

Summary
The convulsant properties of xanthine amine congener (XAC, 8-(4-(2-aminoethyl)-aminocarboxylmethyloxy)phenyl-1,3-dipropylxanthine) are compared to those of caffeine. Male Swiss albino mice were infused with convulsants through a lateral tail vein. Convulsion thresholds (i.e. the amount of convulsants required to elicit convulsions) of 39.8±2.0 mg/kg (n=10) and 109.8±2.3 mg/kg (n=10) were calculated for XAC and caffeine respectively. Pretreatment of animals with the adenosine receptor agonists 2-chloroadenosine, N6-cyclohexyladenosine or 5′-N-ethylcarboxamido-adenosine (1 mg/kg, i.p., 20 minutes prior to infusion) significantly decreased the seizure threshold of both XAC and caffeine. The adenosine uptake blockers, 6-nitrobenzylthioinosine or dipyridamole (0.25 mg/kg, i.p., 20 minutes prior to infusion) did not significantly affect the seizure threshold to either XAC or caffeine. The benzodiazepine agonist diazepam (5 mg/kg, i.p., 20 minutes prior to infusion) significantly increased the seizure threshold to both XAC (p < 0.05) and caffeine (p < 0.01), whereas the benzodiazepine antagonist Ro 15-1788 (10 mg/kg, i.p., 20 minutes prior to infusion) significantly increased the seizure threshold to caffeine (p < 0.01), but not XAC. The results suggest that actions at benzodiazepine receptors may be a tenable hypothesis to explain the convulsant actions of caffeine, but not those of XAC.
Adenosine and a number of pharmacological agents, including the methylxanthines caffeine and theophylline, modulate key physiological functions through interactions at adenosine receptors (1, 2). Such receptors have been termed A1 and A2 on the basis of the ability of agonist compounds to inhibit (A1) or stimulate (A2) adenylate cyclase (3, 4), although it is clear that both subtypes of receptor may be coupled to other second messenger systems (4, 5). A [3H]xanthine amine congener of 1,3-dipropyl-8-phenylxanthine ([3H]XAC) has been recently characterized as a very potent adenosine antagonist with an affinity for adenosine receptors of approximately four orders of magnitude greater than that of caffeine or theophylline (7). Pharmacological studies using unlabelled XAC (8–10), as well as radioligand binding studies using [3H]XAC (7), reveal XAC to have a marked A1 selectivity, although the compound does exhibit some A2 actions (7–11). Indeed [3H]XAC has been successfully employed to radiolabel adenosine A2 receptors of human platelets (11).
Adenosine antagonists, such as caffeine and theophylline have been reported to elicit convulsions in high doses (100–1000 mg/kg, depending on route of administration)(12, 13). The convulsant effect has been presumed to be due to antagonism of adenosine receptors (2). However, caffeine and theophylline also exhibit moderate affinity (Ki 300–500/μM) for benzodiazepine receptors labelled by [3H]-diazepam (14). In addition, seizures induced by caffeine can be blocked by benzodiazepines, such as diazepam and Ro 15-1788 (15, 16), raising the possibility that benzodiazepine receptors may mediate the convulsant actions of methylxanthines.
In view of the much higher affinity and selectivity of XAC for adenosine receptors than caffeine we have examined the convulsant actions of XAC and report a comparison of such actions to those of caffeine.
MATERIALS AND METHODS
Animals and Experimental Design
Naive male Swiss albino mice (25–30g) were employed. Animals were randomly assigned to experimental groups and tested only once. Animals had been maintained on a 12h light /12h dark cycle with ad lib access to food and water.
Drug infusions
Animals were infused with convulsants through a lateral tail vein at a constant flow rate of 313 μl min−1 using a 25 gauge butterfly needle fed from an infusion pump (Harvard 22) as previously described (13). The latency between starting infusion and the onset of convulsions was measured, and from this measurement, the infusion rate, the weight of the animals, and the concentration of the convulsant, the amount of convulsant required to elicit convulsions (convulsion threshold) was calculated. In infusion studies involving a drug pre-treatment, an intra-peritoneal injection of vehicle or compound (4ml/kg) was administered 20 minutes prior to the commencement of infusions.
Materials
XAC was synthesized as described (7), dissolved in 0.1 M acetic acid, and pH was adjusted to 7.0 using sodium hydroxide. Caffeine, theophylline and dipyridamole were obtained from Sigma, St. Louis, Mo. Other xanthines were obtained from Research Biochemicals Inc., Natick, Ma, as were 2-chloroadenosine, N6-cyclohexyladenosine, 5-N-ethylcarboxamidoadenosine and 6-nitrobenzylthioinosine. All benzodiazepines were kindly donated by Hoffman La Roche, Nutley, New Jersey. All reagents were obtained from standard commercial sources.
RESULTS
Initial studies of the convulsant potential of XAC were performed by administering the drug via an i.p. injection. Using this route of administration no convulsant properties of XAC could be demonstrated (seizure threshold > 1000 mg/kg). Subsequent postmortem examination revealed the drug came out of solution in the peritoneal cavity and sequestered locally. Similar observations were observed with i.p. administration of 1,3-dipropyl-8-(2-amino-4-chloro)phenylxanthine (PACPX).
Infusion studies revealed the potent seizure properties of XAC (Table 1). Using this route of administration XAC was more potent as a convulsant than either caffeine or theophylline. PACPX also demonstrated potent convulsant actions, although the dimethyl-sulphoxide (DMSO) vehicle may have affected the convulsant action of PACPX, since the convulsant threshold of XAC (but not pentylenetetrazole, PTZ) was significantly lower when administered in a DMSO vehicle (Table 2).
TABLE 1.
Seizure threshold of XAC and other xanthines
| XANTHINE | SEIZURE THRESHOLD (MG KG−1) |
|---|---|
| Xanthine amine congener (XAC) | 39.8 ± 2.0 (10) |
| 1,3-Dipropyl-8-(2-amino-4-chloro)phenylxanthine (PACPX) | 57.8 ± 2.2 (10)* |
| Caffeine | 161.3 ± 20.4 (10) |
| 3-Isobutyl-1-methylxanthine | 234.8 ± 48.7 (10) |
| Theophylline | 503.1 ± 31.3 (10) |
| 8-Phenyltheophylline | > 1000 (5) |
| 8-Sulphophenyltheophylline | > 1000 (5) |
| Enprofylline (3-propylxanthine) | > 1000 (5) |
| 1,3-dipropyl-8-p-sulphophenyl-xanthine | > 1000 (5) |
Adenosine antagonists were infused through a tail vein using a butterfly needle fed from a constant infusion pump and the seizure threshold for each determined. Values represent the lean ± S. E. mean with the number of animals in parentheses.
The vehicles employed (all compounds were in saline-based vehicles except PACPX which was dissolved in DMSO) were without significant effect except the DMSO vehicle, see also table 2.
TABLE 2.
The effect of DMSO vehicle on the seizure threshold for XAC and PTZ.
| VEHICLE | INFUSED CONVULSANT (SEIZURE THRESHOLD mg/ml) | |
|---|---|---|
| PTZ | XAC | |
| SALINE | 198.3 ± 16.7 (10) | 39.8 ± 2.0 (10) |
| DMSO | 215.3 ± 16.6 (10) | 3.03 ± 0.34 (10)* |
Convulsants were infused through a tail vein using a butterfly needle fed from a constant infusion pump and the seizure threshold for each determined. Values are the mean ± S.E. mean with the number of animals in parentheses.
= p<0.01, compared to XAC in saline vehicle, two-tailed T-test.
The effect of pre-treatment with adenosine agonists and nucleoside uptake blockers on the convulsant threshold to XAC and caffeine was investigated employing relatively high doses of these compounds in order to maximize competitive displacement of the antagonists. The adenosine agonist compounds 2-chloroadenosine, N6-cyclohexyladenosine (CHA) and 5-N-ethyl-carboxamidoadenosine (NECA) all decreased (p’s <0.05) the seizure threshold (i.e. acted as pro-convulsants) to both XAC and caffeine, whilst both nucleoside uptake blockers, 6-nitrobenzylthioinosine (NBI) and dipyridamole (DPR) were without significant effect (Figure 1).
Fig. 1.
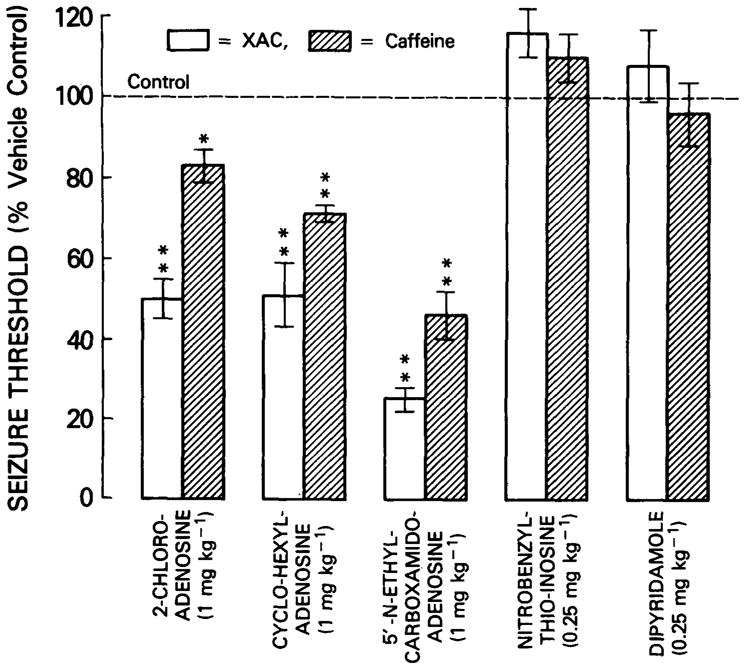
The effect of pretreatment with adenosine agonists and nucleoside uptake blockers on XAC and caffeine seizure threshold. Animals were infused with either XAC or caffeine and the corresponding seizure thresholds computed. Twenty minutes prior to infusion, animals were pretreated with either an adenosine agonist (2-chloroadenosine, CHA or NECA, all 1 mg/kg., i.p. in 1% ethanol saline) or a nucleoside uptake blocker (NBI or DPR, both 0.25 mg/kg., i.p. in 1% ethanol saline). Histogram elements represent the mean ± S.E. mean of 10 animals. Each treatment was compared to a parallel vehicle control group. * = p<0.05, ** = p<0.01, two-tailed T-test.
Pre-treatment of animals with the benzodiazepine agonist, diazepam increased the seizure threshold (i.e. acted as an anticonvulsant) to both XAC (p<0.05) and caffeine (p<0.01), whilst the benzodiazepine antagonist, Ro 15-1788 increased the seizure threshold to caffeine (p<0.01), but not to XAC (Figure 2).
Fig. 2.
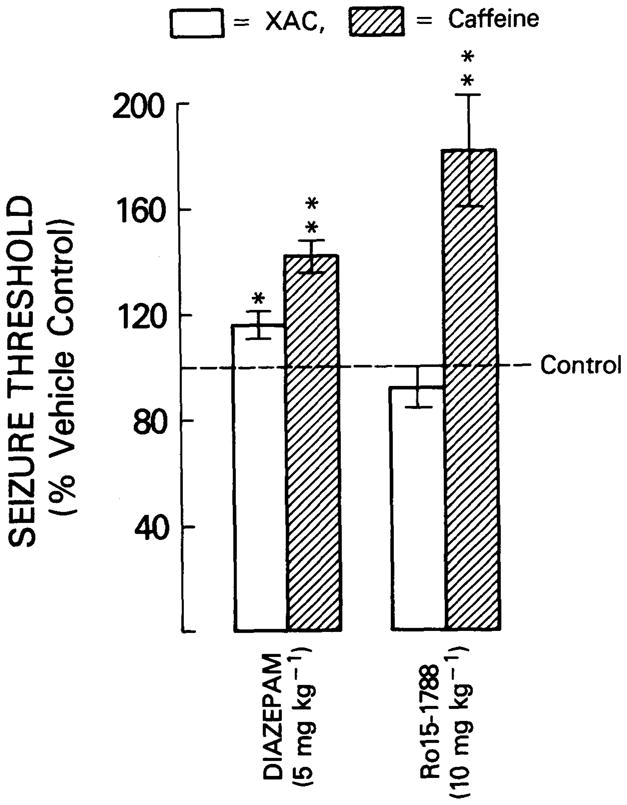
The effect of pretreatment with the benzodiazepine agonist, diazepam, and the benzodiazepine antagonist, Ro 15-1788, on XAC and caffeine seizure threshold. Animals were infused with either XAC or caffeine and the corresponding seizure thresholds computed. Twenty minutes prior to infusion, animals were pretreated with either diazepam (5 mg/kg, i.p., in 1% ethanol saline), Ro 15-1788 (10 mg/kg, i.p., in 1% ethanol saline), or vehicle. Histogram elements represent the mean ± S. E. mean of 10 animals. Each treatment was compared to a parallel vehicle control group. * = p<0.05, ** = p<0.01, two-tailed T-test.
If the convulsant actions of XAC and caffeine are both mediated via adenosine receptors then one may expect caffeine pre-treatment to lower the seizure threshold to XAC. This was not observed. Caffeine, in relatively high doses (approaching the seizure threshold for i.p. administration), did not alter the seizure threshold to XAC. In contrast, the seizure threshold to PTZ was reduced by more than 50% by such caffeine pre-treatment (Figure 3).
Fig. 3.
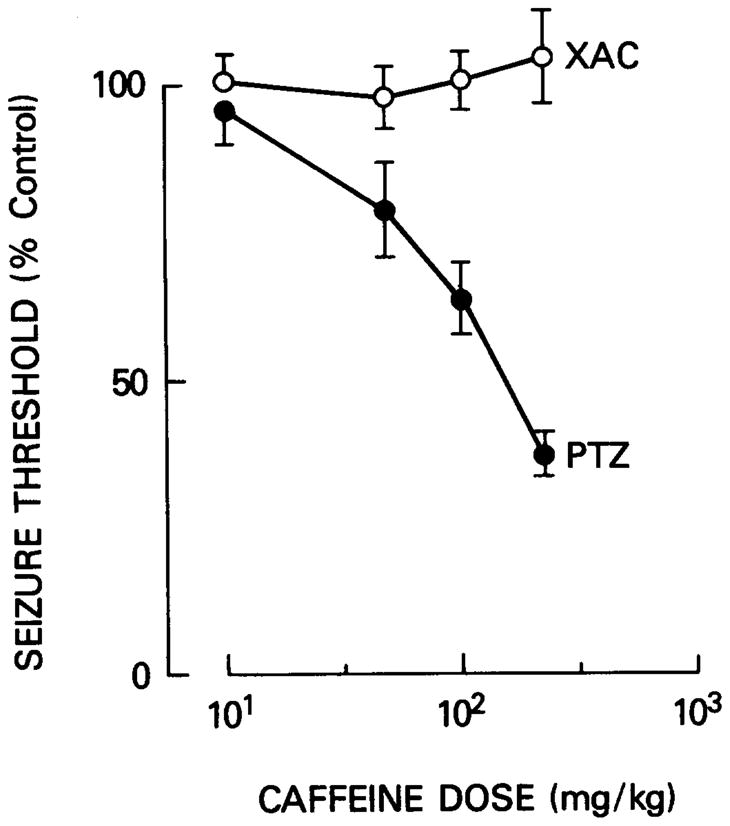
The effect of caffeine pretreatment on XAC and PTZ seizure threshold. Animals were infused with XAC or PTZ and the corresponding convulsion thresholds computed. Twenty minutes prior to infusion, animals were pretreated with caffeine (10–225 mg/kg, i.p., dissolved in saline). Data points represent the mean ± S. E. mean of 10 animals.
DISCUSSION
The very potent A1-adenosine receptor antagonist XAC, which does not exhibit convulsant properties when administered via the intraperitoneal route, is a potent convulsant when administered by intravenous infusion. Indeed it is some 4-fold more potent than caffeine despite the fact that XAC is thought to poorly penetrate the brain (9). In view of the pharmacological selectivity of XAC for adenosine receptors (7), it would appearlikely that the convulsive action may be mediated by antagonism of adenosine receptors. However, the seizure threshold to XAC was not increased by pre-treatment with either adenosine uptake blockers or adenosine agonists. Rather than such data being directly contrary to the above hypothesis, such data more likely highlight two interrelated problems with such an approach: [1] the difficulty of surmounting competitive receptor antagonism with agonists especially when the antagonist (XAC) is used at concentrations far greater than its Ka for the receptor, and [2] the extent of peripheral actions of the agonists (cardiac depression, hypotension, vasodilation), which preclude administration of sufficiently high doses of adenosine agonists to reverse central competitive antagonists.
In view of the potent depressant actions of microiontophoresed adenosine agonists on neuronal activity (17, 18) and the convulsant action of a number of xanthine adenosine antagonists (12, 13, and the present study) the pro-convulsant action of CHA, 2-chloroadenosine and NECA when administered as pre-treatments to XAC or caffeine infusions is surprising. The problem of peripheral actions of adenosine agonists ([2] above) may account for the somewhat paradoxical pro-convulsant actions of these adenosine agonists. For example, agonist-induced vascular changes may increase the subsequent penetration of XAC or caffeine into the brain. However, under some conditions, ‘paradoxical’ effects of adenosine agonists have been observed. Thus, low doses of adenosine agonists have been reported to enhance locomotor activity (19) and after protein kinase C blockade, adenosine agonists have been reported to increase neurotransmitter release (20). Caffeine and XAC, by antagonizing one response to the agonists, may expose excitant actions of the compounds, possibly mediated by a non-xanthine sensitive site.
The benzodiazepine agonist diazepam increased the seizure threshold to both XAC and caffeine, presumably as a result of a general anticonvulsant action. Diazepam has been reported to be effective in reversing a disparate number of convulsants, including caffeine (21). Caffeine-induced seizures have also been reported to be reversed by the benzodiazepine antagonist, Ro 15-1788 (16). The findings of the present study confirm such observations. Whilst Ro 15-1788 and diazepam have been reported to be a moderately effective inhibitors of [3H]-adenosine uptake in synaptosomes (Ki ≃ 10 μM, 22, 23), the present study reveals that far more potent inhibitors of adenosine uptake (NBI and DPR) have no effect on the caffeine seizure threshold, although it is not clear the extent to which the adenosine uptake blockers cross the blood-brain barrier, particularly dipyridamole which has been reported to poorly penetrate the brain (24). In the case of the adenosine agonists, the inhibitory dissociation rate constant, Ki, for caffeine inhibition of radiolabeled benzodiazepine binding to benzodiazepine receptors is 300 μM (14). This, coupled to the fact that caffeine-induced seizures are blocked by Ro 15-1788, a highly selective antagonist for central-type benzodiazepine receptors, suggests that central-type benzodiazepine receptors, rather than adenosine receptors, may principally mediate the convulsant action of caffeine. The results of the present study are consistent with such a hypothesis. In contrast to caffeine, the seizure threshold to XAC is not affected by Ro 15-1788 pre-treatment. Such a finding would suggest that actions at benzodiazepine receptors may not explain the convulsant actions of XAC.
The ability of caffeine pretreatment to lower the seizure threshold to PTZ but not to XAC further suggests that caffeine and XAC may evoke seizures by different mechanisms. The benzodiazepine receptor is envisaged to be part of a GABA receptor/chloride ion channel/benzodiazepine receptor complex at which PTZ may interact to exert its convulsant and anxiogenic actions (25, 26). The reduction of the seizure threshold to PTZ, but not to XAC, by caffeine pretreatment may therefore reflect similar interactions of caffeine and PTZ at the GABA receptor/chloride ion channel/benzodiazepine receptor complex and a lack of interaction of XAC with the complex.
In view of the relative in vitro selectivity of XAC for adenosine receptors, adenosine receptors appear as likely candidates for mediation of the convulsant actions of XAC. Further studies, especially with more selective analogues of XAC may clarify this issue considerably.
References
- 1.STONE TW. Neurosci. 1981;6:523–555. doi: 10.1016/0306-4522(81)90145-7. [DOI] [PubMed] [Google Scholar]
- 2.DUNWIDDIE TV. Int Rev Neurobiol. 1985;27:63–133. doi: 10.1016/s0074-7742(08)60556-5. [DOI] [PubMed] [Google Scholar]
- 3.VAN CALKER D, MULLER M, HAMPRECHT B. J Neurochem. 1979;33:999–1005. doi: 10.1111/j.1471-4159.1979.tb05236.x. [DOI] [PubMed] [Google Scholar]
- 4.LONDOS C, COOPER DMF, WOLF J. Proc Nat Acad Sci USA. 1980;77:2551–2554. doi: 10.1073/pnas.77.5.2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.HAMPRECHT B, VAN CALKER D. Tr Pharmacol Sci. 1985;6:153–154. [Google Scholar]
- 6.FREDHOLM BB, DUNWIDDIE TV. Tr Pharmacol Sci. 1988;9:130–134. doi: 10.1016/0165-6147(88)90194-0. [DOI] [PubMed] [Google Scholar]
- 7.JACOBSON KA, UKENA D, KIRK KL, DALY JW. Proc Nat Acad Sci USA. 1986;83:4089–4093. doi: 10.1073/pnas.83.11.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.COLLIS MG, JACOBSON KA, TOMKINS DM. Br J Pharmacol. 1986;92:69–75. doi: 10.1111/j.1476-5381.1987.tb11297.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.EVONIUK G, JACOBSON KA, SHAMIM MT, DALY JW, WURTMAN RJ. J Pharmacol Exp Ther. 1987;242:882–887. [PMC free article] [PubMed] [Google Scholar]
- 10.CHURCHILL PC, JACOBSON KA, CHURCHILL MC. Arch Int Pharmacodyn Ther. 1987;290:293–301. [PMC free article] [PubMed] [Google Scholar]
- 11.UKENA D, JACOBSON KA, KIRK KL, DALY JW. FEBS Lett. 1986;199:269–274. doi: 10.1016/0014-5793(86)80493-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.RICHIE JM. Central Nervous system stimulants, II. The Xanthines. In: Goodman LS, Gilman A, editors. The Pharmacological Basis of Therapeutics. MacMillan Co; New York: 1975. pp. 367–378. [Google Scholar]
- 13.MORGAN PF, STONE TW. Br J Pharmacol. 1982;77:525–530. doi: 10.1111/j.1476-5381.1982.tb09327.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.MARANGOS PJ, PAUL SM, PARMA AM, GOODWIN FK, SYAPIN P, SKOLNICK P. Life Sci. 1979;24:851–858. doi: 10.1016/0024-3205(79)90369-2. [DOI] [PubMed] [Google Scholar]
- 15.MARANGOS PJ, MARTINO AM, PAUL SM, SKOLNICK P. Psychopharmacol. 1981;72:269–273. doi: 10.1007/BF00431829. [DOI] [PubMed] [Google Scholar]
- 16.VELLUCCI SV, WEBSTER RA. Eur J Pharmacol. 1984;97:289–294. doi: 10.1016/0014-2999(84)90462-x. [DOI] [PubMed] [Google Scholar]
- 17.PHILLIS JW, EDSTROM JP, KOSTOPOULOS GK, KIRKPATRICK JR. Can J Physiol Pharmacol. 1979;57:1289–1310. doi: 10.1139/y79-194. [DOI] [PubMed] [Google Scholar]
- 18.PERKINS MN, STONE TW. Arch Int Pharmacodyn Ther. 1980;246:205–214. [PubMed] [Google Scholar]
- 19.SNYDER SH, KATIMS JS, ANNAU Z, BRUNS RF, DALY JW. Proc Natl Acad sci USA. 1981;78:3260–3264. doi: 10.1073/pnas.78.5.3260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.BRANISTEANU DD, BRANISTEANU DDD, COVIC A, BRAILOIU E, SERBAN DN, HAULICA ID. Neurosci Lett. 1989;98:96–100. doi: 10.1016/0304-3940(89)90380-7. [DOI] [PubMed] [Google Scholar]
- 21.MARANGOS PJ, PAUL SM, GOODWIN FK, SKOLNICK P. Life Sci. 1979;25:1093–1102. doi: 10.1016/0024-3205(79)90130-9. [DOI] [PubMed] [Google Scholar]
- 22.WU PH, PHILLIS JW, BENDER AS. Life Sci. 1981;28:1023–1031. doi: 10.1016/0024-3205(81)90748-7. [DOI] [PubMed] [Google Scholar]
- 23.MORGAN PF, LLOYD HGE, STONE TW. Neurosci Lett. 1983;41:183–188. doi: 10.1016/0304-3940(83)90244-6. [DOI] [PubMed] [Google Scholar]
- 24.SOLLEVI A. Prog Neurobiol. 1986;27:319–349. doi: 10.1016/0301-0082(86)90005-5. [DOI] [PubMed] [Google Scholar]
- 25.ITO M, CHIU TH, ROSENBURG HC. Neurochem Res. 1986;11:637–646. doi: 10.1007/BF00965333. [DOI] [PubMed] [Google Scholar]
- 26.KENT AP, WEBSTER RA. Neuropharmacol. 1986;86:1023–1030. doi: 10.1016/0028-3908(86)90197-8. [DOI] [PubMed] [Google Scholar]