

Abstract
Unclassified sequence variants (UV) arising from clinical mutation screening of cancer susceptibility genes present a frustrating issue to clinical genetics services and the patients that they serve. We created an open-access database holding missense substitutions from the breast and ovarian cancer susceptibility genes BRCA1 and BRCA2. The main inclusion criterion is that each variant should have been assessed in a published work that used the Bayesian integrated evaluation of unclassified BRCA gene variants. Transfer of data on these substitutions from the original publications to our database afforded an opportunity to analyze the missense substitutions under a single model and to remove inconsistencies that arose during the evolution of the integrated evaluation over the last decade. This analysis also afforded the opportunity to re-classify these missense substitutions according to the recently published IARC 5-Class system. From an initial set of 248 missense substitutions, 31 were set aside due to non-negligible probability to interfere with splicing. Of the remaining substitutions, 28 fell into one of the two pathogenic classes (IARC Classes 4 or 5), 174 fell into one of the two non-pathogenic classes (IARC Classes 1 or 2), and 15 remain in IARC Class 3, “Uncertain”. The database is available at <http://brca.iarc.fr/LOVD>.
Keywords: Unclassified variant, UV, VUS, BRCA1, BRCA2, database
INTRODUCTION
The breast and ovarian cancer susceptibility genes BRCA1 and BRCA2 (MIM#s 113705, 600185, respectively) are the most routinely tested genes for cancer predisposition. Under the criteria used in the United States to select patients for testing, between 10% and 15% of subjects who undergo full-sequence BRCA testing are found to carry a clearly pathogenic sequence variant. This pathogenic variant can be either a nonsense variant, a small insertion or deletion variant that creates a frameshift, a larger gene rearrangement, a variant that creates a severe splicing aberration, or a known pathogenic missense substitution. However, more than 5% of patients are found to carry an unclassified variant (UV) – usually either a missense substitution or a variant that falls in the splice junction consensus regions but outside of the canonical GT-AG dinucleotides. Because of ongoing efforts to classify UVs, most former-UVs with frequencies of above 0.1% have now been classified; consequently, the remaining UVs are individually rare. Here, we refer to former-UVs that are now classified as Ex-UVs.
The main method for classifying BRCA gene UVs is called the “integrated evaluation” or the “multifactorial method” (Goldgar et al., 2004; Easton et al., 2007; Goldgar et al., 2008; Spurdle, 2010). The integrated evaluation applies a Bayesian statistical inference to each individual UV analyzed. The evaluation begins with a prior probability in favor of pathogenicity (prior probability) based on sequence analysis (Easton et al., 2007; Tavtigian et al., 2008). Observational data summarized as odds ratios in favor of pathogenicity (likelihood ratios, LR) are used to update the prior, resulting in a posterior probability in favor of pathogenicity (posterior probability). The posterior probability is converted to a qualitative classification via a 5-Class lookup table (Plon et al., 2008). Once this conversion has been made, the variant is considered “classified”. Nonetheless, additional data may result in re-classification; this is especially true for Ex-UVs with an initial classification result of “Class 3, Uncertain”, a possibility that highlights the distinct meanings of “Unclassified” (i.e., the variant has not yet been subjected to an integrated evaluation) and “Uncertain” (i.e., the result from an integrated evaluation was a posterior probability that falls between 0.05 and 0.95) in this field.
Individual elements included in the integrated evaluation have been reviewed recently (Goldgar et al., 2008; Spurdle, 2010), and are summarized briefly below.
1. Prior probability
At the protein level, missense substitutions are initially assessed by their position in the protein. If a substitution falls in one of the domains of BRCA1 or BRCA2 known to harbor missense substitutions that are pathogenic because of missense dysfunction (for BRCA1, the RING and BRCT domains; for BRCA2 the DNA binding domain and perhaps the PALB2 interaction domain), then an in silico assessment of the substitution is performed using a missense substitution analysis program, such as Align-GVGD (Tavtigian et al., 2008), that has been calibrated so that its output can be interpreted as either a probability in favor of pathogenicity or a likelihood ratio. At the mRNA processing level, splice site fitness programs such as MaxEntScan (Yeo and Burge, 2004) could in principle be used to evaluate the probability for a sequence variant to damage a wild-type splice site and/or to create a de novo splice site. However, the splice site program calibration is in progress and has not yet been entirely incorporated into the analysis model.
2. Observational data. Currently, four types of observational data are included in the integrated evaluation
(a) Cosegregation of UVs with cancer phenotype in pedigrees
The cosegregation LR can be calculated when substantial genotyping data from a pedigree(s) in which a UV of interest is present are available. The underlying algorithm is derived from linkage analysis and is evaluated under the hypothesis that the studied variant has the same penetrance as an “average” protein truncating BRCA mutation, compared with the hypothesis that the variant segregates independent of disease in the pedigree(s) under study (Thompson et al., 2003).
(b) Personal and family history
This LR is a comparison of personal and family history between individuals carrying a given UV, those with a demonstrated pathogenic BRCA mutation, and tested individuals from the same population who were found to have wild-type BRCA sequences (aside from known neutral variants) (Easton et al., 2007). Derivation of this LR depended on a large database of mutation data provided by Myriad Genetic Laboratories Inc. The LR will have to be recalibrated in order to be applied to family history data from sources that use patient selection criteria that are different from those used in the United States, or more generally, from sources from which the frequency of pathogenic mutations is not approximately equal to that present in the Myriad data set that was used for the LR derivation.
(c) Co-occurrence with known pathogenic mutations
The basis of this LR is that homozygotes and compound heterozygotes for pathogenic mutations in BRCA1 and BRCA2 are rarer than would be expected from their independent frequencies (Abkevich et al., 2004; Judkins et al., 2005). Reasons behind this phenomenon include that BRCA1-null genotypes are often embryonic lethal; similarly, BRCA2-null genotypes are often embryonic lethal or else can cause Fanconi anemia (Evers and Jonkers, 2006; Howlett 2002; Wagner 2004). The equation for calculating the co-occurrence likelihood ratio is structured as a binomial likelihood ratio based on the probability that an individual in the test population who carries an unclassified neutral variant also carries (in trans) a clearly pathogenic mutation, and an assumed probability that a phenotypically normal individual in the test population who carries an unclassified but actually pathogenic variant also carries (in trans) a deleterious mutation (Goldgar et al., 2004).
(d) Tumor immunohistochemistry and histological grade
The basis of this LR is that some physical tumor characteristics are more common in tumors from carriers of pathogenic BRCA1 or BRCA2 mutations than among tumors from non-carriers (Hofstra et al., 2008). For example, breast tumors from BRCA1 carriers are notably more likely to be negative for the estrogen receptor, progesterone receptor, and HER2/neu than tumors from non carriers; tumors from BRCA2 carriers are slightly more likely to be positive for tubule formation than are tumors from non-carriers. By measuring the frequencies of the possible combinations of such characteristics in the tumors of carriers and non-carriers, Chenevix-Trench et al. and Spurdle et al (Chenevix-Trench et al., 2006; Spurdle et al., 2008) were able to work out empirical LRs for various combinations of tumor characteristics. It is important to note that the individual tumor characteristics such as estrogen receptor status and progesterone receptor status are probably not conditionally independent; consequently, it is inappropriate to measure LRs for each individual characteristic and then use the product of those LRs in an integrated evaluation of a particular UV (Goldgar et al., 2008).
Operationally, a problem faced by testing laboratory staff, clinical geneticists, genetic counselors, and potentially patients is that it is very difficult to go through the literature, identify papers that have used bona fide implementations of the integrated evaluation to assess UVs, and then determine whether a sequence variant reported as a UV – or as one of the clinically used synonyms such as VUS – has actually been classified and is therefore actually an Ex-UV. Focusing on missense substitutions, we have gone through the literature, identified papers that have used bona fide implementations of the integrated evaluation, and cross-referenced the UVs actually assessed. Within the limitations of our current analytic ability, we have extracted appropriate prior probabilities for these missense substitutions, extracted observational LR data for these substitutions, combined them to calculate posterior probabilities, used the posterior probabilities to determine qualitative classifications, and recorded the results in a curated Leiden Open Variant Database (LOVD) (Fokkema et al., 2005).
METHODS
To identify published studies that used bona fide implementations of the integrated evaluation, we used the ISI Web of Knowledge tool to find every paper that cited the original description of this method (Goldgar et al., 2004). Our search, conducted in December 2009, found 90 such papers. We filtered these with the query (BRCA1 or BRCA2 or BRCA) and (UV* or VU* or UCV* or unclassified or uncertain* or unknown* or substitution* or variant* or missense*). We then manually screened abstracts from the remaining 72 papers, resulting in a final set of 15 papers.
1. Missense priors
To automate the process of determining prior probabilities for single nucleotide substitutions to the BRCA gene coding sequences, we wrote a program that performs the following actions:
models every possible single nucleotide substitution from 25 bp before to 25 bp after each coding exon;
gives each substitution HGVS nucleotide nomenclature, HGVS amino acid nomenclature, and BIC nucleotide nomenclature names;
scores each missense substitution with Align-GVGD using curated BRCA1 and BRCA2 protein multiple sequence alignments in which the most diverged sequence is from the sea urchin Strongylocentrotus purpuratus; and
applies the Align-GVGD-specific and position-specific probabilities in favor of pathogenicity defined in Table 3 of Tavtigian et al, 2008 (Tavtigian et al., 2008). The alignment through sea urchin is used for steps (c) and (d) because, of the depths of alignment tested, this depth of alignment gave the clearest resolution in risk prediction between the grades of missense substitutions defined by Align-GVGD (Table 2 and Figure 5 of Tavtigian et al., 2008).
Table 3.
Number and fate of BRCA2 missense substitutions in existing papers by classification
| Previous Classification | Current IARC Class | ||||
|---|---|---|---|---|---|
| Class 1 | Class 2 | Class 3 | Class 4 | Class 5 | |
| Neutral | 66 | 10 | 2 | 0 | 0 |
| Uncertain | 1 | 9 | 9 | 2 | 2 |
| Pathogenic | 0 | 0 | 0 | 0 | 4 |
| Total | 67 | 19 | 11 | 2 | 6 (105) |
Table 2.
Number and fate of BRCA1 missense substitutions in existing papers by classification
| Previous Classification | Current IARC Class | ||||
|---|---|---|---|---|---|
| Class 1 | Class 2 | Class 3 | Class 4 | Class 5 | |
| Neutral | 81 | 5 | 0 | 0 | 0 |
| Uncertain | 1 | 1 | 4 | 2 | 9 |
| Pathogenic | 0 | 0 | 0 | 0 | 9 |
| Total | 82 | 6 | 4 | 2 | 18 (112) |
2. Splicing priors and exclusion
Since work on splicing prior probabilities is in progress, we chose to limit this analysis to missense substitutions and to exclude substitutions with non-negligible probability to cause aberrant splicing (either damaging a wild-type splice site or creating an exonic de novo splice site). First, we set aside substitutions falling in the first three bases or the last three bases of each exon because these often disrupt either the canonical splice donor or the canonical splice acceptor. Second, we scored with MaxEntScan (Yeo and Burge, 2004) all of the de novo splice donors and de novo splice acceptors recorded in the databases of aberrant 3’- and 5’-splice sites (DBASS3 and DBASS5) as of 15 December 2010 (Buratti et al., 2011), as well as the corresponding canonical splice sites that had been skipped in the corresponding genes. DBASS3 (for 3’ splice sites, i.e., splice acceptors) can be found at <http://www.som.soton.ac.uk/research/geneticsdiv/dbass3/>, and DBASS5 (for 5’ splice sites, i.e., splice donors) can be found at <http://www.som.soton.ac.uk/research/geneticsdiv/dbass5/>. Third, we scored with MaxEntScan all possible single nucleotide substitutions to the open reading frames of BRCA1 and BRCA2. We then used the relationship between the scores of spliceogenic sequence variants recorded in DBASS3 and DBASS5 and all possible substitutions in BRCA1 and BRCA2 to determine thresholds below which we could consider the probability to create either a de novo donor or a de novo acceptor to be negligible. Note that MaxEntScan was used for this analysis for three reasons: (1) Houdayer et al. found MaxEntScan to be the single most accurate program for analysis of splice donor sequence variants (Houdayer et al., 2008), (2) we are unaware of evidence that any program is significantly more accurate than MaxEntScan for analysis of splice acceptor sequence variants, and (3) we were able to use the program to analyze all possible single nucleotide substitutions across the coding exons and proximal splice junction regions of BRCA1 and BRCA2.
3. Observational data
For each sequence variant analyzed in the papers that we found to contain methodologically valid integrated evaluations, we combined the prior probabilities described above with the published odds in favor of causality determined from segregation, personal and family history, co-occurrence, and/ or tumor immunohistochemistry to arrive at a posterior probability. For sequence variants assessed in more than one paper, we (i) retained the observational data from the chronologically first result that yielded the most extreme classification, and (ii) checked for examples of conflicting results. We would interpret a variant as having a conflicting result if it received a posterior probability of less than 5% on the basis of one paper’s analysis and a posterior probability of more than 95% on the basis of another paper’s analysis (there were none). Data type point estimates were retained as follows:
Segregation likelihood ratios were retained without modification.
Calculated personal and family history likelihood ratios were retained without modification. If personal and family history data were described but a likelihood ratio not calculated, we used Table 2 from Easton et al. (2007) (Easton et al., 2007) to calculate a likelihood ratio. When data from one individual (or family) fit multiple categories, we used the result from the single most severe category. To avoid issues of non-independence, we did not multiply results from multiple proband categories or multiple family history categories. When appropriate, we did multiply results from the proband category with results from the family history category.
Co-occurrence likelihood ratios were retained without modification.
Immunohistochemistry odds ratios from breast tumors were recalculated from estrogen receptor status, cytokeratin profile (if available) and tumor grade per the analysis underlying Spurdle et al. (Spurdle et al., 2008). Immunohistochemistry odds ratios from ovarian tumors were calculated according to Spearman et al. (Spearman et al., 2008).
For each sequence variant included, the paper that presented these data was recorded as the primary reference, the individual likelihood ratios were included in the database, and the likelihoods multiplied together to obtain the product of likelihood ratios. For each individual sequence variant included, the reference that provided the prior probability was recorded as the secondary reference, the prior was recorded, and the posterior probability calculated from the prior and product of likelihood ratios according to Bayes’ rule. The posterior probability point estimate was then interpreted into a qualitative classification according to the IARC 5-category classifier (Plon et al., 2008).
4. LOVD programming
Our BRCA genes Ex-UV database was built on LOVD v.2.0 Build 22, with several modifications. One shortcoming to manual data entry is the potential for data entry errors. The LOVD system authorizes external modules to work within its pages (such as the embedded modules showmaxdbid, Mutalyzer, and reCaptcha), allowing us to program internal checks to limit such errors. We implemented an LOVD module that, given a BRCA gene nucleotide substitution in HGVS nomenclature, auto-fills the correct exon number, HGVS amino acid substitution designation, and BIC nomenclature nucleotide substitution. The same module calculates posterior probabilities from prior probabilities and likelihood ratios, and then picks IARC class based on the posterior probability. This module is coded in a combination of PHP and Javascript: the PHP part is used to fetch the information to auto-fill and to display data to the user in a pop-up, and Javascript is used to allow the pop-up and the LOVD submission page to communicate with each other. With this combination, one click closes the pop-up and sends the data directly in the relevant fields of the submission page. As the submitter is not required to use these modules, we also wrote a script that finds all entries where the missense, splice, and combined prior are not complete or are incompatible, and that checks that the posterior probability and IARC class are compatible. This feature is only usable by the database administrator and provides a regular check on database integrity. The PHP code for the checker is similar to that of the auto-fill module, the main difference being that this is applied to batches of variants within the database.
We have also modified the LOVD variant display page to change what would by default have been text-only “Primary Reference”, “Secondary Reference” and “IARC class” data columns to hyperlinks. To do this, we modified the source code of the variants list display page to recognize each paper and each “IARC class” text string within the database, and then changed these into HTML hyperlinks. At first glance, the more direct approach would have been to directly save these HTML hyperlinks in the underlying MySQL database fields, allowing the LOVD variant list display page to simply fetch the MySQL database and display the data. However, security features of the LOVD system consider this sort of modification forbidden cross-site scripting. Nonetheless, using hyperlinks both decreases the opportunity for data entry errors and assists users in locating the primary literature associated with the data populating our resource.
To simplify initial population of the BRCA Ex-UV database, we have limited our initial analysis to the set of single nucleotide missense substitutions to the open reading frames of BRCA1 and BRCA2 for which we can provide both sequence analysis based prior probabilities and observational data summarized as LRs in favor of pathogenicity. There are two main routes by which an open reading frame single nucleotide missense substitution can be pathogenic: the underlying nucleotide substitution can interfere with splicing or the missense substitution can interfere with a key element of protein function. Accordingly, a sequence analysis-based prior probability should explicitly incorporate two elements: (1) missense effect on the protein, and (2) nucleotide substitution effect on the splicing. The prior probabilities for missense effect on the protein are based on a combination of position in the proteins and, for substitutions that fall in one of the key domains of the proteins where there is clear evidence that missense loss of function can be pathogenic, an in silico analysis of cross-species sequence conservation and relative missense substitution severity (Easton et al., 2007; Tavtigian et al., 2008). For substitutions falling outside of the key domains and not near a splice junction, the measured prior probability was 0.00 (95% CI 0.00 - 0.04)(Easton et al., 2007). Because a prior of 0.00 cannot be used in a Bayesian calculation, we had used 0.01 for this prior. In the interest of making the priors slightly more conservative, we re-assign this prior to the midpoint of the confidence interval, 0.02. For substitutions falling within the key functional domains but at evolutionary variable positions and assigned the relatively benign score of C0 by our missense analysis program Align-GVGD, the measured prior probability was 0.00 (95% CI 0.00 - 0.06)(Tavtigian et al., 2008). For the same reason as above, we re-assign the prior for this set of substitutions to the midpoint of their confidence interval, 0.03.
We then populated the resulting LOVD with the data defined under subsections 1-3, above. The database is available at <http://brca.iarc.fr/LOVD>.
RESULTS AND DISCUSSION
The LOVD system provides a proven, flexible environment for the creation of locus-specific databases. We designed the BRCA Ex-UV database so that, in the views that list sequence variants, the leftmost seven columns provide the minimal information that a clinician needs in order to know the status of a missense substitution included in the database: the substitution’s name in HGVS and BIC nomenclature, its posterior probability, its classification in the 5-class IARC system, and two literature references on which that classification is based. The “IARC class” link leads to a screenshot of the classification table and summary clinical recommendations from Plon et al., 2008 (Plon et al., 2008). The literature reference links lead to the paper in which the prior probability in favor of pathogenicity is derived and the paper which includes the observational data that contribute to classification.
Scrolling further to the right reveals columns that detail the prior probabilities based on expected missense substitution effect and expected effect on splicing, LRs in favor of pathogenicity for the observational data included in the classification model, a column that will eventually capture functional assay data, and the product of these LRs. By organizing the data in this way, we strive to make the database an accessible and efficient resource for a wide-ranging audience of clinicians, researchers, and laboratory staff.
Most of the empirical prior probability measurements that have been made on BRCA1 and BRCA2 sequence variants actually measure the combination of missense and splice effects. Since the vast majority of substitutions are assigned a prior of 0.02 or 0.03 (previously 0.01) (Easton et al., 2007; Tavtigian et al., 2008), it follows that the splice effects prior for the vast majority of substitutions is ≤0.03. Therefore, we sought to make a sequence analysis-based definition of this subset.
Using MaxEntScan, we scored all of the sequence variants in the DBASS3 and DBASS5 databases that create de novo splice junctions within an exon. In doing so, we recorded the score of the normal sequence at which the de novo splice happens (after mutation), the score of the mutant sequence, and the score of the canonical splice junction that is replaced by the de novo splice event. A table of these scores is available from SVT on request. We then used MaxEntScan to score all possible single nucleotide substitutions to the open reading frames of BRCA1 and BRCA2, both from the point of view of de novo donor and de novo acceptor. The graph in Figure 1 displays the distribution of MaxEntScan donor scores for the de novo donors in the DBASS5 database. The graph also displays the de novo donor scores for all possible single nucleotide substitutions to the segments of BRCA1 and BRCA2 that are both extremely unlikely to harbor a variant that damages a wild type splice junction (i.e., sequence variants falling within 3 bp of a canonical splice junction are excluded) and unlikely to harbor a pathogenic missense substitution (i.e., excluding substitutions falling within the BRCA1 RING or BRCT domains, or the BRCA2 DNA binding domain or PALB2 interaction domain). Concatenated, these gene segments include 79.5% of all possible single nucleotide substitutions to the open reading frames of BRCA1 and BRCA2, and the empirically measured prior probability for substitutions falling in these regions – as an undifferentiated group – is 0.0 (95% confidence interval 0.00-0.04) (Easton et al., 2007). This graph (Figure 1) is also annotated with the position of the 10th, percentile MaxEntScan score for sequence variants recorded in DBASS5 that create de novo splice donors. In other words, 10% of DBASS5 sequence variants reported to create de novo donors have MaxEntScan scores at or below the 10th percentile demarcation. From this annotated graph, it is clear that the distribution MaxEntScan scores for variants recorded in the DBASS5 database that create de novo donors is well resolved from the distribution MaxEntScan scores for BRCA gene substitutions, and that the vast majority of BRCA gene substitutions have MaxEntScan scores that are not indicative of de novo donor creation.
Figure 1.
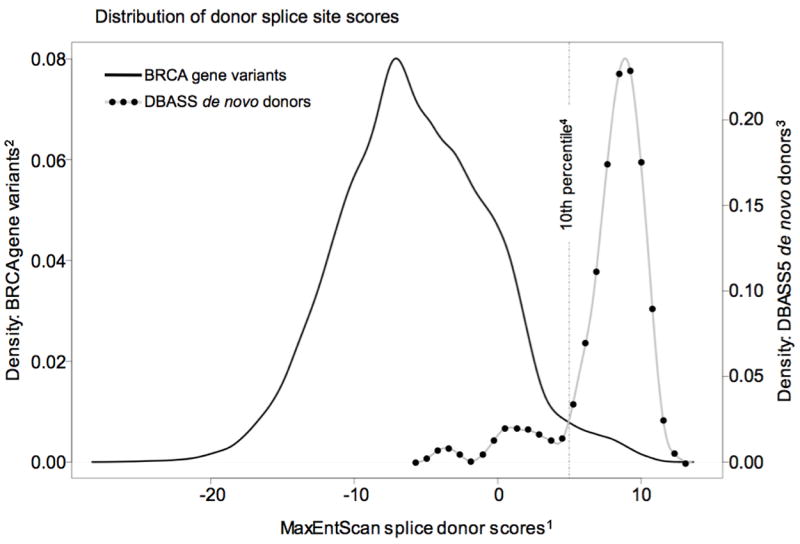
For splice donors, the program MaxEntScan (Yeo and Burge, 2004) scores sequences of length nine nucleotides for their fitness as a splice donor under the assumption of a splice junction following the third nucleotide of the given sequence. Every possible single nucleotide substitution to the coding exons of BRCA1 and BRCA2 was scored nine times, i.e, with the substitution placed from the first to the ninth nucleotide of the program’s scoring window. 1) For each possible substitution, the given MaxEntScan splice donor score is the highest (most fit as a splice donor) of those nine scores. 2) Density: BRCA gene variants is approximately the fraction of MaxEntScan splice donor scores for BRCA variants that fall in an X-axis interval of one MaxEntScan unit; the integral of this curve is exactly 1.00. 3) Density: DBASS5 de novo donors is approximately the fraction of MaxEntScan splice donor scores for de novo donors from the DBASS5 database that fall in an X-axis interval of one MaxEntScan unit; the integral of this curve is exactly 1.00. 4) The 10th percentile is the MaxEntScan score below which fall only 10% of de novo donor mutations recorded in the DBASS5 database. Note that it is a coincidence that the BRCA gene variants curve and DBASS5 curve cross at the 10th percentile demarcation.
For the dataset as a whole, the ratio of DBASS5 de novo donors to possible BRCA gene substitutions is (62 / 37,782)=0.0016. For the <10th percentile subset, this ratio is (7 / 36,836)=0.00019 – almost an order of magnitude lower than for the entire group. The midpoint of the 95% confidence interval of the probability in favor of pathogenicity for single nucleotide substitutions in the dataset as a whole is 0.02. Given these data, the probability that a single nucleotide substitution drawn from the ≤10 th percentile subset will give rise to a pathogenic de novo donor must be ≪0.02 and consequently negligible from the point of view of determining an operationally useful prior probability. Therefore, to avoid having to determine splicing priors for potentially more severe categories at this time, we restrict initial population of the Ex-UV database to exonic substitutions that fall more than three bp from the end of an exon and that reside within the MaxEntScan <10th percentile categories for both de novo donors and de novo acceptors. Thus defined, this dataset still contains >95% of all possible single nucleotide substitutions to the open reading frames of BRCA1 and BRCA2 but nonetheless excludes the vast majority of substitutions that either damage a wild-type splice junction or create a de novo splice junction.
Populating the database. From 12 papers (Chenevix-Trench et al., 2006; Easton et al., 2007; Farrugia et al., 2008; Goldgar et al., 2004; Lovelock et al., 2006; Osorio et al., 2007; Spearman et al., 2008; Spurdle et al., 2008; Sweet et al., 2010; Tavtigian et al., 2006; Tischkowitz et al., 2008; Wu et al., 2005) we found 248 missense substitutions that had been subjected to an integrated evaluation. Two older articles were also used to include data relevant to the assessment of missense substitutions that were widely considered classified before the integrated evaluation was developed (Miki et al., 1994; Wooster et al., 1995). Of these 248 missense substitutions, 10 were set aside because they fall in the first three or last three bp of an exon and consequently have non-negligible prior probabilities to interfere with a canonical splice junction. A further 21 were set aside because their MaxEntScan scores were indicative of non-negligible probability to create a de novo splice junction (Table 1). The remaining 217 substitutions were then included in the LOVD; 112 from BRCA1 (Table 2) and 105 from BRCA2 (Table 3).
Table 1.
Number and status of excluded missense substitutions
| Previous Classification | Reason for exclusion | |||
|---|---|---|---|---|
| Exon ends | Potential de novo splice | |||
| First 3 bp | Last 3 bp | Donor | Acceptor | |
| Neutral | 0 | 3 | 7 | 0 |
| Uncertain | 0 | 5 | 10 | 2 |
| Pathogenic | 0 | 2 | 2 | 0 |
| Total | 0 | 10 | 19 | 2 (31) |
Reassuringly, in moving from the older 3-category classification system to the 5-category IARC classification system (Plon et al., 2008), no sequence variants previously classified as “neutral” were reclassified as Class 4 or 5 (likely pathogenic or definitely pathogenic, respectively). Similarly, no sequence variants previously classified as “pathogenic” were reclassified as Class 2 or 1 (likely not pathogenic or not pathogenic, respectively).
In the next several paragraphs, we describe the fates of the 217 BRCA gene missense substitutions initially included in the LOVD. The section is organized based on the qualitative classification of each variant in the primary reference, i.e., the section headed “previously pathogenic” summarizes the status in the LOVD of the substitutions that were classified as pathogenic in the primary reference. Attention is focused on those substitutions for which there was a discernable change in classification, i.e., from “neutral” to Class 2 or Class 3 or from “uncertain” to a more clinically informative Class.
Previously pathogenic. Of the 13 non-spliceogenic missense substitutions previously classified as pathogenic, all 13 had posterior probabilities of >0.99 and consequently fell into IARC Class 5 (Tables 2 and 3).
Previously neutral. Of the 164 non-spliceogenic missense substitutions previously classified as neutral, 147 had posterior probabilities of <0.001 and consequently fell into IARC Class 1. An additional 15 had posterior probabilities between 0.001 and 0.05; these fell into IARC Class 2.
Two BRCA2 missense substitutions, p.S869L (c.2606C>T) and p.A1170V (c.3509C>T) moved from a published classification of neutral (Chenevix-Trench et al., 2006; Spearman et al., 2008) to IARC Class 3 “uncertain”.
One reason for these changes in previously neutral classifications is that the classification systems employed by Chenevix-Trench et al (2006) and Spearman et al (2008) included loss of heterozygosity at the susceptibility gene locus. These two studies considered loss of the wild-type allele evidence in favor of pathogenicity, lack of LOH modest evidence against pathogenicity, and LOH of the mutant allele stronger evidence against pathogenicity. However, recent evidence throwing doubt on the evidence for LOH as a good predictor of missense substitution pathogenicity in BRCA1 or BRCA2 has led to exclusion of this type of data from the classification model employed here (Hofstra et al., 2008). In Chenevix-Trench et al (2006), LOH data provided odds of pathogenicity of 0.02 for p.S869L. Exclusion of these data is the major reason why the posterior probability for this substitution now rests at 0.104, slightly above the threshold for Class 2. In Spearman et al (2008), LOH data provided odds of pathogenicity of 0.067 for p.A1170V. However, there was a second issue in this analysis. Spearman et al. reported p.A1170V from two apparently unrelated subjects, one breast cancer case and one ovarian cancer case. In combining the resulting data, they used the former sequence analysis based prior probability for this substitution, 0.01, twice. For any given sequence variant, the prior can only be used once in the integrated evaluation. Excluding both LOH data and one instance of the prior removes a factor of 0.0067 in favor of pathogenicity; the posterior probability for this substitution now rests at 0.230.
Of the 40 non-spliceogenic missense substitutions previously classified as uncertain (including “suspect neutral” and “suspect pathogenic” or similarly designated categories), 12 moved to one of the more informative neutral classes, Class 1 or Class 2, and 15 moved to more informative pathogenic classes, Class 4 or Class 5. Thus only 13 of the non-spliceogenic sequence variants previously assigned “uncertain” rest in IARC Class 3 “uncertain”, whereas 27 are now reclassified to one of the two neutral or one of the two pathogenic categories. Below, we outline the informative reclassifications of these 27 substitutions by primary reference, highlighting the reasons behind their reclassification using the integrated evaluation model.
The largest group was a set of 11 missense substitutions analyzed by Easton et al (Easton et al., 2007) that had genetic data of between 23:1 and 350:1 in favor of pathogenicity. In combination with their sequence analysis based prior probabilities, three of these were reclassified as Class 4 (BRCA1 p.M18T and p.M1689R plus BRCA2 p.D3095E) and eight were reclassified as Class 5 (BRCA1 p.T1685A, p.T1685I, p.S1715R, p.G1738R, p.L1764P, p.I1766S, plus BRCA2 p.W2626C and p.T2722R).
A group of four BRCA2 missense substitutions assessed by Spearman et al. (Spearman et al., 2008) are now classified as Class 2. Two of these, p.K1434I and p.A2351G were originally placed in Spearman et al’s “neutral suspected category”; for these, our analysis simply confirms Spearman et al’s. Of the remaining two, p.D1352Y moved into Class 2 because our model excluded LOH data that had provided some evidence in favor of pathogenicity, and p.R2418G moved into Class 2 because our “likely not pathogenic” category extends up to a posterior probability of 0.05 whereas the suspect neutral category used by Spearman et al was more restrictive.
Four substitutions classified as uncertain by Spurdle et al (Spurdle et al., 2008) also move to Class 2. For three of these, BRCA1 p.V920A, BRCA2 p.K607T, and BRCA2 p.S1760A, the published posterior probabilities were within the likely not pathogenic range, but the category did not formally exist when the paper was published. For the fourth, BRCA2 p.Q2858R, the updated prior probability model provides a lower prior probability than that used in Spurdle et al, resulting in a posterior probability in the Class 2 range. The updated prior probability model also moved two BRCA2 missense substitutions assessed by Chenevix-Trench et al, p.R2318Q and p.A2770T from uncertain to Class 2. Additionally, one substitution analyzed by Tavtigian et al. (Tavtigian et al., 2006), BRCA1 p.V772A, moved from uncertain to Class 1 for the same reason.
Three BRCA1 RING domain missense substitutions analyzed by Sweet et al (Sweet et al., 2010), p.L22S, p.C44Y, and p.C44S move from their “likely deleterious” category to Class 5. In each case, the main reason for the upgraded classification is that their paper reported – but did not formally use – summary family history data. We made a conservative interpretation of the given family history data through Table 2 of Easton et al (Easton et al., 2007); these additional data provided odds of between 2:1 and 8:1 in favor of pathogenicity and pushed the substitutions into Class 5.
Finally, two BRCA2 missense substitutions originally classified as uncertain by Farrugia et al (Farrugia et al., 2008) were reclassified: p.N319T moved to Class 1 and p.L2647P moved to Class 4. The underlying basis for reclassification was that Farrugia et al. did not employ a sequence analysis-based prior probability in their classification model. For p.N319T, the prior is 0.02 and for p.L2647P the prior is 0.81 (Tavtigian et al., 2008); calculation of posterior probabilities from these priors and the genetic data provided by Farrugia et al. results in the new classifications.
Currently, it is not possible to directly measure the performance of the integrated evaluation. In time, some re-classifications will certainly occur. Once this happens, it will become possible to estimate the frequency of reclassification from one class to a contradictory class, and it will also become possible to assess the weaknesses of individual elements of the integrated evaluation. Nonetheless, we note that even the maximum possible change in the prior (from 0.02 to 0.81, or vice versa) cannot result in a sequence variant moving from Class 1 or 2 to Class 4 or 5 (or vice versa). Moreover, there are no examples of a Class 1 or Class 2 variant where removing the “most indicative of neutral” individual LR measurement results in a switch to Class 4 or Class 5 (obviously, some variants would move to Class 3, uncertain). Similarly, there are no examples of a Class 4 or Class 5 variant where removing “most indicative of pathogenic” individual LR measurement results in a switch to Class 1 or Class 2.
Moving forward, we intend to make, at minimum, annual updates to this database with results from bona fide integrated evaluations that have been published since the previous update. Beyond updates, we see three obvious directions for the evolution of this database. First, once sequence analysis-based splicing prior probabilities have been worked out, it will become possible to include sequence variants that have non-negligible probability to either damage a canonical splice junction or create a de novo junction. Second, a number of BRCA1 and BRCA2 functional assays have been developed (Couch et al., 2008), but none have been calibrated so that their output can be interpreted as an odds ratio in favor of pathogenicity; once this milestone has been passed, identification of pathogenic missense substitutions should become more efficient. Finally, the Breast Cancer Information Core (BIC) database of variants in BRCA1 and BRCA2 (Szabo et al., 2000) is meant to stand as a compilation of all sequence variants observed in these two genes and does record their classification when available. Accordingly, it will be appropriate to link from the BIC database to our Ex-UV database; as URLs for individual variants in our database can be generated algorithmically from the HGVS name of the variant, this will be quite easy to do.
The BRCA gene Ex-UV LOVD described here houses two of the largest single-gene collections of securely classified missense substitutions. As such, the dataset may contribute to future refinements of in silico missense substitution analysis algorithms. More importantly, it stands as a resource to which clinical cancer geneticists and genetic counselors can refer to see whether specific BRCA1 or BRCA2 missense substitutions observed in their patients have been classified.
Acknowledgments
We would like to thank all of the members of the Breast Information Consortium (BIC) who are not listed as authors for their continuing encouragement. We would like to thank Amanda Spurdle and Frans Hogervorst for help with summarizing potentially spliceogenic sequence variants. This work was supported by the National Cancer Institute, National Institutes of Health (NIH) grant RO1 CA116167 and Canadian Institute for Health Research (CIHR) grant CRN-87521-IC089832. The content of this manuscript does not necessarily reflect the views or policies of the NCI, nor does mention of trade names, commercial products, or organizations imply endorsement by the US Government.
References
- Abkevich V, Zharkikh A, Deffenbaugh AM, Frank D, Chen Y, Shattuck D, Skolnick MH, Gutin A, Tavtigian SV. Analysis of missense variation in human BRCA1 in the context of interspecific sequence variation. J Med Genet. 2004;41:492–507. doi: 10.1136/jmg.2003.015867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratti E, Chivers M, Hwang G, Vorechovsky I. DBASS3 and DBASS5: databases of aberrant 3’- and 5’-splice sites. Nucleic Acids Res. 2011;39(Database issue):D86–91. doi: 10.1093/nar/gkq887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chenevix-Trench G, Healey S, Lakhani S, Waring P, Cummings M, Brinkworth R, Deffenbaugh AM, Burbidge LA, Pruss D, Judkins T, Scholl T, Bekessy A, Marsh A, Lovelock P, Wong M, Tesoriero A, Renard H, Southey M, Hopper JL, Yannoukakos K, Brown M, Easton D, Tavtigian SV, Goldgar D, Spurdle AB. Genetic and histopathologic evaluation of BRCA1 and BRCA2 DNA sequence variants of unknown clinical significance. Cancer Res. 2006;66:2019–2027. doi: 10.1158/0008-5472.CAN-05-3546. [DOI] [PubMed] [Google Scholar]
- Couch FJ, Rasmussen LJ, Hofstra R, Monteiro AN, Greenblatt MS, de Wind N. Assessment of functional effects of unclassified genetic variants. Hum Mutat. 2008;29:1314–1326. doi: 10.1002/humu.20899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Easton DF, Deffenbaugh AM, Pruss D, Frye C, Wenstrup RJ, Allen-Brady K, Tavtigian SV, Monteiro AN, Iversen ES, Couch FJ, Goldgar DE. A Systematic Genetic Assessment of 1,433 Sequence Variants of Unknown Clinical Significance in the BRCA1 and BRCA2 Breast Cancer-Predisposition Genes. Am J Hum Genet. 2007;81:873–883. doi: 10.1086/521032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers B, Jonkers J. Mouse models of BRCA1 and BRCA2 deficiency: past lessons, current understanding and future prospects. Oncogene. 2006;25:5885–5897. doi: 10.1038/sj.onc.1209871. [DOI] [PubMed] [Google Scholar]
- Farrugia DJ, Agarwal MK, Pankratz VS, Deffenbaugh AM, Pruss D, Frye C, Wadum L, Johnson K, Mentlick J, Tavtigian SV, Goldgar DE, Couch FJ. Functional assays for classification of BRCA2 variants of uncertain significance. Cancer Res. 2008;68:3523–3531. doi: 10.1158/0008-5472.CAN-07-1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fokkema IF, den Dunnen JT, Taschner PE. LOVD: easy creation of a locus-specific sequence variation database using an “LSDB-in-a-box” approach. Hum Mutat. 2005;26:63–68. doi: 10.1002/humu.20201. [DOI] [PubMed] [Google Scholar]
- Goldgar DE, Easton DF, Deffenbaugh AM, Monteiro AN, Tavtigian SV, Couch FJ. Integrated evaluation of DNA sequence variants of unknown clinical significance: application to BRCA1 and BRCA2. Am J Hum Genet. 2004;75:535–544. doi: 10.1086/424388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldgar DE, Easton DF, Byrnes GB, Spurdle AB, Iversen ES, Greenblatt MS. Genetic evidence and integration of various data sources for classifying uncertain variants into a single model. Hum Mutat. 2008;29:1265–1272. doi: 10.1002/humu.20897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofstra RM, Spurdle AB, Eccles D, Foulkes WD, de Wind N, Hoogerbrugge N, Hogervorst FB. Tumor characteristics as an analytic tool for classifying genetic variants of uncertain clinical significance. Hum Mutat. 2008;29:1292–1303. doi: 10.1002/humu.20894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houdayer C, Dehainault C, Mattler C, Michaux D, Caux-Moncoutier V, Pages-Berhouet S, d’Enghien CD, Lauge A, Castera L, Gauthier-Villars M, Stoppa-Lyonnet D. Evaluation of in silico splice tools for decision-making in molecular diagnosis. Hum Mutat. 2008;29:975–982. doi: 10.1002/humu.20765. [DOI] [PubMed] [Google Scholar]
- Howlett NG, Taniguchi T, Olson S, Cox B, Waisfisz Q, De Die-Smulders C, Persky N, Grompe M, Joenje H, Pals G, Ikeda H, Fox EA, D’Andrea AD. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297:606–609. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- Judkins T, Hendrickson BC, Deffenbaugh AM, Eliason K, Leclair B, Norton MJ, Ward BE, Pruss D, Scholl T. Application of embryonic lethal or other obvious phenotypes to characterize the clinical significance of genetic variants found in trans with known deleterious mutations. Cancer Res. 2005;65:10096–10103. doi: 10.1158/0008-5472.CAN-05-1241. [DOI] [PubMed] [Google Scholar]
- Lovelock PK, Healey S, Au W, Sum EY, Tesoriero A, Wong EM, Hinson S, Brinkworth R, Bekessy A, Diez O, Izatt L, Solomon E, Jenkins M, Renard H, Hopper J, Waring P, Tavtigian SV, Goldgar D, Lindeman GJ, Visvader JE, Couch FJ, Henderson BR, Southey M, Chenevix-Trench G, Spurdle AB, Brown MA. Genetic, functional, and histopathological evaluation of two C-terminal BRCA1 missense variants. J Med Genet. 2006;43:74–83. doi: 10.1136/jmg.2005.033258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miki Y, Swensen J, Shattuck-Eidens D, Futreal PA, Harshman K, Tavtigian S, Liu Q, Cochran C, Bennett LM, Ding W, Bell R, Rosenthal J, Hussey C, Tran T, McClure M, Frye C, Hattier T, Phelps R, Haugen-Strano A, Katcher H, Yakumo K, Gholami Z, Shaffer D, Stone S, Bayer S, Wray C, Bogden R, Dayananth P, Ward J, Tonin P, Narod S, Bristow PK, Norris FH, Helvering H, Morrison P, Rosteck P, Lai M, Barrett JC, Lewis C, Neuhausen S, Cannon-Albright L, Goldgar D, Wiseman R, Kamb A, Skolnick MH. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994;266:66–71. doi: 10.1126/science.7545954. [DOI] [PubMed] [Google Scholar]
- Osorio A, Milne RL, Honrado E, Barroso A, Diez O, Salazar R, de la Hoya M, Vega A, Benitez J. Classification of missense variants of unknown significance in BRCA1 based on clinical and tumor information. Hum Mutat. 2007;28:477–485. doi: 10.1002/humu.20470. [DOI] [PubMed] [Google Scholar]
- Plon SE, Eccles DM, Easton D, Foulkes WD, Genuardi M, Greenblatt MS, Hogervorst FB, Hoogerbrugge N, Spurdle AB, Tavtigian SV. Sequence variant classification and reporting: recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum Mutat. 2008;29:1282–1291. doi: 10.1002/humu.20880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spearman AD, Sweet K, Zhou XP, McLennan J, Couch FJ, Toland AE. Clinically applicable models to characterize BRCA1 and BRCA2 variants of uncertain significance. J Clin Oncol. 2008;26:5393–5400. doi: 10.1200/JCO.2008.17.8228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurdle AB, Lakhani SR, Healey S, Parry S, Da Silva LM, Brinkworth R, Hopper JL, Brown MA, Babikyan D, Chenevix-Trench G, Tavtigian SV, Goldgar DE. Clinical classification of BRCA1 and BRCA2 DNA sequence variants: the value of cytokeratin profiles and evolutionary analysis--a report from the kConFab Investigators. J Clin Oncol. 2008;26:1657–1663. doi: 10.1200/JCO.2007.13.2779. [DOI] [PubMed] [Google Scholar]
- Spurdle AB. Clinical relevance of rare germline sequence variants in cancer genes: evolution and application of classification models. Curr Opin Genet Dev. 2010;20:315–323. doi: 10.1016/j.gde.2010.03.009. [DOI] [PubMed] [Google Scholar]
- Sweet K, Senter L, Pilarski R, Wei L, Toland AE. Characterization of BRCA1 ring finger variants of uncertain significance. Breast Cancer Res Treat. 2010;119:737–743. doi: 10.1007/s10549-009-0438-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo C, Masiello A, Ryan JF, Brody LC. The breast cancer information core: database design, structure, and scope. Hum Mutat. 2000;16:123–131. doi: 10.1002/1098-1004(200008)16:2<123::AID-HUMU4>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Tavtigian SV, Deffenbaugh AM, Yin L, Judkins T, Scholl T, Samollow PB, de Silva D, Zharkikh A, Thomas A. Comprehensive statistical study of 452 BRCA1 missense substitutions with classification of eight recurrent substitutions as neutral. J Med Genet. 2006;43:295–305. doi: 10.1136/jmg.2005.033878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tavtigian SV, Byrnes GB, Goldgar DE, Thomas A. Classification of rare missense substitutions, using risk surfaces, with genetic- and molecular-epidemiology applications. Hum Mutat. 2008;29:1342–1354. doi: 10.1002/humu.20896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson D, Easton DF, Goldgar DE. A full-likelihood method for the evaluation of causality of sequence variants from family data. Am J Hum Genet. 2003;73:652–655. doi: 10.1086/378100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tischkowitz M, Hamel N, Carvalho MA, Birrane G, Soni A, van Beers EH, Joosse SA, Wong N, Novak D, Quenneville LA, Grist SA, Nederlof PM, Goldgar DE, Tavtigian SV, Monteiro AN, Ladias JA, Foulkes WD. Pathogenicity of the BRCA1 missense variant M1775K is determined by the disruption of the BRCT phosphopeptide-binding pocket: a multi-modal approach. Eur J Hum Genet. 2008;16:820–832. doi: 10.1038/ejhg.2008.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner JE, Tolar J, Levran O, Scholl T, Deffenbaugh A, Sa- tagopan J, Ben-Porat L, Mah K, Batish SD, Kutler DI, Mac- Millan ML, Hanenberg H, Auerbach AD. Germline mutations in BRCA2: shared genetic susceptibility to breast cancer, early onset leukemia and Fanconi anemia. Blood. 2004;103:3226–3229. doi: 10.1182/blood-2003-09-3138. [DOI] [PubMed] [Google Scholar]
- Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, Collins N, Gregory S, Gumbs C, Micklem G, Barfoot R, Hamoudi R, Patel S, Rice C, Biggs P, Hashim Y, Smith A, Connor F, Arason A, Gudmundsson A, Ficenec D, Kelsell D, Ford D, Tonin P, Biship DT, Spur NK, Ponder BAJ, Eeles R, Peto J, Devilee P, Cornelisse C, Lynch H, Narod S, Lenoir G, Egilsson V, Barkadottir RB, Easton DF, Bentley DR, Futreal PA, Ashworth A, Stratton MR. Identification of the breast cancer susceptibility gene BRCA2. Nature. 1995;378:789–792. doi: 10.1038/378789a0. [DOI] [PubMed] [Google Scholar]
- Wu K, Hinson SR, Ohashi A, Farrugia D, Wendt P, Tavtigian SV, Deffenbaugh A, Goldgar D, Couch FJ. Functional evaluation and cancer risk assessment of BRCA2 unclassified variants. Cancer Res. 2005;65:417–426. [PubMed] [Google Scholar]
- Yeo G, Burge CB. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol. 2004;11:377–394. doi: 10.1089/1066527041410418. [DOI] [PubMed] [Google Scholar]