

Abstract
UV light-induced photoproducts are recognized and removed by the nucleotide-excision repair (NER) pathway. In humans, the UV-damaged DNA-binding protein (UV-DDB) is part of a ubiquitin E3 ligase complex (DDB1-CUL4ADDB2) that initiates NER by recognizing damaged chromatin with concomitant ubiquitination of core histones at the lesion. We report the X-ray crystal structure of the human UV-DDB in a complex with damaged DNA and show that the N-terminal domain of DDB2 makes critical contacts with two molecules of DNA, driving N-terminal-domain folding and promoting UV-DDB dimerization. The functional significance of the dimeric UV-DDB [(DDB1-DDB2)2], in a complex with damaged DNA, is validated by electron microscopy, atomic force microscopy, solution biophysical, and functional analyses. We propose that the binding of UV-damaged DNA results in conformational changes in the N-terminal domain of DDB2, inducing helical folding in the context of the bound DNA and inducing dimerization as a function of nucleotide binding. The temporal and spatial interplay between domain ordering and dimerization provides an elegant molecular rationale for the unprecedented binding affinities and selectivities exhibited by UV-DDB for UV-damaged DNA. Modeling the DDB1-CUL4ADDB2 complex according to the dimeric UV-DDB-AP24 architecture results in a mechanistically consistent alignment of the E3 ligase bound to a nucleosome harboring damaged DNA. Our findings provide unique structural and conformational insights into the molecular architecture of the DDB1-CUL4ADDB2 E3 ligase, with significant implications for the regulation and overall organization of the proteins responsible for initiation of NER in the context of chromatin and for the consequent maintenance of genomic integrity.
Keywords: UV damage, ubiquitin-proteosome system, X-ray crystallography
Genome integrity is under constant challenge and various cellular mechanisms exist to maintain DNA fidelity. In human cells, the nucleotide-excision repair (NER) pathway is responsible for the repair of a variety of DNA lesions (1). Although the mechanism of damage detection in chromatin is not well-understood, various studies have identified two principal initiators of the global-genome branch of NER (GG-NER), XPC-human RAD23B (XPC-HR23B) (2), and the UV-damaged DNA-binding protein complex UV-DDB (3–5). UV-DDB is composed of two proteins, a 127-kDa protein (DDB1) and a 48-kDa protein (DDB2) encoded by the DDB1 and DDB2 genes, respectively (6). Mutations in DDB2 cause a cancer prone autosomal recessive disease, xeroderma pigmentosum (XP) complementation group E (XP-E), and are associated with a partial deficiency in GG-NER (7–9). Through the DDB2 subunit, UV-DDB binds avidly to fragments of DNA containing various types of damage, such as UV-induced 6-4 pyrimidine-pyrimidone (6-4PP) and cyclobutane pyrimidine dimers (CPD) (10, 11). Detection of CPD in nontranscribed DNA by XPC is inefficient, indicating that the UV-DDB complex plays a primary and crucial role in the detection and repair of CPD in the context of chromatin (4, 12).
The ubiquitination pathway has recently been shown to play an important regulatory function in the initiation of NER (13, 14). The DDB1 protein is part of the substrate-recruiting module for two closely related types of E3 ligases, the cullins CUL4A and CUL4B, which target proteins for ubiquitination (15, 16). The DDB1-CUL4A complex belongs to a superfamily of cullin-RING ligases (CRL) (17–19), which participate in various aspects of the UV-damage response for maintaining genome stability (20–22). DDB2 is both a binding partner and a substrate receptor for the DDB1-CUL4A-based E3 ligase, DDB1-CUL4ADDB2 (11, 21, 23, 24). Following UV exposure of cells, DDB2 recruits the DDB1-CUL4ADDB2 complex to the site of damaged chromatin, regulating the initiation of GG-NER by modifying core histones around the site of the lesion (13, 24, 25). Available data show a connection between DDB1-CUL4ADDB2 and the monoubiquitination of the core histones (i.e., H2A, H3, and H4) in the cellular response to UV-irradiation (23, 24). Following initial damage recognition, the DDB1-CUL4ADDB2 E3 ligase ubiquitinates XPC and auto-ubiquitinates DDB2 (13), however with different consequences. Ubiquitination stabilizes XPC, increasing its affinity for damaged DNA, whereas polyubiquitination of DDB2 reduces its affinity for damaged DNA and ultimately leads to its degradation (13, 26). This paradoxical UV-dependent degradation of a protein [i.e., DDB2 (27–29)] that is intrinsically involved in the recognition of radiation-induced DNA damage is not fully understood. It has been speculated that this sequence of events is necessary for the accessibility of repair factors at the lesion site—i.e., for reducing the affinity between DDB2 and DNA to facilitate the handover of the damaged DNA from the DDB1-CUL4ADDB2 E3 ligase complex to XPC-Rad23 and for regulation of the cellular response to DNA damage (26, 30). It is currently unknown how DDB2 interacts with the substrate when E3 is anchored to damaged DNA nor how DDB2 targets multiple substrates of various sizes for mono- or polyubiquitination.
Recent progress in understanding the structural basis of NER initiation came from crystal structures of the yeast XPC orthologue Rad4 (31) and of the zebrafish UV-DDB bound to UV-damaged DNA (11). In these structures, these DNA-binding proteins (i.e., XPC, DDB2) appear to recognize conserved perturbations to the DNA topology induced by the lesions. UV-irradiation-induced modifications, such as CPD and 6-4 PP, are believed to disrupt the dynamics and helical topology through DNA bending, altering base-pairing interactions, and widening the major groove, features that are recognized by the NER apparatus through a bidentate recognition process (11, 31–34). The means by which UV-DDB can efficiently scan DNA for damage, while at the same time binding damaged DNA with the highest affinity of any damaged DNA-binding proteins (10, 35) remain unknown. The crystal structures of UV-DDB bound to DNA containing 6-4 PP or an abasic site showed contacts between DDB2 and DNA to be limited to the β-loops, exhibiting largely identical interactions (11).
We report here the crystal structure of full length human UV-DDB bound to damaged DNA, revealing the unique structural motif of the N-terminal helical domain of DDB2. Using biophysical methods of analysis to monitor and characterize the changes in molecular associations and dynamics initiated upon damaged DNA binding, we propose that this helical domain participates in forming the high-affinity binding state of UV-DDB. Mechanistically, the conformational dependence of the N-terminal domain of DDB2 on damaged DNA binding illuminates how UV-DDB can efficiently scan the genome to detect DNA damage, while enabling high-affinity DNA interactions to be formed once damage is detected. In the context of DNA repair, modeling the cullin-RING E3 ligase nucleosome complex on the dimeric UV-DDB-AP24 architecture facilely aligns the numerous molecular components, revealing spatial orientations likely significant in substrate ubiquitination. These results support the role of oligomerization in modulating molecular flexibility, affinities, and specificities in cullin-RING E3 ligase receptor-substrate complexes.
Results
Electron Microscopy and X-ray Crystal Structure Reveal a Dimer of Human UV-DDB in a Complex with Damaged DNA.
EM characterization of the full-length human UV-DDB in the presence of varying amounts of DNA identified solution conditions that stabilized the dimeric state of the complex. A central apyrimidic lesion was generated by introducing a tetrahydrofuran moiety at position 11 (THF11) in a 24-basepair oligodeoxynucleotide (AP24). Several ratios of AP24 were incubated with UV-DDB before EM imaging (Fig. 1) (additional details can be found in SI Appendix). Prior to DNA binding, our negative stain EM studies revealed predominantly spherical particles with a minor fraction exhibiting elliptical profiles (Fig. 1 and SI Appendix, Fig. S1). In the absence of DNA, the distribution of the projected protein surface area yielded a well-defined peak at approximately 3,600 Å2, corresponding to a spherical particle of approximately 70 Å in diameter, consistent with the dimension of a monomer of UV-DDB (i.e., a heterodimer of DDB1 and DDB2; Fig. 1). However, in the presence of damaged DNA substrate (i.e., AP24), a second peak appeared with an area that is consistent with that predicted for dimeric UV-DDB [(DDB1-DDB2)2], approximately 7,200 Å2 (Fig. 1 and SI Appendix, Fig. S1). Notably, particle size distributions shifted dramatically from monomeric to dimeric UV-DDB at a molar ratio of 3 AP24 to 1 UV-DDB (3∶1)—the ratio used for crystallization screening (described below).
Fig. 1.

Visualization and size estimation of UV-DDB particles by negative stain electron microscopy. Representative areas are shown in A without DNA and in B with AP24 oligodeoxynucleotide at a ratio of 1∶3. (Top) Images from electron micrographs and (Bottom) after global and local filtering and thresholding to yield countable particle areas. (C) Histograms collected from micrographs of particle areas for different ratios of UV-DDB to AP24 oligodeoxynucleotide, as indicated, and normalized by particle count (in parentheses). The peak at approximately 36 nm2 evident in the absence of DNA corresponds to a circle of diameter approximately 7 nm that is consistent with a monomer of the UV-DDB1-DDB2 complex. Increasing concentrations of AP24 oligodeoxynucleotide causes the peak shifts to approximately 72 nm2 consistent with a population of dimers. Examples of monomer-sized areas are indicated with arrowheads in A and dimers with double-arrowheads in B.
Crystallization protocols were devised using an analytical approach (36) to systematically identify chemical and additive conditions that stabilized conformational states of UV-DDB in solution. Single crystals of native or selenomethionine (SeMet)-substituted human DDB1 and DDB2 proteins, in complex with the same damaged DNA substrate analogue used in the EM analysis, AP24 (at a molar ratio of 1∶3 UV-DDB∶AP24), were grown. UV-DDB-AP24 crystallized in monoclinic and orthorhombic lattices, depending on the crystallization condition. The unit cell parameters of the orthorhombic (referred to as “ortho-UV-DDB”) crystal form are very similar to the monoclinic (“mono-UV-DDB”) (Table 1), except for a doubling along one axis in the orthorhombic dataset. The early native and anomalous datasets used to phase and refine the dimeric UV-DDB-AP24 complex model were most favorably processed in a monoclinic P21 space group setting. Similarly, the highest resolution dataset used to build missing regions in DDB2, add nucleotides to the DNA substrate, modify loop conformations of DDB1, and fully refine the human UV-DDB-AP24 complex structure was also most favorably processed in monoclinic lattice setting (Table 1, first column). In the final cross-validation stage, orthorhombic data collected from crystals optimized using alternative additive conditions were used to independently verify the overall backbone tracing and subunit configuration of the dimeric UV-DDB-AP24 crystal structure.
Table 1.
Refinement Statistics for the Human UV-DDB Complexes
| UV-DDB-AP24 ‘monomeric form’ | UV-DDB-AP24 ‘orthorhombic dimeric form’ | |
| Bravais Lattice | Monoclinic | Orthorhombic |
| Resolution (Å) | 31.74–2.85 | 41.09–3.22 |
| Rwork/Rfree | 0.22/0.24 | 0.25/0.26 |
| Number of atoms | 13010 | 13010 |
| Protein | 12033 | 12033 |
| Ligand/ion (DNA) | 977 | 977 |
| Water | 0 | 0 |
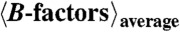 | ||
| All atoms | 35.4 | 39.4 |
| Proteins | 32.2 | 35.2 |
| DNA | 33.5 | 35.7 |
| Water | — | - |
| R.m.s deviations | ||
| Bond lengths (Å) | 0.0052 | 0.0059 |
| Bond angles (°) | 1.363 | 1.012 |
Combinations of bromide and selenomethionine (SeMet) anomalous dispersion methods, in tandem with partial model molecular replacement approach, were applied for initial phasing and refinement. The quality of the early maps was significantly improved by combining the phases calculated from the SeMet heavy atom positions together with phases calculated from the coordinates of human DDB1 and a partial poly-Ala model of the zebrafish DDB2 (residues 100–400; PDB ID code 3EI2). Although the double-stranded (ds) AP24 oligodeoxynucleotide substrate molecule was deliberately omitted from the initial phasing model, strong contiguous densities at the surface of the β-barrel domain of human DDB2 were apparent in solvent-flattened, positively contoured difference Fourier maps, verifying the damaged-DNA bound state of the DDB2 subunit in UV-DDB crystals. Molecular features evident even in the initially phased electron density maps permitted the structure of the central approximately 18 bases in both the damaged and undamaged strands of AP24 molecule to be built according to map densities. Iterative cycles of model building to incorporate the sequence of human DDB2, to adjust regional conformational differences in DDB1 and DDB2, and to extend the AP24 oligodeoxynucleotide molecule gradually improved map and model quality. Once approximatley 90% of the structure of the complete human UV-DDB-AP24 complex was modeled and refined, phase combination utilizing a native monoclinic dataset increased data completeness, intensities, and redundancy of wide angle reflections enhancing the overall data quality, resulting in more distinct electron densities radiating from N-terminal region of DDB2 truncated in the UV-DDB model. Importantly, regions of UV-DDB that were missing or altered in conformation could be progressively modeled as map definitions steadily improved commensurate with data extension to 2.85 Å. Distinct regions of contiguous electron densities radiating from residue 100 of DDB2 allowed additional approximately 80 residues at the N-terminal domain of DDB2 to be traced, monitoring R factors and other statistical factors until refinement converged. To validate the human UV-DDB-AP24 structure we also solved an orthorhombic UV-DDB SeMet dataset to 3.2 Å resolution by ab-initio SAD phasing. The structure of the orthorhombic crystal form independently confirmed the N-terminal-domain fold and subunit organization in the dimeric UV-DDB (refinement statistics for both crystal structures are shown in Table 1). In both monoclinic and orthorhombic datasets, an elongated configuration of the dimer is recapitulated, mirroring the molecular envelope of UV-DDB seen in EM images taken in the presence of damaged DNA. Altogether, the EM and crystal data support the substrate-dependent dimerization of UV-DDB (Fig. 2).
Fig. 2.

Structure of the dimeric human UV-DDB in a complex with damaged DNA. (A) The dimeric UV-DDB subunit organization, shown in ribbon depiction, with each domain colored and labeled accordingly: yellow, DDB2 β-propeller; red, DDB2 N-terminal-α paddle; blue, DDB1 BPA; green, DDB1 BPC; and purple, DDB1 BPB. The 24-bp oligodeoxynucleotide (AP24) contains an abasic lesion site (THF11), with the phosphor-deoxyribose backbone of the damaged strand colored in red and the undamaged strand colored in blue. Each DDB2 subunit is bound to an AP24 oligonucleotide, with DDB2 residues Asn360/Asn360’ straddling the twofold symmetry axis, forming H bonds across the dimer interface. The surface of the Asn360/Asn360’ pair (colored using standard atom convention) is located in a loop spanning two antiparallel β-strands (β-wing). The abasic lesion site in AP24 is marked by the surface mesh drawn around nucleotides THF11/dC12 in their flipped, extra-helical configuration. The β-wing is sandwiched between the two AP24 oligodeoxynucleotides, astride of the twofold axis of rotation relating the monomer subunits in the dimeric DDB2. Residues on the leading β-strand and loop form contacts with the undamaged DNA strand whereas residues on the loop and the retreating β-strand form contacts with the neighboring undamaged DNA strand. Both sets of contacts are predominantly electrostatic in nature, thus largely sequence independent. (B) Same as A but rotated 90 degrees and tilted slightly to show both DNA molecules. (C) Electrostatic potential surfaces of the DDB2 N-terminal domain complement the charge characteristics of the DDB1 BPC domain and the DNA phosphor-deoxyribose interfaces, resulting in favorable electrostatic neutralization. Contacts between residues on the β-wing region form contacts with the DNA bound at its immediate active site and with the neighbouring DNA molecule bound to the second monomer of DDB2 in the dimer. Extensive interactions between residues on the N-terminal-helical domain (α-paddle) and the neighboring DNA molecule augment the intermolecular associations, contributing to the high affinity of damaged DNA binding. (D) The skewed positioning of the DNA binding surface can now be understood in terms of the DDB2 dimer interface, located adjacent to the DNA binding site, at a loop bridging blades 6 and 7 of the β-propeller (β-wing) of DDB2. To accommodate the steric constraints imposed through dimerization along with DNA binding, the two adjacent sites are positioned diametrically across one face of the molecular surface of DDB2, readily seen in the dimeric DDB2-DNA (AP24) complex.
Crystal Structure of the Dimeric UV-DDB Complex.
DDB1 is a large tri-β-propeller substrate adaptor protein. Following nomenclature defined previously (19), the DDB1 β-propeller domains are denoted as BPA, BPB, and BPC, with a C-terminal helical domain referred to as CTD (37). The structure of the human DDB2 substrate receptor is composed of a large seven-bladed WD40 β-propeller domain (residues 103–421), preceded by an N-terminal domain (residues 1–102) (Fig. 2 A and B) (11). The dimeric DDB2 forms the core of the UV-DDB complex, with a twofold axis located close to blade 6 of the major seven-bladed β-propeller domain of DDB2 (Fig. 2 A and B; yellow), the conserved WD40 structural motif.
Distinct Topological Motifs Mediate Associations Between DDB1 and DDB2.
In the human UV-DDB dimeric complex structures, the previously missing N-terminal helical domain of DDB2 (11) has been built by modeling into experimentally phased electron density maps. The N-terminal region preceding the β-propeller domain of DDB2 is composed of approximately the first 102 residues and topologically distinguished by predominantly helical features. The first 66 residues fold into three helical segments arranged into a triangular topology (α-paddle, in red, Fig. 2 A–C), followed by an extended helix-turn-helix (residues 67–102) that inserts into the BPA-BPC double-propeller cleft (Fig. 2 A and B). The variations in conformation and domain organization in the dimeric relative to the monomeric states of UV-DDB are primarily centred at the DDB2 component.
The interface formed between BPA-BPC double propellers of DDB1 displays significant hydrophobic characteristics, concentrated mainly on the surface of the BPA domain (SI Appendix, Fig. S2A; hydrophobic surfaces in white, defined by a dotted yellow oval) facing the cleft where the β-propeller domain of DDB2 docks. Upon initial complex formation, extensive hydrophobic contacts are formed between residues on the BPA domain and aliphatic loop residues extending from the β-propeller domain of DDB2 (SI Appendix, Fig. S2B; hydrophobic surfaces in white, defined by a dotted blue rectangle). In contrast, interactions between the BPC domain of DDB1 (SI Appendix, Fig. S2A; negative electrostatic surfaces in red, positive in blue, defined by a dotted violet oval) and the N-terminal-α-helical region preceding the β-propeller domain of DDB2 are largely electrostatic in nature (SI Appendix, Fig. S2B; negative electrostatic surfaces in red, positive in blue, defined by a dotted green rectangle). The helical topology of the N-terminal domain of DDB2 is important for aligning clusters of acidic/basic residues on surfaces facing the BPA domain, enabling charge complementation at the intermolecular interface (SI Appendix, Fig. S2C). The α-paddle helical fold of DDB2 segregates aromatic residues to the opposite face of the N-terminal domain of DDB2, juxtaposing hydrophobic patches to form favorable contacts to the BPC domain of DDB1. The helical conformation of the N-terminal domain of DDB2 is a key topological feature that enables the precise spatial alignment of residues at intermolecular interfaces, mediating multiple molecular associations in the context of the complete complex.
Damaged DNA Binding Induces Helical Folding of the N-terminal Domain of DDB2.
Another novel structural feature found in the dimeric UV-DDB-AP24 complex is at the region encompassing residues 356–370 of DDB2, which forms a well-ordered loop that extends from blade 6 of the seven-bladed β-propeller domain of DDB2, forming a fold defined herein as β-wing (Fig. 2 A–C). The closest contacts between two DDB2 subunits within the dimer are at the β-wing regions of DDB2, centred at Asn360. The twofold axis is located between two Asn360 side chains, which form of H-bonds with favorable geometries and distances [Fig. 2A; 2.8 Å between neighboring twofold related Oδ1 (red) and Nδ2 (blue) atoms of Asn360].
In the dimeric DDB2, the β-wing loops are sandwiched between two DNA molecules, with β-wing residues forming contacts to both the DNA immediately bound and to its neighboring DNA bound to the second β-propeller domain (SI Appendix, Fig. S3). At one end of the β-wing turn, an electrostatic network stabilizes the undamaged DNA strand immediately opposite the lesion (SI Appendix, Fig. S2A; defined by a dotted yellow oval). Bonds between the β-wing residues to the DNA are predominantly electrostatic in nature, contacting anionic phospho-deoxyribose backbone atoms of the DNA, similar in nature to those formed by the N-terminal domain of DDB2. In comparison, contacts formed at the lesion site between the insertion loop of DDB2 and the damaged strands are nucleobase specific. Explicit contacts centred within a 3-nucleotide window on the duplex DNA are limited by atomistic and spatial constraints dictated by the DNA topology and chemical functionality of DDB2’s insertion loop residues.
Located in the N-terminal-α-paddle domain of DDB2 are numerous arginines, lysines, glutamates, aspartates, and glutamines, aligned by the helical topology along a face of the α-paddle, presenting charged residues (Arg46, Arg47, Asp51; shown as sticks, Fig. 2C) to the phospho-deoxyribose backbone of the DNA. Additional charged residues contributed by the adjacent β-propeller domain of DDB2 further enhance the highly cationic electrostatic surface of the N-terminal domain of DDB2 (SI Appendix, Fig. S2B; positive electrostatic surfaces shown in blue). These form complementary electrostatic molecular interfaces for binding both the DNA and the BPC domain of DDB1 (SI Appendix, Fig. S2 A–C). As noted earlier, the helical motif of the N-terminal domain of DDB2 is important for aligning charged residues on one face of the helices and projecting hydrophobic residues on the other, enabling these to simultaneously interact with the hydrophobic BPA domain of DDB1 and the highly anionic nature of the DNA phosphor-deoxyribose backbone as well as the largely cationic surface features of the BPC domain of DDB1.
The dimer interface of DDB2 modulates multiple intermolecular contacts, providing a structural rationale for the remarkably high binding affinities to damaged DNA found in biochemical studies of UV-DDB (10, 35, 38). In the current studies, the 24-bp oligodeoxynucleotide (AP24) is substantially longer than the 14-and 16-bp oligodeoxynucleotides used in the earlier structural studies, affording unique insight to the interactions beyond those formed directly at the lesion site. Four distinct networks of contacts are formed between DDB2 and the DNA. At the lesion site, DDB2 residues (His333, Phe334, Gln335, His336, represented by orange spheres and rectangles; SI Appendix, Fig. S3 A–C) insert at the abasic site, resulting in flipping of the immediate upstream nucleotide, which is stabilized in an extra-helical conformation through a second group of DDB2 contacts (yellow spheres; SI Appendix, Fig. S3 A–C). Beyond the lesion, interactions between DDB2 and the deoxyribose-phosphate backbone atoms upstream (green spheres, rectangles; SI Appendix, Fig. S3 A–C) and downstream (purple spheres, rectangles; SI Appendix, Fig. S3 A–C) on the damaged DNA strand further serve to clamp the DNA. The distinct nature and multiple levels of interactions found between the DNA molecule and DDB2 in the crystal structure are highly suggestive that the additive contributions from the multiple sites of contacts are a major factor for the high overall avidity of binding exhibited by UV-DDB for damaged DNA.
DNA Binding Promotes Dimerization of the DDB1-DDB2 Heterodimer (DDB1-DDB2).
To further probe the oligomeric states of UV-DDB under physiologically relevant solution conditions, atomic force microscopy (AFM) was used to characterize the molecular topology of UV-DDB and to monitor substrate-induced changes in intermolecular interactions. AFM-derived volumes have been used extensively in studies examining the oligomeric states of multi-component complexes and to ascertain the nature of protein–protein interactions of globular proteins (39, 40). For our AFM studies, 517-bp PCR fragments were produced as the undamaged DNA substrate and the fragments were subjected to UV-irradiation to generate the damaged DNA (41, 42). AFM analyses of UV-DDB in the presence of undamaged DNA (SI Appendix, Fig. S4 A and B), UV-damaged DNA (Fig. 3), and in the absence of substrate binding (Fig. 3) found clearly distinguishable changes in the oligomeric states of UV-DDB. To quantitate the volumes and molecular mass derived from the AFM data, a standard curve was generated using proteins with well-defined oligomeric states, shapes, and molecular masses (additional experimental details are provided in SI Appendix, SI Materials and Methods). Converting the apparent AFM volumes of the UV-DDB molecules in the absence of DNA to molecular mass using the standard curve (SI Appendix, Fig. S5) shows that the peak at approximately 66 ± 10 nm3 (three depositions) corresponds to a protein with a molecular mass of 184 ± 23 kDa, a value consistent with the size of monomeric UV-DDB (i.e., heterodimeric DDB1-DDB2, with a combined theoretical molecular mass of 175 kDa). A second peak at approximately 190 nm3, corresponding to a molecular mass of approximately 505 kDa, represents a complex mixture of higher order oligomeric states (Fig. 3C and SI Appendix, Fig. S5). Consistent with the EM results, no distinct peak at a volume corresponding to a dimer of UV-DDB [(DDB1-DDB2)2] is found in the absence of DNA.
Fig. 3.

AFM imaging shows that damaged DNA binding promotes the dimerization of the DDB1-DDB2 heterodimer. (A) A representative surface plot of UV-DDB (50 nM) in the absence of DNA. The thin and wide white arrows point to molecules consistent with the size of the UV-DDB monomer (DDB1-DDB2 heterodimer) and trimer of UV-DDB, respectively. (B) Representative surface plot of UV-DDB (50 nM) in the presence of UV-irradiated 517 bp PCR fragments (25 nM). The yellow and red arrows point to dimeric UV-DDB [(DDB1-DDB2)2] binding to one and two molecules of duplex DNA, respectively. (C) AFM volume analysis of free UV-DDB (n = 1,160). (D) AFM volume analysis of UV-DDB on one strand (gray bars, n = 339) and two strands (black bars, n = 79) of duplex DNA. The images in A and B are at 500 nm × 500 nm and 3 nm in height. (Bottom) The dashed lines (C, free in solution, and D, bound to DNA) represent Gaussian fits to the data. Field view images of UV-DDB binding to separate DNA molecules (E) or two different regions of the same DNA molecule (F). The images are at 300 nm × 300 nm and 2 nm in height.
When UV-DDB was incubated with UV-irradiated DNA at a molar ratio of 2∶1, approximately 96% of the UV-DDB molecules were found to be bound to DNA molecules (Fig. 3 B and D and SI Appendix, Fig. S4C). In addition, among all of the UV-DDB molecules bound to DNA, 18% of the molecules bound to two DNA molecules simultaneously. These binding events included both middle to middle (Fig. 3 E and F) and end to middle sites (Fig. 3B, red arrow) on two separate DNA molecules. The AFM-derived volume of UV-DDB bound to one molecule of DNA is Gaussian centred at approximately 133 nm3 (Fig. 3D; gray histogram) which is consistent with the size of dimeric UV-DDB [(DDB1-DDB2)2]. The AFM volume of UV-DDB simultaneously binding to two molecules of DNA is approximately 139 nm3, which is slightly larger than UV-DDB binding to only one molecule (Fig. 3D; black histogram). These oligomeric states and substrate interactions found from the AFM analysis of UV-DDB in the presence of damaged DNA contrasts dramatically to those found when UV-DDB is in the presence of undamaged DNA substrate.
The interactions found between UV-DDB and the undamaged DNA fragment from the AFM analysis indicates that UV-DDB binds undamaged DNA to a significantly reduced extent (approximately 37% of the total UV-DDB was bound to DNA) compared to those formed when UV-damaged DNA is present (96%) (SI Appendix, Fig. S4C). These AFM results are consistent with those found earlier by EMSA analysis, which showed that small but measureable amounts of UV-DDB bound undamaged DNA (38, 43). Analysis of the volumes of these nonspecific UV-DDB complexes observed on DNA indicated that a majority (approximately 75%) * of the complexes were monomeric consisting of only one molecule of DDB1 and DDB2 (SI Appendix, Fig. S4B). These volume measurements are in striking contrast to the AFM-derived volume of UV-DDB when bound to UV-irradiated PCR fragments, inducing volume changes in UV-DDB that were consistent with a dimeric UV-DDB [(DDB1-DDB2)2] bound to damaged DNA.
While EM and AFM studies involved different sample preparation procedures and different criteria for evaluating size (projected area vs. volume), these complementary techniques can provide insights into the molecular topologies, organization, and nature of interactions in multi-component complexes. Compounding the innate methods-related differences described above, the sizes of the damaged DNA substrates used in the EM and AFM studies represented different lesions types (THF in AP24 versus UV-induced lesions in 517-bp PCR fragments). Yet the EM and AFM studies provided corroborating data verifying that the shorter abasic site mimic (AP24) induced similar dimerization upon damaged DNA binding as found when UV-DDB bound the 517-bp UV-irradiated DNA fragments. Notably, the remarkable agreement between the dimensional values and molecular profile obtained by the negative-stained EM, AFM, and derived from the X-ray diffraction crystal structure supports our proposal that UV-DDB dimerizes as a function of damaged-DNA binding. To summarize, EM and AFM imaging revealed that (i) in solution, UV-DDB exists as a monomer (composed of DDB1-DDB2 heterodimer) and no significant dimer population of UV-DDB [(DDB1-DDB2)2] was observed; (ii) binding to damaged DNA promotes the dimerization of UV-DDB, which can simultaneously bind to two DNA molecules; (iii) the organization and dimensions of the dimeric UV-DDB-damaged DNA complex found in the AFM and EM analyses are consistent with those found in the crystal structure of the dimeric complex. It is important to note that whereas the DDB1-CUL4ADDB2 ubiquitin ligase complexed to a nucleosome modeled according to the dimeric architecture captured in our crystal structure leads to a surprisingly rational organization of the individual molecular components (Fig. 4), the dimeric state does not necessarily constrain the number of lesions that can be simultaneously bound nor inform about the number of lesions required to induce dimerization; but the assumption that a single lesion can induce the dimeric form is reasonable.
Fig. 4.

Model of DDB1-CUL4ADDB2 ubiquitin ligase complexed to a nucleosome. (A) Modeling of the complex with CUL4A-Rbx (gray, light blue) onto the dimeric UV-DDB2 (domains colored as in Fig. 2); the region defined by two adjacent AP24 oligodeoxynucleotides (AP24-1 & AP24-2, in orange) used for the docking of a nucleosome; (B) Docking of the nucleosome in the dimeric UV-DDB, showing the fit of one AP24-1 (in orange) relative to the nucleosome (in blue); (C) fit of the nucleosome onto both oligomers showing that the distance between the two oligonucleotides can readily accommodate the nucleosome molecule (second DNA molecule, AP24-2, shown in orange) with minor adjustments of the second DDB2 component, as needed. The dimeric scaffold accommodates the numerous proteins that transiently assemble and disassemble on the DDB1-CUL4ADDB2 ubiquitin ligase complex at the vicinity of the lesion site in the subsequent DNA repair process. The dimeric architecture also spatially aligns the various molecular subunits in the reactions monoubiquitinylating histones and polyubiquitinylating substrate receptors (i.e., DDB2) for proteasomal degradation and verified by docking the E2 ubiquitin transferase enzyme onto the DDB1-CUL4ADDB2 ubiquitin ligase complex, resulting in E2 bridging distances to histones. In this figure, the histone and E2 proteins are omitted for clarity.
Dimeric UV-DDB Binds Damaged DNA with Approximately Fourfold Higher Affinity than in the Monomeric State.
The role of oligomerization in mechanisms involving DCAF proteins and, more specifically, the dimerization of DDB2, has been proposed earlier (44, 45). In our study, numerous lines of structural evidence from crystallography, EM, and AFM results, when combined with earlier published results (10, 44) coherently implicates the involvement of dimeric UV-DDB [(DDB1-DDB2)2] in mediating molecular interactions at specific stages along the DNA repair pathway. To experimentally validate the premise derived from the structural results, that dimerization mediates DNA-binding activities, dynamic light scattering (DLS) and surface plasmon resonance (SPR) were used to characterize the interactions between monomeric and dimeric UV-DDB to AP24. Unlike EMSA or DNA footprinting gel-based assays that require labeling and are end points measurements that do not allow for kinetic analysis, DLS and SPR monitor molecular interactions in real time, permitting delineation of concentration dependencies and other solution effects on molecular interactions.
Characterization by DLS clearly shows that the binding of damaged DNA results in the formation of a distinct, monodisperse state of UV-DDB, with dimensions in agreement with the EM and AFM values for the dimeric UV-DDB [(DDB1-DDB2)2] (SI Appendix, Tables S2 and S4). Furthermore, dimerization of UV-DDB is readily promoted by binding AP24, even at dilute protein concentrations, whereas in the absence of damaged DNA binding, a mixture of monomers and dimers is found even at high protein concentrations. The kinetics and binding affinities of UV-DDB to AP24 differ in the monomeric versus dimeric states, according to the SPR data, which clearly show that the dimerization of UV-DDB is stimulated by damaged DNA binding and that, moreover, the kinetics of both the association and dissociation steps are modified, resulting in > four-fold† enhancement in the damaged DNA binding affinities in dimeric compared to the monomeric states (SI Appendix, Table S3). The combination of data from AFM, EM, and biophysical analysis (SI Appendix, Tables S2–S4) presented here supports the biological relevance of the dimeric state of UV-DDB as revealed in the crystal structure.
Discussion
Our comprehensive study has elucidated the structure of dimeric UV-DDB in a complex with damaged DNA, utilizing a combination of structural, biophysical, and biochemical approaches that collectively support the pivotal role that dimerization plays in modulating intermolecular associations and in organizing the architecture of the multi-component cullin-RING E3 ligase receptor-substrate complexes. The dimeric UV-DDB structure presented in this paper provides the first high-resolution views of a cullin-RING E3 ligase receptor-substrate complex captured in a high-affinity state, with direct mechanistic and functional implications.
Dimer Interface is Adjacent to the Damaged DNA Binding Site in DDB2.
The primary damaged DNA binding site is located at the narrow end of the β-propeller, opposite to the DDB1 interaction surface in DDB2 (Fig. 2). The DNA spans the surface of the DDB2, surprisingly offset from the center of the seven-bladed β-propeller. This offset can now be explained in terms of the constraints arising from the dimerization interface. To accommodate these constrains along with DNA binding, the two neighboring binding sites are located diametrically across a molecular face of the β-propeller domain of DDB2, leading to the offset. The location of the dimer interface on the same molecular surface as the DNA binding site allows for cooperativity between DNA binding and dimerization (SI Appendix).
A High-Affinity DNA Binding Motif is Formed by Dimerization of UV-DDB.
Given the overwhelming binding preference exhibited by UV-DDB to UV-induced lesions, a mechanism based on substrate-driven conformational folding of the N-terminal-domain of DDB2 would permit specificities and binding affinities to be tuned, optimizing interactions according to the specific chemical nature of the lesion site. This mechanism ensures that high affinity interactions are formed only when damage is found. Multiple unique sites of DNA contacts are found in the dimeric UV-DDB, interactions that are absent in the monomeric state and mediated by the N-terminal-α-helical paddle and the β-wing regions of DDB2.
The α-paddle helical fold adopted by the N-terminal domain of DDB2 aligns residues so contacts to the DNA immediately bound and to a neighboring DNA molecule are formed in tangent, promoting the dimerization of UV-DDB. The β-wing loop of DDB2 is located at the interface of two DNA molecules within the dimeric UV-DDB. These interactions independently augment DNA contacts but when analyzed together resemble a “winged helix” motif that has been found in numerous DNA-binding proteins (46). Analogous to those found in other winged-helix DNA binding proteins, the β-wings in UV-DDB form direct contacts to the backbone atoms of the DNA. Additionally, the conformation and the apparent function of the β-wings of DDB2 in dimeric UV-DDB resemble those shown in the transcription factors, Ets-1, and the tripartite factor X, RFX, by linking and modulating nucleotide binding affinities with dimerization (47, 48). In these winged-helix proteins, exposed patches of hydrophobic residues are displayed, causing conformational changes to present new protein–protein interaction surfaces and inducing dimerization as a function of nucleotide binding (SI Appendix). Thus, the N-terminal domain of DDB2 modulates molecular affinities, independently and in conjunction with its β–wing domain, while further coordinating dimerization. The specific α-paddle helical motif enables multi-molecular contacts to be formed without perturbing interactions with DDB1 and, presumably, without blocking the subsequent binding of proteins involved in DNA repair. The dimeric DDB1-CUL4ADDB2 ubiquitin ligase complexed to a nucleosome, modeled according to the molecular architecture of the UV-DDB-AP24 complex (Fig. 4), demonstrates that the multi-component complex can be accommodated within the dimeric framework, providing additional support for the plausibility that dimerization of UV-DDB regulates and modulates association to DNA lesions.
Dimerization Accommodates Spatial Constraints for Substrate Ubiquitination.
Recently, the concept that dimerization is the key molecular determinant in enabling interactions with the vast and diverse set of proteins targeted by CRLs and their complexes has gained prominence. The dimeric state would be advantageous to monomeric E3 in targeting proteins of different sizes and in regulating auto-ubiquitination of the substrate receptor. The functional importance of CRL dimerization is supported by the observation that mutations of substrate-recognition regions retain their dimerization properties but act in a dominant-negative fashion, in vivo (45). Formation of higher-order oligomers can be initiated by receptor association or through another E3 component (49).
Consideration of the holocomplex containing the E3 ligase indicates that the molecular architecture of the DDB1-CUL4ADDB2 complex should complement the ubiquitination machinery in the assembled state. The cullin subunit is an elongated moiety in all cases, consisting of a long stalk and a globular domain RING finger adapter protein, RBX1, which docks through an intermolecular β-sheet, forming a two-subunit catalytic core that recruits the ubiquitin-conjugating enzyme, E2. It is recognized that the cullin subunit (e.g., CUL4A/4B, CUL1, CUL5) serves as a rigid scaffold in organizing the various substrates for ubiquitination after complex formation. The distinct structural motif displayed by various cullin complexes results in a distance of over 100 Å between the RBX-E2-Ub proteins relative to the substrate protein (45), which poses a question as to how activated ubiquitin bridges the 100-Å gap. However, dimerization in conjunction with domain flexibility found in the dimeric UV-DDB complex, described here, appears to address the multitude of proteins and chemical variability while permitting the dynamic adjustments needed during the poly-ubiquitination reaction.
In the context of the holo-complex with CUL4A, the dimeric UV-DDB structure seems to be optimized to meet the spatial requirements of the elongated cullin architecture. Modeling the DDB1-CUL4ADDB2 E3 complex by superimposing CUL4A-RBX onto the BPB-domain of DDB1 in the dimeric UV-DDB complex shows that the 100-Å distance is readily bridged by the activated ubiquitin-E2 moiety (E2-Ub). Particularly striking is the overall alignment when the nucleosome is oriented onto the dimeric architecture, using the AP24 with the dimeric UV-DDB as the reference for placement of the nucleotide backbone (Fig. 4). The specific BPB domain conformation of the DDB1 subunit captured in the dimeric UV-DDB would present the activated Ub (CUL4A-RBX1-E2 –Ub complex) within 10 Å of several lysines of DDB2 that are candidate sites for auto-ubiquitination. The model also indicates that the resulting complex would additionally position the histone proteins, which are also ubiquitinated, to E2-Ub. Consequently, dimerization of the substrate-recognition subunits, as exemplified by UV-DDB, further supplements regulatory, fine-tuning activities so that a spectrum of ubiquitination can be moderated, possibly permitting the simultaneous modification of multiple substrates (e.g., XPC, histones and DDB2) and/or mono- versus poly-ubiquitination of substrates (e.g., poly-Ub of DDB2 and mono-Ub of H2A). The structural and biochemical findings reported here provide compelling evidence for the dimeric state as a critical organizational unit of UV-DDB. The dimeric associations found in the UV-DDB may be representative of those formed in other complexes based on the DDB1-CUL4 ligase platform.
Functional Implications of the Dimeric State of UV-DDB.
The preference for binding to UV-damaged DNA by UV-DDB was verified by AFM analysis, which found significantly less binding of UV-DDB to an undamaged 517 bp PCR fragment, results consistent with the high specificity reported for UV-DDB (10, 35, 38, 43). The AFM analysis also shows the dimeric state of UV-DDB, bridging two duplexes of UV-damaged DNA, under conditions that reproduced UV-DDB’s specificity for damaged DNA.
The functional significance of the dimeric state can be assessed by comparing the molecular regions identified as structurally significant to those reported by other approaches. In the dimeric UV-DDB complex structure, the β-wing represents the area forming closest intermolecular contacts and the functional importance of this region has strong genetic support. Four DDB2 variants, formed by alternative splicing, were identified in Hela cells (44). The D1 variant, with deletion of residues 153–341 that excluded part of the β-propeller domain of DDB2, but preserved Asn360 and the β-wing, could form dimers with DDB2-WT and itself. The variant D2, containing only the first 156 amino acid residues, could not form the dimeric complex. Interestingly, DDB2 splice variants are dominant negative inhibitors of NER when expressed in HeLa cells (44). The deleterious effects of these splice variants are difficult to reconcile from the respective locations of the residues or segments according to the structure of the monomeric UV-DDB complex. However, these resides map to regions at the β-wing of DDB2 in our dimeric structure of the UV-DDB-damaged DNA complex, residues centrally positioned to bridge both molecules of DNA in the dimeric configuration. Thus, residues located in the vicinity of the dimer interface (e.g., on the β-wing region according to the UV-DDB-AP24 dimer configuration in the crystal structure) may function in signalling the substrate complexed state of DDB2, leading to cooperative enhancement of DNA binding affinities upon stimulating helical folding of the N-terminal domain of DDB2.
Our studies suggest that the transition between disordered to ordered folding of the N-terminal domain of DDB2 may be intimately related to modulating the intermolecular associations formed subsequent to those primary contacts formed immediately upon docking of DDB2 to DDB1 and upon the binding of damaged DNA to DDB2 at the substrate binding site. Through a series of fine-tuning steps, secondary intermolecular contacts are formed between the damaged DNA substrate to DDB2 (i.e., at the β-wing and N-terminal domain of DDB2 to damaged DNA ± 4 nucleotides from the lesion site) (SI Appendix, Fig. S3C) and DDB1 to DDB2 (at the interface between the BPC domain of DDB1 to the N-terminal domain of DDB2) (SI Appendix, Fig. S3). These molecular interactions allow the orientation of the individual subunits within the multiprotein complex to be adjusted so that the plethora of reactions catalyzed by the DDB1-CUL4ADDB2 multiprotein complex can be accommodated for the monoubiquitination of histones, and polyubiquitination of DDB2, ultimately leading to DNA repair.
Regulation by oligomerization has been speculated for other proteins involved in binding various states of DNA. Our extensive studies commenced with the crystal structure elucidation, revealing the dimeric state of the UV-DDB-AP24 complex, and expanded to in-depth, multi-dimensional biophysical and structural characterization of the substrate-binding dependencies, are consistent with inducing distinct dimeric states of UV-DDB. These different lines of analysis consistently point to the roles played by dimerization and localized conformational changes in protein subunits which together modulate conformation of the multi-component E3 ligase complex and influence catalytic efficiencies of specific reactions. The iterative cycles involve discrete modification of subunit intermolecular contacts that propagates to the overall complex and permits a spectrum of activities to be generated, centered on dimerization that additionally reduces spatial and molecular constraints while increasing the range of subunits and reactions that can be accommodated.
The binding of UV-damaged DNA initiates conformational changes at the N-terminal domain of DDB2, inducing helical folding in the context of the bound DNA to promote dimerization of the UV-DDB-substrate complex, to ensure that high affinity contacts are formed only when damage is found in DNA. This temporal and spatial interplay between domain ordering and dimerization provides an elegant molecular rationale for DDB2’s enhanced UV-damaged DNA selectivity (10, 38). Based on the additional extensive contacts formed by the dimeric UV-DDB, oligomerization can modulate substrate affinities on multiple levels, serving to allosterically regulate the substrate-receptor complex.
XP-E Mutations Disrupt Key Intermolecular Contacts in Dimeric UV-DDB.
The mutations found in XP-E patients have been mapped to their locations on the human UV-DDB complex structure (SI Appendix, Fig. S6; key amino acid mutations shown in space-filling depictions). Genetic mutations identified in XP-E patients (7, 9) are at residues that form either direct or key bridging interactions with the oligodeoxynucleotides (K244, D307) or DDB1 (R273, L350). Perturbation of these contacts is highly detrimental because these mediate both direct and secondary interactions with the DNA or DDB1. The effects for two of the mutations highlight the significance of the N-terminal helical domain and the dimer interface of DDB2. The L350P mutant would significantly perturb the stability of the DDB1-DDB2 complex as L350 aligns the long N-terminal-α-paddle of DDB2 that inserts into the BPA-BPC domain cleft of DDB1. The position of L350 is central to an aliphatic cluster at the DDB1 BPC interface with DDB2, so that mutation would cooperatively disrupt multiple associations. The position of D307 is at the DDB2 dimer interface, close to the β-wing loop, disturbing DNA binding as well as dimer formation. Notably, the mutated residues identified in XP-E patients are at sites in DDB2 that mediate multiple contacts, with the detrimental consequences amplified due to disruption of correlated interactions.
Conclusions
We describe here the 2.85-Å dimeric structure of the full-length human UV-DDB (DDB1-DDB2)2 in a complex with damaged DNA. This new structure revealed the importance of the N-terminal 102 residues of DDB2 in mediating interactions with DDB1 and damaged DNA. The remarkable agreement on the molecular topology between the negative-stained EM, AFM, and crystal structure results (Figs. 1–4), further validated by DLS and SPR analysis, collectively supports the distinct dimeric state formed by UV-DDB upon binding damaged DNA. Taken together these multiple lines of evidence strongly support the existence of higher oligomeric states of UV-DDB, in vivo. These findings have direct regulatory and functional implications.
The dimeric UV-DDB acts as a molecular scaffold for aligning multiple protein partners, during the complex and dynamic process of damaged DNA detection and repair. DDB1-CUL4ADDB2 assists in transfer of ubiquitin from the E2 to the histones and repair proteins at the site of a lesion. UV-DDB thus has a unique role for the initiation of NER in the context of chromatin. The structural flexibility of the N-terminal domain of DDB2 suggests that this domain is conformationally adaptable, its precise domain-fold driven by substrate binding. This structural malleability in receptors enables recognition of a wide array of diverse protein and nucleic acid substrates. DDB2 presumably forms complexes with multiple substrates, including histones and XPC in addition to damaged DNA, displaying a range of binding affinities to different chemical lesions found in UV-irradiated DNA. We surmise that DDB2’s selectivity—its ability to distinguish subtle discrete differences in chemical moieties within the framework of a nucleosome—is related to its domain-fold adaptability. Considered all together, substrate-induced N-terminal-domain folding endows molecular and conformational adaptability, features that are further enhanced and optimized by dimerization.
Our findings regarding the significance of the dimeric state of the UV-DDB-AP24 agree remarkably well with reports on other DCAF-family proteins, providing a molecular scaffold for integrating the assorted biochemical, genetic, and cellular observations into a coherent mechanism directing NER. A central tenet evolving from these multiple lines of evidence is the pivotal role that oligomerization plays in modulating specificities and affinities of associations in multi-component macromolecular complexes and, consequently, controlling rates of reactions. Recently, the concept of dimerization as a key molecular determinant in enabling recognition and interactions between the diverse set of proteins targeted by the family of cullin-RING E3 Ub ligases, has gained prominence. However, experimental evidence for the existence of dimeric substrate-receptor complexes has been largely missing. We believe that our structural and imaging studies of UV-DDB provide such experimental support.
The E3 ligase architecture derived from our crystal structure of the dimeric UV-DDB indicates that dimerization is a means of modulating intermolecular association parameters in cullin-RING E3 Ub ligase systems and is likely generalizable to other multi-component complexes with similar modular molecular architecture as UV-DDB. Homo-oligomerization is a fundamental step, allowing the individual components to be aligned in the context of the holo-complex, so that a multitude of reaction parameters can be spatially accommodated. Thus, a wide spectrum of functions can be regulated globally as biomolecular components dynamically assemble and disassemble along the NER and ubiquitin proteasome system pathways. Our study suggests that the rates of DNA binding and the high affinities for damaged DNA are a consequence of optimizing molecular associations in the holo-complex, an intrinsically basic mechanism for controlling substrate and protein–protein interactions, yet having profound effects on the overall efficiency of DNA repair.
Materials and Methods
Protein Expression and Purification.
Native and SeMet-substituted proteins were expressed in Sf9 cells and purified as previously published (10) (SI Appendix).
Synthetic oligodeoxynucleotides, sequences, methods of purification, and analysis of the oligodeoxynucleotides described in this study are provided in SI Appendix.
Electron Microscopy.
Uranyl acetate stained UV-DDB samples were prepared on grids and imaged on film in an FEI Tecnai T12 microscope operating at 120 kV and magnification of 30,000×. Micrographs were digitized with a Nikon Super CoolScan 9000 scanner and processed with the ImageJ software (50) to remove background variations, including uneven depth of stain and thickness of the carbon support film, and to estimate the particle size distributions (SI Appendix).
AFM Sample Preparation and Imaging.
UV-DDB was incubated with nondamaged or UV-irradiated 517 bp PCR fragments and diluted 1∶5- to 1∶10-fold before deposition. All images were collected using a MultiMode V microscope (Veeco Instruments). Images were captured at a scan size of 1 μm × 1 μm, a scan rate of 2–4 Hz, a target amplitude of 0.3 V and a resolution of 512 × 512 pixels (additional experimental details and statistical analysis of AFM images are provided in SI Appendix).
Crystallography.
Purified native UV-DDB mixed in a 1∶3 molar ratio with AP24 oligodeoxynucleotides were prepared and immediately used in crystallization screening setups. Preliminary small crystals of the UV-DDB, with damaged DNA bound verified by gel electrophoresis, were obtained but diffracted weakly to 8 Å. Further optimization using additive screening protocols (36) generated single crystals in both monoclinic and orthorhombic lattices, and seeding eventually produced crystals that diffracted to 2.85–3.25 Å. A combination of a selenomethionine anomalous phasing approach in combination with partial model phasing yielded initial electron density maps clearly defining solvent and macromolecular boundaries. Solvent flattening and histogram matching improved the preliminary electron density maps (SI Appendix, Fig. S7), verifying the dimeric composition of the asymmetric unit. Iterative cycles of model building, rigid-body, molecular dynamics, simulated annealing, and grouped-B factor refinement monitoring Rwork and Rfree values throughout, improved model accuracy and map quality, permitting the DDB2 subunit to be traced and the AP24 oligodeoxynucleotide molecule built into its electron densities. The asymmetric unit is comprised of the full-length sequence of the human DDB1 (residues 1–1140), human DDB2 (residues 20–421), and 24-bp oligodeoxynucleotide duplex containing a central abasic site, refined to Rwork/Rfree values of 0.22/0.24 (monoclinic) and 0.25/0.26 (orthorhombic). Data processing and refinement statistics are shown in (SI Appendix, Table S1), including details related to crystallization, data processing, structure determination, and refinement for both the monoclinic and orthorhombic forms of UV-DDB.
Dynamic Light Scattering (DLS) Analysis.
A 20-μL UV-DDB sample was passed through a 0.2-μm filtering assembly into the sample chamber of a DynaPro (Wyatt Technology) molecular-sizing instrument equipped with a Plate Reader (Protein Solutions). Data collection and analysis utilized Dynamics 6.0 software package, as originally described (51). The particle sizes of UV-DDB, measured at six different protein concentrations were determined in the presence and absence of damaged oligodeoxynucleotide, AP24 (SI Appendix, Tables S2 and S4). Statistical analysis and additional details are included in SI Appendix.
Surface Plasmon Resonance (SPR).
Immobilization of UV-DDB onto CM5 chip surfaces used standard EDC/NHS-mediated amine coupling procedures (52), using concentrations determined by the DLS results. The association and dissociation phases for the interaction of AP24 to UV-DDB were monitored on a BIAcore 3000 System (GE Healthcare), allowing the rates and binding affinities to be determined as a function of AP24 concentration. Data analysis (BIAevaluation software version 4.1) applied a Langmuir binding model to calculate the kinetics and affinity constants for the binding of AP24 to the monomeric UV-DDB (SI Appendix, Table S3).
Supplementary Material
ACKNOWLEDGMENTS.
We thank Drs. John Rosenberg and Richard Wood for helping with the design of the AP oligodeoxynucleotides and Dr. Angela Gronenborn for early discussions. We appreciate the preliminary UV-DDB characterization done by Dr. Inja Byeon and the assistance of Dr. Jinwoo Ahn and Thomas Vu on early oligodeoxynucleotide purification. We thank Dr. Alexander Makhov for assisting with the electron microscopy and image analysis. We thank the staff members at the General Medicine and Cancer Institutes Collaborative Access Team (GM/CA-CAT) and the Southeast Regional Collaborative Access Team (SER-CAT), both at the Advanced Photon Source, Argonne National Laboratory, for access and technical assistance. Financial contributions toward the structural and biophysical methods applied in this work were provided by National Institutes of Health (NIH) Grants AI76121, GM82251, and GM66466 (to J.I.Y.). Financial support for this work were provided by NIH Grant ES019566 (to B.V.H.), the Commonwealth of Pennsylvania, Department of Health (to J.I.Y. and J.F.C.), and the Pittsburgh Foundation (to V.R.O.). GM/CA-CAT has been funded in whole or in part with federal funds from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Sciences (Y1-GM-1104). Use of the Advanced Photon Source is supported by the US Department of Energy, Office of Science, and Office of Basic Energy Sciences, under Contract W-31-109-Eng-38.
Note Added in Proof.
While this paper was under review a study appeared revealing the crystal structure of a single complex consisting of DDB1-DDB2-CUL4A-RBX1 (CRL4ADDB2) bound to a 12 bp DNA duplex containing a tetrahydrofuran (THF) lesion. While this structure differs significantly from the dimeric structure of UV-DDB bound to DNA in our study, their new structure helps explain the ubiquitin ligase substrate flexibility in damage recognition in chromatin (53).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
See Author Summary on page 16408 (volume 109, number 41).
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 4E54 and 4E5Z).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1110067109/-/DCSupplemental.
*Percentage value calculated from the integration of the number of molecules under two Gaussian fits after deconvoluting the peaks shown in (SI Appendix, Fig. S4B), Inset.
†Determined by accounting for the presence of both monomeric and dimeric states, using the mass distributions found from the DLS measurements, conducted using identical protein concentrations and experimental conditions, and estimating respective contributions of monomeric and dimeric states to the kinetic and affinity parameters calculated from the SPR data.
References
- 1.Friedberg EC, et al. DNA Repair and Mutagenesis. 2nd Ed. Washington, DC: ASM Press; 2005. pp. 1–1164. [Google Scholar]
- 2.Sugasawa K, et al. Xeroderma pigmentosum group C protein complex is the initiator of global genome nucleotide excision repair. Mol Cell. 1998;2:223–232. doi: 10.1016/s1097-2765(00)80132-x. [DOI] [PubMed] [Google Scholar]
- 3.Wittschieben BO, Wood RD. DDB complexities. DNA Repair (Amst) 2003;2:1065–1069. doi: 10.1016/s1568-7864(03)00113-7. [DOI] [PubMed] [Google Scholar]
- 4.Moser J, et al. The UV-damaged DNA binding protein mediates efficient targeting of the nucleotide excision repair complex to UV-induced photo lesions. DNA Repair (Amst) 2005;4:571–582. doi: 10.1016/j.dnarep.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 5.Wakasugi M, et al. DDB accumulates at DNA damage sites immediately after UV irradiation and directly stimulates nucleotide excision repair. J Biol Chem. 2002;277:1637–1640. doi: 10.1074/jbc.C100610200. [DOI] [PubMed] [Google Scholar]
- 6.Dualan R, et al. Chromosomal localization and cDNA cloning of the genes (DDB1 and DDB2) for the p127 and p48 subunits of a human damage-specific DNA binding protein. Genomics. 1995;29:62–69. doi: 10.1006/geno.1995.1215. [DOI] [PubMed] [Google Scholar]
- 7.Rapic-Otrin V, et al. True XP group E patients have a defective UV-damaged DNA binding protein complex and mutations in DDB2 which reveal the functional domains of its p48 product. Hum Mol Genet. 2003;12:1507–1522. doi: 10.1093/hmg/ddg174. [DOI] [PubMed] [Google Scholar]
- 8.Itoh T, Mori T, Ohkubo H, Yamaizumi M. A newly identified patient with clinical xeroderma pigmentosum phenotype has a non-sense mutation in the DDB2 gene and incomplete repair in (6-4) photoproducts. J Invest Dermatol. 1999;113:251–257. doi: 10.1046/j.1523-1747.1999.00652.x. [DOI] [PubMed] [Google Scholar]
- 9.Nichols AF, Ong P, Linn S. Mutations specific to the xeroderma pigmentosum group E Ddb-phenotype. J Biol Chem. 1996;271:24317–24320. doi: 10.1074/jbc.271.40.24317. [DOI] [PubMed] [Google Scholar]
- 10.Wittschieben BO, Iwai S, Wood RD. DDB1-DDB2 (xeroderma pigmentosum group E) protein complex recognizes a cyclobutane pyrimidine dimer, mismatches, apurinic/apyrimidinic sites, and compound lesions in DNA. J Biol Chem. 2005;280:39982–39989. doi: 10.1074/jbc.M507854200. [DOI] [PubMed] [Google Scholar]
- 11.Scrima A, et al. Structural basis of UV DNA-damage recognition by the DDB1-DDB2 complex. Cell. 2008;135:1213–1223. doi: 10.1016/j.cell.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fitch ME, Nakajima S, Yasui A, Ford JM. In vivo recruitment of XPC to UV-induced cyclobutane pyrimidine dimers by the DDB2 gene product. J Biol Chem. 2003;278:46906–46910. doi: 10.1074/jbc.M307254200. [DOI] [PubMed] [Google Scholar]
- 13.Takedachi A, Saijo M, Tanaka Ke. DDB2 complex-mediated ubiquitylation around DNA damage is oppositely regulated by XPC and Ku and contributes to the recruitment of XPA. Mol Cell Biol. 2010;30:2708–2723. doi: 10.1128/MCB.01460-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hannah J, Zhou P. Regulation of DNA damage response pathways by the cullin-RING ubiquitin ligases. DNA Repair (Amst) 2009;8:536–543. doi: 10.1016/j.dnarep.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Higa LA, et al. CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nat Cell Biol. 2006;8:1277–1283. doi: 10.1038/ncb1490. [DOI] [PubMed] [Google Scholar]
- 16.Guerrero-Santoro J, et al. The cullin 4B-based UV-damaged DNA-binding protein ligase binds to UV-damaged chromatin and ubiquitinates histone H2A. Cancer Res. 2008;68:5014–5022. doi: 10.1158/0008-5472.CAN-07-6162. [DOI] [PubMed] [Google Scholar]
- 17.Petroski MD, Deshaies RJ. Function and regulation of cullin-RING ubiquitin ligases. Nat Rev Mol Cell Biol. 2005;6:9–20. doi: 10.1038/nrm1547. [DOI] [PubMed] [Google Scholar]
- 18.Zheng N, et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416:703–709. doi: 10.1038/416703a. [DOI] [PubMed] [Google Scholar]
- 19.Angers S, et al. Molecular architecture and assembly of the DDB1-CUL4A ubiquitin ligase machinery. Nature. 2006;443:590–593. doi: 10.1038/nature05175. [DOI] [PubMed] [Google Scholar]
- 20.Higa LA, et al. L2DTL/CDT2 interacts with the CUL4/DDB1 complex and PCNA and regulates CDT1 proteolysis in response to DNA damage. Cell Cycle. 2006;5:1675–1680. doi: 10.4161/cc.5.15.3149. [DOI] [PubMed] [Google Scholar]
- 21.Groisman R, et al. The ubiquitin ligase activity in the DDB2 and CSA complexes is differentially regulated by the COP9 signalosome in response to DNA damage. Cell. 2003;113:357–367. doi: 10.1016/s0092-8674(03)00316-7. [DOI] [PubMed] [Google Scholar]
- 22.Higa LA, Zhang H. Stealing the spotlight: CUL4-DDB1 ubiquitin ligase docks WD40-repeat proteins to destroy. Cell Div. 2007;2:5. doi: 10.1186/1747-1028-2-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang H, et al. Histone H3 and H4 ubiquitylation by the CUL4-DDB-ROC1 ubiquitin ligase facilitates cellular response to DNA damage. Mol Cell. 2006;22:383–394. doi: 10.1016/j.molcel.2006.03.035. [DOI] [PubMed] [Google Scholar]
- 24.Kapetanaki MG, et al. The DDB1-CUL4ADDB2 ubiquitin ligase is deficient in xeroderma pigmentosum group E and targets histone H2A at UV-damaged DNA sites. Proc Natl Acad Sci USA. 2006;103:2588–2593. doi: 10.1073/pnas.0511160103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luijsterburg MS, et al. Dynamic in vivo interaction of DDB2 E3 ubiquitin ligase with UV-damaged DNA is independent of damage-recognition protein XPC. J Cell Sci. 2007;120:2706–2716. doi: 10.1242/jcs.008367. [DOI] [PubMed] [Google Scholar]
- 26.Sugasawa K, et al. UV-induced ubiquitylation of XPC protein mediated by UV-DDB-ubiquitin ligase complex. Cell. 2005;121:387–400. doi: 10.1016/j.cell.2005.02.035. [DOI] [PubMed] [Google Scholar]
- 27.El-Mahdy MA, et al. Cullin 4A-mediated proteolysis of DDB2 protein at DNA damage sites regulates in vivo lesion recognition by XPC. J Biol Chem. 2006;281:13404–13411. doi: 10.1074/jbc.M511834200. [DOI] [PubMed] [Google Scholar]
- 28.Rapic-Otrin V, McLenigan MP, Bisi DC, Gonzalez M, Levine AS. Sequential binding of UV DNA damage binding factor and degradation of the p48 subunit as early events after UV irradiation. Nucleic Acids Res. 2002;30:2588–2598. doi: 10.1093/nar/30.11.2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nag A, Bondar T, Shiv S, Raychaudhuri P. The xeroderma pigmentosum group E gene product DDB2 is a specific target of cullin 4A in mammalian cells. Mol Cell Biol. 2001;21:6738–6747. doi: 10.1128/MCB.21.20.6738-6747.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alekseev S, et al. Cellular concentrations of DDB2 regulate dynamic binding of DDB1 at UV-induced DNA damage. Mol Cell Biol. 2008;28:7402–7413. doi: 10.1128/MCB.01108-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Min JH, Pavletich NP. Recognition of DNA damage by the Rad4 nucleotide excision repair protein. Nature. 2007;449:570–575. doi: 10.1038/nature06155. [DOI] [PubMed] [Google Scholar]
- 32.Maillard O, Camenisch U, Clement FC, Blagoev KB, Naegeli H. DNA repair triggered by sensors of helical dynamics. Trends Biochem Sci. 2007;32:494–499. doi: 10.1016/j.tibs.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 33.Maillard O, Camenisch U, Blagoev KB, Naegeli H. Versatile protection from mutagenic DNA lesions conferred by bipartite recognition in nucleotide excision repair. Mutat Res. 2008;658:271–286. doi: 10.1016/j.mrrev.2008.01.007. [DOI] [PubMed] [Google Scholar]
- 34.Camenisch U, et al. Two-stage dynamic DNA quality check by xeroderma pigmentosum group C protein. EMBO J. 2009;28:2387–2399. doi: 10.1038/emboj.2009.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hwang BJ, Chu G. Purification and characterization of a human protein that binds to damaged DNA. Biochemistry. 1993;32:1657–1666. doi: 10.1021/bi00057a033. [DOI] [PubMed] [Google Scholar]
- 36.Yeh JI, Beale SI. Calorimetric approaches to characterizing effects of additives on protein crystallization. Cryst Growth Des. 2007;7:2134–2139. [Google Scholar]
- 37.Li T, Chen X, Garbutt KC, Zhou P, Zheng N. Structure of DDB1 in complex with a paramyxovirus V protein: Viral hijack of a propeller cluster in ubiquitin ligase. Cell. 2006;124:105–117. doi: 10.1016/j.cell.2005.10.033. [DOI] [PubMed] [Google Scholar]
- 38.Batty D, Rapic’-Otrin V, Levine AS, Wood RD. Stable binding of human XPC complex to irradiated DNA confers strong discrimination for damaged sites. J Mol Biol. 2000;300:275–290. doi: 10.1006/jmbi.2000.3857. [DOI] [PubMed] [Google Scholar]
- 39.Yang Y, Wang H, Erie DA. Quantitative characterization of biomolecular assemblies and interactions using atomic force microscopy. Methods. 2003;29:175–187. doi: 10.1016/s1046-2023(02)00308-0. [DOI] [PubMed] [Google Scholar]
- 40.Wang H, Yang Y, Erie DA. Characterization of protein-protein interactions using atomic force microscopy. In: Schuck P, editor. Protein Interactions Biophysical Approaches for the Study of Complex Reversible Systems. Vol. 5. Springer Science+Business Media, LLC; 2007. pp. 39–78. (Protein Reviews). [Google Scholar]
- 41.Kad NM, Wang H, Kennedy GG, Warshaw DM, Van Houten B. Collaborative dynamic DNA scanning by nucleotide excision repair proteins investigated by single-molecule imaging of quantum-dot-labeled proteins. Mol Cell. 2010;37:702–713. doi: 10.1016/j.molcel.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang H, Tessmer I, Croteau DL, Erie DA, Van Houten B. Functional characterization and atomic force microscopy of a DNA repair protein conjugated to a quantum dot. Nano Lett. 2008;8:1631–1637. doi: 10.1021/nl080316l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fujiwara Y, et al. Characterization of DNA recognition by the human UV-damaged DNA-binding protein. J Biol Chem. 1999;274:20027–20033. doi: 10.1074/jbc.274.28.20027. [DOI] [PubMed] [Google Scholar]
- 44.Inoki T, et al. Human DDB2 splicing variants are dominant negative inhibitors of UV-damaged DNA repair. Biochem Biophys Res Commun. 2004;314:1036–1043. doi: 10.1016/j.bbrc.2004.01.003. [DOI] [PubMed] [Google Scholar]
- 45.Merlet J, Burger J, Gomes JE, Pintard L. Regulation of cullin-RING E3 ubiquitin-ligases by neddylation and dimerization. Cell Mol Life Sci. 2009;66:1924–1938. doi: 10.1007/s00018-009-8712-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rhodes D, Burley SK. Protein–nucleic acid interactions. Curr Opin Struct Biol. 2000;10:75–77. doi: 10.1016/s0959-440x(99)00052-4. [DOI] [PubMed] [Google Scholar]
- 47.Nekrep N, et al. Mutations in a winged-leix DNA-binding motif causes atypical bare lymphocyte syndrome. Nat Immunol. 2002;3:1075–1081. doi: 10.1038/ni840. [DOI] [PubMed] [Google Scholar]
- 48.Lamber EP, et al. Regulation of the transcription factor Ets-1 by DNA-mediated homo-dimerization. EMBO J. 2008;27:2006–2017. doi: 10.1038/emboj.2008.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bosu DR, Kipreos ET. Cullin-RING ubiquitin ligases: Global regulation and activation cycles. Cell Div. 2008;3:7. doi: 10.1186/1747-1028-3-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Abramoff MD, Magelhaes PJ, Ram SJ. Image Processing with ImageJ. Biophotonics Int. 2004;11:36–42. [Google Scholar]
- 51.Moradian-Oldak J, Leung W, Fincham AG. Temperature and pH-dependent supramolecular self-assembly of amelogenin molecules: A dynamic light-scattering analysis. J Struct Biol. 1998;122:320–327. doi: 10.1006/jsbi.1998.4008. [DOI] [PubMed] [Google Scholar]
- 52.Murphy M, Jason-Moller L, Bruno J. Using biacore to measure the binding kinetics of an antibody-antigen interaction. Curr Protoc Protein Sci. 2006;19:1–17. doi: 10.1002/0471142301.ps1914s45. [DOI] [PubMed] [Google Scholar]
- 53.Fischer ES, et al. The molecular basis of CRL4DDB2/CSA ubiquitin ligase architecture, targeting, and activation. Cell. 2011;147:1024–1039. doi: 10.1016/j.cell.2011.10.035. [DOI] [PubMed] [Google Scholar]
