

Abstract
Antibody diversification requires the DNA deaminase AID to induce DNA instability at immunoglobulin (Ig) loci upon B cell stimulation. For efficient cytosine deamination, AID requires single-stranded DNA and needs to gain access to Ig loci, with RNA pol II transcription possibly providing both aspects. To understand these mechanisms, we isolated and characterized endogenous AID-containing protein complexes from the chromatin of diversifying B cells. The majority of proteins associated with AID belonged to RNA polymerase II elongation and chromatin modification complexes. Besides the two core polymerase subunits, members of the PAF complex, SUPT5H, SUPT6H, and FACT complex associated with AID. We show that AID associates with RNA polymerase-associated factor 1 (PAF1) through its N-terminal domain, that depletion of PAF complex members inhibits AID-induced immune diversification, and that the PAF complex can serve as a binding platform for AID on chromatin. A model is emerging of how RNA polymerase II elongation and pausing induce and resolve AID lesions.
In B cells, antibody diversity is created via two DNA instability mechanisms (Rajewsky, 1996). In the first, RAG1/2 mediate antigen-independent V(D)J recombination, and in the second, activation-induced deaminase (AID) drives antigen-dependent Ig diversification. The latter includes somatic hypermutation (SHM), Ig gene conversion (iGC), and class switch recombination (CSR). SHM and iGC induce variable (V) region diversification via templated and nontemplated DNA mutations (Di Noia and Neuberger, 2007), whereas CSR recombines DNA constant (C) switch regions, resulting in IgM to IgG, IgA, or IgE isotype switching (Stavnezer et al., 2008). Mechanistically, SHM, iGC, and CSR are initiated by the DNA deaminase AID, which deaminates cytosine (dC) residues to uracil (dU) on single-stranded DNA (ssDNA; Petersen-Mahrt, 2002, 2005; Bransteitter et al., 2003; Chaudhuri et al., 2003). At the genetic level, deamination causes a change in base recognition, as uracil is read as thymine during replication. At the biochemical level, reformation of double-stranded DNA (dsDNA) causes an alteration of DNA structure, resulting in a dU:dG lesion, which in turn activates DNA repair pathways resulting in mutated or otherwise altered chromosomes.
Because of the high oncogenic potential of AID, understanding how DNA deaminases are regulated at the target site is one of the most important aspects in the field of DNA editing and Ig diversification; however, little is known about the protein complexes and mechanisms involved. Mechanistically, AID requires ssDNA as a substrate, and although several chromatin alteration events could lead to ssDNA formation, transcription at the Ig locus is required for SHM and CSR. The rate of transcription correlates with the rate of SHM (Peters and Storb, 1996), and germline transcription through the switch and the constant region precedes CSR (Stavnezer-Nordgren and Sirlin, 1986). Interaction of AID with CTNNBL1 (Conticello et al., 2008; Ganesh et al., 2011) demonstrated an association with RNA processing. More recently, though, direct links between AID and mRNA transcription were demonstrated. It was shown that CSR required the basal transcription factor SUPT5H (Pavri et al., 2010) and its associated factor SUPT4H (Stanlie et al., 2012), the transcription-associated chromatin modifier FACT complex (Stanlie et al., 2010), and histone chaperon SUPT6H (Okazaki et al., 2011), whereas AID activity during CSR was enhanced by components of the RNA-processing exosome (Basu et al., 2011).
To delineate the biochemical link of RNA pol II transcription to AID-induced Ig diversification, and to further characterize the AID interactome, we developed a novel biochemical approach: we C-terminally tagged the endogenous AID protein in Ig diversifying cells with a FLAG or a FLAG/Myc epitope (Pauklin et al., 2009), and we adapted a recently developed method for isolation of chromatin-bound protein complexes (Aygün et al., 2008). This method allowed, for the first time, the identification and characterization of proteins that are associated with AID on chromatin in their physiological environment. The majority of the identified proteins (FACT complex, SUPT5H, SUPT6H, RNA polymerase-associated factor (PAF) complex, RPB1A, RPB1B, and DNA topo I) are involved in RNA processing, chromatin remodeling, exosome processing, and RNA pol II transcription elongation/pausing. We identified a direct interaction of AID (the N-terminal domain) with PAF1, and by using knockdown experiments, we could demonstrate physiological importance of the PAF complex for Ig class switching and recruitment of AID to the Ig locus. A model of how this complex could influence AID efficacy at the Ig locus will be discussed.
RESULTS
To determine the composition of the protein complexes that interact with AID on chromatin in B cells undergoing Ig receptor diversification, we developed cell lines in which endogenous AID was tagged with epitope-peptides at the C terminus (Pauklin et al., 2009). In the chicken B cell lymphoma DT40, which continuously undergoes AID-dependent diversification of the Ig locus, AID was tagged with either 3xFLAG peptides (3F) or the combination of 3xFLAG peptide, 2xTEV cleavage sites, and 3xMyc peptides (3FM). This yielded expression of tagged AID to levels that were comparable to endogenous amounts. Although it is known that the C terminus of AID plays an important role in subcellular localization, we could not detect a significant change in AID relocalization or immune diversification activity caused by the monoallelic C-terminal tags (unpublished data).
Chromatin AID is part of a multimeric complex
Because AID is predominantly localized in the cytoplasm (Rada et al., 2002; Brar et al., 2004; Ito et al., 2004; McBride et al., 2004), and only limited amounts can be identified within the nucleus, we grew 1–2 × 1010 AID-3FM cells for biochemical analysis. Cell lysates were subfractionated into cytoplasm, nucleoplasm, and chromatin fractions. We focused on chromatin-bound AID, which we estimated to be <2% of total AID-3FM (unpublished data). The isolated chromatin fraction was further separated using a Superdex 200 column for size exclusion chromatography (SEC), thereby determining the size of the AID-associated protein complex bound to chromatin (Fig. 1 a). AID was identified as part of a 200-kD protein complex (120–300 kD based on standard proteins), whereas only a minor fraction of AID eluted at its theoretical monomeric size of 27 kD (Fig. 1 a). This demonstrated that AID isolated from chromatin under physiological conditions is part of a large heteromeric complex.
Figure 1.

Chromatin-bound AID is in a multimeric complex. (a) Elution profile of size exclusion chromatography. The chromatin fraction from DT40 AID-3FM cells was loaded onto a Superdex 200 column. (top) The elution profile of standard proteins is plotted in the graph. Red circle, theoretical elution volume of AID-3FM (29 kD). (bottom) Eluted fractions (elution volume indicated, milliliters) were analyzed by Western blot probing for the presence of AID-3FM, PAF1, LEO1, CTR9, and CDC73. (b) Analysis of filtered peptide hits. The most abundant peptides identified were grouped using gene network software analysis, and groups are displayed in a pie chart as percentage of total peptides (75) that could be assigned to each complex. A full table of chromatin peptides is listed in Fig. S1.
The PAF complex associates with AID on chromatin in Ig diversifying cells
To identify proteins associated with chromatin-bound AID, we performed FLAG peptide immunoprecipitations (IPs), followed by one-dimension SDS-PAGE and mass spectrometry identification (Fig. S1). We obtained 1,319 peptide identities (Ids), corresponding to 391 proteins from AID-3FM cells. Mass spectrometric analysis of IPs from cytoplasmic, nucleoplasmic, and chromatin fractions of a control cell line (expressing AID without a tag) served as a control peptide Id database. Using this database, we eliminated 366 of the 391 proteins (>15-fold enrichment; all AID-interacting proteins are listed in Fig. S1). When we submitted the protein Ids into the Ingenuity Systems Pathway Analysis gene network software, we obtained a potential interacting network containing >80% of all isolated peptides (Fig. S2). The majority of the AID-associated proteins from the chromatin fraction were part of mRNA processing. Aside from the core RNA pol II subunits, we identified the core PAF complex (RNA polymerase-associated factor; PAF1, CTR9, LEO1), FACT complex (SSRP1, SUPT16H), SUPT5H, SUPT6H, and DNA topo I (Fig. 1 b). These factors play a direct role in RNA pol II pausing/restarting and elongation, as well as in chromatin modification and exosome processing. Furthermore, an additional 20 peptides comprised proteins involved in RNA metabolism (splicing-associated factors and RNA helicase). The high percentage (54%) of peptides that are part of the same biological process (early mRNA biogenesis), and which co-isolate with AID, indicated that our isolation and analysis procedure had identified key AID-interacting proteins at the chromatin level from immune-diversifying cells. Consistent with this, several of the proteins that we identified (RNA pol II, SUPT5H, SUPT6H, FACT complex, and DNA topo I) have been previously described to play a role in SHM and CSR. It is important to note that the chicken genome is not fully characterized and annotated, and thus the number of proteins we have identified may be underestimated.
Our mass spectrometry analysis of the AID chromatin interactome showed PAF1, CTR9, and LEO1 as AID-associated proteins on chromatin. They form part of the PAF complex, a RNA pol II–associated complex that promotes elongation (Kim et al., 2010) by recruiting enzymes for histone H2 monoubiquitination and other co-transcriptional chromatin marks (Jaehning, 2010). We could verify the associations of AID by analyzing the chromatin FLAG-IP for PAF1 (two different antibodies), LEO1, CDC73 (also known as HRPT2 or parafibromin), as well as confirm SUPT5H and SUPT6H (Fig. 2 a). The AID-association with SUPT5H (Fig. 2 a), although technically difficult, was further confirmed by multiple large scale FLAG immunopurification and mass spectrometry, in which SUPT5H association was identified in three out of five experiments (and SUPT6H and PAF1 were identified in each IP). In conclusion, our work has for the first time identified and verified AID-associated complexes on chromatin in diversifying B cells.
Figure 2.

PAF complex and RNA pol II–associated proteins coIP with AID in Ig diversifying cells. (a) Verification of DT40 AID-3F associations. DT40 chromatin fractions were immunoprecipitated via FLAG and eluted complexes were probed by Western-blotting: anti-PAF1 (two antibodies), anti-LEO1, anti-CDC73, anti-SUPT5H, anti-SUPT6H, anti-FLAG, and anti-AID antibodies. Input lysates from both cell lines are shown on the left (input); parental DT40 cell line (DT40) served as negative control for FLAG-IPs from chromatin fractions. The anti-AID antiserum was unable to detect chicken AID at physiological levels (not shown). (b) Nuclear extracts from CH12 cells stably expressing AIDFLAG-HA were immunopurified via anti-FLAG and the following controls: murine IgGs (mIgG) and rabbit IgG (rIgG). The isolated complex was probed with the indicated antibodies. Input represents 1% of material used for IP. Self (ctrl) refers to an IP using the indicated antibody on the left. ND, not determined. Molecular weight in kilodaltons are indicated. Data are representative of three independent experiments.
The PAF complex associates with AID in CSR-competent murine B cells
To determine whether the identified associations between AID and RNA pol II-associated factors observed in DT40 cells is also present in murine CSR-proficient cells, we performed a coIP experiment from nuclear extracts of CH12 B cells expressing tagged AID (AIDFlag-HA; Jeevan-Raj et al., 2011). Consistent with the DT40 analysis (Fig. 2 a), PAF1, LEO1, CTR9, SUPT5H, and SUPT6H could be identified to associate with AID (Fig. 2 b). Moreover in a reciprocal experiment, in which PAF1, LEO1, CTR9, SUPT5H, SUPT6H, and RNA polymerase II were precipitated, we identified AID in all IPs performed (unpublished data). This indicated that the identified AID associations were present in both DT40 and CH12 cells, thereby establishing a potential biochemical link between V region diversification (DT40 cells) and CSR (CH12 cells).
AID associates with the PAF complex via PAF1
To further characterize the PAF complex association with AID, we used immunoblot analysis of the chromatin SEC fractions (Fig. 1 a), and demonstrated that PAF1, LEO1, and CTR9 co-migrate in a large (>400-kD) complex (Fig. 1 a, lanes 1–3), with the peak trailing fractions overlapping with the AID peak (Fig. 1 a, lane 5). Although AID did not fully co-migrate in the same peaks, the data indicated that the classical PAF complex was present in DT40 and partially associated with AID on chromatin. It was therefore likely that AID interacted with one of the components of the PAF complex rather than with each individual member.
We coexpressed AID with individual PAF members in E. coli and monitored binding by coIP and Western blot analysis (Fig. 3 a). This approach avoided possible eukaryotic bridging proteins being present in the assay and was likely to identify direct interaction. The cloned (human) cDNAs were FLAG tagged and coexpressed with untagged human AID from the same plasmid. FLAG-PAF1 was co-isolated in AID immunoprecipitates, whereas CDC73 (Fig. 3 a), SSRP1 (not depicted), and LEO1 (not depicted) did not show robust association. The PAF1–AID association was specific (Fig. 3 a, lanes 4–6) and did not occur in the absence of AID-specific antisera (Fig. 3 a, lanes 7–9). A reciprocal IP experiment was also performed (unpublished data), verifying the AID–PAF1 association. To confirm the possible direct interaction between AID and PAF1, we performed classical pull-down analysis with recombinant AID and in vitro–produced PAF1. As shown in Fig. 3 b, PAF1 associated with AID but not APOBEC2, a member of the AID/APOBEC deaminase family. We also attempted to identify AID and SUPT5H association in the Escherichia coli and in vitro translation assays, but unlike the robust PAF1 association, were unable to demonstrate significant co-isolations (unpublished data).
Figure 3.

AID–PAF complex association via PAF1. (a) FLAG-PAF1 or FLAG-CDC73 were expressed alone (lanes 1, 4, 7, and 11) or from the same plasmid as AID (lanes 2, 3, 5, 6, 8, 9, 12, and 13) in E. coli. PAF1 is processed as a shortened (50 kD) fragment in bacteria. E. coli lysates were used for IP with anti-FLAG (lanes 1–3), anti-AID antibodies (lanes 4–6), or anti-MYC control antibody (lanes 7–9) and precipitates were analyzed in Western blots using anti-FLAG. AID expression in the lysates is shown in a separate blot on the right. Expressed protein and tag are indicated above lanes. (b) Pull-down assays were performed with 35S-labeled, in vitro–translated PAF1 and recombinant AID (lanes 1 and 2) or APOBEC2 (APO2; lanes 3 and 4) purified from E. coli. Pull downs were performed using anti-AID (lanes 1 and 3) or control antibody anti-Myc (lane 2 and 4) and analyzed on SDS-PAGE, followed by autoradiography. Assays performed with APOBEC2 protein (lanes 3 and 4) served as controls. A 10% Input is shown in lane 5. (c, left) PathDetect HLR cells transfected with GAL4-AID and GAL4-AIDE58Q were analyzed 30 h after transfection for transcription by luminometer (in triplicate). Data are representative of one of two independent experiments. (c, right) 30 h after transfection, chromatin was isolated and subjected to ChIP with anti-PAF1 or anti-LEO1 antibodies. Purified precipitated DNA was analyzed with qPCR (in triplicates), amplifying the 5′ part of the luciferase gene. Data are representing one of two independent experiments. Mock transfected cells (-) served as references, and the control IgG was set to 1. (d, top) A scheme representing the AID/APOBEC2 chimera. The numbers indicate the amino acids of AID that are replaced in each chimera by corresponding APOBEC2 amino acids. (d, bottom) Myc-tagged PAF1 was coexpressed with GFP-AID, GFP-APOBEC2, or GFP-AID/APOBEC2 chimera A–D (Conticello et al., 2008) in HEK293T cells. GFP-proteins were immunopurified (anti-GFP) and IPs were probed with anti-MYC and anti-GFP antibodies (middle and bottom). For input, 5% of the lysate used for IP was analyzed for MYC-PAF1 expression (top).
To demonstrate that the AID–PAF1 association can provide a functional consequence in mammalian cells, we used a transcription reporter assay. PathDetect HeLa luciferase reporter (HLR) cells harbor a luciferase transgene in their genome that can be activated by the PKA-phosphorylated CREB transcription factor. The presence of GAL4-binding sites (UAS) within the promoter allows for monitoring the effect of GAL4-fusion proteins on transcription. When GAL4 fusions of AID or AID mutant (E58Q) protein were transiently transfected, luciferase activity was enhanced nearly sixfold (Fig. 3 c, left). PAF1 and LEO1 chromatin IP (ChIP) of the transfected cells demonstrated that endogenous PAF1 and LEO1 were recruited to the locus upon AID expression. (Fig. 3 c, right), further underlying a more direct association between AID and the PAF complex.
Mapping the domain of AID that fostered this association was demonstrated by the use of AID-APOBEC2 chimeras, which substitute corresponding APOBEC2 peptide regions in place of AID peptide regions (Conticello et al., 2008). GFP-tagged AID, APOBEC2, or AID/APOBEC2 chimera proteins were coexpressed with Myc-peptide tagged human PAF1 in HEK293T cells and subjected to coIP. While IPs of AID and chimeras C and D showed co-purification of PAF1, APOBEC2 and chimera A and B failed to isolate PAF1 (Fig. 3 d), suggesting that the N-terminus of AID is responsible for the PAF1 association.
The PAF complex is required for functional CSR
Our finding that RNA pol II elongation factors associate with AID on chromatin, along with the previously established link of transcription being essential for SHM and CSR, provides an insight into the mechanism of AID activity at Ig loci. To determine the biological relevance of the PAF complex in CSR, we undertook knockdown experiments in murine B cells. CH12 cells were transduced with retrovirus-expressing shRNAs specific for the different subunits of the PAF complex. Transduced cells were stimulated in vitro, and their capacity to undergo CSR to IgA was determined by flow cytometry (Fig. 4, a and b). As controls we used shRNAs specific for AID and SUPT5H, together with a nontarget shRNA control. Knockdown efficiencies were determined by qRT-PCR (Fig. 4 c). Consistent with previous results (Pavri et al., 2010), we found that knockdown of AID and SUPT5H resulted in a significant reduction of CSR efficiency (Fig. 4, a and b). Knockdown of PAF1, LEO1, and CTR9 resulted in a similar reduction in the efficiency of CSR, which ranged from 31 to 35% (Fig. 4 b, gray bars), thus indicating the involvement of the PAF complex in CSR. No effects on viability, as determined by Topro-3 staining, were observed (not depicted). CDC73 depletion showed a reduction in CSR, but the change was not as significant as that of the other PAF complex members. To verify the retrovirus shRNA knockdown effects on the PAF complex and possibly enhance the efficacy, we developed a lentivirus-based system. Although the overall switching efficiency was reduced even in the control samples, the lentiviral caused effect was much more pronounced, with a LEO1 knockdown reducing switching by >70% (Fig. 4 b and not depicted). This enhanced CSR inhibition by LEO1, can be explained, in part, by the more pronounced reduction of the target mRNA (Fig. 4c). Importantly, although the knockdown did not lead to a complete loss of the target, biological changes in CSR were observed.
Figure 4.

Knockdown of PAF complex members impairs CSR. (a) CH12 cells were retrovirally transduced with shRNAs for AID, PAF1, LEO1, CTR9, CDC73, SUPT5H, or a respective nontarget. After stimulation, IgA surface expression was monitored by flow cytometry, representative plots are shown. Numbers within the FACS plots indicate the percentage of IgA-positive cells. (b) The mean percentage (+ SD) of CSR in stimulated cells that were RV-shRNA transduced (gray bars) or LV-shRNA transduced (black bars), relative to the nontarget shRNA control (white bars) set to 100% from three independent experiments, is shown. The difference (Δ) in CSR efficiency between nontarget and target shRNA knockdown is shown below. Significance of a decrease versus nontarget is indicated by the following p-values: *, P < 0.05; **, P < 0.01; ***, P < 0.001, based on Student’s t test. Quantitative RT-PCR for AID, PAF1, LEO1, CTR9, CDC73, and SUPT5H transcripts (c), μ germline transcript (d), α germline transcript (e), and AID transcripts from cells transduced with RV-shRNA (gray bars) or LV-shRNA (black bars) shown relative to the corresponding nontarget shRNA control (f; white bars). Results are from three independent experiments. Transcript Ct values were normalized to CD79b or HPRT mRNA abundance and are presented relative to the levels in the nontarget shRNA negative control, set to 1. *, P < 0.05; **, P < 0.01; ***, P < 0.001, based on Student’s t test.
As the PAF complex is part of the RNA pol II transcription machinery, the knockdown of its individual subunits could have broader influences on the cell than just altering AID’s function at the IgH locus during CSR. We thus monitored the effect of knockdown on switch region transcription and AID expression. Although transcription at the donor switch region was not affected by the knockdown of any of the PAF complex subunits (Fig. 4 d), we found that knockdown of PAF1 and CTR9 resulted in altered levels of germline transcription at the acceptor switch region (Fig. 4 e). Furthermore, knockdown of PAF1, CTR9, CDC73, and SUPT5H resulted in a significant reduction in the level of AID mRNA (Fig. 4 f). Importantly, however, knockdown of LEO1 did not reduce AID mRNA expression (Fig. 4 f), nor reduce the levels of germline transcripts (Fig. 4, d and e), yet CSR was significantly reduced (Fig. 4, a and b); a finding that was confirmed with the lentivirus system. Because reduction in the expression of mismatch repair and base excision repair proteins, like UNG and MSH2/MSH6, could also explain the observed reduction in CSR, we monitored their expression level (by qRT-PCR) after knockdown of AID, PAF1, and LEO1. We were unable to identify any significant changes in mRNA levels (unpublished data).
Reducing the expression of the PAF complex proteins induced a loss in CSR, thereby identifying the PAF complex as a key component during Ig diversification. The observation that the core PAF protein LEO1 knockdown reduced CSR threefold, whereas not altering the expression of key transcript units, indicated that the PAF complex (or at least LEO1) plays a direct role in regulating AID function at the chromatin target.
PAF is present on the functional Ig allele of DT40 independently of AID
As a complex associated with active transcription, the PAF complex is present on numerous genes. To determine whether the PAF complex is recruited to an active Ig locus, we performed ChIP from DT40 chromatin using antibodies specific for PAF1 and LEO1 (Fig. 5). As in most B cells, in DT40 there is a strong allelic exclusion bias with only one of the two Ig light chain (lambda) alleles being active. By designing specific primers for the active (R, rearranged) and inactive (UR, unrearranged) allele (Fig. 5, schematic), we could identify PAF1 and LEO1 to be specifically located at the active allele. The PAF1 and LEO1 occupancy near the C domain (which is present on both alleles) was analogous to that of the previously described SUPT5H, and indicated a presence of the PAF complex outside of AID-targeted regions. This also led us to investigate if AID presence was necessary for PAF complex presence at the Ig locus, and we performed the same ChIP in AID-deficient DT40 cells (Harris et al., 2002). We found that PAF1 and LEO1 occupancy at the rearranged allele was not disrupted, and was even increased, by AID-deficiency (Fig. 5), indicating an AID-independent function for the loading of the PAF complex proteins to Ig loci. We conclude that the PAF complex could serve as a binding platform for AID.
Figure 5.

PAF complex presence at the active Ig allele independent of AID. DT40 ChIP was performed as described in the Materials and methods section. (top) Schematics of the R (rearranged) and UR (unrearranged) lambda alleles in DT40 cells. PCR amplifications are shown as lines: 1, rearranged V/J region allele; 2, 3, and 4, unrearranged V/J region allele; 5, both C region alleles. WT, parental DT40; aid −/−, AID KO has previously been described (Harris et al., 2002). ChIP was performed using anti-PAF1 (gray bars) and anti-LEO1 (white bars), and qPCR results (in triplicate) were compared with IgG control ChIP (black bars set to 1). One of two representative experiments is shown.
AID presence at Sμ is impaired by LEO1 knockdown
If the PAF complex can serve as a site for AID association at Ig loci, then reducing PAF expression should alter AID’s occupancy at an Ig locus. To determine whether AID recruitment to the Sμ switch region is dependent on LEO1, ChIP experiments using an anti-AID antibody (Pavri et al., 2010) on unstimulated or stimulated transduced CH12 cells were performed (Jeevan-Raj et al., 2011). Before analysis, the cells had been transduced with a lentivirus expressing a shRNA specific for LEO1, AID, or a nontarget and were sorted for enhanced GFP expression. AID occupancy at the Sμ switch region was significantly reduced in LEO1 knockdown cells when compared with the nontarget control shRNA (Fig. 6). AID-ChIP signal was specific, as there was no significant difference in AID occupancy between unstimulated CH12 cells (not expressing AID) and stimulated cells expressing an shRNA specific for AID. We conclude that AID binding to Sμ is impaired by LEO1 knockdown. This result indicates that the functional mechanism of the PAF complex (at least LEO1) is to allow for AID to reside at an Ig locus during immune diversification.
Figure 6.
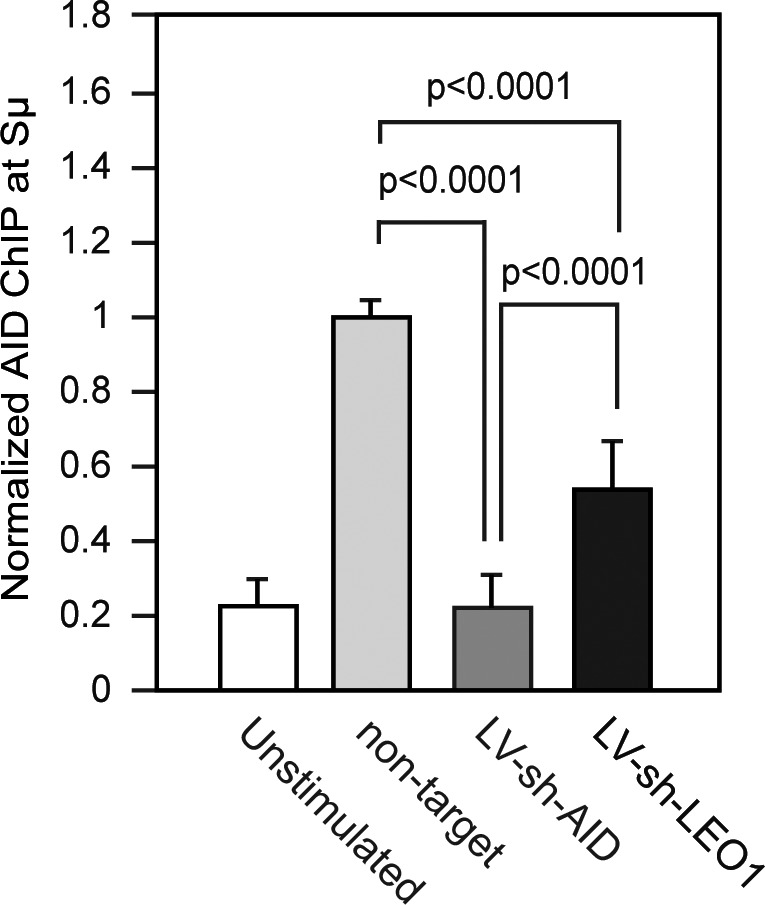
AID binding to Sμ is impaired after LEO1 knockdown. Unstimulated and/or stimulated CH12 cells were transduced with a lentivirus expressing shRNAs specific for AID, LEO1, or a nontarget shRNA control. AID occupancy at the Sμ switch region was ChIP analyzed using anti-AID antibodies. Normalized AID-ChIP data from three independent experiments assayed with two different primer sets is shown. For each sample, AID-ChIP values were normalized to the input control. AID-ChIP signal in cells expressing a nontarget shRNA control was set to 1. P-values, two-tailed Student’s t test.
DISCUSSION
Transcription has long been associated with AID-induced immune diversification. Early transgenic work demonstrated that the removal of the Ig promoter or enhancer elements abolished SHM (Betz et al., 1994). Mutation distribution across the V region of Ig genes indicated that AID-induced mutations are initiated 100–150 bp downstream of the transcription start site (TSS), and continue for ∼1,500–2,000 bp (Rada et al., 2002). Recent work has identified a similar AID-induced mutation profile across non-Ig genes (Liu et al., 2008), although the extent and frequency of SHM on these non-Ig genes was much more restricted. This indicated that although transcription is crucial, location and chromatin configuration also play a significant role, whereas sequence alone does not.
Several AID-associated proteins have been identified, some of which are linked directly to RNA processing (Conticello et al., 2008; Pavri et al., 2010; Stanlie et al., 2010; Basu et al., 2011; Okazaki et al., 2011), whereas others are important for subcellular localization (Patenaude et al., 2009; Maeda et al., 2010) or substrate accessibility (Chaudhuri et al., 2003). After fractionating B cells undergoing Ig diversification, we focused on the chromatin-bound AID and its physiological interactome (Fig. 1), which consisted of RNA pol II core (RNA pol II sub unit 1A and 2A) and associated proteins (SUPT5H), splicing factors (SF3A and 3B, Prp6, PrP4), RNA helicases, chromatin modifiers (SUPT6H, SSRP1 and SUPT16H), and an RNA pol II elongation complex (PAF complex; PAF1, LEO1, CTR9, CDC73). We verified these associations in DT40 and CH12F3 cells (Fig. 2, a and b), and demonstrated that PAF1 was the likely AID-interacting subunit within the PAF complex (Fig. 3). The biological significance of the AID–PAF complex association was shown by LEO1 knockdown in induced CH12 cells, where we observed reduced CSR without reducing AID or Ig transcript levels (Fig. 4). Mechanistically, at the Ig locus, the presence of the PAF complex (Fig. 5) enhanced AID occupancy (Fig. 6).
Transcription-coupled AID function
Genetically, transcription has been linked to SHM and CSR (Stavnezer-Nordgren and Sirlin, 1986; Peters and Storb, 1996), whereas an AID RNA pol II association has subsequently been implicated (Nambu et al., 2003). During SHM and CSR, mutations do not occur until after promoter escape (>100 bp downstream of the start site), and because of this the processing of RNA is likely a mechanistic link to AID activity. This was confirmed by the discoveries of an association between the following: AID and CTNNBL1, a protein of the splicing machinery, which occurs concomitantly during RNA pol II elongation (Conticello et al., 2008); AID and PTBP2, a splicing protein (Nowak et al., 2011); AID and SUPT5H, a protein known to associate with paused and elongating RNA pol II (Pavri et al., 2010); AID and SUPT4H, a factor known to associate with SUPT5H (Stanlie et al., 2012); AID and SUPT6H, a histone chaperone (Okazaki et al., 2011); CSR and SET1, a methyl-transferase for H3K4me3 (Stanlie et al., 2010); CSR and the FACT complex, a chromatin-modifying complex during RNA processing (Stanlie et al., 2010). Because of the involvement of the various RNA biogenesis and chromatin modification proteins in AID-induced Ig diversification, one cannot exclude the possibility that some of these factors serve multiple roles in directly controlling AID at the Ig locus, in changing the chromatin state of the Ig locus through the regulation of key factors, and in influencing the pathway and resolution of AID-lesions based on altered chromatin states.
The RNA pol II C-terminal domain (CTD) tail, which is temporally and spatially modified, serves as a platform for co-transcriptional mRNA maturation and chromatin modification. The PAF complex helps to set the right co-transcriptional chromatin marks, itself serving as a docking platform for the H2B ubiquitination machinery, as well as for setting H3K4me3 marks (Jaehning, 2010). H3K4me3 serves as an important mark in CSR (Wang et al., 2009; Stanlie et al., 2010), but is generally restricted to the 5′ end of a gene, and replaced by H3K36me3 toward the 3′ end of the gene. Both of these marks are induced upon transcriptional activation of S-regions (Wang et al., 2009), but at these loci, the H3K4me3 domain is extended, whereas onset of H3K36me3 is pushed back toward the 3′ end. This correlates roughly with the cease of mutational load/AID activity in C regions (Wang et al., 2009). Our ChIP data in DT40 confirm that the machineries required to set the various marks are also skewed along the transcription unit during Ig diversification (Fig. 5). This data also confirms that occupancy by AID-associated factors does not equate to AID occupancy, given that the gross SUPT5H and RNA pol II occupancy profile is not altered for several hundred base pairs, extending into the C region (Pavri et al., 2010), and not all stalled genes are target for AID binding or mutation (Yamane et al., 2011). Furthermore, AID has been associated with TSS of non-Ig genes (Yamane et al., 2011), yet no functional relevance (e.g., AID-induced mutations) has been identified at these locations. Therefore, the current data of linking the early transcriptional events to AID association provides further insight into the establishment of 5′ boundary-marks of SHM, whereas the understanding of molecular mechanism for the 3′ boundary remains less clear.
Overall, our work now provides the biochemical (and physiological) foundation for the aforementioned AID associations, while at the same time providing the molecular link (PAF complex) between early transcription elongation, marked by SUPT5H/SUPT4H, and downstream extended chromatin modifications dependent on FACT (SSRP1 and SUPT16H), SET1, and SUPT6H (Pavri et al., 2006; Fleming et al., 2008; Chen et al., 2009; Jaehning, 2010; Selth et al., 2010). A possible order of events at the Ig locus (Fig. 7) would entail the following: RNA pol II pausing after promoter escape and phosphorylation of its CTD tail; binding of the SUPT4H–SUPT5H complex to RNA pol II; recruitment of PAF complex to the holocomplex and initiation of histone modifications near the pause site (H2B mono-ubiquitination by the BRE1/RAD6 complex serves as a platform for SET1 complex for H3K4 trimethylation) and phosphorylation of CTD and SUPT4H–SUPT5H complex by pTEFb; concomitant association of AID to the PAF–SUPT5H–RNA pol II complex, FACT complex recruitment and chromatin remodeling, SUPT6H association to the restarting polymerase; elongating/pausing transcription for enhanced AID resident time at Ig locus, RNA biogenesis, opening of chromatin and DNA for AID accessibility, recruitment of DNA repair factors to initiate SHM and CSR; hyperphosphorylation of the CTD, loss of AID association, and completion of RNA synthesis.
Figure 7.

Model of the AID-linked transcriptional events at the Ig locus. Factors not currently identified to associate with AID or SHM/CSR are in white circles; other factors are indicated in the legend.
As mentioned above, several of the proposed proteins have been demonstrated to either associate with AID and/or play a role during Ig diversification. The identification of the nucleosome modifiers SUPT6H and FACT at the Ig locus, the demonstration that histone H3K4 trimethylation is necessary for CSR (Stanlie et al., 2010), and the correlation of H2Bser14 phosphorylation (Odegard and Schatz, 2006), H4K20 methylation (Schotta et al., 2008), H3 acetylation (Kuang et al., 2009; Wang et al., 2009), and H3K9 trimethylation (Chowdhury et al., 2008; Jeevan-Raj et al., 2011; Kuang et al., 2009; Wang et al., 2009) with Ig diversification indicates that the interplay of transcription and chromatin modification during AID-induced Ig diversification, although complex, is beginning to be unraveled. Although our data suggest that the predominant function of the PAF complex during SHM is to provide a site for AID association, we cannot exclude the possibility that reduced PAF activity also alters nucleosome marks needed for the resolution of AID-induced lesions, but more detailed future analysis may.
SHM versus CSR
The AID–PAF complex and AID-SUPT5H interactions were isolated from DT40 cells, which undergo SHM as well as gene conversion, but do not undergo CSR. Past work has implicated histone modification during SHM, but detailed understanding is still lacking, whereas H3K4me3 seems to play an important role during CSR (Stanlie et al., 2010). Our isolation of most of the required components for setting this mark during transcription would imply a similar requirement during V region diversification. Furthermore, we also identified PAF interactions from cells undergoing CSR. On the other hand, there have been indications that SUPT6H (Okazaki et al., 2011), SUPT4H (Stanlie et al., 2012), and the FACT components (Stanlie et al., 2010) have different functionality during SHM and CSR, but detailed analysis from knock-outs and the endogenous SHM or CSR loci need to confirm the exact mechanisms.
Conclusion
Our work has provided biochemical and genetic insight into understanding the association of AID to the Ig locus. Our novel approach to isolate physiological AID-containing protein complexes only from chromatin has identified a new component, the PAF complex, as well as biochemically verified the significance of previously identified complexes (SUPT5H, SUPT6H, and FACT) in AID biology. Furthermore, our data extends the current model of AID gaining access to DNA by stalled RNA polymerase II to a more complex model, where AID is intimately and specifically linked with RNA pol II in the phase of pausing and elongation, surrounded by a specific chromatin environment defined by histone modification cascades.
The finding that AID interacts with the PAF and the RNA pol II elongation complexes is somewhat reminiscent of a model put forth by Peters and Storb (1996), where an unknown mutator (now known to be AID) would bind to initiating RNA pol II and travel along with the machinery during transcription elongation.
MATERIALS AND METHODS
Plasmids, cell lines, and antibodies.
Plasmids were constructed using standard PCR and molecular biology techniques; sequences are available upon request. Tagging AID exon 5 in DT40 has been previously described (Pauklin et al., 2009), with the following modification: instead of a 3xFLAG-2xTEV-3xMyc tagged AID construct (AID-3FM), we also generated AID-3F (3xFLAG). Expression plasmids for GFP-AID, GFP-Apobec2, and GFP-AID/Apobec2 chimera A-D were obtained from the Neuberger Laboratory (Conticello et al., 2008). A CMV promoter-driven MYC-PAF1 expression vector was obtained by cloning the human PAF1 cDNA into pcDNA™3.1/myc-His (Invitrogen). For a complete list of antibodies used in this study please see Table S3.
Chromatin AID-3FM and AID-3F isolation.
Isolation was based on a previously described method (Aygün et al., 2008), with modifications. 1–2 × 1010 DT40 cells (Pauklin et al., 2009) were collected by centrifugation at 1,200 rpm 4°C for 10 min, and cell pellets were washed twice with 50 ml cold 1xPBS. Cytoplasmic lysis: 5 times packed cell volume (∼1 µl PCV = 106 cells) of Hypotonic Lysis Buffer (HLB; 10 mM Tris HCl [pH 7.5], 2 mM MgCl2, 3 mM CaCl2, and 0.32 M sucrose, protease inhibitor cocktail [Roche], and phosphatase inhibitor cocktail [Roche]) was added to the cell pellet, resuspended gently, and incubated for 12 min on ice. To the swollen cells, 10% Triton X-100 was added to a final concentration of 0.3%. The suspension was mixed and incubated for 3 min on ice, centrifuged for 5 min at 1,000 g at 4°C, and the supernatants (cytoplasmic fraction) were collected. Nuclear pellets were washed once in HLB + 0.3% Triton X-100, resuspended in 2xPCV LB-T (LB - 50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 50 mM KCl, 2 mM MgCl2, 1 mM EDTA, 10% Glycerol, protease inhibitor cocktail [Roche], phosphatase inhibitor cocktail [Roche], and 0.3% Triton X-100), and dounce homogenized with 30 strokes. The samples were incubated with gentle agitation for 30 min at 4°C and ultracentrifuged at 33,000 g for 30 min at 4°C. The pellets were dounce homogenized until resistance was lost in 2xPCV LB-TB (LB-T + 150 U/ml Benzonase [VWR International]). The samples were incubated at room temperature for 30 min and ultracentrifuged as before. The supernatants (chromatin fraction) were subjected to a preclearing step with agarose beads before adding M2-affinity beads (Sigma-Aldrich) for IP. For PAF complex analysis, NaCl and KCl were doubled to give a final concentration of 300 mM, and the Triton X-100 concentration increased to 0.5%. For Western analysis of input, lysates between 0.5 and 3% of total lysate was loaded per lane.
Size exclusion chromatography.
Chromatin extract of DT40 was prepared as described above. 1 ml of extract was loaded onto a Superdex 200 10/300 GL column, which had been equilibrated in LB and calibrated with standard proteins using Äkta Explorer (GE Healthcare). Fractions were collected at 1-ml volume steps using a 0.5 ml flow-rate, concentrated, and analyzed by Western blot.
FLAG-IP.
Anti-FLAG M2 affinity beads (Sigma-Aldrich) were washed and equilibrated in LB-T. For chromatin fractions, 100 µl of M2 beads per 5 × 109 DT40 cells were incubated for 3–4 h with the chromatin at 4°C on a rotator and collected for 3 min at 300× g at 4°C. Beads were washed 5 times in 25× bead volume of LB-TF (LB-T supplemented with 0.5–1 µg/ml 1xFLAG peptide N-DYDDDDK-C) and once with LB at 4°C for 10 min, followed by two elution steps in 4× bead volume of EB (LB + 500 µg/ml 3xFLAG peptide N-MDYKDHDGDYKDHDIDYKDDDDK-C); first for 1 h at room temperature, and then over night at 4°C.
Mass spectrometry.
Polyacrylamide gel slices (1–2 mm) containing IP-purified proteins were prepared for mass spectrometric analysis using the Janus liquid handling system (PerkinElmer). In brief, the excised protein gel pieces were placed in a well of a 96-well microtiter plate and de-stained with 50% vol/vol acetonitrile and 50 mM ammonium bicarbonate, and then reduced with 10 mM DTT and alkylated with 55 mM iodoacetamide. After alkylation, proteins were digested with 6 ng/µL trypsin (Promega) overnight at 37°C. The resulting peptides were extracted in 2% vol/vol formic acid, 2% vol/vol acetonitrile. The digest was analyzed by nano-scale capillary LC-MS/MS using a nanoAcquity UPLC (Waters) to deliver a flow of ∼300 nL/min. A C18 Symmetry 5 µm, 180 µm × 20 mm µ-Precolumn (Waters), trapped the peptides before separation on a C18 BEH130 1.7 µm, 75 µm × 100 mm analytical UPLC column (Waters). Peptides were eluted with a gradient of acetonitrile. The analytical column outlet was directly interfaced via a modified nano-flow electrospray ionization source, with a linear quadrupole ion trap mass spectrometer (LTQ XL/ETD, Thermo Fisher Scientific). LC-MS/MS information was collected using a data dependent analysis procedure. MS/MS scans were collected using an automatic gain control value of 4 × 104 and a threshold energy of 35 for collision induced dissociation. LC-MS/MS data were then searched against a protein database (UniProt Knowledge Base) using the Mascot search engine program (Matrix Science; Perkins et al., 1999). Database search parameters were set with a precursor tolerance of 1.0 D and a fragment ion mass tolerance of 0.8 D. One missed enzyme cleavage was allowed and variable modifications for oxidized methionine, carbamidomethyl cysteine, phosphorylated serine, threonine and tyrosine were included. MS/MS data were validated using the Scaffold program (Proteome Software, Inc.; Keller et al., 2002). All data were additionally interrogated manually.
Western blotting.
The antibodies used are shown in Table S3. Samples were prepared using standard procedures. Proteins were fractionated using NuPage Bis-Tris gels (Invitrogen) or homemade 10% PAA gels and transferred to Immobilon-P membranes (Millipore).
Nuclear extracts and coIP in murine B cells.
Nuclear extracts were prepared using standard techniques from CH12F3 cells stably expressing AIDFLAG-HA (Jeevan-Raj et al., 2011). coIPs and Western blot analyses were performed as previously described (Jeevan-Raj et al., 2011). For antibodies used, please see Table S3.
In vitro translation and coIP.
AID-His–tagged protein was expressed in E. coli and purified as previously described (Coker et al., 2006). 35S-labeled PAF1 was expressed using the TnT T7 Coupled Reticulocyte Lysate System (IVT) according to the manufacturer’s instructions (Promega). Labeled protein mixture was mixed with 100 ng of AID or 300 ng of APOBEC2 protein for 1 h at room temperature and for 30 min at 4°C. Proteins were isolated by anti-AID (hAnp52-1; Conticello et al., 2008) or anti-Myc (9E10) coupled to Sepharose beads for 1 h at 4°C, washed 5 times in 1× TBS-T (50 mM Tris, pH 8.0, 300 mM NaCl, 1% Triton X-100, 2.5 mM TCEP, 2% BSA, and protease inhibitor [Roche]), resuspended in SDS-PAGE loading buffer, and separated on 12% Bis-Tris polyacrylamide gels (Invitrogen). Gels were dried, exposed, and analyzed using a Fuji Imaging system.
E. coli coIP.
cDNAs of PAF1 and CDC73 were fused to a C-terminal FLAG tag in a pET DUET derivative coexpressing untagged human AID. Plasmids were transformed into BL21-CODONPLUS (DE3)-RIL cells (Stratagene), and protein expression was induced at 16°C with 1 mM IPTG in the presence of 0.1 mM ZnCl2 (3 h). Cells were sonicated in TBS-T, debris were pelleted at 19,000 g, and IPs were performed using Sepharose-coupled anti-AID hAnp52-1 or anti-FLAG M2 antibodies. After five washes with TBS-T, the immunoprecipitates were analyzed by Western blot, using polyclonal anti-AID (Abcam) and monoclonal anti-FLAG (M2-HRP; Sigma-Aldrich) antibodies.
HeLa PathDetect analysis.
The Stratagene PathDetect HLR Cell Line and GAL4-CREB and PKA expression vectors were purchased from Agilent. This HeLa-based Luciferase Reporter cell line contains a single locus with integrated synthetic minimal promoter and five yeast GAL4-binding sites (UAS) driving expression of the luciferase gene. Plasmids expressing GAL4-CREB, PKA, and GAL4-AID were transfected using Lipofectamine2000 (Invitrogen), and luciferase activity (in triplicate) was monitored 24–48 h after transfection according to the Luciferase Assay System manual (Promega). ChIP analysis using anti-PAF1, anti-LEO1, and a control IgG were done as follows: cells were cross-linked with 1% formaldehyde, nuclei were isolated and lysed in sonication buffer (1% SDS, 50 mM Tris HCl, pH 8.0, and 10 mM EDTA). After sonication in a BioRuptor, fragmented chromatin was diluted and incubated with antibodies or control IgGs over night. Collected protein–DNA complexes were purified and analyzed by qPCR (in triplicate). ChIP data were normalized to the input signal for each chromatin sample, and control ChIPs were set to 1. For antibodies used, please see Table S3. For oligonucleotides used, please see Table S1. Two independent experiments were performed with one representative shown.
coIP in HEK293T cells.
HEK293T cells were transfected with a plasmid expressing MYC-PAF1. 12 h after transfection cells were pooled to guarantee equal expression of MYC-PAF1, and split to allow a second transfection (12 h later) with expression plasmids for GFP-AID, GFP-APOBEC2, or GFP-AID/APO2 chimera. 24 h after second transfection, cells were lysed (lysis buffer: 50 mM Tris HCl, pH 8.0, 150 mM NaCl, 0.04% SDS, 1% NP-40), and GFP-protein expression in the lysates was estimated by scanning aliquots of a dilution series of the lysates with a Typhoon Scanner. Equal GFP and protein amounts were subjected to IP with anti-GFP at 4°C on, immunoprecipitates were collected with protein-A/G–Sepharose beads, and beads were washed and analyzed by Western blotting.
shRNA knockdown.
Retroviral knockdown was done as follows: vectors containing shRNAs specific for SUPT5H, PAF1, LEO1, CTR9, CDC73, and the nontarget shRNA control were purchased from OriGene (Table S2). The hairpin sequence for AID (5′-ACCAGTCGCCATTATAATGCAA-3′) was cloned into the LMP retroviral vector (Open Biosystems). CH12 cells were transduced as previously described (Barreto et al., 2003). Transduced cells were selected with 0.5 µg/ml puromycin for 1–5 d before induction and sorting. Lentiviral knockdown was done as follows: The hairpin sequences for AID, PAF1, LEO1, and the nontarget shRNA control were cloned into the pLKO.1-puro-CMV-TurboGFP lentiviral vector (Sigma-Aldrich). Lenti-X 293T cells (Takara Bio Inc.) were transfected with vectors to produce the virus. 2 d later, CH12F3 cells were spin-infected with viral supernatants supplemented with 10 µg/ml polybrene (Santa Cruz Biotechnology). Cells were selected for 5 d with 1 µg/ml puromycin before induction. Hairpin sequences used are listed in Table S2.
Cell culture and flow cytometry.
Retrovirally transduced CH12 cells were cultured with 5 ng/ml IL-4 (Sigma-Aldrich), 0.3 ng/ml TGF-β (R&D Systems), monoclonal 200 ng/ml anti-CD40 antibody (eBioscience), and 0.5 µg/ml puromycin and analyzed after 48–72 h for CSR (IgM to IgA) by flow cytometry, as previously described (Robert et al., 2009).
Real-time quantitative RT-PCR.
RNA and cDNA were prepared using standard techniques. qPCR was performed in triplicates using SYBR Green JumpStart Taq ReadyMix (Sigma-Aldrich) and a LightCycler 480 (Roche). Transcript quantities were calculated relative to standard curves and normalized to CD79b or HPRT mRNA. For primers see Table S1.
ChIP from DT40 and CH12 cells.
In brief, DT40 cells were treated and analyzed as for the ChIP in the HeLa PathDetect analysis section. For antibodies used please see Table S3. For oligonucleotides used please see Table S1. Two independent experiments were performed, with one representative shown. For quantitative AID-ChIP from shRNA knockdowns: CH12 cells were transduced with a lentivirus expressing shRNAs specific for AID, LEO1 and a nontarget control. Cells were stimulated for 48 h and sorted for enhanced GFP expression using a FACS Aria II (BD) and/or FACSVantage SE (BD) cell sorters before ChIP analysis. Cells were cross-linked with 1% formaldehyde for 10 min. Chromatin was prepared and immunoprecipitated with an anti-AID antibody (Pavri et al., 2010) and analyzed by quantitative PCR as previously described (Jeevan-Raj et al., 2011). Raw data were normalized to the input signal for each sample. AID-ChIP signal in cells expressing a nontarget shRNA control was assigned an arbitrary value of 1. Statistical analysis was performed using a two-tailed Student’s t test.
Online supplemental material.
Fig. S1 is a schematic of how the isolation and analysis of the AID-associated complex was undertaken and a table of peptide Ids. Fig. S2 shows the AID interactome. Table S1 shows primer sequences. Table S2 lists shRNA sequences. Table S3 lists antibodies used in this study. Online supplemental material is available at http://www.jem.org/cgi/content/full/jem.20112145/DC1.
Supplementary Material
Acknowledgments
We would like to thank the members of the Petersen-Mahrt and Reina-San-Martin laboratories for discussions; Cancer Research UK (CRUK) cell services for performing DT40 growth; Michel Nussenzweig for the anti-AID antibody and Anna Gazumyan for advice on AID-ChIP; Gudrun Bachmann and Dafne Solera for help in the generation of cell lines; Claudine Ebel for cell sorting; and Jesper Svejstrup for discussion and critical reading of the manuscript.
K.L. Willman, S. Pauklin, G. Rangam, M.T. Simon, S. Maslen, and M. Skehel were supported by CRUK. S. Pauklin was supported in part by SA Archimedes-Estonian Foundation of European Union Education and Research. K.-M. Schmitz is supported by a Marie Curie FP 7 fellowship. S. Milosevic was supported by la Ligue Contre le Cancer, France. B. Reina-San-Martin is an AVENIR-INSERM young investigator. This work was supported by grants to B. Reina-San-Martin from the Agence Nationale pour la Recherche (ANR-07-MIME-004-01) and the Institut National de la Santé et de la Recherche Médicale (INSERM), and to S.K. Petersen-Mahrt from Istituto FIRC di Oncologia Molecolare, Italy (IFOM) and CRUK.
The authors have no conflicting financial interests.
Author contributions: K.L. Willman, S. Milosevic, S. Pauklin, K.-M. Schmitz, G. Rangam, M.T. Simon, I. Robert, V. Heyer, and E. Schiavo performed experiments. S. Pauklin performed the initial AID on chromatin fractionation and isolation. S. Maslen and M. Skehel performed mass spectrometry analysis. K.L. Willman, S. Milosevic, K.-M. Schmitz, B. Reina-San-Martin, and SKPM analyzed the data. K.-M. Schmitz, K.L. Willman, S. Milosevic, B. Reina-San-Martin, and S.K. Petersen-Mahrt wrote the paper. K.L. Willman, SM, S. Pauklin, K.-M. Schmitz, B. Reina-San-Martin, and S.K. Petersen-Mahrt designed the experiments. S.K. Petersen-Mahrt conceived the approach.
Footnotes
Abbreviations used:
- AID
- activation-induced deaminase
- ChIP
- chromatin IP
- CSR
- class switch recombination
- CTD
- C-terminal domain (of RNA pol II)
- iGC
- Ig gene conversion
- IP
- immunoprecipitation
- PAF
- RNA polymerase-associated factor
- qRT-PCR
- quantitative RT-PCR
- SEC
- size exclusion chromatography
- SHM
- somatic hypermutation
- TSS
- transcription start site
References
- Aygün O., Svejstrup J., Liu Y. 2008. A RECQ5-RNA polymerase II association identified by targeted proteomic analysis of human chromatin. Proc. Natl. Acad. Sci. USA. 105:8580–8584 10.1073/pnas.0804424105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barreto V., Reina-San-Martin B., Ramiro A.R., McBride K.M., Nussenzweig M.C. 2003. C-terminal deletion of AID uncouples class switch recombination from somatic hypermutation and gene conversion. Mol. Cell. 12:501–508 10.1016/S1097-2765(03)00309-5 [DOI] [PubMed] [Google Scholar]
- Basu U., Meng F.L., Keim C., Grinstein V., Pefanis E., Eccleston J., Zhang T., Myers D., Wasserman C.R., Wesemann D.R., et al. 2011. The RNA exosome targets the AID cytidine deaminase to both strands of transcribed duplex DNA substrates. Cell. 144:353–363 10.1016/j.cell.2011.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betz A.G., Milstein C., González-Fernández A., Pannell R., Larson T., Neuberger M.S. 1994. Elements regulating somatic hypermutation of an immunoglobulin kappa gene: critical role for the intron enhancer/matrix attachment region. Cell. 77:239–248 10.1016/0092-8674(94)90316-6 [DOI] [PubMed] [Google Scholar]
- Bransteitter R., Pham P., Scharff M.D., Goodman M.F. 2003. Activation-induced cytidine deaminase deaminates deoxycytidine on single-stranded DNA but requires the action of RNase. Proc. Natl. Acad. Sci. USA. 100:4102–4107 10.1073/pnas.0730835100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brar S.S., Watson M., Diaz M. 2004. Activation-induced cytosine deaminase (AID) is actively exported out of the nucleus but retained by the induction of DNA breaks. J. Biol. Chem. 279:26395–26401 10.1074/jbc.M403503200 [DOI] [PubMed] [Google Scholar]
- Chaudhuri J., Tian M., Khuong C., Chua K., Pinaud E., Alt F.W. 2003. Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature. 422:726–730 10.1038/nature01574 [DOI] [PubMed] [Google Scholar]
- Chen Y., Yamaguchi Y., Tsugeno Y., Yamamoto J., Yamada T., Nakamura M., Hisatake K., Handa H. 2009. DSIF, the Paf1 complex, and Tat-SF1 have nonredundant, cooperative roles in RNA polymerase II elongation. Genes Dev. 23:2765–2777 10.1101/gad.1834709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury M., Forouhi O., Dayal S., McCloskey N., Gould H.J., Felsenfeld G., Fear D.J. 2008. Analysis of intergenic transcription and histone modification across the human immunoglobulin heavy-chain locus. Proc. Natl. Acad. Sci. USA. 105:15872–15877 10.1073/pnas.0808462105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coker H.A., Morgan H.D., Petersen-Mahrt S.K. 2006. Genetic and in vitro assays of DNA deamination. Methods Enzymol. 408:156–170 10.1016/S0076-6879(06)08010-4 [DOI] [PubMed] [Google Scholar]
- Conticello S.G., Ganesh K., Xue K., Lu M., Rada C., Neuberger M.S. 2008. Interaction between antibody-diversification enzyme AID and spliceosome-associated factor CTNNBL1. Mol. Cell. 31:474–484 10.1016/j.molcel.2008.07.009 [DOI] [PubMed] [Google Scholar]
- Di Noia J.M., Neuberger M.S. 2007. Molecular mechanisms of antibody somatic hypermutation. Annu. Rev. Biochem. 76:1–22 10.1146/annurev.biochem.76.061705.090740 [DOI] [PubMed] [Google Scholar]
- Fleming A.B., Kao C.-F., Hillyer C., Pikaart M., Osley M.A. 2008. H2B ubiquitylation plays a role in nucleosome dynamics during transcription elongation. Mol. Cell. 31:57–66 10.1016/j.molcel.2008.04.025 [DOI] [PubMed] [Google Scholar]
- Ganesh K., Adam S., Taylor B., Simpson P., Rada C., Neuberger M. 2011. CTNNBL1 is a novel nuclear localization sequence-binding protein that recognizes RNA-splicing factors CDC5L and Prp31. J. Biol. Chem. 286:17091–17102 10.1074/jbc.M110.208769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris R.S., Sale J.E., Petersen-Mahrt S.K., Neuberger M.S. 2002. AID is essential for immunoglobulin V gene conversion in a cultured B cell line. Curr. Biol. 12:435–438 10.1016/S0960-9822(02)00717-0 [DOI] [PubMed] [Google Scholar]
- Ito S., Nagaoka H., Shinkura R., Begum N., Muramatsu M., Nakata M., Honjo T. 2004. Activation-induced cytidine deaminase shuttles between nucleus and cytoplasm like apolipoprotein B mRNA editing catalytic polypeptide 1. Proc. Natl. Acad. Sci. USA. 101:1975–1980 10.1073/pnas.0307335101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaehning J.A. 2010. The Paf1 complex: platform or player in RNA polymerase II transcription? Biochim. Biophys. Acta. 1799:379–388 10.1016/j.bbagrm.2010.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeevan-Raj B.P., Robert I., Heyer V., Page A., Wang J.H., Cammas F., Alt F.W., Losson R., Reina-San-Martin B. 2011. Epigenetic tethering of AID to the donor switch region during immunoglobulin class switch recombination. J. Exp. Med. 208:1649–1660 10.1084/jem.20110118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A., Nesvizhskii A.I., Kolker E., Aebersold R. 2002. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 74:5383–5392 10.1021/ac025747h [DOI] [PubMed] [Google Scholar]
- Kim J., Guermah M., Roeder R.G. 2010. The human PAF1 complex acts in chromatin transcription elongation both independently and cooperatively with SII/TFIIS. Cell. 140:491–503 10.1016/j.cell.2009.12.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuang F.L., Luo Z., Scharff M.D. 2009. H3 trimethyl K9 and H3 acetyl K9 chromatin modifications are associated with class switch recombination. Proc. Natl. Acad. Sci. USA. 106:5288–5293 10.1073/pnas.0901368106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M., Duke J.L., Richter D.J., Vinuesa C.G., Goodnow C.C., Kleinstein S.H., Schatz D.G. 2008. Two levels of protection for the B cell genome during somatic hypermutation. Nature. 451:841–845 10.1038/nature06547 [DOI] [PubMed] [Google Scholar]
- Maeda K., Singh S.K., Eda K., Kitabatake M., Pham P., Goodman M.F., Sakaguchi N. 2010. GANP-mediated recruitment of activation-induced cytidine deaminase to cell nuclei and to immunoglobulin variable region DNA. J. Biol. Chem. 285:23945–23953 10.1074/jbc.M110.131441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride K.M., Barreto V., Ramiro A.R., Stavropoulos P., Nussenzweig M.C. 2004. Somatic hypermutation is limited by CRM1-dependent nuclear export of activation-induced deaminase. J. Exp. Med. 199:1235–1244 10.1084/jem.20040373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambu Y., Sugai M., Gonda H., Lee C.-G., Katakai T., Agata Y., Yokota Y., Shimizu A. 2003. Transcription-coupled events associating with immunoglobulin switch region chromatin. Science. 302:2137–2140 10.1126/science.1092481 [DOI] [PubMed] [Google Scholar]
- Nowak U., Matthews A.J., Zheng S., Chaudhuri J. 2011. The splicing regulator PTBP2 interacts with the cytidine deaminase AID and promotes binding of AID to switch-region DNA. Nat. Immunol. 12:160–166 10.1038/ni.1977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odegard V.H., Schatz D.G. 2006. Targeting of somatic hypermutation. Nat. Rev. Immunol. 6:573–583 10.1038/nri1896 [DOI] [PubMed] [Google Scholar]
- Okazaki I.-M., Okawa K., Kobayashi M., Yoshikawa K., Kawamoto S., Nagaoka H., Shinkura R., Kitawaki Y., Taniguchi H., Natsume T., et al. 2011. Histone chaperone Spt6 is required for class switch recombination but not somatic hypermutation. Proc. Natl. Acad. Sci. USA. 108:7920–7925 10.1073/pnas.1104423108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patenaude A.M., Orthwein A., Hu Y., Campo V.A., Kavli B., Buschiazzo A., Di Noia J.M. 2009. Active nuclear import and cytoplasmic retention of activation-induced deaminase. Nat. Struct. Mol. Biol. 16:517–527 10.1038/nsmb.1598 [DOI] [PubMed] [Google Scholar]
- Pauklin S., Sernández I.V., Bachmann G., Ramiro A.R., Petersen-Mahrt S.K. 2009. Estrogen directly activates AID transcription and function. J. Exp. Med. 206:99–111 10.1084/jem.20080521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavri R., Zhu B., Li G., Trojer P., Mandal S., Shilatifard A., Reinberg D. 2006. Histone H2B monoubiquitination functions cooperatively with FACT to regulate elongation by RNA polymerase II. Cell. 125:703–717 10.1016/j.cell.2006.04.029 [DOI] [PubMed] [Google Scholar]
- Pavri R., Gazumyan A., Jankovic M., Di Virgilio M., Klein I., Ansarah-Sobrinho C., Resch W., Yamane A., Reina San-Martin B., Barreto V., et al. 2010. Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell. 143:122–133 10.1016/j.cell.2010.09.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins D.N., Pappin D.J., Creasy D.M., Cottrell J.S. 1999. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 20:3551–3567 [DOI] [PubMed] [Google Scholar]
- Peters A., Storb U. 1996. Somatic hypermutation of immunoglobulin genes is linked to transcription initiation. Immunity. 4:57–65 10.1016/S1074-7613(00)80298-8 [DOI] [PubMed] [Google Scholar]
- Petersen-Mahrt S. 2005. DNA deamination in immunity. Immunol. Rev. 203:80–97 10.1111/j.0105-2896.2005.00232.x [DOI] [PubMed] [Google Scholar]
- Petersen-Mahrt S.K., Harris R.S., Neuberger M.S. 2002. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature. 418:99–103 10.1038/nature00862 [DOI] [PubMed] [Google Scholar]
- Rada C., Jarvis J.M., Milstein C. 2002. AID-GFP chimeric protein increases hypermutation of Ig genes with no evidence of nuclear localization. Proc. Natl. Acad. Sci. USA. 99:7003–7008 10.1073/pnas.092160999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajewsky K. 1996. Clonal selection and learning in the antibody system. Nature. 381:751–758 10.1038/381751a0 [DOI] [PubMed] [Google Scholar]
- Robert I., Dantzer F., Reina-San-Martin B. 2009. Parp1 facilitates alternative NHEJ, whereas Parp2 suppresses IgH/c-myc translocations during immunoglobulin class switch recombination. J. Exp. Med. 206:1047–1056 10.1084/jem.20082468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schotta G., Sengupta R., Kubicek S., Malin S., Kauer M., Callén E., Celeste A., Pagani M., Opravil S., De La Rosa-Velazquez I.A., et al. 2008. A chromatin-wide transition to H4K20 monomethylation impairs genome integrity and programmed DNA rearrangements in the mouse. Genes Dev. 22:2048–2061 10.1101/gad.476008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selth L.A., Sigurdsson S., Svejstrup J.Q. 2010. Transcript Elongation by RNA Polymerase II. Annu Rev Biochem. [DOI] [PubMed] [Google Scholar]
- Stanlie A., Aida M., Muramatsu M., Honjo T., Begum N.A. 2010. Histone3 lysine4 trimethylation regulated by the facilitates chromatin transcription complex is critical for DNA cleavage in class switch recombination. Proc. Natl. Acad. Sci. USA. 107:22190–22195 10.1073/pnas.1016923108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanlie A., Begum N.A., Akiyama H., Honjo T. 2012. The DSIF subunits Spt4 and Spt5 have distinct roles at various phases of immunoglobulin class switch recombination. PLoS Genet. 8:e1002675 10.1371/journal.pgen.1002675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavnezer J., Guikema J.E., Schrader C.E. 2008. Mechanism and regulation of class switch recombination. Annu. Rev. Immunol. 26:261–292 10.1146/annurev.immunol.26.021607.090248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavnezer-Nordgren J., Sirlin S. 1986. Specificity of immunoglobulin heavy chain switch correlates with activity of germline heavy chain genes prior to switching. EMBO J. 5:95–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L., Wuerffel R., Feldman S., Khamlichi A.A., Kenter A.L. 2009. S region sequence, RNA polymerase II, and histone modifications create chromatin accessibility during class switch recombination. J. Exp. Med. 206:1817–1830 10.1084/jem.20081678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane A., Resch W., Kuo N., Kuchen S., Li Z., Sun H.-W., Robbiani D.F., McBride K., Nussenzweig M.C., Casellas R. 2011. Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nat. Immunol. 12:62–69 10.1038/ni.1964 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.