

Abstract
The sugarcane root endophyte Trichoderma virens 223 holds enormous potential as a sustainable alternative to chemical pesticides in the control of sugarcane diseases. Its efficacy as a biocontrol agent is thought to be associated with its production of chitinase enzymes, including N-acetyl-ß-D-glucosaminidases, chitobiosidases and endochitinases. We used targeted gene deletion and RNA-dependent gene silencing strategies to disrupt N-acetyl-ß-D-glucosaminidase and endochitinase activities of the fungus, and to determine their roles in the biocontrol of soil-borne plant pathogens. The loss of N-acetyl-ß-D-glucosaminidase activities was dispensable for biocontrol of the plurivorous damping-off pathogens Rhizoctonia solani and Sclerotinia sclerotiorum, and of the sugarcane pathogen Ceratocystis paradoxa, the causal agent of pineapple disease. Similarly, suppression of endochitinase activities had no effect on R. solani and S. sclerotiorum disease control, but had a pronounced effect on the ability of T. virens 223 to control pineapple disease. Our work demonstrates a critical requirement for T. virens 223 endochitinase activity in the biocontrol of C. paradoxa sugarcane disease, but not for general antagonism of other soil pathogens. This may reflect its lifestyle as a sugarcane root endophyte.
Introduction
Sugarcane is an economically important crop that is grown in more than 100 tropical countries, contributing not only to economic growth and development but also to global energy security in the form of sugarcane-fermented bioethanol. In 2010, the sugarcane sector contributed US$50 billion to the gross domestic product of Brazil, equivalent to almost 2.4% of the entire economy [1]. ‘Pineapple’ disease of sugarcane is caused by the soil-borne fungus Ceratocystis paradoxa (Dade) and is a devastating disease that causes complete loss of sugarcane setts, and occurs in almost all countries where sugarcane is grown. The disease affects sugarcane in the first week of planting and can reduce the germination of setts by up to 47% and subsequent cane yields by 31–35% [2]. Control of the disease is considered a priority especially where soil inoculum levels are high. Indeed, procedures that favour germination of buds and emergence of young shoots can dramatically improve crop yields. At present, systemic fungicides are used to protect against C. paradoxa, but chemical control is expensive and environmentally damaging. Sustainable methods of disease control are therefore urgently needed.
Biological control using beneficial soil microorganisms represents a sustainable means by which soil-borne pathogens can be controlled and Trichoderma species have been proposed as credible alternatives to environmentally-damaging chemicals in the control of plant diseases [3], [4]. Soil applications of Trichoderma harzianum, for example, have been found to antagonise C. paradoxa leading to increased germination of sugarcane setts and improvements in cane yields [2]. In previous work, we have demonstrated that the sugarcane root endophyte T. virens 223 is an effective antagonist of the pathogen [5], but the mechanisms of biocontrol of sugarcane pineapple disease by the fungus are currently poorly understood. Trichoderma virens is considered one of the most effective biological control agents studied to date [6], and its ability to control plant diseases has been attributed to its production of anti-fungal compounds [7]–[11] and induction of plant disease resistance [12], [13]. The contribution of chitinolytic enzymes to the anti-fungal activity of T. virens is less well understood, but chitinolytic enzymes are now regarded as key components of biocontrol by Trichoderma species.
Chitin is an integral component of the hyphal cell walls of fungal plant pathogens [14], and its enzymatic degradation allows subsequent colonization of host tissues by Trichoderma species [3], [15]. Chitinases are separated into different groups depending on their modes of action [16]. While N-acetyl-ß-D-glucosaminidases (EC 3.2.1.52) belong to glycoside hydrolase (GH) family 20 and catalyze the release of terminal, non-reducing N-acetylglucosamine (GlcNAc) residues from chitin, fungal endochitinases (EC 3.2.1.14) are members of GH family 18 and catalyze hydrolysis of the ß-1,4 linkages in chitin and chito-oligomers [16]. Although several Trichoderma chitinases have been purified and characterized [9], [17]–[24], and some of the corresponding genes cloned [24]–[32], there have been conflicting reports regarding the roles of these enzymes in biological control activities of Trichoderma species. Many studies have, for instance, ascribed biocontrol efficacies to chitinase production [9], [30], [33]–[38], while others have shown that biocontrol activities are governed by mechanisms other than chitinase production [39]–[42].
Recently, genome analysis of the T. virens strain Gv29-8 has shown that the fungus possesses two N-acetyl-ß-D-glucosaminidase-encoding genes and one additional hypothetical protein, distantly related to N-acetyl-ß-hexosaminidase [43]. In contrast, T. virens Gv29-8 contains 36 chitinase-encoding genes, the highest number yet reported in a fungal genome [43]. This high degree of redundancy therefore makes investigations of the involvement of chitinase in T. virens biocontrol very challenging. To date, only one endochitinase gene (T. harzianum cht42 = T. harzianum ech42 = T. virens ech1) has been functionally characterized. The endochitinase gene cht42 of T. harzianum was over-expressed and disrupted, resulting in enhanced and reduced biocontrol, respectively, of the root pathogen Rhizoctonia solani [44]. Transgenic cotton plants expressing the T. virens cht42 endochitinase gene were also shown to be more resistant to disease [15], [45], [46]. Despite these studies, the role of chitinolytic enzymes in the biocontrol capability of T. virens remains unclear.
In this study, we set out to investigate the role of N-acetyl-ß-D-glucosaminidases and endochitinase enzymes in the biological control of ‘pineapple’ disease of sugarcane by T. virens 223. We used targeted gene deletion analysis to study the function of two N-acetyl-ß-D-glucosaminidase-encoding genes and RNA-dependent gene silencing to impair the activity of the expanded subgroup A chitinase gene family of T. virens. We report that the biocontrol efficacy of T. virens is unaffected by loss of N-acetyl-ß-D-glucosaminidase activity, but that impairing endochitinase activity by gene silencing resulted in dramatically reduced fitness of T. virens as a biocontrol agent of sugarcane pineapple disease. Significantly, the ability of T. virens to control diseases caused by other soil-borne fungi was unaffected. When considered together, this indicates that endochitinases play a critical function in T. virens as a biocontrol agent of sugarcane disease but not as a general antagonist of other soil pathogens. This may reflect its lifestyle as a sugarcane-specific root endophyte.
Materials and Methods
Fungal Strains and Culture Conditions
The sugarcane root endophyte Trichoderma virens strain Tv.223 (GenBank accession number GQ495269) has been previously described [5], [47], [48]. Ceratocystis paradoxa, the causal agent of sugarcane pineapple disease, was supplied by Centro de Tecnologia Canavieira. The pathogen Sclerotinia sclerotiorum (GenBank accession number FJ984493) was obtained from the University of Exeter culture collection [49] and the Rhizoctonia solani strain was obtained from the Genetics of Microorganisms Laboratory (Department of Genetics/ESALQ, University of São Paulo) culture collection. Fungi were grown on potato dextrose agar (PDA, Difco) at 26°C under a 16 h photoperiod of fluorescent light.
DNA Extraction, Amplification and Sequencing of Tv.223 Chitinase Genes
Genomic DNA of T. virens strain 223 was prepared according to the method of [50]. PCR primers were designed for amplification of the two N-acetyl-ß-D-glucosaminidase-encoding genes Tvnag1 and Tvnag2 and the three endochitinase-encoding genes Tvech1, Tvech2 and Tvech3 (Table 1) using the T. virens Gv29-8 v2.0 genome available at the Joint Genome Institute (JGI) Genome Portal (http://genome.jgi-psf.org/TriviGv29_8_2/TriviGv29_8_2.home.html) as reference. PCR products were purified with the UltraClean PCR Clean-up Kit (MoBio Laboratories Inc.) and sequenced at the Human Genome Research Center, University of São Paulo, Brazil, using an automated ABI 3730 DNA Analyser (Applied Biosystems). The gene fragments were assembled using the CAP3 Sequence Assembly Program [51] and the chitinolytic function was confirmed by a BLAST search for similar sequences and also an InterPro [52] search for chitinolytic enzyme motif signatures. Trichoderma virens strain 223 genes sequenced in this work were deposited at GenBank under accession numbers JQ066769 to JQ066773.
Table 1. Details of PCR primers used in this study.
| Primer name | Sequence 5′-3′ | Target |
| Tvech1-Fw | Cagcacagaagtggcaagcttgaa | Tvech1 gene |
| Tvech1-Rv | acttggtacacacacgaattcacc | Tvech1 gene |
| Tvech1-Rv2 | tccgttgtaagtctggccaatacc | Tvech1 gene |
| Tvech2-Fw | ccatctcaagtaaacatgtcggga | Tvech2 gene |
| Tvech2-Rv | gggtgcaaacaacaagtaagcctc | Tvech2 gene |
| Tvech2-Rv2 | ttgtgggttatagcagtagctgga | Tvech2 gene |
| Tvech3-Fw | gaccagaacctactttgaaggctt | Tvech3 gene |
| Tvech3-Rv | gcctcgcagaaccatacaagacaa | Tvech3 gene |
| Tvech3-Rv2 | ttgcccagatacagtatcccagct | Tvech3 gene |
| Tvnag1-Fw | cgtagtagtgctattgccatcgct | Tvnag1 gene |
| Tvnag1-Fw2 | gatactacccagacgttcaagccg | Tvnag1 gene |
| Tvnag1-Rv | tcccggcttctcttaatccatacc | Tvnag1 gene |
| Tvnag1-Rv2 | acggccgcagtccaagtactataa | Tvnag1 gene |
| Tvnag2-Fw | Tcgtgtggcctgaccttgtcaact | Tvnag2 gene |
| Tvnag2-Fw2 | tcttcttcaagcatagctcaggca | Tvnag2 gene |
| Tvnag2-Rv | taccacactgcatactttgttccg | Tvnag2 gene |
| Tvnag2-Rv2 | ataaccttgaatccagcctccgcg | Tvnag2 gene |
| HEX1_Fw | ccggttgctgccgaaaccttcaat | Tvnag2 LF |
| HEX1.M13F_Rev | gtcgtgactgggaaaaccctggcggttgccggttgaggagtgtttcgg | Tvnag2 LF |
| HEX2.M13R_Fw | tcctgtgtgaaattgttatccgctccgattacgcagctgtggtgtagt | Tvnag2 RF |
| HEX2_Rev | ggccgaaatgatgaccctttgcgg | Tvnag2 RF |
| Chito2_50.1 | gcttcgactgctactgtactccgt | Tvnag1 LF |
| Chito2_m13F | gtcgtgactgggaaaaccctggcggctgaaagccaatgcggcaatcgc | Tvnag1 LF |
| Chito2_m13R | tcctgtgtgaaattgttatccgctcgctgttcacctaaggggagagtc | Tvnag1 RF |
| Chito2_30.1 | aacagcatacactgggaagactcg | Tvnag1 RF |
| HY split | ggatgcctccgctcgaagta | hph gene |
| YG split | cgttgcaagacctgcctgaa | hph gene |
| IL split | tctggttgtattctcaggac | ilv gene |
| LV split | cataccaagcatgtgcagtg | ilv gene |
| M13 F | cgccagggttttcccagtcacgac | binding site of pUC/M13 forward sequencing primer |
| M13 R | agcggataacaatttcacacagga | binding site of pUC/M13 reverse sequencing primer |
| 1F.endochi.XhoI | cgccaaatctagtctcgagct | Tvech2 gene |
| 2R.endochi.HindIII | aagcttacccctccccgatgccattat | Tvech2 gene |
| 3F.endochi.KpnI | ggtacccgccaaatctagtctcgagct | Tvech2 gene |
| 4R.endochi.SphI | gcatgcacccctccccgatgccattat | Tvech2 gene |
| 1F.SpeI.ToxA | actagtcatggaggagttctgtacgcgc | ToxA promoter |
| 2R.fusion.ToxA | agcagctcgagactagatttggcggacctatattcattcaatgtcagc | ToxA promoter |
| 3F.fusion.ToxA | gctgacattgaatgaatataggtccgccaaatctagtctcgagctgct | Tvech2 gene |
| Hex probe F | accagacggtccaagttacctaca | Tvnag2 gene |
| Hex probe R | tgtttgccttatactcgtcgcctc | Tvnag2 gene |
| Chito2 probe F | cgtgctcttcattgaccaggctgt | Tvnag1 gene |
| Chito2 probe R | gctcggagtcgttgacgttgagct | Tvnag1 gene |
| Hyg probe 2F | aagcctgaactcaccgcgacgtct | hph gene |
| Hyg probe 3R | tgctggggcgtcggtttccactat | hph gene |
| Endochi probe1F | tgagcttcctcggcaaatccgtgg | Tvech1 gene |
| Endochi probe1R | agcagaatccgtgttgaagggagt | Tvech1 gene |
| Endochi probe2F | gggagacggctatcgttcagttgc | Tvech2 gene |
| Endochi probe2R | cgaagatgccattctcccacgacc | Tvech2 gene |
| Endochi probe3F | gctcctttgcgtcgttttggccgt | Tvech3 gene |
| Endochi probe3R | gtgtcgccacggtcagaaggtagt | Tvech3 gene |
LF – left flank.
RF – right flank.
Targeted Deletion of Chitinase Genes and Construction of pSilent-1 Silencing Vectors
Two different strategies were used for generation of chitinase-deficient mutants. Targeted gene deletion by the split-marker method [53] was used for deletion of the two N-acetyl-ß-D-glucosaminidase-encoding genes, Tvnag1 and Tvnag2. In addition, a double mutant (ΔTvnag1ΔTvnag2) was generated in which both N-acetyl-ß-D-glucosaminidase-encoding genes were deleted in a single strain. The constructs were obtained using fusion PCR from two rounds of PCR reactions (Figure 1). Selectable marker primers and gene-specific primers used to make the constructs are listed in Table 1.
Figure 1. Targeted gene deletion of the N-acetyl-ß-D-glucosaminidase-encoding genes nag1 and nag2 by using the split-marker strategy.

Schematic representations of vector construction are shown in (a) for the single knockout strains, and (b) for the double knockout strains (a) Primers 1F/2R and 3F/4R amplify 1kb target gene flanking sequences. Primers 2R and 3F are hybridised once the 5′ ends complement the M13F and M13R sequences, respectively. For nag1, the primer pairs Chito2_50.1/Chito2_M13F and Chito2_M13R/Chito2_30.1 were used and, for nag2, the primer pairs HEX1_Fw/HEX1.M13F_Rev and HEX2.M13R_Fw/HEX2_Rev were used. Primer pairs M13F/HYsplit and M13R/YGsplit amplify the ‘HP’ and ‘PH’ marker fragments, respectively. Two separate PCR reactions (1F/HYsplit) and (YGsplit/4R) fuse the flanking sequences to the 5′ ‘HP’ or 3′ ‘PH’ fragments of the hygromycin resistance gene hph. Similar steps were used for double knockout mutant generation (b), using a nag2 deletion mutant for transformation with a second selectable marker ilv1 bestowing resistance to sulfonylurea. In this case, the primer pairs used in the first round PCR reactions were Chito2_M13R/Chito2_30.1 and Chito2_50.1/Chito2_M13F for amplification of the 1 kb nag1 flanking sequences and ILsplit/M13R and M13F/LVsplit for amplification of the ‘IL’ and ‘LV1’ fragments, respectively. Second round PCR reactions were performed with the primer pairs Chito2_30.1/ILsplit and LVsplit/Chito2_50.1. Primer sequences are shown in Table 1. (c), Putative ?nag2 transformants. The lane indicated by Wt consists of wild type Tv.223 DNA and lanes 1–14 contain DNA of putative ?nag2 knockouts. The ?Tvnag2 mutant in lane 10 (confirmed by the absence of a band) was selected for nag2 loss-of-function studies and for the generation of ?Tvnag1?Tvnag2 mutants. (d) Putative ?nag1 transformants. The lane indicated by Wt consists of wild type Tv.223 DNA and lanes 1–13 contain DNA of putative ?nag1 knockouts. The ?Tvnag1 mutant in lane 5 (confirmed by the absence of a band) was selected for nag1 loss-of-function studies. (e) Putative ?Tvnag1?Tvnag2 transformants. Lane 1 contains DNA of the ?Tvnag2 mutant (lane 10 in (c)) used for the generation of the double mutants. Lane 2 contains DNA of the ?Tvnag1 mutant from lane 5 in (d). Lanes 3–12 contain DNA of putative double mutants. The double mutant, ?Tvnag1?Tvnag2, in lane 3 was selected for loss-of-function studies.
Given the redundancy of endochitinase-encoding genes in the T. virens genome, we used conditional gene silencing by RNA interference (RNAi) [54] as a strategy for obtaining simultaneous silencing of all endochitinase-encoding genes. Selection of target chitinase genes to be silenced was based on a similarity analysis of the eight T. virens subgroup A chitinases (data not shown), in which we selected the three most similar gene sequences, the chitinases Tvech1, Tvech2 and Tvech3. Two silencing vectors (pSilent-endochi and pSilent-ToxA) (Figure 2) were constructed using the silencing vector pSilent-1 as a template. The pSilent-1 vector carries a hygromycin resistance cassette and a transcriptional unit allowing double-stranded RNA expression caused by splicing of a cutinase gene intron from Magnaporthe oryzae [54]. For construction of gene silencing vectors, a 491-bp fragment from Tvech2 was targeted for silencing. This fragment represents the most conserved region shared by the three chitinase gene sequences and was amplified from Tv.223 genomic DNA with two pairs of primers: 1F.endochi.XhoI +2R.endochi.HindIII and 3F.endochi.KpnI +4R.endochi.SphI (Table 1). The PCR fragments (1F2R and 3F4R) were eluted from the gel (Wizard® SV Gel and PCR Clean-up System, Promega) and cloned into the plasmid pGEM-T (Promega). Plasmid DNA from bacterial colonies containing pGEM-T-1F2R and pGEM-T-3F4R were digested with the restriction enzymes XhoI + HindIII and KpnI + SphI, respectively. The resulting 491-bp fragments contained the restriction sites required for cloning into pSilent-1 in both sense and antisense orientations. For constructing the silencing vector pSilent-endochi (7.9 kb), the 1F2R fragment was cloned into the XhoI site of the vector pSilent-1 (6.9 kb) downstream of the trpC promoter. Subsequently, the fragment 3F4R was inserted into the SphI site downstream of the cutinase gene intron. The pSilent-ToxA (7.0 kb) vector was constructed by cloning the 3F4R fragment into the SphI site of the vector pSilent-1, and then replacing the trpC promoter (1.6 kb) by the fusion fragment (ToxA+1F2R) at the SpeI restriction site. The fusion fragment was generated by two rounds of PCR reactions; in the first round the bacterial promoter ToxA and the 1F2R fragment were amplified using the primer pairs 1F.SpeI.ToxA +2R.fusion.ToxA and 3F.fusion.ToxA +2R.endochi.HindIII, respectively; in the second round PCR the first round products and the 1F.SpeI.ToxA +2R.endochi.HindIII primer pair were used to amplify a 1 kb fragment containing ToxA promoter linked to the 1F2R fragment. This fragment was eluted from the gel (Wizard® SV Gel and PCR Clean-up System, Promega) and cloned in the plasmid pGEM-T (Promega).
Figure 2. RNA-mediated silencing vectors and Southern blot analysis.
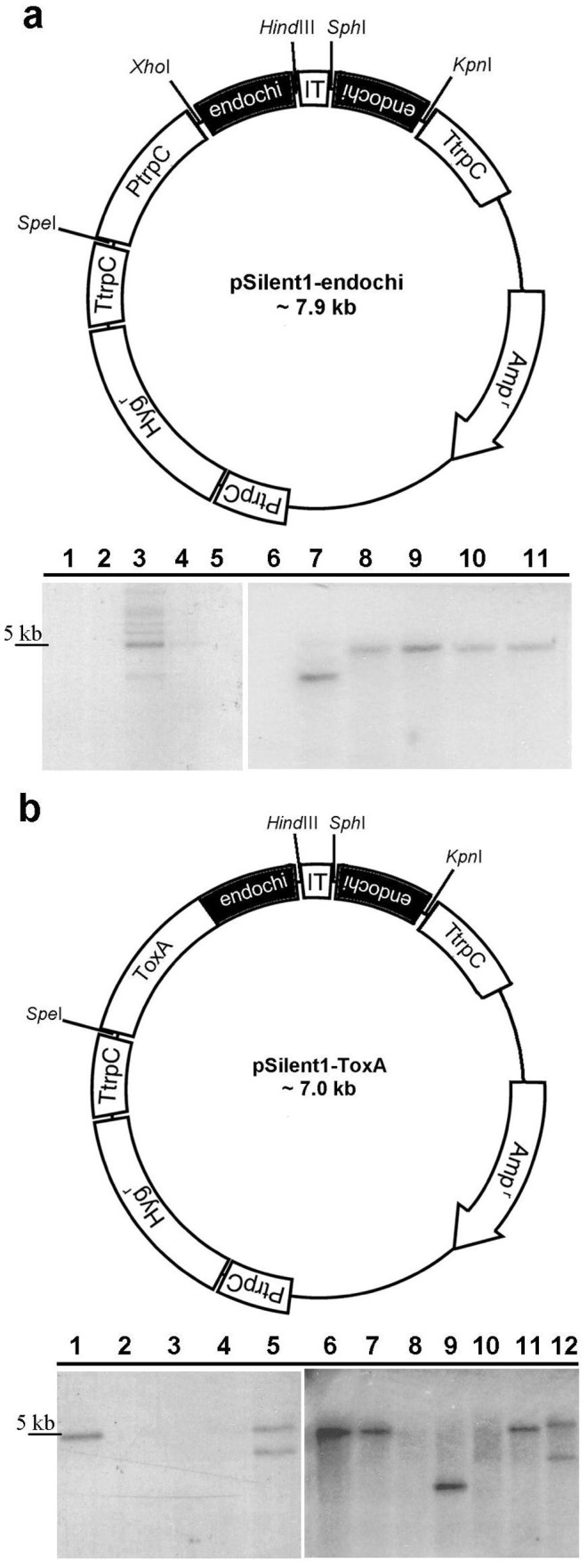
(a) Schematic representation of the vector pSilent1-endochi, derived from pSilent-1 (Nakayashiki et al., 2005) by two asymmetric clonings of a 491 bp Tvech2 fragment. (b) Schematic representation of the vector pSilent1-ToxA, derived from pSilent-1 (Nakayashiki et al., 2005) by replacement of the trpC promoter by ToxA followed by two asymmetric clonings of a 491 bp Tvech2 fragment. Ampr, ampicillin-resistant gene; Hygr, hygromycin-resistant gene; endochi, 491 bp Tvech2 fragment; IT, intron 2 of the cutinase (CUT) gene from Magnaporthe oryzae; PtrpC, Aspergillus nidulans trpC promoter; and TtrpC, A. nidulans trpC terminator. Southern blots are show below the vector maps. Genomic DNA was digested with SacI + KpnI and probed with a 1 kb fragment amplified from the hygromycin cassette with the primers Hyg probe 2F/Hyg probe 3R. In (a): lane 1 contains DNA of the wild type strain Tv.223 and lanes 2–11 contain DNA of the putative transformants S.pNO6::hph, S.pNO8::hph (used in the loss-of-function studies), S.pNO15::hph, S.pNO23::hph, S.pNO28::hph, S.pNO3::hph, S.pNO5::hph, S.pNO9::hph, S.pNO13::hph and S.pNO31, respectively. In (b): lane 1 contains DNA of the transformant S.pOf28::hph (used in the loss-of-function studies), lane 2 contains the DNA of the wild type strain Tv.223 and lanes 3–12 contain DNA of the putative transformants S.pOf31::hph, S.pOf32::hph, S.pOf39::hph, S.pOf11::hph, S.pOf21::hph, S.pOf17::hph, S.pOf36::hph, S.pOf40::hph, S.pOf15::hph and S.pOf8::hph, respectively. The presence of a band confirms insertion of the vector.
Fungal Transformations
Preparation of protoplasts and polyethylene glycol-mediated transformation of T. virens 223 was performed as described previously [55]. For double gene deletion selection, the protoplasts were plated onto 0.8 M sucrose BDCM (yeast nitrogen base without amino acids (Difco), 1.7 g l−1, asparagine 2 g l−1, NH4NO3 1 g l−1, glucose, 10 g l−1, pH to 6.0 with Na2HPO4), grown for 24 h at 26°C in the dark, and then covered with a 15 ml BDCM overlay containing 300 µg ml−1 of sulfonylurea (Chlorimuron ethyl, Chem Service). For all other transformations, the protoplasts were plated onto PDA containing 0.8 M sucrose, grown for 24 h at 26°C in the dark and covered with a 15 ml PDA overlay containing 900 µg ml−1 of hygromycin B (Calbiochem). Putative transformants were transferred to fresh selective medium and allowed to sporulate. Single spores were isolated from each putative transformant and grown in potato dextrose broth (PDB, Difco) to allow production of mycelia for DNA extraction [50]. The gene deletion events were identified by Southern blot analysis using, as a probe, a 1-kb fragment amplified with primers that anneal within the gene cassette. The insertion of the gene silencing vectors (pSilent1-endochi and pSilent1-ToxA) into the Tv.223 genome was confirmed by Southern blot using, as a probe, a 1-kb fragment amplified from the hygromycin cassette. The primers used are listed in Table 1. Gel electrophoresis, restriction enzyme digestions and DNA gel blot hybridizations were performed according to standard procedures [56]. Probes were radiolabelled with P32 using the random primer method [57].
Gene Silencing
The T. virens wild type strain Tv.223 and twelve putative silenced mutants (transformed with the pSilent-endochi or the pSilent-ToxA silencing vectors) were subjected to five rounds of subculturing on PDA, following by subculturing on PDA containing hygromycin. Stable transformants were grown in 100 ml of PDB, inoculated with 1×108 conidia m l−1, for 6 d at 26°C with shaking (120 rpm) and transferred to 100 ml of chitin minimal medium (CMM; crab shell chitin (Sigma), 10 g l−1 NaNO3, 6 g l−1, KCl, 0.5 g l−1, KH2PO4 1.5 g l−1, MgSO4.7 H2O 0.5 g l−1, ZnSO4 0.001 g l−1, FeSO4 0.001 g l−1, thiamine 0.001%, biotin 0.000025%, pH to 6.5 with NaOH). After 24 h, the mycelium was collected and RNA isolated using the LiCl-RNA protocol. The RNA blots were performed using standard procedures [56] and probed with three different fragments, corresponding to 1-kb bands amplified from the coding regions of each chitinase gene (see Table 1 for primers sequences).
Chitinase Assays
Spores of the T. virens wild type strain Tv.223 and five mutant strains (?Tvnag1, ?Tvnag2, ?Tvnag1?Tvnag2, S.pNO8::hph, S.pOf28::hph) were used to inoculate PDB (1×106 conidia ml−1) and cultures were incubated for 4 d at 26°C with shaking (120 rpm), prior to transfer to CMM. After 48 h, the mycelium was separated from the culture fluids by centrifugation at 10,000 g for 10 min at 4°C. Supernatant fluids were collected and stored at −20°C prior to chitinase assays. There were three replicates per treatment and negative control samples consisted of uninoculated medium only. The total protein concentration in the extracts was measured according to the Bradford method [58]. The N-acetyl-ß-D-glucosaminidase, chitobiosidase and chitotriosidase activities were measured by using enzymatic hydrolysis of the substrates 4-Nitrophenyl N-acetyl-ß-D-glucosaminide, 4-Nitrophenyl N,N’-diacetyl-ß-D-chitobioside or 4-Nitrophenyl ß-D-N,N’,N’’-triacetylchitotriose, respectively. The substrate hydrolysis releases p-nitrophenol (4-nitrophenol), which upon ionization in basic pH, can be measured colorimetrically at 405 nm. The enzymatic assays were performed according to the instructions provided in a commercial Chitinase Assay Kit (Sigma).
Phenotypic Characterisation of Transformants
T. virens Tv.223 and the five chitinase mutants were examined for hyphal growth and spore production. Plugs of mycelium (2 mm diameter) were removed from the margins of actively growing colonies and were placed in the centre of PDA plates. Plates were incubated at 25°C under a 16 h photoperiod of fluorescent light and colony diameters were measured after 12, 24, 36 and 48 h. Spore production was determined by removing three fungal discs (5 mm diameter) from 5-day-old cultures, placing them in sterile dH2O, vortexing for 1 min, and counting conidia with a haemocytometer. Each treatment was replicated three times, and the entire experiment was repeated twice.
Biological Control Assays
The biological control capabilities of Tv.223 and the five chitinase-deficient mutants were conducted using three different plant pathogens. These were Ceratocystis paradoxa (the causal agent of sugarcane pineapple disease), Sclerotinia sclerotiorum (a plurivorous soil-borne damping-off pathogen) and Rhizoctonia solani (a pre-emergence damping-off strain of the pathogen). Wheat bran inoculum was prepared by inoculating an autoclaved mixture of 10 g wheat bran and 30 ml dH2O with five plugs of mycelium (5 mm diameter), and allowing growth for 5 days at 25°C. Eight g of 5-day-old inoculum was mixed thoroughly with 300 g of growing medium. Sugarcane (Saccharum sp.) setts (cv. SP80–1842), with one bud each, were planted in Basaplant compost inoculated with both T. virens and the pathogen C. paradoxa and were evaluated weekly for percentage germination. Each treatment had 12 replicates and the entire experiment was repeated twice. Lettuce seeds (Lactuca sativa, cv. Webb’s Wonderful) were planted in square plates containing an autoclaved peat preparation (1 l sphagnum moss peat (Shamrock) and 400 ml dH2O) inoculated with bran inoculum of T. virens and the pathogen S. sclerotiorum. Each treatment consisted of 3 replicate plates each containing 25 lettuce seeds, and percentage germination was determined after 10 d. Bean seeds (Phaseolus vulgaris L, cv. IAC-Alvorada) were planted in Basaplant compost inoculated with T. virens and the pathogen R. solani, and percentage germination was determined after 20 d. Each treatment was replicated six times, with five seeds each replicate and the entire experiment was repeated twice. All the bioassays included controls that consisted of plants grown in uninoculated soils.
Data Analysis
Statistical analysis of experiments was performed by the general linear models procedures of Statistical Analysis Systems (SAS Institute Inc., Cary, NC, USA). Mean separations were conducted using the means procedure of SAS. Percentage germination data from biocontrol assays were normalised by transformation using arc sin−1 function before statistical analysis.
Results
Generation of Chitinase-deficient Mutants of T. virens
We set out to functionally characterize the role of chitinolytic enzyme activity in the biocontrol fungus T. virens strain 223. Analysis of its genome sequence revealed the presence of five genes, including Nag1 and Nag2, which encode N-acetyl-ß-D-glucosaminidases, and three genes that putatively encode endochitinases; ech1, ech2, and ech3. Strain 223 was transformed with specific constructs (Figure 1a) to generate deletion mutants in which the N-acetyl-ß-D-glucosaminidase-encoding genes nag1 or nag2 were replaced by a 1.4 kb cassette, comprising the hph gene which bestowed resistance to the antibiotic hygromycin B. Hygromycin-resistant transformants were identified by Southern blot (Figure 1c,d) and stable transformants selected. A single nag1 deletion mutant (lane 5 in Figure 1d) was selected for further study, and is referred to hereafter as ?Tvnag1. A nag2 deletion mutant (lane 10 in Figure 1c), hereafter referred to as ?Tvnag2, was generated and then used to produce a double mutant (hereafter referred to as ?Tvnag1?Tvnag2), in which the nag1 gene was replaced by a 2.8 kb cassette comprising the ilv1 gene which bestowed resistance to the selectable marker sulfonylurea (Figure 1b).
To carry out silencing of the endochitinase gene family, T. virens 223 was transformed with two different gene silencing vectors, pSilent-endochi and pSilent-ToxA, which differed only in their promoters (Figure 2a,b). Insertion of the vectors was confirmed by Southern blot analysis (Figure 2a,b) and gene silencing evaluated by assessing levels of expression of the three T. virens chitinase genes Tvech1, Tvech2 and Tvech3 by Northern blot analysis (Figure 3). The results showed different consequences from insertion of the silencing vectors in the T. virens genome. Total elimination, reductions, no change or even increases in chitinase mRNA transcript levels were observed in the transformants investigated (Figure 3). Only two of the transformants, S.pOf28::hph and S.pNO8::hph, were stable after five consecutive rounds of subculturing on PDA and hygromycin. S.pOf28::hph exhibited complete elimination of the Tvech1 transcript and reduction in the Tvech2 transcript compared to the wild type control. S.pNO8::hph exhibited reductions in Tvech1 and Tvech3 transcripts, but no reduction in Tvech2 mRNA. Replacement of the trpC promoter (in the pSilent1-endochi vector) by ToxA promoter (in the pSilent1-ToxA vector) did not lead to a higher efficiency in the silencing of the endochitinase genes (Figure 3).
Figure 3. Northern blot analysis.

The effects of insertion of the silencing vectors pSilent1-endochi and pSilent1-ToxA on the expression of three chitinase genes (Tvech1, Tvech2 and Tvech3) in T. virens transformants are shown in (a) detection of Tvech1 transcripts, (b) detection of Tvech2 transcripts and, (c) detection of Tvech3 transcripts. Transformants containing pSilent1-endochi vector have the prefix S.pNO and the ones containing pSilent1-ToxA have the prefix S.pOf. Twenty micrograms of total RNA were separated on formaldehyde-agarose gels, transferred to nylon membranes and probed with a 1 kb gene-specific fragment. Equal loading of total RNA was estimated by ethidium bromide staining of rRNA.
Enzyme Activities of Chitinase-deficient Mutants
To test the chitinolytic activities of the five chitinase mutants (?Tvnag1, ?Tvnag2, ?Tvnag1?Tvnag2, S.pNO8::hph, S.pOf28::hph) and the wild type strain Tv.223 enzymatic hydrolysis of chitinase substrates was carried out. Comparison of the chitinase activities of the mutants to Tv.223 showed that the enzyme disruption strategies were successful in generating mutants lacking different chitinolytic activities (Figure 4). Activities of N-acetyl-ß-D-glucosaminidase were significantly reduced in the single ?Tvnag1 and ?Tvnag2 mutants, and completely eliminated in the double knockout ?Tvnag1?Tvnag2 (Figure 4). No changes were observed in the N-acetyl-ß-D-glucosaminidase activities of the knock-down mutants S.pNO8::hph and S.pOf28::hph compared to the wild type strain. Chitobiosidase activities were significantly reduced in the ?Tvnag1 and ?Tvnag2 single mutants, and completely eliminated in the double mutant ?Tvnag1?Tvnag2. The mutants ?Tvnag1 and ?Tvnag1?Tvnag2 exhibited significant reductions in their endochitinase activities, and while a reduction in endochitinase activity was also found in the ?Tvnag2 mutant, the decrease was not significant when compared to Tv.223. Endochitinase activities were significantly reduced in the two knock-down mutants, with an approximately 50% decrease in both S.pNO8::hph and S.pOf28::hph (Figure 4). N-acetyl-ß-D-glucosaminidase and chitobiosidase activities were unchanged in the mutant S.pNO8::hph. N-acetyl-ß-D-glucosaminidase activity was similarly unchanged in the mutant S.pOf28::hph, but there was a slight (albeit significant) reduction in chitobiosidase activity.
Figure 4. Detection of chitinolytic activities in culture filtrates of the T. virens strains.

N-acetyl-ß-D-glucosaminidase, chitobiosidase and endochitinase activities of the wild type strain Tv.223 and the five chitinase-deficient mutants ?Tvnag1, ?Tvnag2, ?Tvnag1?Tvnag2, S.pNO8::hph, and S.pOf28::hph were determined by enzymatic hydrolysis of the chitinase-specific substrates 4-Nitrophenyl N-acetyl-ß-D-glucosaminide, 4-Nitrophenyl N,N’-diacetyl-ß-D-chitobioside and 4-Nitrophenyl ß-D-N,N’,N’’-triacetylchitotriose, respectively. Substrate hydrolysis releases p-nitrophenol which, upon ionization in basic pH, can be measured colorimetrically at 405 nm. Protein concentrations of the extracts were adjusted to 20 µg.mL−1 prior to enzyme assays. Histograms are the means of three replicate values with standard deviations. One unit of the activity releases 1.0 µmole of p-nitrophenol from the appropriate substrate per minute at pH 4.8 and 37°C. Letters denote the results of a t-test for comparison of means. Bars with different letter(s) are significantly different at 95% confidence level.
Growth Rates and Sporulation of Chitinase-deficient Mutants
Hyphal development and sporulation of mutants was determined prior to biocontrol assays, to ensure that all strains had similar growth characteristics. The results showed no evident abnormalities in the mutants based on hyphal growth and sporulation efficiencies. All of the mutants showed similar colony morphology, growth rates, and conidiation compared to each other and to the wild type strain Tv.223 (Table 2).
Table 2. Colony diameters and conidiation of wild type Tv.223 and the chitinase-deficient mutants.
| Strain | Colony diameter (cm)* | Conidiation (spores ml−1) | |||
| 12 h | 24 h | 36 h | 48 h | ||
| Tv.223 | 1.87 | 4.03 | 5.52 | 6.57 | 4.88×107 |
| ?Tvnag1 | 1.82 | 4.03 | 5.50 | 6.60 | 5.09×107 |
| ?Tvnag2 | 1.82 | 4.03 | 5.53 | 6.67 | 3.97×107 |
| ?Tvnag1?Tvnag2 | 2.03 | 4.13 | 5.58 | 6.63 | 4.36×107 |
| S.pNO8::hph | 1.75 | 4.00 | 5.53 | 6.73 | 5.09×107 |
| S.pOf28::hph | 1.85 | 4.03 | 5.50 | 6.60 | 3.84×107 |
Each figure is the mean diameter of three replicate cultures.
Based on ANOVA (P<0.05), there were no significant differences in hyphal growth and conidiation between strains.
Biocontrol Fitness of Chitinase-deficient Mutants
In order to assess whether the loss of chitinase activities altered the biocontrol capabilities of the mutants, each strain was tested for its ability to control plant diseases caused by different soil-borne pathogens. Planting sugarcane setts in soil infested with the sugarcane pathogen C. paradoxa led to a significant reduction (P<0.05) in the germination of plants, a trend followed throughout the 6 week sampling period. In week 2, when the buds started to germinate, there were significant reductions in germination of setts treated with the pathogen alone and with mixed populations of the pathogen and the T. virens mutants ?Tvnag1?Tvnag2, S.pNO8::hph or S.pOf28::hph, indicating a loss of biocontrol fitness in these mutants (Figure 5a). This effect was more persistent over the 6 week period for the mutants S.pNO8::hph and S.pOf28::hph. At week 6, germination percentages for these mutants were significantly reduced compared to wild type-treated setts. Furthermore, while not significantly different, germination percentages were also lower in the S.pNO8::hph + C. paradoxa and S.pOf28::hph + C. paradoxa treated setts than in setts treated with C. paradoxa only (Figure 5a). In contrast to sugarcane, lettuce infection studies showed that none of the chitinase mutants exhibited reduced capacities to control the pre-emergence damping-off pathogen S. sclerotiorum, when compared to the wild type strain Tv.223 (Figure 5b). Microcosms inoculated with S. sclerotiorum only exhibited total inhibition of lettuce seed germination, whereas microcosms inoculated with Tv.223 or the mutants and the pathogen showed various degrees of germination (from 69.3% up to 86.7%), all of which were not statistically different when compared to the control (Figure 5b). In addition to being able to protect lettuce seeds from disease, treatment with the wild type strain resulted in a higher percentage of germination when compared to the control treatment (Figure 5b). Strikingly, tests with the T. virens mutants and the pathogen R. solani, showed that none of the chitinase-deficient mutants had reduced fitness in protecting bean seeds from the pathogen when compared to the wild type strain (Figure 5c). Treatment with Tv.223 and the chitinase mutants resulted in statistically similar rates of germination (Figure 5c). However, it was found that while Tv.223 protected bean seeds from the pathogen, it was unable to restore germination rates comparable to that of the pathogen-free control experiment.
Figure 5. Biological control of soil-borne pathogens.

(a) Biocontrol of pineapple disease of sugarcane caused by the soil-borne pathogen Ceratocystis paradoxa. Germination of sugarcane setts was determined in compost infested with the pathogen C. paradoxa and in mixed-species microcosms containing the pathogen and chitinase-deficient mutants. Percentage germination was measured weekly, over a 6 week period. The control consisted of sugarcane setts planted in uninfested compost. Each point is the mean of twelve replicates and percentages were converted to arc sin−1 values for statistical analysis by t-test. Points with different letters are significantly different at 95% confidence level, considering each week separately. (b) Biocontrol of pre-emergence damping-off disease of lettuce caused by the pathogen Sclerotinia sclerotiorum, Germination of lettuce seeds was determined in peat-based microcosms infested with the pathogen S. sclerotiorum and in mixed-species microcosms containing the pathogen and chitinase-deficient mutants. Percentage germination was measured 10 days after sowing. The control consisted of lettuce seeds planted in uninfested peat. Each point is the mean of three replicates (each consisting of 25 lettuce seed) and percentages were converted to arc sin−1 values for statistical analysis by Tukey test for comparison of means. Histograms with different letters are significantly different at 99% confidence level. (c) Biocontrol of bean rot caused by the pathogen Rhizoctonia solani. Germination of bean seeds was determined in compost infested with the pathogen R. solani and in mixed-species microcosms containing the pathogen and chitinase-deficient mutants. Percentage germination was measured 20 days after sowing. The control consisted of bean seeds planted in uninfested compost. Each point is the mean of six replicates and percentages were converted to arc sin−1 values for statistical analysis by Tukey test for comparison of means. Histograms with different letters are significantly different at 95% confidence level.
Discussion
The relevance of chitinolytic enzymes to the biological control activities of Trichoderma spp. remains controversial, with conflicting evidence regarding their importance in plant disease control. The chitinolytic system of T. atroviride is the most comprehensively investigated system to date, although the chitinase genes of T. virens have also previously been described [24], [43], and one of the chitinases (T. harzianum chit42 = T. harzianum ech42 = T. virens ech1) studied in detail [9], [44]. In the present work, we investigated the involvement of N-acetyl-ß-D-glucosaminidases (GH family 20) and subgroup A chitinases (GH family 18) in the biocontrol fitness of a sugarcane endophytic strain of T. virens (strain Tv.223). Our objective was to determine whether these enzymes contributed to its fitness as a biocontrol agent of sugarcane pineapple disease caused by the soil-borne pathogen Ceratocystis paradoxa. Furthermore, we aimed to determine whether loss of chitinase activities affected its ability to control root diseases caused by other soil-borne pathogens. The presence of only two N-acetyl-ß-D-glucosaminidase-encoding genes in the T. virens genome meant that a targeted gene deletion strategy could be employed to generate single and double gene deletion mutants lacking N-acetyl-ß-D-glucosaminidase activities. In contrast, T. virens possesses 8 subgroup A chitinases and a total of 36 chitinase genes [43]. This redundancy in chitinase-encoding genes meant that an alternative strategy was needed for disruption of endochitinase activities. To this end, we used RNA interference [54] to disrupt endochitinase production by silencing the endochitinase gene Tvech2.
Using the split-marker method [53], we generated single (?Tvnag1 or ?Tvnag2) and double (?Tvnag1?Tvnag2) deletion mutants lacking N-acetyl-ß-D-glucosaminidase activities. Using enzyme activity assays, we showed that the single deletion mutants had significantly reduced N-acetyl-ß-D-glucosaminidase activities, with total elimination in the double mutant. Elimination of both nag1 and nag2 genes in the ?Tvnag1?Tvnag2 double mutant resulted in concomitant, and significant, loss of chitobiosidase and endochitinase activities compared to the wild-type strain. While simultaneous loss of chitobiosidase and endochitinase activities was also exhibited in the single deletion mutants, loss of chitobiosidase activities was more pronounced in both mutants compared to endochitinase activities. Loss of chitobiosidase and endochitinase activities as a consequence of N-acetyl-ß-D-glucosaminidase disruption is in keeping with other studies that showed that N-acetyl-ß-D-glucosaminidase activity in T. atroviride and T. hamatum is essential for the induction of other chitinase activities [36], [49].
Pineapple disease of sugarcane is characterized by a reduction in the germination of sugarcane setts, leading to patchy establishment of plants. We therefore chose germination assays of different host plants (sugarcane, lettuce and bean) to quantify the biocontrol capabilities of the chitinase mutants during antagonistic interactions with soil-borne, root-infecting, pathogens. The different pathobiologies of the three pathogens were reflected in the lengths of the germination assays, with bean and lettuce seeds taking substantially less time to germinate and emerge than sugarcane setts when exposed to their respective pathogens.
Despite the loss of chitinase production in the N-acetyl-ß-D-glucosaminidase-deficient mutants, their biocontrol activities against the sugarcane ascomycete pathogen C. paradoxa, and the plurivorous ascomycete and basidiomycete pathogens Sclerotinia sclerotiorum and Rhizoctonia solani respectively, were largely unaltered compared to the wild-type strain Tv.223. Despite an initial lag in germination of sugarcane setts in mixed populations of C. paradoxa and the double mutant ?Tvnag1?Tvnag2, there was no significant difference in plant establishment compared to the untreated control by the end of the sampling period. Similarly, there was no significant reduction in abilities of the mutants to control lettuce and bean diseases. The dispensable role of N-acetyl-ß-D-glucosaminidase to biocontrol activity was proposed previously [16] and our work supports this suggestion. Our findings are also consistent with a recent study by [59] that showed that the biocontrol efficacy of T. atroviride was unaffected by single and double deletions of the nag1 and nag2 genes.
Using RNAi, we successfully silenced the three Tv.223 endochitinase genes Tvech1, Tvech2 and Tvech3. Different levels of mRNA accumulation were exhibited among the mutants, varying from total absence of target mRNA transcripts to no change or even an increase in mRNA accumulation in some mutants. Simultaneous silencing of Tvech1, Tvech2 and Tvech3 genes was detected in approximately 17% of the mutants analyzed, whereas half of the mutants exhibited silencing of two of the endochitinases. Co-silencing of genes [60]–[62] seems likely to be correlated, to some extent, with the degree of sequence similarity to the target gene.
Despite initial success with the gene silencing strategy, it was found that the majority of mutants selected were unstable. Only two of the mutants were found to be stable following sequential rounds of selection for antibiotic resistance. The two stable mutants, S.pNO8::hph and S.pOf28::hph, that differed only in the promoter employed, showed differing levels of mRNA silencing. While S.pOf28::hph exhibited total elimination of Tvech1 expression and a reduction in Tvech2 mRNA, S.pNO8::hph exhibited only slight reductions in Tvech1 and Tvech3 transcript levels. However, this is consistent with previous reports that show that knock-down mutants generated by RNA silencing, often show different levels of gene silencing [54], [62]. Despite this, and considering the presence of a further five subgroup A chitinases and a total of 36 chitinase genes in the T. virens genome [43], both of the mutants showed a 50% reduction in endochitinase activities.
The gene silencing strategy enabled loss of endochitinase activity on biocontrol fitness of Tv.223 to be determined in the absence of extensive reductions in expression of N-acetyl-ß-D-glucosaminidase and chitobiosidase activities. There was no effect of gene silencing on N-acetyl-ß-D-glucosaminidase and chitobiosidase production in S.pNO8::hph and only slight, albeit significant, reduction in chitobiosidase activity in S.pOf28::hph. Thus any reduction in biocontrol fitness of these two mutants could be largely attributed to their loss of endochitinase activities. Consequently, while phenotypic analysis of the mutants revealed no evident changes in hyphal growth or sporulation, their abilities to control pineapple disease of sugarcane were found to be significantly impaired, while biocontrol of S. sclerotinia and R. solani was unaffected.
Our findings add additional support to the importance of endochitinases in the biocontrol activities of Trichoderma species. Reduced biocontrol fitness as a consequence of endochitinase disruption has been described in T. virens and T. atroviride gene deletion strains [44], [63]. Furthermore, increased biocontrol activities have been shown in T. atroviride and T. virens through overexpression of endochitinases [35], [44], [64] and expression of T. atroviride endochitinase during mycoparasitism further demonstrates its role during antagonistic interactions [65].
The specific loss of biocontrol fitness of the Tv.223 endochitinase-silenced mutants in the C. paradoxa-sugarcane pathosystem may reflect its lifestyle as a sugarcane root endophyte. Notwithstanding this, it is clear that endochitinases contribute to its fitness as biocontrol agent of sugarcane disease, but not as an antagonist of other soil-borne plant pathogens. Furthermore, N-acetyl-ß-D-glucosaminidases do not appear to contribute to its fitness as a biocontrol agent of C. paradoxa disease or plant diseases caused by other soil-borne pathogens.
Acknowledgments
We gratefully acknowledge Dr Michael Kershaw for molecular biology assistance, Dr Darren Soanes for assistance with bioinformatics, and Nick Tongue, Dr Ana Lilia Martínez Rocha, Dr Lauren Ryder and Dr Romain Huguet for their help with laboratory experiments. We thank Sabrina M. Chabregas (Centro de Tecnologia Canavieira) for supplying the sugarcane plants and Ceratocystis paradoxa.
Funding Statement
This work was supported by FAPESP (grant no. 2002/14143-3) and CAPES. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Hollanda E (2010) Trade and Environment Review. United Nations: 68–80. [Google Scholar]
- 2. Talukder M, Begum F, Azad M (2007) Management of pinapple disease of sugarcane through biological means. J Agric Rural Dev 5: 79–83. [Google Scholar]
- 3. Harman GE, Howell CR, Viterbo A, Chet I, Lorito M (2004) Trichoderma species - opportunistic, avirulent plant symbionts. Nat Rev Microbiol 2: 43–56. [DOI] [PubMed] [Google Scholar]
- 4. Verma M, Brar SK, Tyagi RD, Surampalli RY, Valéro JR (2007) Antagonistic fungi, Trichoderma spp.: panoply of biological control. Biochem Eng J 37: 1–20. [Google Scholar]
- 5.Romão A (2010) Análise da comunidade fúngica associada à cana-de-açúcar e estudo da interação Trichoderma-virens-planta hospedeira [Ph.D]. Brazil: Escola Superior de Agricultura Luiz de Queiroz, University of São Paulo. 268 p. [Google Scholar]
- 6. Howell CR (2006) Understanding the mechanisms employed by Trichoderma virens to effect biological control of cotton diseases. Phytopathology 96: 178–180. [DOI] [PubMed] [Google Scholar]
- 7. Tu JC (1980) Gliocladium virens, a destructive mycoparasite of Sclerotinia sclerotiorum . Phytopathology 70: 670–674. [Google Scholar]
- 8. Howell CR (1982) Effect of Gliocladium virens on Pythium ultimum, Rhizoctonia solani, and damping-off of cotton seedlings. Phytopathology 72: 496–498. [Google Scholar]
- 9. Di Pietro A, Lorito M, Hayes CK, Broadway RM, Harman GE (1993) Endochitinase from Gliocladium virens: isolation, characterization and synergistic antifungal activity in combination with gliotoxin. Phytopathology 83: 308–313. [Google Scholar]
- 10. Wilhite SE, Lumsden RD, Straney DC (1994) Mutational analysis of gliotoxin production by the biocontrol fungus Gliocladium virens in relation to suppression of Pythium damping-off. Phytopathology 84: 816–821. [Google Scholar]
- 11. Howell CR, Puckhaber LS (2005) A study of the characteristics of “P” and “Q” strains of Trichoderma virens to account for differences in biological control efficacy against cotton seedling diseases. Biol Control 33: 217–222. [Google Scholar]
- 12. Yedidia I, Shoresh M, Kerem Z, Benhamou N, Kapulnik Y, et al. (2003) Concomitant induction of systemic resistance to Pseudomonas syringae pv. lachrymans in cucumber by Trichoderma asperellum (T-203) and accumulation of phytoalexins. Appl Environ Microbiol 69: 7343–7353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hanson LE, Howell CR (2004) Elicitors of plant defense responses from biocontrol strains of Trichoderma virens . Phytopathology 94: 171–176. [DOI] [PubMed] [Google Scholar]
- 14.Peberdy JF (1990) Fungal cell walls – a review. In: Kuhn PJ, Trinci APJ, Jung MJ, Goosey MW, editors. Biochemistry of cell walls and membranes in fungi. Berlin: Springer-Verlag. 5–30. [Google Scholar]
- 15.Lorito M (1998) Chitinolytic enzymes ands their genes. In: Harman GE, Kubicek CP, editors. Trichoderma and Gliocladium: Enzymes, biological control and commercial application London: Taylor and Francis. 73–99. [Google Scholar]
- 16. Seidl V (2008) Chitinases of filamentous fungi: a large group of diverse proteins with multiple physiological functions. Fungal Biol Rev 22: 36–42. [Google Scholar]
- 17. Ulhoa CJ, Peberdy JF (1991) Purification and characterization of an extracellular chitobiase from Trichoderma harzianum . Curr Microbiol 23: 285–289. [Google Scholar]
- 18. de La Cruz J, Hidalgo-Gallego A, Lora JM, Benitez T, Pintor-Toro JA, et al. (1992) Isolation and characterization of three chitinases from Trichoderma harzianum . Eur J Biochem 206: 859–867. [DOI] [PubMed] [Google Scholar]
- 19. Ulhoa CJ, Peberdy JF (1992) Purification and some properties of the extracellular chitinase produced by Trichoderma harzianum . Enzyme Microb Technol 14: 236–240. [Google Scholar]
- 20. Harman GE, Hayes CK, Lorito M, Broadway RM, Di Pietro A, et al. (1993) Chitinolytic enzymes of Trichoderma harzianum: purification of chitobiosidase and endochitinase. Phytopathology 83: 313–318. [Google Scholar]
- 21. Lorito M, Hayes CK, Di Pietro A, Woo SL, Harman GE (1994) Purification, characterization, and synergistic activity of a glucan 1,3-ß-glucosidase and an N-acetyl-ß-glucosaminidase from Trichoderma harzianum . Phytopathology 84: 398–405. [Google Scholar]
- 22. Haran S, Schickler H, Oppenheim A, Chet I (1995) New components of the chitinolytic system of Trichoderma harzianum. . Mycol Res 99: 441–446. [Google Scholar]
- 23. El-Katatny MH, Gudelj M, Robra KH, Elnaghy MA, Gübitz GM (2001) Characterization of a chitinase and an endo-ß-1,3-glucanase from Trichoderma harzianum Rifai T24 involved in control of the phytopathogen Sclerotium rolfsii . Appl Microbiol Biotechnol 56: 137–143. [DOI] [PubMed] [Google Scholar]
- 24. Kim DJ, Baek JM, Uribe P, Kenerley CM, Cook DR (2002) Cloning and characterization of multiple glycosyl hydrolase genes from Trichoderma virens . Curr Genet 40: 374–384. [DOI] [PubMed] [Google Scholar]
- 25. Carsolio C, Gutiérrez A, Jiménez B, Van Montagu M, Herrera-Estrella A (1994) Characterization of ech-42, a Trichoderma harzianum endochitinase gene expressed during mycoparasitism. Proc Nat Acad Sci U S A 91: 10903–10907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. García I, Lora JM, Cruz J, Benítez T, Llobell A, et al. (1994) Cloning and characterization of a chitinase (CHIT42) cDNA from the mycoparasitic fungus Trichoderma harzianum . Curr Genet 27: 83–89. [DOI] [PubMed] [Google Scholar]
- 27. Hayes CK, Klemsdal S, Lorito M, Di Pietro A, Peterbauer C, et al. (1994) Isolation and sequence of an endochitinase-encoding gene from a cDNA library of Trichoderma harzianum . Gene 138: 143–148. [DOI] [PubMed] [Google Scholar]
- 28. Draborg H, Kauppinen S, Dalbøge H, Christgau S (1995) Molecular cloning and expression in S. cerevisiae of two exochitinases from Trichoderma harzianum . Biochem Mol Biol Int 36: 781–791. [PubMed] [Google Scholar]
- 29. Limón MC, Lora JM, García I, Cruz J, Llobell A, et al. (1995) Primary structure and expression pattern of the 33-kDa chitinase gene from the mycoparasitic fungus Trichoderma harzianum . Curr Genet 28: 478–483. [DOI] [PubMed] [Google Scholar]
- 30. Peterbauer CK, Lorito M, Hayes CK, Harman GE, Kubicek CP (1996) Molecular cloning and expression of the nag1 gene (N-acetyl-ß-D-glucosaminidase-encoding gene) from Trichoderma harzianum P1. Curr Genet 30: 325–331. [DOI] [PubMed] [Google Scholar]
- 31. Viterbo A, Haran S, Friesem D, Ramot O, Chet I (2001) Antifungal activity of a novel endochitinase gene (chit36) from Trichoderma harzianum Rifai TM. FEMS Microbiol Lett 200: 169–174. [DOI] [PubMed] [Google Scholar]
- 32. Viterbo A, Montero M, Ramot O, Friesem D, Monte E, et al. (2002) Expression regulation of the endochitinase chit36 from Trichoderma asperellum (T. harzianum T-203). Curr Genet 42: 114–122. [DOI] [PubMed] [Google Scholar]
- 33. Schirmböck M, Lorito M, Wang YL, Hayes CK, Arisan-Atac I, et al. (1994) Parallel formation and synergism of hydrolytic enzymes and peptaibol antibiotics, molecular mechanisms involved in the antagonistic action of Trichoderma harzianum against phytopathogenic fungi. Appl Env Microbiol 60: 4364–4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Carsolio C, Benhamou N, Haran S, Cortes C, Gutierrez A, et al. (1999) Role of the Trichoderma harzianum endochitinase gene, ech42, in mycoparasitism. Appl Env Microbiol 65: 929–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Limón MC, Pintor-Toro JA, Benitez T (1999) Increased antifungal activity of Trichoderma harzianum transformants that overexpress a 33-kDa chitinase. Phytopathology 89: 254–261. [DOI] [PubMed] [Google Scholar]
- 36. Brunner K, Peterbauer CK, Mach RL, Lorito M, Zeilinger S, et al. (2003) The Nag1 N-acetylglucosaminidase of Trichoderma atroviride is essential for chitinase induction by chitin and of major relevance to biocontrol. Curr Genet 43: 289–295. [DOI] [PubMed] [Google Scholar]
- 37. De Marco JL, Valadares-Inglis M, Felix C (2004) Purification and characterization of an N-acetylglucosaminidase produced by a Trichoderma harzianum strain which controls Crinipellis perniciosa . Appl Microbiol Biotechnol 64: 70–75. [DOI] [PubMed] [Google Scholar]
- 38. Lu Z, Tombolini R, Woo S, Zeilinger S, Lorito M, et al. (2004) In vivo study of Trichoderma-pathogen-plant interactions, using constitutive and inducible green fluorescent protein reporter systems. Appl Env Microbiol 70: 3073–3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Howell CR (1987) Relevance of mycoparasitism in the biological control of Rhizoctonia solani by Gliocladium virens . Phytopathology 77: 992–994. [Google Scholar]
- 40. Roberts DP, Lumsden RD (1990) Effect of extracellular metabolites from Gliocladium virens on germination of sporangia and mycelial growth of Pythium ultimum . Phytopathology 80: 461–465. [Google Scholar]
- 41. Zimand G, Elad Y, Chet I (1991) Biological control of Botrytis cinerea by Trichoderma spp. Phytoparasitica 19: 252–253. [Google Scholar]
- 42.Caron J (1993) Isolement et caractérisation de divers isolats de Trichoderma comme agent de lutte biologique contre la moisissure grise (Botrytis cinerea) dans la production de la fraise Québec: Université Laval. [Google Scholar]
- 43. Kubicek C, Herrera-Estrella A, Seidl-Seiboth V, Martinez D, Druzhinina I, et al. (2011) Comparative genome sequence analysis underscores mycoparasitism as the ancestral life style of Trichoderma . Genome Biol 12: R40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Baek J-M, Howell CR, Kenerley CM (1999) The role of an extracellular chitinase from Trichoderma virens Gv29–8 in the biocontrol of Rhizoctonia solani . Curr Genet 35: 41–50. [DOI] [PubMed] [Google Scholar]
- 45. Bolar JP, Norelli JL, Wong KW, Hayes CK, Harman GE, et al. (2000) Expression of endochitinase from Trichoderma harzianum in transgenic apple increases resistance to apple scab and reduces vigor. Phytopathology 90: 72–77. [DOI] [PubMed] [Google Scholar]
- 46. Emani C, Garcia JM, Lopata-Finch E, Pozo MJ, Uribe P, et al. (2003) Enhanced fungal resistance in transgenic cotton expressing an endochitinase gene from Trichoderma virens . Plant Biotechnol J 1: 321–336. [DOI] [PubMed] [Google Scholar]
- 47.Romão AS, Araújo WL (2007) Efeito do cultivo de cana-de-açúcar geneticamente modificada sobre a comunidade fúngica associada. In: Costa-Maia L, Malosso E, Yano-Melo AM, editors. Micologia: avanços no conhecimento. Recife: UFPE. 150–159. [Google Scholar]
- 48.Romão AS, Sebastianes FLS, Fávaro LCL, Araújo WL (2008) Agrobacterium-mediated transformation of the biological control fungus Trichoderma virens. In: XII International Congress of Mycology, Istanbul, Turkey. [Google Scholar]
- 49. Ryder L, Harris B, Soanes D, Kershaw M, Talbot N, et al. (2012) Saprotrophic competitiveness and biocontrol fitness of a genetically-modified strain of the plant-growth-promoting fungus Trichoderma hamatum GD12. Microbiol 158: 84–97. [DOI] [PubMed] [Google Scholar]
- 50. Raeder U, Broda P (1985) Rapid preparation of DNA from filamentous fungi. Lett Appl Microbiol 1: 17–20. [Google Scholar]
- 51. Huang X, Madan A (1999) CAP3: A DNA sequence assembly program. Genome Res 9: 868–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hunter S, Apweiler R, Attwood T, Bairoch A, Bateman A, et al.. (2009) InterPro: the integrative protein signature database. Nucleic Acids Res 37 (Database Issue): D211–D215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Catlett NL, Lee B, Yoder OC, Turgeon BG (2003) Split-marker recombination for efficient targeted deletion of fungal genes. Fungal Genet Newsl 50: 9–11. [Google Scholar]
- 54. Nakayashiki H, Hanada S, Quoc NB, Kadotani N, Tosa Y, et al. (2005) RNA silencing as a tool for exploring gene function in ascomycete fungi. Fungal Genet Biol 42: 275–283. [DOI] [PubMed] [Google Scholar]
- 55. Baek JM, Kenerley CM (1998) The arg2 gene of Trichoderma virens: cloning and development of a homologous transformation system. Fungal Genet Biol 23: 34–44. [DOI] [PubMed] [Google Scholar]
- 56.Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: A laboratory manual. New York: Cold Spring Harbour Laboratory Press. 1659 p. [Google Scholar]
- 57. Feinberg AP, Vogelstein B (1983) A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Anal Biochem 132: 6–13. [DOI] [PubMed] [Google Scholar]
- 58. Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248–254. [DOI] [PubMed] [Google Scholar]
- 59. Lopez-Mondejar R, Catalano V, Kubicek C, Seidl V (2009) The beta-N-acetylglucosaminidases NAG1 and NAG2 are essential for growth of Trichoderma atroviride on chitin. FEBS J 276: 5137–5148. [DOI] [PubMed] [Google Scholar]
- 60. Liu H, Cottrell TR, Pierini LM, Goldman WE, Doering TL (2002) RNA interference in the pathogenic fungus Cryptococcus neoformans . Genetics 160: 463–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mouyna I, Henry C, Doering TL, Latge JP (2004) Gene silencing with RNA interference in the human pathogenic fungus Aspergillus fumigatus . FEMS Microbiol Lett 237: 317–324. [DOI] [PubMed] [Google Scholar]
- 62. Nguyen QB, Kadotani N, Kasahara S, Tosa Y, Mayama S, et al. (2008) Systematic functional analysis of calcium-signalling proteins in the genome of the rice-blast fungus, Magnaporthe oryzae, using a high-throughput RNA-silencing system. Mol Microbiol 68: 1348–1365. [DOI] [PubMed] [Google Scholar]
- 63. Woo SL, Donzelli B, Scala F, Mach R, Harman GE, et al. (1999) Disruption of the ech42 (endochitinase-encoding) gene affects biocontrol activity in Trichoderma harzianum P1. Mol Plant Microbe Interact 12: 419–429. [Google Scholar]
- 64. Limon M, Chacon M, Mejias R, Delgado-Jarana J, Rincon A, et al. (2004) Increased antifungal and chitinase specific activities of Trichoderma harzianum CECT 2413 by addition of a cellulose binding domain. Appl Microbiol Biotechnol 64: 675–685. [DOI] [PubMed] [Google Scholar]
- 65. Zeilinger S, Galhaupa C, Payera K, Woob SL, Macha RL, et al. (1999) Chitinase gene expression during mycoparasitic interaction of Trichoderma harzianum with its host. Fungal Genet Biol 26: 131–140. [DOI] [PubMed] [Google Scholar]