not using these samples for statistical inference for the original data set, only for illustrating chance events in the sampling of data leading to different estimability problems.
Figure 5.
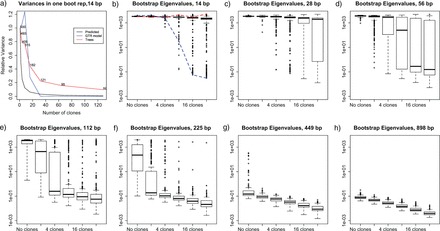
Nonparametric bootstrap distribution of the first eigenvalue of the variance–covariance matrix of the r-th cloned posterior distribution, for r=1, 2, 4, 8, 16, 32 [panels b) to h)], when the resampled replicates are 14, 28, 56,112,225,449, and 898 bp long. Panel a) depicts the rate of decrease of the first eigenvalue of the posterior distribution sample variance for the continuous parameters (in blue) and the trees (in red, see explanation of Figure 4 in the text), and compared with the theoretical rate decrease of 1/r (in black). In panel b), the changes in the first eigenvalue for two other data sets also of size 14 bp are followed with a red and with a blue dotted line. In one of the cases (marked with a blue line), the size of the first eigenvalue decreases sharply as the number of clones increases. In the other case (red line), it does not.
In the upper leftmost panel of Figure 5, we show the results of the analysis of only one of the 250 randomly drawn data sets of length 14 bp. For that particular data set, we ran DC with 1, 2, 4, 8, 16, 32, 64, and 128 clones. We then plotted together the rate of decrease of the first eigenvalue of the posterior distribution sample variance for the continuous parameters (in blue) to the theoretical rate decrease of 1/r (in black). Once the first eigenvalue size begins to decrease, it does so at the expected rate of 1/r, thus indicating that the continuous model parameters are identifiable (although the size of the confidence intervals for such parameter is expected to be relatively high). The variance of the posterior distribution of topologies, however, seems to stabilize at a nonnull quantity. Furthermore, the number of topologies in the posterior sample of trees does not drop down below 59. These results strongly suggest that although the evolutionary model parameters may be identifiable, the topology itself may not be estimable for such low sample size.
Discussion
Non-identifiability of parameters has long been known in the statistical literature Rothenberg (1971) and expositions like Rannala's paper Rannala (2002) have helped to introduce to the phylogenetics community the important inferential problems it poses. Here, we have shown how to use data cloning as an unequivocal, easy to implement tool to detect the presence of nonidentifiability.
Practitioners only need to clone their sequence alignment (see Supplementary Material to see examples) and run the cloned alignment using a Bayesian software pertinent to the problem at hand. As the number of clones r increases, the posterior mean of the identifiable model parameters should stabilize numerically, whereas their posterior variance decreases toward 0 like the function 1/r. Non-identifiable parameters, if present, would be exposed because their cloned posterior variance does not decrease to 0, and rather, it would appear to stabilize at a nonnull quantity. Hence, the value of r times the variance of the marginal r-th posterior distribution for these parameters, for increasing r, would also appear as ever increasing.
Theoreticians now have a powerful tool to detect nonidentifiability while investigating complex modeling scenarios, where getting closed-form expressions for the r-th posterior distribution is not as straightforward as in the exponential model cases shown here. Even if a great deal of thought is invested on the model formulation, (McCulloch and Searle 2001), (Yang and Rannala 2006), and (Lele et al. 2010) show that it is relatively easy for nonidentifiability to be introduced inadvertently in a model.
Nonidentifiability and weak estimability also can be distinguished early on in the DC process. In a WE scenario, as the number of clones is increased, the rate of change of the DC variance does not change as strongly as in the NI case. Indeed, because the variance of the cloned posterior distribution does not converge to 0 when NI is present as the number of clones increases, Wald's asymptotic variance, computed as r times the empirical variance of the posterior distribution, will be an ever increasing quantity. In both cases (WE and NI), in the beginning of the cloning process, the mean of the cloned posterior distribution is changing. Also, in both cases, it is possible that at the beginning of the cloning process the variance of the cloned posterior distribution increases instead of it diminishing (see Figure 2, two rightmost panels of the lower row). Then, in both cases, the graph of the DC standard deviation as a function of r will seem monotonically increasing. However, in the NI case, the slope of such graph changes much faster than in the WE case. This fact may be used as an early diagnostic tool to ascertain the particular estimability scenario, although more research regarding this topic in phylogenetic examples is needed.
Although we show examples of the use of DC to recognize NI, WE, SE, and INE, we do not include an example of the application of DC and NI to phylogenetics. Our results call for a variety of topics for further research. In particular, DC may be especially effective to check the estimability of parameters in complex hierarchical models where it is very difficult to elicit prior distributions, such as the recent species tree/gene tree models (Liu and Pearl 2007).
It is unquestionably true that hierarchical models, like the ones solved through reversible MCMC in a Bayesian context (Evans and Sullivan 2012; Green 1995; Hastie and Green 2012; Huelsenbeck et al. 2004), have enormously increased the scope and complexity of modeling in phylogenetics. However, neither hierarchical models nor MCMC should be automatically associated with Bayesian inference (see Felsenstein 2004; Hastie and Green 2012; Lele and Dennis 2009; Robert and Casella 2004). As stated earlier, DC can yield likelihood inference from an initial Bayesian formulation for hierarchical models (Lele et al. 2010), thus a natural avenue for further research is evaluating how, and if at all, current Bayesian software for fitting hierarchical phylogenetic models can be used to yield samples from the appropriate cloned posterior. A requirement for this to happen is that the implementation must allow the replication of both, the data and the latent structure. However, this article is only concerned with informing practitioners and theoreticians about the possibility of using DC to assess parameter identifiability, reveal weak estimability, and quantify the effect of priors. The extent and applicability of DC as an inferential tool for complex model structures is a topic for further research.
If DC detects nonestimability, it could be because the parameters are truly NI and no additional data will help, or it could be that some aspect of the data prevent estimation, but estimation would be possible with other data sets (INE). We believe that these cases could be distinguished through suitable additions of small random perturbations to the data, which is analogous to a technique known as the “Infinitesimal Jackknife” (Efron and Tibshirani 1993). Further research should clarify if this is a useful approach. If so, this could allow practitioners wishing to determine if it is worthwhile to collect more data to resolve a difficult phylogenetic problem. For instance, it might be possible to use DC as a tool to distinguish between the case where adding data will never result in the resolution to a bifurcating tree (i.e., when a hard polytomy is present) and a situation where the addition of data would effectively resolve the problem of estimating the divergence times of all the lineages in the tree such that each node has only two immediate descending branches (we note however that in general, for DC to converge on the true parameter value, the support of the prior distribution must include such value). Accordingly, in order to resolve a polytomy problem, the MCMC implementation used must include 0 as a plausible prior branch length (Braun and Kimball 2001; Edwards 2009; Lewis et al. 2005).
DC is a computer-intensive numerical device to find the ML estimates and thus, by nature, shares some similarity with other computer-intensive methods in phylogenetics. While cloning the data, the analyst must honor the structure of the data. For instance, if one is dealing with time series data, one replicated data set is composed of an entire time series of points (Lele et al. 2007; Ponciano et al. 2009). It is important to realize, however, that in doing so, the quality of the inferences is not artificially boosted by performing the cloning. The data copies are only used to get the ML estimate, and if the data are weakly informative, the ML estimate and its precision will likewise have poor statistical properties (Figure 2). Both, DC and the bootstrap seek an estimate of the sampling distribution of the ML estimate. However, the point of cloning the data is to retrieve the ML estimates when the likelihood function is not tractable. While the bootstrap is also an inferential tool, it seeks to describe the properties of the ML estimates, given that the ML estimates are obtainable. If DC is fast enough to perform for a particular problem, one could use parametric bootstrap to estimate the sampling distribution of the ML estimates by computing them using DC for each bootstrap sample. Finally, DC bears some resemblance with simulated annealing, and this has been discussed elsewhere (Lele et al. 2007; Robert and Casella 2004).
DC can also be used as a tool to examine the influence of the priors. To a great extent, Bayesian statistical analysis in phylogenetics has gained popularity over the years as an alternative to the problem of maximizing difficult likelihood functions, rather than as a method to incorporate prior knowledge. The process of learning about a particular evolutionary process while taking into account a priori information has been successfully carried by means of subjective Bayesianism (e.g., Huelsenbeck et al. 2008). However, when it is not clear at all how to elicit priors, as in the case of hierarchical models (Lele and Dennis 2009), DC can be used as an effective tool to accurately assess the influence of the prior distributions on the estimation results. If the data set happens to be very informative, the prior distribution will carry a small weight and the posterior mode and the ML estimate should not differ by much. Different data sets of the same size can carry vastly different amounts of information (see Figure 1S in Online Appendix 3, second panel from left to right, upper row). A practitioner does not know how informative a particular data set is a priori and the cloning process could be used to easily reveal by how much do the ML estimates and the Bayesian Maximum A Posteriori (MAP) estimates differ. Such exercise should expose the extent to which any given prior distribution is really “non-informative” (e.g., (Brown et al. 2010).
When applied to phylogenetic inference, DC motivates at least two important statistical questions. First, because we cannot use ML asymptotic theory in the discrete tree space, the results plotted in Figures 3 and 4 show that, potentially, we could do all of our tree-related inference in the BHV distance space using profile likelihoods and then transfer it back to the tree space. However, we do not know if the inference would be transformation invariant. A strong indication that invariance may indeed occur is the fact that the SS-BHV total distance decreases like 1/r, just like the predicted decrease of the variance of the cloned posterior distribution, for continuous parameters. Hence, just as (Chakerian and Holmes 2010) suggested, this sum of squares can be used as a measure of the variance of the posterior distribution in tree-distance space. Second, because implementing DC is just like implementing a standard Bayesian analysis, except that with more data, running a cloning process can be computationally expensive and difficult to tune. Recent work shows, however, that when the statistical problem involves computationally expensive posterior densities, efficient sampling methods can be implemented (Bliznyuk et al. 2011) and DC can potentially be used in conjunction with such methodology to further improve the posterior sampling efficiency. Because DC uses MCMC, it inherits all the technical difficulties associated with any MCMC implementation. One of these is finding an adequate mixing of the Markov Chain so that the parameter space is suitably explored. Note however that, when multi-modality in the likelihood function occurs, as the number of clones increase in DC, the multiple modes become less and less important while the peakedness of the consistent mode increases (Lele et al. 2007). Because smaller peaks can be more and more difficult to escape from (Rannala et al. 2012), on a practical note, we strongly suggest that when multiple independent chains are run, convergence to the same set of posterior trees for large r and all the chains should be sought after.
Phylogenetics has clearly come to an age where fundamental biological questions are posed and answered by means of the language of stochastic processes and computer-intensive, model-based inference. Relying extensively on modern computationally techniques requires vigilant examination of the very tools we use to learn from the natural world. Data cloning, as a technique, has grown from being estimation centered (Lele et al. 2007), to include hypothesis-testing and model-selection (Lele et al., 2010; Ponciano et al., 2009) and this study demonstrate its virtues as a diagnostic tool. Further, we show here that despite the peculiarities of the tree topology estimation problem—the presence of an inherently complex discrete parameter—DC can serve as a powerful yet easy to implement tool to diagnose the quality of statistical inference in phylogenetics. Thus, data cloning has an enormous potential to help both, practitioners and theoreticians alike.
Supplementary Material
Supplementary material, including data files and/or online-only appendices, can be found at http://datadryad.org and in the Dryad data repository (DOI:10.5061/dryad.rr6400b4).
Acknowledgments
We thank the Associate Editor, Cécile Ané, Subhash Lele, Paul Joyce, Darin Rokyta, Mary Christman, Robert D. Holt, Emily Lemmon, and Alan Lemmon for useful comments and insights. Avinash Ramu helped to run the chloroplast and primate data sets and Julie Allen assisted during the preparation of figures for final submission.
Appendix 1
Here we derive the expected value and variance of the r-th marginal posterior for λ1 in Rannala's second model, along with the r-th covariance and correlation between the parameters λ1 and λ2 and the limit results involving those quantities stated in the main text. To do so, we need to find first the marginal posterior of λ1 by integrating the joint posterior distribution over the second parameter, λ2:
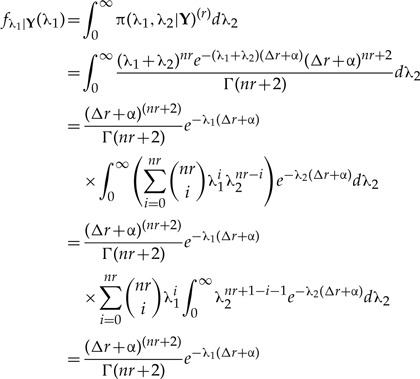 |
(A.1) |
where besides algebraic cancelations, in the step before the last we used the fact that
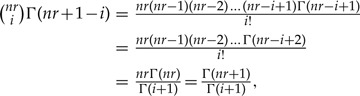 |
Once a closed expression for the r-th marginal posterior for λ1 is found, it is straightforward to compute the expected value and variance [Equations (1) and (2)] of such distribution from their definition (see for instance Rice 1995, chapter 4). Accordingly, the first moment is found to be
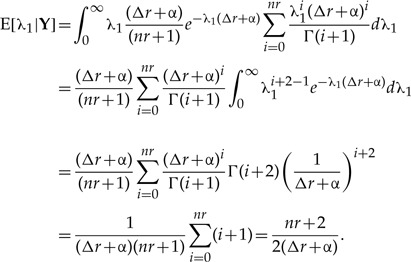 |
(A.2) |
The variance is found by subtracting the squared first moment from the second moment, that is:
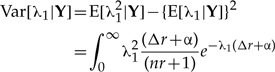 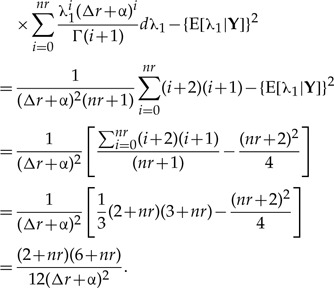
|
(A.3) |
The covariance between λ1 and λ2 is in turn
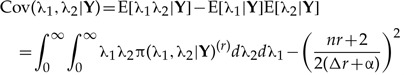 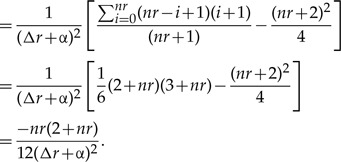
|
(A.4) |
Noting that marginally, the r-th posterior for λ1 and λ2 are identical and therefore their variances are the same and equal to Equation (2) in the main text and applying the definition of correlation we may use the expressions above to compute the r-th posterior correlation between the parameters:
| (A.5) |
Finally, we leave to the interested reader to verify that in order to show that 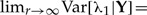 it suffices to develop both the numerator and the denominator, simplify, and divide both quantities by r elevated to its highest exponent in the fraction. The limit of E[λ1|Y] as r→∞ is found in the same way.
it suffices to develop both the numerator and the denominator, simplify, and divide both quantities by r elevated to its highest exponent in the fraction. The limit of E[λ1|Y] as r→∞ is found in the same way.
Appendix 2
Here we derive the mean and variance of the r-th marginal posterior distribution for λ1 under the third model, where extra information about this parameter is added through an extra sample of size m from an exponential distribution with parameter λ1. Thus, the total sample size is n+m. To derive the first two moments of this marginal posterior distribution, first note that the joint posterior distribution with r=1 (i.e., the standard bayesian posterior) of these parameters is:
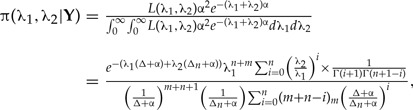 |
(A.6) |
where the notation (x)θ=x(x−1)(x−2)…(x−θ+1) represents the Pochhammer symbol, or falling factorial. To go from the first line to the second one in the above equation we used the binomial expansion of (a+b)n (where a, b are constants and n is an integer), various laborious algebraic simplifications and three simple mathematical facts:
The closed expression for the r-th joint posterior distribution is very similar to the standard joint posterior distribution above:
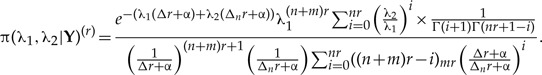 |
(A.7) |
By integrating this joint distribution over λ2 we find the marginal r-th posterior distribution of the parameter λ1, with which we compute the first two moments and the variance of λ1 as a function of the number of clones, much in the same way as what is shown in Appendix 1. These moments are found to be
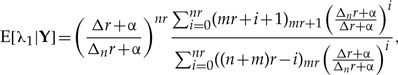 |
(A.8) |
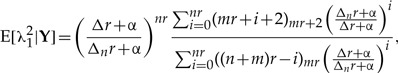 |
(A.9) |
and the variance is
| (A.10) |
These quantities were then computed and compared with the empirical mean and variance from MCMC samples of the r-th posterior distribution as the number of clones increases (Figure 2).
Funding
Support for this project was provided by funds from the College of Liberal Arts and Sciences at the University of Florida to J.M.P.
References
- Abdo Z., Minin V., Joyce P., Sullivan J. Accounting for uncertainty in the tree topology has little effect on the decision-theoretic approach to model selection in phylogeny estimation. Mol. Biol. Evol. 2005;22:691–703. doi: 10.1093/molbev/msi050. [DOI] [PubMed] [Google Scholar]
- Akashi H., Gojobori T. Metabolic efficiency and amino acid composition in the proteomes of Escherichia coli and Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 2002;99:3695–3700. doi: 10.1073/pnas.062526999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alfaro M., Holder M. The posterior and the prior in bayesian phylogenetics. Annu. Rev. Ecol. Evol. Syst. 2006;37:19–42. [Google Scholar]
- Allman E., Ané C., Rhodes J. Identifiability of a Markovian model of molecular evolution with gamma-distributed rates. Adv. Appl. Probab. 2008;40:229–249. [Google Scholar]
- Allman E., Rhodes J. The identifiability of tree topology for phylogenetic models, including covarion and mixture models. J. Comput. Biol. 2006;13:1101–1113. doi: 10.1089/cmb.2006.13.1101. [DOI] [PubMed] [Google Scholar]
- Allman E., Rhodes J. Identifying evolutionary trees and substitution parameters for the general Markov model with invariable sites. Math. Biosci. 2008;211:18–33. doi: 10.1016/j.mbs.2007.09.001. [DOI] [PubMed] [Google Scholar]
- Billera L., Holmes S., Vogtmann K. Geometry of the space of phylogenetic trees. Adv. Appl. Math. 2001;27:733–767. [Google Scholar]
- Bliznyuk N., Ruppert D., Shoemaker C. Efficient interpolation of computationally expensive posterior densities with variable parameter costs. J. Comput. Graph. Stat. 2011;20:636–655. doi: 10.1198/jcgs.2011.09212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borcard D., Gillet F., Legendre P. Numerical Ecology with R. Springer; 2011. [Google Scholar]
- Brandley M., Leaché A., Warren D., McGuire J. Are unequal clade priors problematic for Bayesian phylogenetics? Syst Biol. 2006;55:38–146. doi: 10.1080/10635150500431155. [DOI] [PubMed] [Google Scholar]
- Braun E., Kimball R. Polytomies, the power of phylogenetic inference, and the stochastic nature of molecular evolution: a comment on Walsh et al. (1999) Evolution. 2001;55:1261–1263. doi: 10.1111/j.0014-3820.2001.tb00647.x. [DOI] [PubMed] [Google Scholar]
- Brown J., Hedtke S., Lemmon A., Lemmon E. When trees grow too long: investigating the causes of highly inaccurate Bayesian branch-length estimates. Syst. Biol. 2010;59:145–161. doi: 10.1093/sysbio/syp081. [DOI] [PubMed] [Google Scholar]
- Cartwright R., Lartillot N., Thorne J. History can matter: non-Markovian behavior of ancestral lineages. Syst. Biol. 2011;60:276–290. doi: 10.1093/sysbio/syr012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai J., Housworth E. On Rogers' proof of identifiability for the GTR + Γ + I model. Syst. Biol. 2011;60:713–718. doi: 10.1093/sysbio/syr023. [DOI] [PubMed] [Google Scholar]
- Chakerian J., Holmes S. Computational tools for evaluating phylogenetic and hierarchical clustering trees. 2010 doi: 10.1080/10618600.2012.640901. Arxiv preprint arXiv:1006.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J. Full reconstruction of Markov models on evolutionary trees: identifiability and consistency. Math. Biosci. 1996;137:51–73. doi: 10.1016/s0025-5564(96)00075-2. [DOI] [PubMed] [Google Scholar]
- Chojnowski J., Braun E. Turtle isochore structure is intermediate between amphibians and other amniotes. Integ. Comp. Biol. 2008;48:454–462. doi: 10.1093/icb/icn062. [DOI] [PubMed] [Google Scholar]
- Chojnowski J., Franklin J., Katsu Y., Iguchi T., Guillette L., Kimball R., Braun E. Patterns of vertebrate isochore evolution revealed by comparison of expressed mammalian, avian, and crocodilian genes. J. Mol. Evol. 2007;65:259–266. doi: 10.1007/s00239-007-9003-2. [DOI] [PubMed] [Google Scholar]
- Doucet A., Godsill S., Robert C. Marginal maximum a posteriori estimation using Markov Chain Monte Carlo. Stat. Comput. 2002;12:77–84. [Google Scholar]
- Drummond A., Ho S., Phillips M., Rambaut A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006;4:e88. doi: 10.1371/journal.pbio.0040088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberly L., Carlin B. Identifiability and convergence issues for Markov Chain Monte Carlo fitting of spatial models. Stat. Med. 2000;19:2279–2294. doi: 10.1002/1097-0258(20000915/30)19:17/18<2279::aid-sim569>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Edwards S. Is a new and general theory of molecular systematics emerging? Evolution. 2009;63:1–19. doi: 10.1111/j.1558-5646.2008.00549.x. [DOI] [PubMed] [Google Scholar]
- Efron B., Tibshirani R. An introduction to the bootstrap. Vol. 57. Chapman & Hall/CRC; 1993. [Google Scholar]
- Evans J., Sullivan J. Generalized mixture models for molecular phylogenetic estimation. Syst. Biol. 2012;61:12–21. doi: 10.1093/sysbio/syr093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Felsenstein J. Cases in which parsimony or compatibility methods will be positively misleading. Syst. Biol. 1978;27:401–410. [Google Scholar]
- Felsenstein J. Parsimony in systematics: biological and statistical issues. Annu. Rev. Ecol. Syst. 1983;14:313–333. [Google Scholar]
- Felsenstein J. Inferring phytogenies. Sunderland (MA): Sinauer Associates; 2004. [Google Scholar]
- Fisher D. Stratocladistics: integrating temporal data and character data in phylogenetic inference. Annu. Rev. Ecol. Syst. 2008;39:365–385. [Google Scholar]
- Gelfand A., Sahu S. Identifiability, improper priors, and Gibbs sampling for generalized linear models. JASA. 1999;94:247–253. [Google Scholar]
- Green P. Reversible jump markov chain monte carlo computation and bayesian model determination. Biometrika. 1995;82:711–732. [Google Scholar]
- Hastie D. I., Green P. J. Model choice using reversible jump Markov Chain Monte Carlo. Stat. Neerland. 2012 doi: 10.1111/j.1467-9574.2012.00516.x. [Google Scholar]
- Hayasaka K., Gojobori T., Horai S. Molecular phylogeny and evolution of primate mitochondrial DNA. Mol. Biol. Evol. 1988;5:626–644. doi: 10.1093/oxfordjournals.molbev.a040524. [DOI] [PubMed] [Google Scholar]
- Hendy M., Penny D. A framework for the quantitative study of evolutionary trees. Syst. Biol. 1989;38:297–309. [Google Scholar]
- Hillis D., Bull J., White M., Badgett M., Molineux I. Experimental phylogenetics: generation of a known phylogeny. Science. 1992;255:589–592. doi: 10.1126/science.1736360. [DOI] [PubMed] [Google Scholar]
- Hillis D., Huelsenbeck J., Cunningham C. Application and accuracy of molecular phylogenies. Science. 1994;264:671–677. doi: 10.1126/science.8171318. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck J., Joyce P., Lakner C., Ronquist F. Bayesian analysis of amino acid substitution models. Philos. T. Roy. Soc. B. 2008;363:3941–3953. doi: 10.1098/rstb.2008.0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck J., Larget B., Alfaro M. Bayesian phylogenetic model selection using reversible jump Markov Chain Monte Carlo. Mol. Biol. Evol. 2004;21:1123–1133. doi: 10.1093/molbev/msh123. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck J., Larget B., Swofford D. A compound Poisson process for relaxing the molecular clock. Genetics. 2000;154:1879–1892. doi: 10.1093/genetics/154.4.1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck J., Rannala B. Frequentist properties of Bayesian posterior probabilities of phylogenetic trees under simple and complex substitution models. Syst. Biol. 2004;53:904–913. doi: 10.1080/10635150490522629. [DOI] [PubMed] [Google Scholar]
- Jacquier E., Johannes M., Polson N. MCMC Maximum Likelihood for latent state models. J. Econometrics. 2007;137:615–640. [Google Scholar]
- Jobson R., Qiu Y. Amino acid compositional shifts during streptophyte transitions to terrestrial habitats. J. Mol. Evol. 2011:1–11. doi: 10.1007/s00239-010-9416-1. [DOI] [PubMed] [Google Scholar]
- Kim J. Slicing hyperdimensional oranges: the geometry of phylogenetic estimation. Mol. Phylogenet. Evol. 2000;17:58–75. doi: 10.1006/mpev.2000.0816. [DOI] [PubMed] [Google Scholar]
- Kuk A. Automatic choice of driving values in monte carlo likelihood approximation via posterior simulations. Stat. Comput. 2003;13:101–109. [Google Scholar]
- Lele S., Allen K. On using expert opinion in ecological analyses: a frequentist approach. Environmetrics. 2006;17:683–704. [Google Scholar]
- Lele S., Dennis B. Bayesian methods for hierarchical models: are ecologists making a Faustian bargain. Ecol. Appl. 2009;19:581–584. doi: 10.1890/08-0549.1. [DOI] [PubMed] [Google Scholar]
- Lele S., Dennis B., Lutscher F. Data cloning: easy maximum likelihood estimation for complex ecological models using Bayesian Markov chain Monte Carlo methods. Ecol. Lett. 2007;10:551–563. doi: 10.1111/j.1461-0248.2007.01047.x. [DOI] [PubMed] [Google Scholar]
- Lele S., Nadeem K., Schmuland B. Estimability and likelihood inference for generalized linear mixed models using data cloning. JASA. 2010;105:1617–1625. [Google Scholar]
- Lewis P., Holder M., Holsinger K. Polytomies and Bayesian phylogenetic inference. Syst. Biol. 2005;54:241–253. doi: 10.1080/10635150590924208. [DOI] [PubMed] [Google Scholar]
- Lindley D. Bayesian statistics, a review. 2. Society for Industrial and Applied Mathematics; 1972. [Google Scholar]
- Lindley D. The philosophy of statistics. J. Roy. Stat. Soc. D. 2000;49:293–337. [Google Scholar]
- Liu L. BEST: Bayesian estimation of species trees under the coalescent model. Bioinformatics. 2008;24:2542–2543. doi: 10.1093/bioinformatics/btn484. [DOI] [PubMed] [Google Scholar]
- Liu L., Pearl D. Species trees from gene trees: reconstructing Bayesian posterior distributions of a species phylogeny using estimated gene tree distributions. Syst. Biol. 2007;56:504–514. doi: 10.1080/10635150701429982. [DOI] [PubMed] [Google Scholar]
- Maddison W. Gene trees in species trees. Syst. Biol. 1997;46:523–536. [Google Scholar]
- Matsen F., Steel M. Phylogenetic mixtures on a single tree can mimic a tree of another topology. Syst. Biol. 2007;56:767–775. doi: 10.1080/10635150701627304. [DOI] [PubMed] [Google Scholar]
- McCulloch C., Searle S. Generalized linear and mixed models. New York: John Wiley & Sons; 2001. [Google Scholar]
- Moore M., Soltis P., Bell C., Burleigh J., Soltis D. Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. U.S.A. 2010;107:4623–4628. doi: 10.1073/pnas.0907801107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossel E., Vigoda E. Phylogenetic MCMC algorithms are misleading on mixtures of trees. Science. 2005;309:2207–2209. doi: 10.1126/science.1115493. [DOI] [PubMed] [Google Scholar]
- Naylor G., Brown W. Amphioxus mitochondrial dna, chordate phylogeny, and the limits of inference based on comparisons of sequences. Syst. Biol. 1998;47:61–76. doi: 10.1080/106351598261030. [DOI] [PubMed] [Google Scholar]
- Nye T. Principal components analysis in the space of phylogenetic trees. Ann. Stat. 2011;39:2716–2739. [Google Scholar]
- Owen M. Ph.D. thesis. Cornell University; 2008. Distance computation in the space of phylogenetic trees. [Google Scholar]
- Owen M., Provan J. A fast algorithm for computing geodesic distances in tree space. IEEE/ACM Trans. Comput. Biol. Bioinformatics. 2011;8:2–13. doi: 10.1109/TCBB.2010.3. [DOI] [PubMed] [Google Scholar]
- Pickett K., Randle C. Strange bayes indeed: uniform topological priors imply non-uniform clade priors. Mol. Phylogenet. Evol. 2005;34:203–211. doi: 10.1016/j.ympev.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Ponciano J., Taper M., Dennis B., Lele S. Hierarchical models in ecology: confidence intervals, hypothesis testing, and model selection using data cloning. Ecology. 2009;90:356–362. doi: 10.1890/08-0967.1. [DOI] [PubMed] [Google Scholar]
- Rannala B. Identifiability of parameters in MCMC Bayesian inference of phylogeny. Syst. Biol. 2002;51:754–760. doi: 10.1080/10635150290102429. [DOI] [PubMed] [Google Scholar]
- Rannala B., Zhu T., Yang Z. Tail paradox, partial identifiability, and influential priors in Bayesian branch length inference. Molecular Biology and Evolution. 2012;29:325–335. doi: 10.1093/molbev/msr210. [DOI] [PubMed] [Google Scholar]
- Ripplinger J., Sullivan J. Does choice in model selection affect maximum likelihood analysis? Syst Biol. 2008;57:76–85. doi: 10.1080/10635150801898920. [DOI] [PubMed] [Google Scholar]
- Robert C. Prior feedback: a Bayesian approach to maximum likelihood estimation. Comput. Statist. 1993;8:279–294. [Google Scholar]
- Robert C. The Bayesian choice: from decision-theoretic foundations to computational implementation. Springer; 2007. [Google Scholar]
- Robert C., Casella G. Monte Carlo statistical methods. Springer; 2004. [Google Scholar]
- Rogers J. On the consistency of maximum likelihood estimation of phylogenetic trees from nucleotide sequences. Syst. Biol. 1997;46:354–357. doi: 10.1093/sysbio/46.2.354. [DOI] [PubMed] [Google Scholar]
- Rogers J. Maximum likelihood estimation of phylogenetic trees is consistent when substitution rates vary according to the invariable sites plus gamma distribution. Syst. Biol. 2001;50:713–722. doi: 10.1080/106351501753328839. [DOI] [PubMed] [Google Scholar]
- Ronquist F., Huelsenbeck J., van der Mark P. MrBayes 3.1 manual. School of Computer Science. Florida State University; 2005. [Google Scholar]
- Rothenberg T. Identification in parametric models. Econometrica J. Econometric Soc. 1971:577–591. [Google Scholar]
- Schwartz R., Mueller R. Branch length estimation and divergence dating: estimates of error in bayesian and maximum likelihood frameworks. BMC Evol. Biol. 2010;10:5. doi: 10.1186/1471-2148-10-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A., Haussler D. Phylogenetic estimation of context-dependent substitution rates by maximum likelihood. Mol. Biol. Evol. 2004;21:468–488. doi: 10.1093/molbev/msh039. [DOI] [PubMed] [Google Scholar]
- Sprott D. Statistical inference in Science. Springer; 2000. [Google Scholar]
- Sullivan J., Abdo Z., Joyce P., Swofford D. Evaluating the performance of a successive-approximations approach to parameter optimization in maximum-likelihood phylogeny estimation. Mol. Biol. Evol. 2005;22:1386–1392. doi: 10.1093/molbev/msi129. [DOI] [PubMed] [Google Scholar]
- Sullivan J., Joyce P. Model selection in phylogenetics. Annu. Rev. Ecol. Evol. Syst. 2005:445–466. [Google Scholar]
- Sullivan J., Swofford D. Should we use model-based methods for phylogenetic inference when we know that assumptions about among-site rate variation and nucleotide substitution pattern are violated? Syst. Biol. 2001;50:723–729. doi: 10.1080/106351501753328848. [DOI] [PubMed] [Google Scholar]
- Thorne J., Kishino H., Painter I. Estimating the rate of evolution of the rate of molecular evolution. Mol. Biol. Evol. 1998;15:1647–1657. doi: 10.1093/oxfordjournals.molbev.a025892. [DOI] [PubMed] [Google Scholar]
- Wald A. Asymptotic properties of the maximum likelihood estimate of an unknown parameter of a discrete stochastic process. Ann. Math. Stat. 1948;19:40–46. [Google Scholar]
- Walker A. On the asymptotic behavior of posterior distributions. J. Roy. Stat. Soc. B. 1969;31:80–88. [Google Scholar]
- Yang Z., Rannala B. Branch-length prior influences Bayesian posterior probability of phylogeny. Syst. Biol. 2005;54:455–470. doi: 10.1080/10635150590945313. [DOI] [PubMed] [Google Scholar]
- Yang Z., Rannala B. Bayesian estimation of species divergence times under a molecular clock using multiple fossil calibrations with soft bounds. Mol. Biol. Evol. 2006;23:212–226. doi: 10.1093/molbev/msj024. [DOI] [PubMed] [Google Scholar]
- Zwickl D., Holder M. Model parameterization, prior distributions, and the general time-reversible model in Bayesian phylogenetics. Syst. Biol. 2004;53:877–888. doi: 10.1080/10635150490522584. [DOI] [PubMed] [Google Scholar]