

Abstract
A 204 residue covalent-dimer vascular endothelial growth factor, with full mitogenic activity, was made by one-pot native chemical ligation from three unprotected peptide segments. The covalent structure of synthetic VEGF was confirmed by precise mass measurement, and the three dimensional structure of the synthetic protein molecule was determined by high resolution X-ray crystallography. Robust, practical synthetic access to biologically active VEGF will aid in the development of novel anti-angiogenic therapeutics.
Keywords: chemical protein synthesis, VEGF, one-pot ligation, X-ray structure, HUVEC proliferation
Vascular endothelial growth factor (VEGF), also known as VEGF-A, is the principal endothelial cell-specific mitogen that induces embryonic and somatic angiogenesis.[1–4] VEGF also plays an important role in tumor angiogenesis, and in proliferative retinopathy.[5] In animal models of pathological conditions, it has been shown that over-expression of VEGF stimulates the generation of new blood vessels and thus accelerates tumor growth and the progression of the neovascular form of age-related macular degeneration.[2,6–7]
VEGF is a homodimer and exists in multiple isoforms, each containing two identical polypeptide chains derived from alternative splicing of exons in a single mRNA.[8] VEGF165 is the most abundant naturally occurring form.[9] All the isoforms of VEGF share a common N-terminus domain of ~115 residues;[10] it is this domain that interacts with the VEGFR1 and VEGFR2 receptors and causes the biological response. Targeting the N-terminal domain of VEGF with specific inhibitors of its function has been shown to have broad therapeutic potential for a variety of indications. Monoclonal antibody therapeutics that target VEGF are already approved for cancer and age-related macular degeneration.[11–13]
VEGF is a member of the cystine knot growth factor protein family in which two disulfide bridges connect backbone segments of the protein to form a ring structure through which a third disulfide bond interpenetrates.[14] VEGF itself is a covalent homodimeric protein with three intramolecular disulfides in each polypeptide chain, and with two interchain disulfide bonds covalently joining the two identical monomers.[14] The resulting VEGF molecule has a two-fold axis of symmetry and thus has two receptor binding regions in the one protein.[15–16] It has previously been shown that a truncated variant, VEGF (8-109), binds to the KDR with wild type affinity and stimulates a full biological response.[15] VEGF (8-109) has been the subject of extensive structural and biophysical studies including the determination of its X-ray structure.[14]
Robust, practical synthetic access to biologically active VEGF will enable novel approaches to the development of novel anti-angiogenic therapeutics. Here we report a highly optimized total synthesis of human VEGF (8-109), by a one-pot, three segment native chemical ligation strategy.[17–19] Folding of the synthetic polypeptide chain with concomitant disulfide bond formation gave a VEGF protein molecule with full biological activity. The structure of the covalent homodimeric protein molecule was confirmed by high resolution X-ray crystallography.
The amino acid sequence of the VEGF (8-109) target polypeptide chain is shown in Figure 1A, and contains eight cysteine residues. In our initial synthetic design, we explored a potentially simple approach that made use of a single native chemical ligation,[17] at Arg49-Cys50, of two unprotected peptide segments of almost equal size for the assembly of the 102 residue polypeptide chain. However, unsatisfactory purity of the 1-49-thioester segment prompted us to use the three segment native chemical ligation strategy shown in Figure 1B for the total synthesis of VEGF. In the optimized synthetic design, the N-terminal half of the full-length 102 amino acid polypeptide chain was divided into two synthetic peptide segments. The N-terminal cysteine residue of the middle segment was introduced as a 1,3-thiazolidine-4-R-carboxo- moiety in order to prevent reaction of the N-terminal Cys with the thioester in the same peptide.[18] To avoid excessive product losses from multiple intermediate purification steps, we used one-pot sequential native chemical ligations, together with a single final purification of the full-length 102 residue ligation product.[18] The purified full-length polypeptide was folded with concomitant formation of disulfides to give the homodimeric synthetic VEGF protein molecule.
Figure 1.

Total chemical synthesis of VEGF. (A) Target amino acid sequence; (B) Synthetic strategy for the total chemical synthesis of VEGF. One-pot sequential native chemical ligations[18] from three peptide segments is used to make the 102 amino acid residue polypeptide chain; subsequent folding/disulfide bond formation gives the desired covalent dimer VEGF protein molecule. [R = -CH2-CH2-SO3H]
The three synthetic peptides (peptide-thioester (1); Thz-peptide-thioester (2); Cys-peptide (3)) were prepared by the Boc chemistry SPPS using the in situ neutralization protocol,[20] as detailed in the Supporting Information. Based on the synthetic strategy shown in Figure 1B, the C-terminal peptide segment Cys50-Asp102 (3) [1.7 mM] was reacted with Thz19-Arg49-αCOSR (2) [1.5 mM] under standard native chemical ligation conditions (aqueous 6M Gu.HCl, 0.1 M Na2HPO4, at pH 7.0, 100 mM 4-carboxymethyl-thiophenol (MPAA),[21] 50 mM TCEP.HCl) to give the ligation product Thz19-Asp102 (4). Without purification, the thiazolidine (Thz19-) was then converted to Cys19- by addition of 60 mM methoxylamine.HCl at pH 4.0.[22] After the conversion of Thz- to Cys- was complete (16 h, product 5), the peptide-thioester segment (Gly1-Tyr18-αCOSR, 1) [2 mM] was added to the same reaction mixture, and the pH adjusted to 6.8; within 2 h, native chemical ligation gave near-quantitative conversion to the target polypeptide 6. Analytical data for all steps in the synthesis of the full-length polypeptide are shown in Figure 2. After HPLC purification, the target polypeptide 6 was obtained in 48% overall yield based on limiting starting peptide segment 2. The reverse phase HPLC profile of the product 102 residue polypeptide is shown in Figure 3A. The unfolded monomeric full-length polypeptide 6 was characterized by HPLC-electrospray ionization mass spectrometry (LC-MS) (Figure 3B), and had the expected mass: 11932.2±0.7 Da (mean of the four most abundant charge states); calculated mass 11932.5 Da (average isotope composition).
Figure 2.

Analytical LC-MS data for the total chemical synthesis of VEGF. Analytical HPLC profiles (λ = 214 nm) are shown, together with online electrospray ionization mass spectrometry (LC-MS) data (inset) corresponding to each major product. (A) Native chemical ligation between Thz19-Arg49 –αCOSR (2, 15 mg, 3.37 μmol, 1.53 mM) and Cys50̃Asp102̃ COOH (3, 22 mg, 3.66 μmol, 1.66 mM) at t < 2 min. 7 is the intermediate MPAA-exchanged product; (B) Native chemical ligation reaction after 1 h: 4 is the ligation product; (C) Crude reaction mixture after Thz– to Cys– conversion using 60 mM methoxylamine. HCl at pH 4: formation of the desired Cys– product 5 was confirmed by a mass decrease of 12 Da; (D) One-pot native chemical ligation of Gly1-Tyr18 –αCOSR (1, 10.4 mg, 4.39 μmol, 2 mM) and Cys19̃Asp102̃ COOH at t = 0 min; (E) Crude reaction mixture after 2 h at pH = 6.8: 6 is the ligation product Gly1-Asp102-COOH [overall yield (after purification) = 19.3 mg, 1.6 μmol, 48% based on limiting peptide segment 2]. Analytical HPLC was performed using a linear gradient (10̃54%) of buffer B in buffer A over 22 min (buffer A = 0.1% trifluoroacetic acid (TFA) in water; buffer B = 0.08% TFA in acetonitrile) on a C-3 (Agilent), 4.6 °— 150 mm column at 40 °C (flow rate = 1 mL/min). R = -CH2-CH2-SO3H and asterisk (*) corresponds to MPAA.
Figure 3.

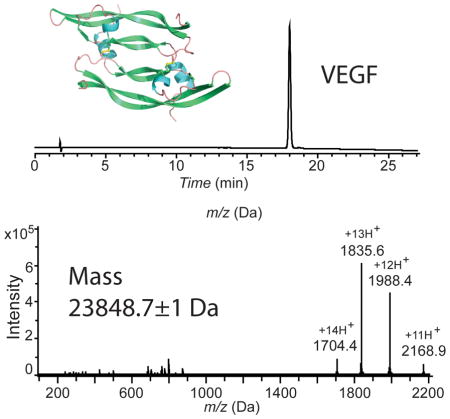
HPLC and mass spectrometry characterization of synthetic VEGF. (A) Analytical HPLC profile (λ = 214 nm) of the purified unfolded 102 amino acid polypeptide [Gly1-Asp102-COOH]; (B) LC-MS of purified unfolded 102 amino acid polypeptide [Gly1-Asp102-COOH]; observed mass: 11932.2±0.7 Da (average of the four most abundant charge states), calculated mass: 11932.54 Da (average isotope); (C) Analytical HPLC profile (λ = 214 nm) of the purified synthetic VEGF protein. Folded VEGF showed a 2.8 min earlier retention time shift compared to the reduced polypeptide; (D) Direct infusion electrospray ionization MS of the synthetic VEGF protein: observed mass: 23848.7±1 Da (mean of the three most abundant charge states), calculated mass: 23849.1 Da (average isotope composition). A decrease of 15.7 Da from twice the monomer mass confirmed the formation of eight disulfides. Note the narrow distribution of the charge states in the folded VEGF dimer compared with the unfolded monomer. Analytical HPLC was performed using a linear gradient (10̃54%) of buffer B in buffer A over 22 min (buffer A = 0.1% trifluoroacetic acid (TFA) in water; buffer B = 0.08% TFA in acetonitrile) on a C-3 (Agilent), 4.6 ×150 mm column at 40 °C (flow rate = 1 mL/min).
In its folded form, VEGF (8-109) exists as a covalent homodimer of two identical 102 residue polypeptide chains, with six intra-chain disulfide bonds and two inter-chain disulfides connecting the two monomer polypeptides.[14] These disulfide bridges are crucial for the correct folding and structural integrity of functional VEGF.[23] Reduced polypeptide 6 was folded at pH 8.4 in 0.15 M aqueous Gu.HCl containing a [Glutathione]reduced/ [Glutathione]oxidized (2mM/0.4 mM) redox couple.[24] The completion of the folding was revealed by the near-quantitative formation after 5 days of a product that eluted earlier than the reduced (i.e. unfolded) polypeptide monomer in analytical reverse phase HPLC (Figure 3C). Such a shift to an earlier retention time is typically observed upon folding of disulfide-containing globular proteins, because of burial of the hydrophobic residues in the core of the folded protein molecule. Formation of 8 disulfide bonds in the synthetic VEGF protein molecule was confirmed by measurement of the mass of this early-eluting peak (below). Reverse phase HPLC purification gave synthetic VEGF in 45% yield based on the amount of 6 used.
Attempts to determine the mass of the folded VEGF synthetic protein by LC-MS were unsuccessful. Under the conditions of direct infusion into the MS from the analytical HPLC column, no ion current was observed for the UV-detected peak of the purified synthetic product. Poor ionization under electrospray injection conditions is typical of tightly folded disulfide-crosslinked globular proteins dissolved in water-acetonitrile-0.1% TFA.[25] The purified synthetic protein was dissolved in a solvent {1% acetic acid in methanol-water (80:20v/v)} that is more conducive to electrospray ionization of protein molecules,[26] and was characterized by direct infusion electrospray ionization mass spectrometry (Figure 3D). The synthetic protein had an observed mass of 23848.7±1.2 Da (mean of the three most abundant charge states), calculated mass: 23849.1 Da (average isotope composition). The observed decrease of 15.7 Da, compared with twice the mass of the 102 residue polypeptide chain, was consistent with the formation of eight disulfide bonds and consequent loss of 16 protons in the synthetic VEGF protein molecule.
VEGF contains 204 amino acid residues and has a total of 34 protonation sites (His, Lys, Arg side chains, plus 2 x N-terminal amino groups). Yet, the positive ion electrospray ionization MS of the synthetic protein exhibited only a narrow distribution of charge states, centered around plus 13H+ with protonation states ranging from 11+ to 14+ (Figure 3D). In contrast, the reduced and unfolded synthetic VEGF polypeptide chain with 102 amino acid residues contained a total of 17 protonation sites (His, Lys, Arg side chains, plus an N-terminus amino group) and its positive ion electrospray ionization mass spectrum was dominated by highly charged ions with a maximum around plus 11H+ with protonation states ranging from 8+ to 13+ in the LC-MS spectrum (Figure 3B). Such a dramatic effect on the charge state distribution in folded versus unfolded protein has been reported previously and is still a matter of debate. Several different explanations have been offered by different investigators.[25–31] The most plausible rationale for the low number of added protons in the folded protein molecule under electrospray ionization conditions was suggested by Fenn.[31] He noted that it is “the surface of the charged droplet that determines the nature and amount of the charge on a departing ion… in its compact configuration, a molecule has a smaller surface area in contact with the solution than when it is unfolded. Consequently, less work may be required to remove it from the droplet, so that it could lift off with fewer charges than when it is unfolded.”
The circular dichroism spectrum of an aqueous solution of the folded VEGF revealed the presence of beta-sheet and alpha-helix secondary structural elements, as shown in the SI (Figure S5).
The structure of the synthetic VEGF protein molecule was determined by X-ray crystallography. Synthetic VEGF was crystallized at a protein concentration of 2.5 mg/mL from 0.2 M NH4OAc, 0.1 M BIS-TRIS (pH 5.5) and 45% (v/v) (±)-2-methyl-2,4-pentanediol. Synchrotron radiation diffraction data were collected to 1.85 Å resolution from a single crystal at the Advanced Photon Source, Argonne National Laboratory. The synthetic VEGF structure was solved in the monoclinic space group C2 by molecular replacement using the reported X-ray structure (PDB accession code 2VPF) as a search model. The synthetic VEGF structure was refined to a crystallographic R-factor of 18.0% (R-free 22.3%) using the program Phenix.[32] X-ray data collection and the refinement statistics are summarized in Table S1.
The X-ray structure of the chemically synthesized VEGF protein reported here is identical, within experimental uncertainty, to the previously reported X-ray structure of recombinant VEGF (8-109)[14]: 96 C-alpha atoms of the 102-residue monomer unit can be superimposed with a root-mean-square-deviation (r.m.s.d.) of only 0.7 Å (Figure 4C). The X-ray structure of the chemically synthesized VEGF protein consists of an N-terminal α-helix that folds on top of the second monomer, followed by an anti-parallel four-stranded β-sheet forming the central part of the molecule. In addition, there is a short α-helical segment followed by a loop, and the second β-strand is positioned between the first and the third β-strand. Figure 4B shows a representative 2Fo-Fc electron density map contoured at 1 sigma encompassing the intermolecular disulfide interface. The asymmetric unit contained three VEGF molecules: i.e. it contained six crystallographically independent copies of the folded VEGF polypeptide chain, in the form of three covalent homodimers. Figure 4A shows a cartoon representation of the synthetic protein structure. Superposition of the six independent copies of VEGF monomers revealed very similar structures in the core region as expected, but significant deviation was observed in one of the loop regions (Met71-Ser88) which is a part of the receptor binding site of the VEGF protein molecule (Figure S6). Different copies of the molecule represent different snapshots of the loop movement, with a largest deviation of 6.7 Å at His79.
Figure 4.

X-ray structure of chemically synthesized VEGF. (A) Cartoon representation of the experimentally determined structure of the synthetic protein molecule; (B) Sigma A-weighted 2Fo-Fc electron density map of the VEGF contoured at 1σ encompassing the intermolecular disulfide bridge between the two monomer units; (C) Superposition of the polypeptide chain backbones from the crystal structure of the chemically synthesized VEGF reported here (red) and the reported crystal structure (PDB 2VPF) of the recombinant VEGF (8-109) monomer (blue) with 96 residues aligned with r.m.s.d. = 0.7 Å.
The synthetic VEGF protein had full mitogenic activity as demonstrated by the human umbilical vein endothelial cell (HUVEC) proliferation bioassay (Figure 5). The ED50 of 4.6 ng/mL is within the typical ED50 range observed for human VEGF-165 (i.e. 1–6 ng/mL).
Figure 5.
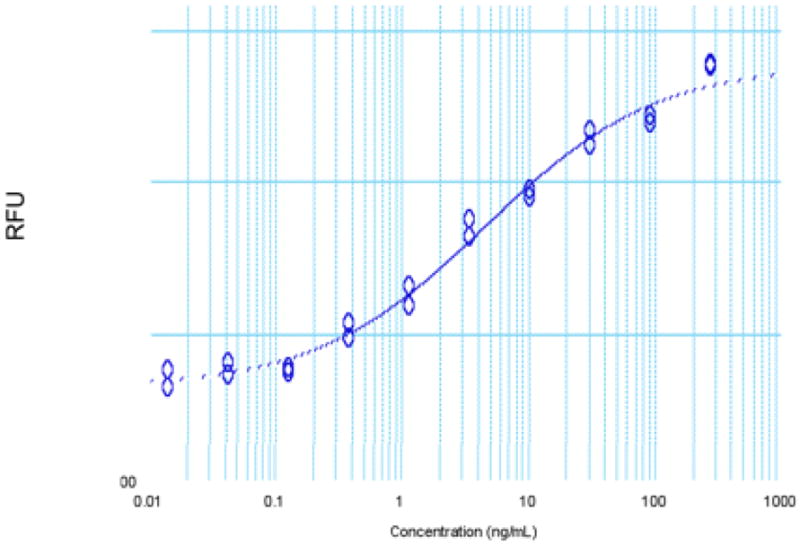
Human umbilical vein endothelial cell (HUVEC) proliferation assay: ED50 observed for the chemically synthesized VEGF (8-109) is 4.6 ng/mL; typical ED50 range for human VEGF-165 is 1–6 ng/mL.
We have developed a robust and reproducible total chemical synthesis of the homodimeric VEGF protein molecule. The synthesis reported here is one of only a handful of reported total syntheses of protein molecules of this size.[33] At 204 amino acid residues this is the largest protein molecule prepared by one-pot native chemical ligation; an overall yield of ~22% was obtained, based on the peptide building blocks used. Folding of proteins that contain large numbers of disulfides is not always straightforward; the near-quantitative formation of eight native disulfide bonds in folded synthetic VEGF is notable. Precise mass measurement was consistent with the expected covalent structure of the synthetic protein. Furthermore, synthetic VEGF showed full biological activity and was characterized by high resolution X-ray crystallography. Synthetic access to this important protein molecule previously available only from natural and recombinant sources is an essential first step in novel approaches to the development of improved anti-angiogenic agents.
Supplementary Material
Acknowledgments
This research was supported by the Office of Science (BER), U.S. Department of Energy, Grant No. DE-FG02 07ER64501 to S.B.H.K. and by the National Institutes of Health, Grant No. R01 GM075993 to S.B.H.K. Use of NE-CAT beamline 24-ID at the Advanced Photon Source is supported by award RR-15301 from the National Center for Research Resources at the National Institutes of Health. Use of the Advanced Photon Source is supported by the U.S. Department of Energy, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. The authors thank Dr. Kay Perry, Dr. Kanagalaghatta Rajashankar, and Mr. Somnath Mukherjee for useful discussions.
Footnotes
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Ferrara N. Breast Cancer Res Treat. 1995;36:127–137. doi: 10.1007/BF00666035. [DOI] [PubMed] [Google Scholar]
- 2.Folkman J. Nat Med. 1995;1:27–31. doi: 10.1038/nm0195-27. [DOI] [PubMed] [Google Scholar]
- 3.Dvorak HF, Brown LF, Detmar M, Dvorak AM. Am J Pathol. 146:1029–1039. [PMC free article] [PubMed] [Google Scholar]
- 4.Neufeld G, Tessler S, Gitay-Goren H, Cohen T, Levi BZ. Prog Growth Factor Res. 1994;5:89–97. doi: 10.1016/0955-2235(94)90019-1. [DOI] [PubMed] [Google Scholar]
- 5.Aiello LP, et al. N Engl J Med. 1994;331:1480–1487. doi: 10.1056/NEJM199412013312203. [DOI] [PubMed] [Google Scholar]
- 6.Lopez PF, Sippy BD, Lambert HM, Thach AB, Hinton DR. Invest Ophthalmol Vis Sci. 1996;37:855–868. [PubMed] [Google Scholar]
- 7.Kvanta A, Algvere PV, Berglin L, Seregard S. Invest Ophthalmol Vis Sci. 1996;37:1929–1934. [PubMed] [Google Scholar]
- 8.Kowanetz M, Ferrara N. Clin Cancer Res. 2006;12:5018–22. doi: 10.1158/1078-0432.CCR-06-1520. [DOI] [PubMed] [Google Scholar]
- 9.Vaisman N, Gospodarowicz D, Neufeld G. J Biol Chem. 1990;265:19461–19466. [PubMed] [Google Scholar]
- 10.Houck KA, Ferrara N, Winer J, Cachianes G, Li B, Leung DW. Mol Endocrinol. 1991;5:1806–1814. doi: 10.1210/mend-5-12-1806. [DOI] [PubMed] [Google Scholar]
- 11.Presta LG, Chen H, O’Connor SJ, et al. Cancer Res. 1997;57:4593–9. [PubMed] [Google Scholar]
- 12.Ferrara N. Endocr Rev. 2004;25:581–611. doi: 10.1210/er.2003-0027. [DOI] [PubMed] [Google Scholar]
- 13.Ferrara N, Damico L, Shams N, Lowman H, Kim R. Retina. 2006;26:859–70. doi: 10.1097/01.iae.0000242842.14624.e7. [DOI] [PubMed] [Google Scholar]
- 14.Muller YA, Christinger HW, Keyt BA, De Vos AM. Structure. 1997;5:1325–1338. doi: 10.1016/s0969-2126(97)00284-0. [DOI] [PubMed] [Google Scholar]
- 15.Muller YA, Li B, Christinger HW, Wells JA, Cunningham BC, De Vos AM. Proc Natl Acad Sci. 1997;94:7192–7197. doi: 10.1073/pnas.94.14.7192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wiesmann C, Fuh G, Christinger HW, Eigenbrot C, Wells JA, De Vos AM. Cell. 1997;91:695–704. doi: 10.1016/s0092-8674(00)80456-0. [DOI] [PubMed] [Google Scholar]
- 17.Dawson PE, Muir TW, Clarklewis I, Kent SBH. Science. 1994;266:776–779. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 18.Bang D, Kent SBH. Angew Chem Int Ed. 2004;43:2534–2538. doi: 10.1002/anie.200353540. [DOI] [PubMed] [Google Scholar]
- 19.Kent SBH. Chem Soc Rev. 2009;38:338–351. doi: 10.1039/b700141j. [DOI] [PubMed] [Google Scholar]
- 20.Schnölzer M, Alewood P, Jones A, Alewood D, Kent SBH. Int J Pept Res Ther. 2007;13:31–44. [Google Scholar]
- 21.Johnson ECB, Kent SBH. J Am Chem Soc. 2006;128:7140–7141. doi: 10.1021/ja058377y. [DOI] [PubMed] [Google Scholar]
- 22.Villain M, Vizzavona J, Rose K. Chem Biol. 2001;8:673–679. doi: 10.1016/s1074-5521(01)00044-8. [DOI] [PubMed] [Google Scholar]
- 23.Muller YA, Heiring C, Misselwitz R, Welfle K, Welfle H. J Biol Chem. 2002;277:43410–43416. doi: 10.1074/jbc.M206438200. [DOI] [PubMed] [Google Scholar]
- 24.Zisch AH, Lutolf MP, Ehrbar M, Raeber GP, Rizzi SC, Davies N, Schmökel H, Bezuidenhout D, Djonov V, Zilla P, Hubbell JA. FASEB J. 2003;17:2260–2. doi: 10.1096/fj.02-1041fje. [DOI] [PubMed] [Google Scholar]
- 25.Loo JA, Edmonds CG, Udseth HR, Smith RD. Anal Chem. 1990;62:693–698. doi: 10.1021/ac00206a009. [DOI] [PubMed] [Google Scholar]
- 26.Chowdhury SK, Katta V, Chait BT. J Am Chem Soc. 1990;112:9012–9013. [Google Scholar]
- 27.Katta V, Chait BT. J Am Chem Soc. 1991;113:8534–8535. 21. [Google Scholar]
- 28.Grandori R. J Mass Spectrom. 2003;38:11–15. doi: 10.1002/jms.390. [DOI] [PubMed] [Google Scholar]
- 29.Prakash H, Mazumdar S. J Am Soc Mass Spectrom. 2005;16:1409–1421. doi: 10.1016/j.jasms.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 30.Konermann L, Simmons DA. Mass Spectrom Rev. 2003;22:1–26. doi: 10.1002/mas.10044. [DOI] [PubMed] [Google Scholar]
- 31.Fenn JB. J Am Soc Mass Spectrom. 1993;4:524–535. doi: 10.1016/1044-0305(93)85014-O. [DOI] [PubMed] [Google Scholar]
- 32.Adams PD, et al. Acta Cryst D. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kumar KSA, Bavikar SN, Spasser L, Moyal T, Ohayon S, Brik A. Angew Chem Int Ed. 2011 May 18; doi: 10.1002/anie.201101920. Article first published online. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.