

Abstract
Excitotoxic neuronal death is mediated in part by NMDA receptor-induced activation of NOX2, an enzyme that produces superoxide and resultant oxidative stress. It is not known, however, whether the superoxide is generated in the intracellular space, producing oxidative stress in the neurons responding to NMDA receptor activation, or in the extracellular space, producing oxidative stress in neighboring cells. We evaluated these alternatives by preparing cortical neuron cultures from p47phox−/− mice, which are unable to form a functional NOX2 complex, and transfecting the cultures at low density with GFP-tagged p47phox to reconstitute NOX2 activity in widely scattered neurons. NMDA exposure did not induce oxidative stress or cell death in the nontransfected, p47-phox−/− cultures, but did produce oxidative stress and neuronal death in neurons surrounding the transfected, NOX2-competent neurons. This cell-to-cell spread of NMDA-induced oxidative injury was blocked by coincubation with either superoxide dismutase or the anion channel blocker 4′-diisothiocyanostilbene-2,2′-disulphonate, confirming superoxide anion as the mediating oxidant. In neurons plated on a preexisting astrocyte layer, NMDA induced oxidative stress in both the neurons and the astrocytes, and this was also prevented by superoxide dismutase. These findings show that activation of NMDA receptors on one neuron can lead to oxidative stress and cell death in neighboring neurons and astrocytes by a process involving the extracellular release of superoxide by NOX2.
Introduction
Sustained activation of NMDA-type glutamate receptors leads to excitotoxic neuronal death (Choi, 1988). An important but poorly understood aspect of excitotoxicity is its propensity to spread from one cell to another. This process may contribute to the propagation of injury in acute neurological disorders such as stroke, brain trauma, and seizures (Faden et al., 1989; Meldrum, 1993; During et al., 2000), and also in more slowly progressive disorders such as amyotrophic lateral sclerosis, Alzheimer's disease, and Huntington's disease (Lipton, 2004; Rothstein, 2009; Milnerwood and Raymond, 2010).
Neuronal production of the gaseous signaling molecules, nitric oxide and superoxide, is recognized as a crucial step linking NMDA receptor activation to cell death (Lafon-Cazal et al., 1993; Dawson, 1995; Pacher et al., 2007; Brennan et al., 2009; Forder and Tymianski, 2009; Girouard et al., 2009). Neither superoxide nor nitric oxide is intrinsically highly toxic, but these molecules avidly combine to form peroxynitrite and other substances that readily react with lipids, DNA, and other cell constituents (Pacher et al., 2007). Nitric oxide is produced in a subset of neurons in response to NMDA receptor activation, and it then passes freely across cell membranes (Bredt et al., 1991; Sattler et al., 1999; Garthwaite, 2008). The origin and distribution of superoxide is less clear. NMDA-induced superoxide was originally thought to originate from mitochondria, as a consequence of mitochondrial calcium uptake (Dykens, 1994; Dugan et al., 1995). However, it was more recently demonstrated that neurons express the superoxide-generating enzyme NADPH oxidase (Sorce and Krause, 2009), and manipulations that block NADPH oxidase activity effectively block NMDA-induced superoxide production (Brennan et al., 2009; Girouard et al., 2009; Guemez-Gamboa et al., 2011).
The NADPH oxidase isoform NOX2 is the predominate form expressed by neurons (Sorce and Krause, 2009). NMDA-induced NOX2 activation is calcium-dependent and occurs in many, if not all, neurons with NMDA receptors (Brennan et al., 2009; Girouard et al., 2009). NOX2 is composed of two membrane-bound subunits, p22phox and gp91, and three cytosolic subunits, p47phox, p67phox and p40phox. Phosphorylation of p47phox leads to assembly of the active complex at cell membranes (Dang et al., 2001; Bedard and Krause, 2007). Evidence suggests that NOX2 produces superoxide at intracellular membranes in some cell types and at the plasma membrane in others (Jesaitis et al., 1990; Bedard and Krause, 2007; Prosser et al., 2011), and its functional location in neurons is uncertain. The distinction is important because, unlike nitric oxide, superoxide does not readily cross cell membranes, and superoxide production at the cell surface provides a mechanism for propagating NMDA-induced oxidative stress to neighboring cells. Here we show that activated NOX2 releases superoxide to the extracellular space and produces oxidative injury in neighboring neurons and astrocytes.
Materials and Methods
Reagents were purchased from Sigma-Aldrich unless otherwise specified. Experiments were performed in accordance with protocols approved by the San Francisco Veterans Affairs Medical Center Animal Studies subcommittee. C57BL/6 wild-type and p47phox−/− mice were purchased from Simonsen and Jackson Laboratories, respectively. The p47phox−/− mice were maintained as homozygotes, using breeder stock that was backcrossed to WT C57BL/6 mice after every eight generations.
Cultures.
Neuronal monocultures were prepared from the cortices of embryonic day 14 mice of either sex and plated in 24-well culture plates or on poly-d-lysine-coated glass coverslips. After 1 d in culture, 10 μm cytosine arabinoside was added for 24 h to prevent glial proliferation. The neurons were subsequently maintained with serum-free NeuroBasal medium (Invitrogen) containing 5 mm glucose, at 37°C in a 5% CO2 atmosphere. These cultures contain 95% neurons, 5% astrocytes, and no detectable microglia (Kauppinen and Swanson, 2005). Neuron–astrocyte cocultures were prepared by plating neurons onto a preexisting astrocyte monolayer, as previously described (Alano et al., 2010).
Transfection.
Transfections with green fluorescent protein (GFP) and GFP-tagged p47phox (gifts from L. Terada, University of Texas Southwestern, Dallas, TX) were done with p47phox−/− neurons on day 6 or 7 in vitro. Culture medium was replaced with Opti-MEM (Invitrogen) that contained 0.3% Lipofectamine (Invitrogen) and 400 ng of DNA construct. This medium was changed back to NeuroBasal medium after 4 h incubation.
Experimental procedures.
Neurons were used for experiments at day 9–12 in vitro. Studies were initiated by washing the cultures into a balance salt solution (BSS) containing 137 mm NaCl, 5.3 mm KCl, 1.2 mm CaCl2, 0.8 mm MgSO4, 0.4 mm KH2PO4, 0.3 mm Na2HPO4, 5 mm glucose, and 10 mm 1,4-piperazinediethanesulfonate buffer, pH 7.2. Bovine erythrocyte superoxide dismutase type 2 (SOD) was prepared in BSS, and heat-inactivated SOD was prepared by heating to 90°C for 5 min. Solutions containing superoxide (KO2) were prepared immediately before use by adding 1 g of KO2 to 2 ml of DMSO to produce a saturated solution of KO2 (Reiter et al., 2000). The mixture was centrifuged at 345 × g to pellet the undissolved KO2, and 10 μl of the DMSO/KO2 solution was added to culture wells containing 400 μl of BSS. The superoxide anion was liberated in the aqueous solution to create an initial superoxide concentration of ∼40 μm in the medium (Hawkins et al., 2007).
Superoxide detection.
Cellular superoxide was assessed by evaluating the oxidation of dihydroethidium to fluorescent ethidium species (Eth) (Bindokas et al., 1996; Brennan et al., 2009) and by evaluating formation of 4-hydroxynonenal (4HNE) (Esterbauer et al., 1991; Uchida, 2003). For studies of Eth formation, 5 μm dihydroethidum was added to the BSS 15 min before the addition of KO2 or NMDA, and photographs were obtained 30 min after. In wild-type, nontransfected cultures, the photographs were obtained from three randomly selected fields per culture well. In p47phox−/− cultures transfected with GFP or p47phox-GFP, the 125 × 175 μm photographic fields were centered on GFP-expressing neurons randomly selected in each culture well (because there was negligible Eth or 4HNE signal distant from these cells). More than 850 cells were evaluated in each of the experimental conditions. Eth fluorescence images were obtained using 510–550 excitation and >580 nm emission filters. Neurons were considered Eth-positive when cell body fluorescence exceeded background fluorescence by 50% or more. In studies comparing degree of Eth fluorescence to distance from transfected process, the percentage change of Eth fluorescence in each neuronal soma was measured over the 30 min NMDA incubation period. For studies in astrocyte–neuron cocultures, Fura fluorescence was used to identify cell bodies and processes after loading with 4 μm Fura-2 AM for 25 min. Images were acquired with 380 nm excitation and >510 nm for Fura-2, interleaved with 535 nm excitation/610 nm emission for imaging of Eth.
Immunostaining for 4HNE was performed in cells fixed with 4% formaldehyde. After treatment with blocking buffer (2% goat serum, 0.2% Triton-X, 0.2% bovine serum albumin), cultures were incubated with 1:500 rabbit anti-4HNE (Abcam) overnight at 4°C. Fluorescent donkey anti-rabbit IgG (Invitrogen) was used to visualize primary antibody binding. Quantification and analysis of 4HNE staining were done the same way as for Eth fluorescence. Superoxide (or superoxide metabolites) was confirmed as the oxidant species by the absence of NMDA-induced Eth and 4HNE formation in p47phox−/− neurons, which lack functional NOX2.
Cell death.
Cultures used for cell death assays were washed free of NMDA or KO2 after 30 min, replaced in the 37°C incubator, and evaluated 24 h later. Dead cells were identified by their failure to exclude trypan blue (0.05%) or propidium iodide (1 μg/ml; Calbiochem) from cell soma. Photographs were taken of three randomly selected fields per well in wild-type cultures or three randomly selected fields containing GFP-positive neurons in the transfected cultures, and the total number of live and dead cells was counted in each photograph. More than 900 cells were evaluated in each of the experimental conditions.
Statistical analyses.
The n for each experiment denotes the number of independent culture preparations evaluated. The treatment conditions were evaluated in quadruplicate culture wells in each experiment. Data are presented as means ± SEM. Statistical significance was evaluated using one-way ANOVA with either Dunnett's test, for comparing multiple groups against a single experimental or control group, or the F statistic, for testing slope of a regression line.
Results
To determine whether activation of NMDA receptors in one neuron produces oxidative stress in neighboring neurons, we prepared cultures of p47phox−/− neurons, which are unable to form functional NOX2 complexes. The cultures were transfected at low density with GFP-tagged p47phox to reconstitute functional NOX2 in ∼10% of the neurons in each culture well. Subsequent incubation with NMDA induced formation of the lipid peroxidation product 4HNE exclusively in neurons located around the GFP-tagged, p47phox-expressing neurons. 4HNE formation was negligible in cultures not exposed to NMDA, and in control cultures transfected with GFP alone (Fig. 1A,B).
Figure 1.
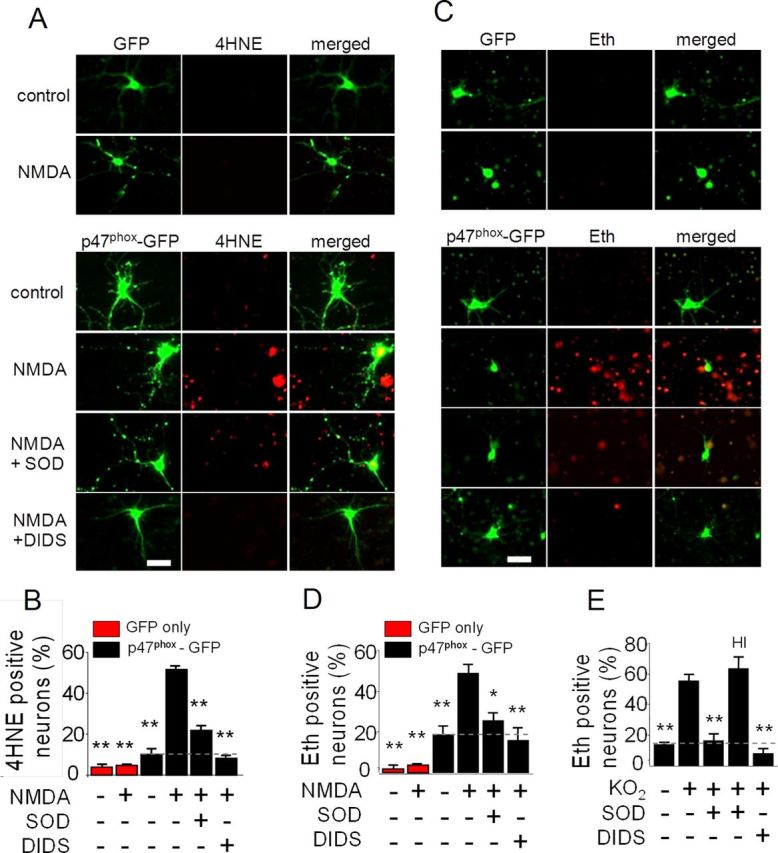
Activation of neuronal NMDA receptors produces superoxide-mediated oxidative stress in neighboring neurons. A, GFP expression and 4HNE immunostaining in neuron cultures. p47phox−/− neurons were transfected at low efficiency with GFP alone as controls (upper rows) or with GFP-labeled p47phox to reconstitute NADPH oxidase activity (lower rows). Merge images show 4HNE formation primarily in nontransfected neurons. Cultures were treated with 100 μm NMDA for 30 min or with medium exchanges alone. Cultures were additionally treated with SOD (1000 U) or DIDS (20 μm), where indicated. Scale bar, 50 μm. B, Quantification of 4HNE. **p < 0.01 versus NMDA-treated, p47phox-GFP transfected cultures (n = 3). C, GFP expression and oxidized Eth fluorescence in neuron cultures. Cultures were incubated with 5 μm dihydroethidium and otherwise treated as in A. Merge images show Eth formation primarily in nontransfected neurons. Scale bar, 50 μm. D, Quantification of Eth. *p < 0.05 and **p < 0.01 versus the NMDA-treated, p47phox-GFP transfected cultures (n = 4). E, Effects of DIDS and SOD on Eth fluorescence in neuron cultures assessed 30 min after treatment with exogenous superoxide (40 μm KO2). Cultures were treated with 20 μm DIDS, 1000 U SOD, or heat-inactivated SOD (HI) as indicated. **p < 0.01 versus KO2 alone (n = 4).
To confirm that this oxidative stress was caused by superoxide, studies were also performed in which SOD was added to the culture medium. The addition of SOD markedly reduced 4HNE formation (Fig. 1A,B). A similar reduction was observed with the anion blocker 4,4′-diisothiocyanostilbene-2,2′-disulphonate (DIDS), which blocks the entry of superoxide anion (O2·−) into cells through anion channels (Lynch and Fridovich, 1978; Hawkins et al., 2007).
Studies were additionally performed using the oxidation of dihydroethidium to Eth as a second, independent measure of oxidative stress. Results of these studies were quantitatively similar to the 4HNE studies: NMDA-induced Eth formation was observed only in the vicinity of neurons transfected with GFP-tagged p47phox, and the Eth signal in these surrounding neurons was reduced by the presence of either SOD or DIDS in the medium (Fig. 1C,D). Higher concentrations of SOD and DIDS produced no further reductions in Eth or 4HNE formation (data not shown). Exogenously added superoxide, in the form of KO2, was used confirm reliability of the superoxide probes and efficacy of SOD and DIDS in these preparations. Wild-type neurons exposed to KO2 showed increased Eth fluorescence, and this increase was completely blocked by DIDS and SOD at the concentrations used throughout these studies (Fig. 1E).
The likelihood that any given neuron would exhibit NMDA-induced oxidative stress in the transfected cultures increased with its proximity to a GFP-tagged, p47phox -expressing neuronal process, as quantified in Figure 2A. This relationship also held for the transfected cells themselves, of which only 28 ± 9% became Eth-positive. The anatomical relationships between transfected cells and oxidative stress were better seen with 4HNE staining, which unlike the Eth signal is not confined to the nucleus. Figure 2B shows an example of a transfected cell that remained 4HNE-negative after NMDA exposure while many nontransfected cells around it formed 4HNE. The high-power view of this panel shows GFP-positive processes of the neurons contacting cell bodies and processes of the 4HNE-positive neurons.
Figure 2.
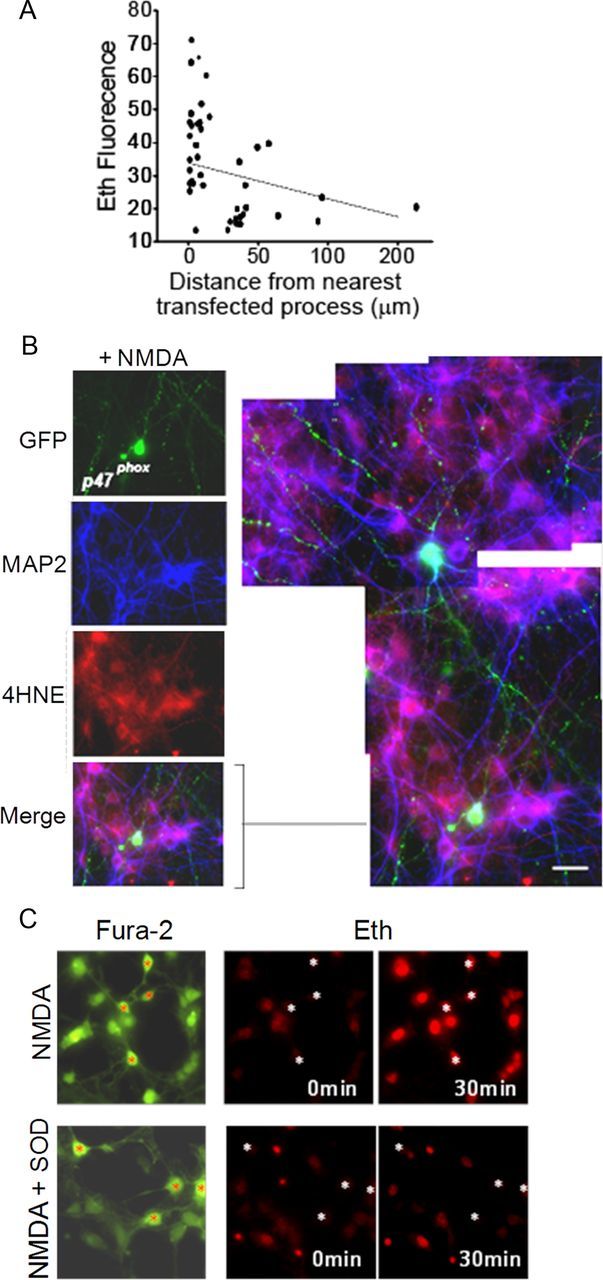
NMDA-induced oxidative stress correlates with proximity to NOX2-competent neurons. A, Scatter plot showing percentage change in neuronal soma Eth fluorescence versus distance to nearest visualized p47phox-positive neuronal process. Cultures prepared as in Figure 1A. Dashed line shows linear regression (r2 = 0.16, p < 0.01 versus a zero slope). B, Cultures prepared and treated as in Figure 1A, in which neuronal processes are labeled with MAP2 (blue). The expanded and enlarged view of the merged panel shows 4HNE-positve neuronal bodies and processes in proximity to processes of p47phox-reconstituted neurons. Scale bar, 10 μm. C, Cocultures of wild-type neurons and astrocytes in which Fura-2 (green) identifies cell bodies and processes, and Eth fluorescence (red) identifies nuclei of cells with oxidative stress. The angulated, process-bearing neuronal soma are identified in each panel by red or white asterisks. NMDA incubations (100 μm) produced oxidative stress in all of the neurons and astrocytes, and this effect was blocked in cultures co-incubated with SOD. Representative of n = 4 experiments, all with similar results.
We also evaluated NMDA-induced oxidative stress in neurons plated onto a confluent layer of astrocytes, given the importance of astrocytes in neuronal development and synapse formation (Pfrieger and Barres, 1997). Technical issues precluded neuronal transfection in these preparations, so studies were performed with wild-type neurons on astrocytes. In these cultures, NMDA produced oxidative stress in essentially all of the neurons and, interestingly, also in the underlying astrocytes (Fig. 2C). The presence of SOD blocked Eth formation in both the wild-type neurons and astrocytes (Fig. 2C).
To determine whether cell-to-cell movement of superoxide also contributes to cell death in excitotoxicity, p47phox−/− cultures were transfected with GFP-labeled p47phox as above, and treated with 100 μm NMDA for 30 min with or without the addition of SOD or DIDS to the medium. Neuronal death was evaluated 24 h later and was observed predominately in the vicinity of the transfected neurons with reconstituted NOX2 function (data not shown). The cell death was blocked by both SOD and DIDS, and transfection with p47phox did not significantly increase neuron death in the absence of NMDA exposure (Fig. 3A). NMDA-induced neuronal death in wild-type cultures was also reduced by both SOD and DIDS (Fig. 3B), thus excluding the possibility that neuronal death caused by superoxide release is an experimental artifact of transfection.
Figure 3.

NMDA-induced superoxide production causes death of neighboring neurons. A, p47phox−/− neurons were transfected with GFP alone (red bars) or with GFP-labeled p47phox (black bars) and treated as in Figure 1. Neuronal death in the vicinity of GFP-positive cells was evaluated 24 h later by propidium iodide staining. **p < 0.01 versus the NMDA-treated, p47phox-GFP transfected group (n = 3). B, Wild-type neurons were treated as in A, and neuronal death was evaluated by trypan blue staining 24 h later. **p < 0.01 versus the NMDA-treated group (n = 3).
Discussion
Superoxide generated by neuronal NOX2 contributes to neuronal death induced by glutamate, NMDA, ischemia, hypoglycemia, and other insults (Suh et al., 2007, 2008; Brennan et al., 2009; Girouard et al., 2009; Guemez-Gamboa et al., 2011; Yoshioka et al., 2011). However, it has not been resolved whether the superoxide generated by neuronal NOX2 produces oxidative stress selectively in the neurons in which it is formed, or predominantly in neighboring neurons. The former would be expected if the superoxide were produced by mitochondria or NOX2 within neurons, while the latter would be expected if the superoxide were produced by NOX2 on the cell surface and released into the extracellular space. Results presented here support the latter scenario. Neuronal NMDA receptor activation produced oxidative stress and death in neighboring neurons, and the cell-to-cell spread of both oxidative stress and death were blocked by SOD and DIDS.
Key to these studies was the use of cultures in which NOX2 competent neurons were scattered among p47phox−/− neurons that cannot assemble a functional NOX2 complex. Prior studies with cultures of uniformly p47phox−/− neurons showed that NMDA induces normal NMDA-induced calcium transients in these cells, but not oxidative stress or neuronal death (Brennan et al., 2009). Here, neurons lacking the p47phox subunit likewise displayed negligible NMDA-induced oxidative stress or cell death, unless they were near transfected, NOX2-competent neurons. The cell-to-cell transmission of oxidative stress was blocked by either SOD or DIDS, thereby confirming that the oxidant species is superoxide anion (or an anionic superoxide metabolite).
Surprisingly, the NOX2-positive neurons in the transfected cultures were often themselves unaffected by oxidative stress. Our findings do not provide an explanation for this, but the effects of DIDS in these studies suggests that the distribution of superoxide-permeable anion channels might be a crucial factor determining where superoxide can enter cells, and thus where superoxide-mediated oxidative stress occurs.
One limitation to the transfection approach is that neurons expressing GFP-labeled p47phox could manifest protein overexpression or nonphysiological localization of p47phox and superoxide production. However, p47phox is present in the cytosol until phosphorylated, and the sites of superoxide production are dictated by the location of membrane-tethered NOX2 catalytic subunits rather than by p47phox (Dang et al., 2001; Bedard and Krause, 2007). Additionally, cultures of nontransfected, wild-type neurons similarly exhibited NMDA-induced oxidative stress and cell death that were suppressed by the addition of SOD or DIDS to the medium.
A second limitation is that, for technical reasons, astrocytes were only sparsely present in the transfection preparations; a factor that could lead to aberrant neuronal maturation. However, cultures using wild-type neurons plated on astrocyte layers also exhibited NMDA-induced oxidative stress. Moreover, the astrocytes in these cultures exhibited SOD-suppressible oxidative stress, providing additional support for superoxide as an extracellular mediator.
Together, these studies show that the extracellular production of superoxide by NOX2 can contribute to the propagation of excitotoxic injury in cell culture. This injury is presumably dependent on concomitant production of nitric oxide, given that blocking production of either molecule blocks cell death (Lafon-Cazal et al., 1993; Dawson, 1995; Pacher et al., 2007). Unlike nitric oxide, superoxide has a very short half-life and diffusion distance in brain and does not readily cross lipid membranes (Pacher et al., 2007). The local production of superoxide could thus be a determining factor in the local propagation of excitotoxic injury. Although the physiological role of neuronal NOX2 has not been firmly established, evidence suggests that neuronal superoxide signaling contributes to the intercellular signaling required for synaptic plasticity (Massaad and Klann, 2011). The unique vulnerability of brain to excitotoxic and propagating cell death could thus be considered an exaggerated, pathological manifestation of this normal intercellular signaling process.
Footnotes
This work was supported by NIH Grant 2 RO1 NS041421 (R.A.S.) and the U.S. Department of Veterans Affairs. We thank Colleen Hefner for expert technical assistance.
References
- Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, Swanson RA. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. J Neurosci. 2010;30:2967–2978. doi: 10.1523/JNEUROSCI.5552-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Bindokas VP, Jordán J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. J Neurosci. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredt DS, Glatt CE, Hwang PM, Fotuhi M, Dawson TM, Snyder SH. Nitric oxide synthase protein and mRNA are discretely localized in neuronal populations of the mammalian CNS together with NADPH diaphorase. Neuron. 1991;7:615–624. doi: 10.1016/0896-6273(91)90374-9. [DOI] [PubMed] [Google Scholar]
- Brennan AM, Suh SW, Won SJ, Narasimhan P, Kauppinen TM, Lee H, Edling Y, Chan PH, Swanson RA. NADPH oxidase is the primary source of superoxide induced by NMDA receptor activation. Nat Neurosci. 2009;12:857–863. doi: 10.1038/nn.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi DW. Glutamate neurotoxicity and diseases of the nervous system. Neuron. 1988;1:623–634. doi: 10.1016/0896-6273(88)90162-6. [DOI] [PubMed] [Google Scholar]
- Dang PM, Fontayne A, Hakim J, El Benna J, Périanin A. Protein kinase C zeta phosphorylates a subset of selective sites of the NADPH oxidase component p47phox and participates in formyl peptide-mediated neutrophil respiratory burst. J Immunol. 2001;166:1206–1213. doi: 10.4049/jimmunol.166.2.1206. [DOI] [PubMed] [Google Scholar]
- Dawson VL. Nitric oxide: role in neurotoxicity. Clin Exp Pharmacol Physiol. 1995;22:305–308. doi: 10.1111/j.1440-1681.1995.tb02005.x. [DOI] [PubMed] [Google Scholar]
- Dugan LL, Sensi SL, Canzoniero LM, Handran SD, Rothman SM, Lin TS, Goldberg MP, Choi DW. Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-D-aspartate. J Neurosci. 1995;15:6377–6388. doi: 10.1523/JNEUROSCI.15-10-06377.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- During MJ, Symes CW, Lawlor PA, Lin J, Dunning J, Fitzsimons HL, Poulsen D, Leone P, Xu R, Dicker BL, Lipski J, Young D. An oral vaccine against NMDAR1 with efficacy in experimental stroke and epilepsy. Science. 2000;287:1453–1460. doi: 10.1126/science.287.5457.1453. [DOI] [PubMed] [Google Scholar]
- Dykens JA. Isolated cerebral and cerebellar mitochondria produce free radicals when exposed to elevated CA2+ and Na+: implications for neurodegeneration. J Neurochem. 1994;63:584–591. doi: 10.1046/j.1471-4159.1994.63020584.x. [DOI] [PubMed] [Google Scholar]
- Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- Faden AI, Demediuk P, Panter SS, Vink R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science. 1989;244:798–800. doi: 10.1126/science.2567056. [DOI] [PubMed] [Google Scholar]
- Forder JP, Tymianski M. Postsynaptic mechanisms of excitotoxicity: involvement of postsynaptic density proteins, radicals, and oxidant molecules. Neuroscience. 2009;158:293–300. doi: 10.1016/j.neuroscience.2008.10.021. [DOI] [PubMed] [Google Scholar]
- Garthwaite J. Concepts of neural nitric oxide-mediated transmission. Eur J Neurosci. 2008;27:2783–2802. doi: 10.1111/j.1460-9568.2008.06285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girouard H, Wang G, Gallo EF, Anrather J, Zhou P, Pickel VM, Iadecola C. NMDA receptor activation increases free radical production through nitric oxide and NOX2. J Neurosci. 2009;29:2545–2552. doi: 10.1523/JNEUROSCI.0133-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guemez-Gamboa A, Estrada-Sánchez AM, Montiel T, Páramo B, Massieu L, Morán J. Activation of NOX2 by the stimulation of ionotropic and metabotropic glutamate receptors contributes to glutamate neurotoxicity in vivo through the production of reactive oxygen species and calpain activation. J Neuropathol Exp Neurol. 2011;70:1020–1035. doi: 10.1097/NEN.0b013e3182358e4e. [DOI] [PubMed] [Google Scholar]
- Hawkins BJ, Madesh M, Kirkpatrick CJ, Fisher AB. Superoxide flux in endothelial cells via the chloride channel-3 mediates intracellular signaling. Mol Biol Cell. 2007;18:2002–2012. doi: 10.1091/mbc.E06-09-0830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesaitis AJ, Buescher ES, Harrison D, Quinn MT, Parkos CA, Livesey S, Linner J. Ultrastructural localization of cytochrome b in the membranes of resting and phagocytosing human granulocytes. J Clin Invest. 1990;85:821–835. doi: 10.1172/JCI114509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppinen TM, Swanson RA. Poly(ADP-ribose) polymerase-1 promotes microglial activation, proliferation, and matrix metalloproteinase-9-mediated neuron death. J Immunol. 2005;174:2288–2296. doi: 10.4049/jimmunol.174.4.2288. [DOI] [PubMed] [Google Scholar]
- Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature. 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- Lipton SA. Failures and successes of NMDA receptor antagonists: molecular basis for the use of open-channel blockers like memantine in the treatment of acute and chronic neurologic insults. NeuroRx. 2004;1:101–110. doi: 10.1602/neurorx.1.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch RE, Fridovich I. Permeation of the erythrocyte stroma by superoxide radical. J Biol Chem. 1978;253:4697–4699. [PubMed] [Google Scholar]
- Massaad CA, Klann E. Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid Redox Signal. 2011;14:2013–2054. doi: 10.1089/ars.2010.3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meldrum BS. Excitotoxicity and selective neuronal loss in epilepsy. Brain Pathol. 1993;3:405–412. doi: 10.1111/j.1750-3639.1993.tb00768.x. [DOI] [PubMed] [Google Scholar]
- Milnerwood AJ, Raymond LA. Early synaptic pathophysiology in neurodegeneration: insights from Huntington's disease. Trends Neurosci. 2010;33:513–523. doi: 10.1016/j.tins.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfrieger FW, Barres BA. Synaptic efficacy enhanced by glial cells in vitro. Science. 1997;277:1684–1687. doi: 10.1126/science.277.5332.1684. [DOI] [PubMed] [Google Scholar]
- Prosser BL, Ward CW, Lederer WJ. X-ROS signaling: rapid mechano-chemo transduction in heart. Science. 2011;333:1440–1445. doi: 10.1126/science.1202768. [DOI] [PubMed] [Google Scholar]
- Reiter CD, Teng RJ, Beckman JS. Superoxide reacts with nitric oxide to nitrate tyrosine at physiological pH via peroxynitrite. J Biol Chem. 2000;275:32460–32466. doi: 10.1074/jbc.M910433199. [DOI] [PubMed] [Google Scholar]
- Rothstein JD. Current hypotheses for the underlying biology of amyotrophic lateral sclerosis. Ann Neurol. 2009;65(Suppl 1):S3–S9. doi: 10.1002/ana.21543. [DOI] [PubMed] [Google Scholar]
- Sattler R, Xiong Z, Lu WY, Hafner M, MacDonald JF, Tymianski M. Specific coupling of NMDA receptor activation to nitric oxide neurotoxicity by PSD-95 protein. Science. 1999;284:1845–1848. doi: 10.1126/science.284.5421.1845. [DOI] [PubMed] [Google Scholar]
- Sorce S, Krause KH. NOX enzymes in the central nervous system: from signaling to disease. Antioxid Redox Signal. 2009;11:2481–2504. doi: 10.1089/ars.2009.2578. [DOI] [PubMed] [Google Scholar]
- Suh SW, Gum ET, Hamby AM, Chan PH, Swanson RA. Hypoglycemic neuronal death is triggered by glucose reperfusion and activation of neuronal NADPH oxidase. J Clin Invest. 2007;117:910–918. doi: 10.1172/JCI30077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh SW, Shin BS, Ma H, Van Hoecke M, Brennan AM, Yenari MA, Swanson RA. Glucose and NADPH oxidase drive neuronal superoxide formation in stroke. Ann Neurol. 2008;64:654–663. doi: 10.1002/ana.21511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida K. 4-Hydroxy-2-nonenal: a product and mediator of oxidative stress. Prog Lipid Res. 2003;42:318–343. doi: 10.1016/s0163-7827(03)00014-6. [DOI] [PubMed] [Google Scholar]
- Yoshioka H, Niizuma K, Katsu M, Okami N, Sakata H, Kim GS, Narasimhan P, Chan PH. NADPH oxidase mediates striatal neuronal injury after transient global cerebral ischemia. J Cereb Blood Flow Metab. 2011;31:868–880. doi: 10.1038/jcbfm.2010.166. [DOI] [PMC free article] [PubMed] [Google Scholar]