


Abstract
The transcription factor THAP1 (THanatos Associated Protein 1) has emerged recently as the cause of DYT6 primary dystonia, a type of rare, familial and mostly early-onset syndrome that leads to involuntary muscle contractions. Many of the mutations described in the DYT6 patients fall within the sequence-specific DNA-binding domain (THAP domain) of THAP1 and are believed to negatively affect DNA binding. Here, we have used an integrated approach combining spectroscopic (NMR, fluorescence, DSF) and calorimetric (ITC) methods to evaluate the effect of missense mutations, within the THAP domain, on the structure, stability and DNA binding. Our study demonstrates that none of the mutations investigated failed to bind DNA and some of them even bind DNA stronger than the wild-type protein. However, some mutations could alter DNA-binding specificity. Furthermore, the most striking effect is the decrease of stability observed for mutations at positions affecting the zinc coordination, the hydrophobic core or the C-terminal AVPTIF motif, with unfolding temperatures ranging from 46°C for the wild-type to below 37°C for two mutations. These findings suggest that reduction in population of folded protein under physiological conditions could also account for the disease.
INTRODUCTION
Torsion dystonias refer to a variety of movement disorders that are associated with dysfunction in central nervous system (CNS) regions controlling movement. Twenty monogenic sub-types of hereditary dystonia have been characterized, including eight primary torsion dystonia forms where dystonia is the only clinical manifestation (1). Of these primary torsion dystonias, only two have been linked to mutations in genes, namely DYT1 and DYT6 (see (2) for reviews). The DYT1 gene has been for several years the only one gene identified as causing autosomal dominant primary torsion dystonia. DYT1 dystonia is caused by a single mutation in the TOR1A gene, resulting in the deletion of a single glutamate residue of the encoded torsinA protein (3). In 2009, the THAP1 gene has been identified as a second gene causing primary torsion dystonia (DYT6) (4). Since then, a typical phenotype characteristic of DYT6-affected individuals has emerged. Similar to DYT1, the DYT6 dystonia symptoms tend to start early. However, unlike DYT1, the cases rarely start in the leg. Onset most often begins in an arm and in the majority of the cases, symptoms spread to involve multiple body regions with segmental, multifocal or generalized dystonia (5). The THAP1 gene encodes a transcription factor (THanatos Associated Protein 1 (THAP1)) of 213 residues with a conserved DNA-binding domain (family-designating THAP domain) at its N-terminus (amino acids 1–81), a central proline-rich region (amino acids 90–110), and a large coiled-coil domain including a short predicted nuclear localization signal (nuclear localization signal (NLS), amino acids 146–162) at its C-terminus. The NLS domain has been shown to interact with prostate apoptosis response-4 protein (Par-4), evidencing a role of THAP1 in apoptosis (6). Moreover, the THAP1 protein is linked to the pRb (retinoblastoma protein)/E2F cell-cycle pathway and functions as an endogenous physiological regulator of endothelial cell proliferation by controlling a series of pRb/E2F cell-cycle-specific target genes (7). In vivo, THAP1 associates with the RRM1 promoter, a critical pRb/E2F target gene activated at the G1/S transition and required for S-phase DNA synthesis (7). An optimal range of THAP1 expression level is required for THAP1 to finely control EC proliferation and cell-cycle progression (7). Furthermore, THAP1 recruits the host-cell factor-1 (HCF-1) on cell-cycle specific promoters, supporting a critical role of this transcription factor in cell-cycle regulation (8).
The DNA-binding activity of THAP1 occurs at the N-terminal THAP domain, which is a highly conserved motif with a CCCH signature (residues C5, C10, C54 and H57) providing ligands for zinc coordination, four invariant residues (P26, W36, F58 and P78) and a C-terminal AVPTIF motif (9). Single-point mutations of the CCCH signature and of the four invariant residues abrogate DNA binding (9). The THAP domain (residues 1–81 in THAP1) adopts a βαβ fold containing an atypical long loop-helix-loop (L2-H1-L3) insertion between the two anti-parallel β-strands (10). The THAP domain from human THAP1 specifically recognizes a consensus THAP-binding site (THABS) consisting of the 5′TxxG/TGGCA3′ sequence (9). We have previously shown that other THAP domains do not bind specifically to the THABS motif (10). Specific DNA recognition by the THAP domain of human THAP1 is achieved by insertion of the double-stranded β-sheet and the N-terminal loop into the DNA major groove, giving direct contacts with invariant GGCA bases of the THABS motif, while the C-terminal loop points toward the minor-groove (11), adopting a bipartite DNA-recognition strategy shared with the drosophila transposase (12).
The transcription factor THAP1 has joined recently the family of dystonia disorders, following the discovery of two distinct heterozygous mutations (one frame-shift and one missense) in the THAP1 gene in patients with primary torsion dystonia, DYT6, in Amish-Mennonite families (4). As these mutations resulted in truncated or substituted THAP domain, it has been proposed that they would negatively affect DNA binding, causing transcriptional dysregulation of THAP1 target genes (4). Subsequently, the number of dystonia-related mutations identified in the THAP1 gene has increased significantly and, to date, more than 50 mutations, in several genetically different populations have been reported. The sequence variations are mainly missense mutations or frame-shift mutations, mostly in the heterozygous state with one normal copy in the allele, producing an autosomal dominant disease with reduced penetrance of about 60% (5). Though the mutations are spread over the whole gene, affecting the N-terminal DNA-binding domain, the NLS or the C-terminal coiled-coil (4,13–18), many of them (more than 30) cluster within the DNA-binding domain, most of which cause early-onset dystonias (4,13–15,17–21). Some synonymous mutations affecting the THAP domain have also been discovered in adult-onset dystonia patients, but the role of these mutations in THAP1 function remains unclear (22). A functional link between the two primary dystonia sub-types, DYT6 and DYT1 was proposed recently, following the discovery of direct regulation of the human core promoter TOR1A (DYT1) activity by THAP1, that could be mediated by a THABS site present in TOR1A (23,24). Therefore, the mutations described in DYT6 patients and which affect the THAP domain have been proposed to disrupt DNA binding, decreasing repression of the TOR1A gene expression (23).
So far, there is no obvious genotype/phenotype relationship noted in THAP1 and the molecular mechanisms by which THAP1 mutants cause DYT6 dystonia are not known. The UMD (Universal Mutation Database)-THAP1 locus specific database has recently compiled up-to-date available informations about the mutations in the THAP1 gene, providing a classification of pathogenicity (20).
The biophysical studies conducted here focus on a panel of missense mutations reported in the DNA-binding domain of human THAP1. Based on the 3D structure of the THAP domain of THAP1 interacting with its natural DNA target (11) and on our capacity to form and assess experimentally DNA/substituted THAP complexes, we discuss the impact of various mutations on the structural integrity and thermostability of the mutant proteins relative to the wild-type protein. We have examined the thermodynamic signatures of DNA-binding by the wild-type THAP domain and the DYT6-associated mutant proteins. No obvious correlation between the thermostability and the DNA binding activity of the missense mutants was found. Many of the mutations investigated do not strongly decrease DNA binding and some of them even lead to stronger DNA binding. The most striking effect is the decrease of stability observed for most of the mutations. In summary, our biochemical and biophysical data provide insights into the probable structural and functional defects caused by the DYT6-mutations in the THAP domain and should help to improve predictions of pathogenicity.
MATERIALS AND METHODS
Sample preparation
The wild-type THAP domain of human THAP1 (Met1-Phe81) was amplified by PCR and cloned in-frame with a C-terminal His-tag into a modified pET-26 plasmid as already described (10). The point-mutants (S6F, Y8C, G9C, N12K, D17G, S21T, P26R, R29Q, A39T, F81L) were amplified by PCR using specific primers containing the corresponding mutations and sub-cloned as NdeI-XhoI fragments into modified pET-26 plasmid. Recombinant THAP domains (wild-type and mutant proteins) were produced as His-tag fusion proteins in Escherichia coli BL21(DE3). Cells were grown at 37°C either in LB medium or minimal (M9) medium containing 15NH4Cl and 15N celtone to produce unlabeled or isotopically 15N uniformly labeled proteins, respectively. To improve the production of the P26R recombinant mutant protein, a lower growth temperature (16°C) was also tried. All the proteins were purified at 4°C following the same protocol as previously described (10,11). The 16-bp DNA duplexes were reconstituted by mixing equimolar amounts of oligonucleotides (Eurofin MWG Operon, Germany), 5′dGCTTGTGTGGGCAGCG3′ and 5′dCGCTGCCCACACAAGC3′ (RRM1 DNA probe) that were then heated to 75°C prior to slowly cooling. The same protocol was followed to reconstitute the 14-bp non-specific DNA fragment (dGATTTGCATTTTAA/dTTAAAATGCAAATC).
NMR measurements
Nuclear magnetic resonance (NMR) experiments were recorded at 298 K on a Bruker Avance 600 MHz spectrometer equipped with a cryogenic probe (Bruker Biospin, Germany). Two-dimensional 1H-15N HSQC spectra were collected for each protein sample dissolved in 50 mM deuterated Tris (pH 6.8), 100 mM NaCl and 2 mM DTT. Spectra were processed with TopSpin (Bruker Biospin) and analyzed using CARA (25). The correct folding was assessed from the chemical shift dispersion on a 2D 1H-15N HSQC spectrum. The peak-picking of the 2D 1H-15N HSQC spectrum of the wild-type protein (Met1-Phe81) was based on the assignment of the 15N and HN backbone proton resonances of the THAP domain including a double mutation C62SC67S (BMRB code 16 485). The 15N and HN backbone resonances for each mutant were clearly identified based on the assignment of the wild-type protein. Overall chemical shift perturbations (CSPs) were calculated from the 15N and HN chemical shift changes according to: Δδ = {(ΔδHN)2 + (ΔδN × 0.154)2}1/2 (10). Similarly, 2D 1H-15N HSQC spectra were collected for the NMR DNA-protein samples prepared in 50 mM deuterated Tris (pH 6.8), 30 mM NaCl and 2 mM DTT.
Differential scanning fluorimetry
The stability of wild-type and mutant proteins was assessed using a SYPRO® Orange unfolding temperature (Tm) test performed on a CFX 96 real-time polymerase chain reaction detection system. Data were analyzed using the CFX Manager Software (Bio-Rad Laboratories, USA). The purified proteins were diluted to concentrations ranging from 70 µM to 150 µM in a buffer consisting of 50 mM Hepes pH 7.2, 100 mM KCl, 1 mM MgCl2 and 250 µM tris(2-carboxyethyl)phosphine (TCEP) with 1:1000 SYPRO® Orange (Invitrogen Life Technologies, USA). The proteins were submitted to the heating cycle from 20 to 70°C with a ramp rate of 0.3°C per min. The fluorescence of the SYPRO® Orange dye was followed at 580 nm as a function of temperature. The Tm value was estimated from the transition midpoint of the fluorescence curve, which corresponds to temperature at which half of the protein population is unfolded.
Isothermal titration calorimetry
Isothermal titration calorimetry (ITC) experiments were conducted at 25°C on a Microcal ITC200 instrument (Microcal GE Healthcare, UK). Buffer screening was performed to optimize the quality of the experimental ITC data. Suitable buffer consisted of 20 mM Hepes pH 7.2, 100 mM KCl, 1 mM MgCl2 and 250 µM TCEP. To ensure minimal buffer mismatch, protein and DNA samples were dialyzed against the same buffer. Experiments consisted of a series of 20 x2 µl injections of DNA (150–200 µM) into the protein (15–20 µM) containing thermostatic cell (initial delay of 60 s, duration of 4 s and spacing of 180 s). Duplicates or triplicates experiments were measured systematically. According to previous NMR data indicating that the THAP domain binds DNA as a monomer (11), the corrected binding isotherms were fitted for a single-site binding model using non-linear least squares analysis to obtain values of equilibrium binding constant (Ka), stoichiometry and enthalpy changes (ΔH) associated with DNA binding.
Fluorescence measurements
Fluorescence spectra were recorded on a PTI Model QM-4 spectrofluorimeter (Photon Technology International, USA) at 25°C equipped with a thermo-electrically temperature-controlled cell holder (quartz cuvette, 1 cm × 1 cm). The single tryptophan was excited at 295 nm and emission was recorded at 324 nm. Experiments were performed with initial protein concentration of 0.5 µM in a 4 ml buffer volume, with a buffer identical to that used for differential scanning fluorimetry (DSF) and ITC experiments. Each DNA duplex (100 µM) dissolved in the same buffer was progressively added to the protein sample with protein:DNA ratios ranging from 1:0 to 1:6.5, with a final protein dilution <1%. For data analysis, the observed fluorescence intensities were normalized, relating to the initial fluorescence intensity and the dissociation constants were determined using non-linear least squares analysis with GOSA software (26), using the following equation: 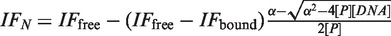 with α = [P] +[DNA] + Kd. IFN is the normalized fluorescence intensity, IFfree and IFbound, are the normalized fluorescence intensities for the unbound and DNA-bound forms, respectively. [P] and [DNA] are the protein and DNA concentrations, respectively.
with α = [P] +[DNA] + Kd. IFN is the normalized fluorescence intensity, IFfree and IFbound, are the normalized fluorescence intensities for the unbound and DNA-bound forms, respectively. [P] and [DNA] are the protein and DNA concentrations, respectively.
RESULTS
Structural location of the variants
The 10 DYT6-associated mutations investigated in the present work (S6F, Y8C, G9C, N12K, D17G, S21T, P26R, R29Q, A39T and F81L) correspond to a representative panel of missense mutations in the THAP DNA-binding domain that have been reported by several genetic studies (4,13–15,19,21). Figure 1 shows the location of the studied mutations on the structure of the wild-type THAP domain of THAP1. In a previous study, we showed that the THAP domain of THAP1, adopts a βαβ topology consisting of a short antiparallel β-sheet (β1, F22-K24; β2, S52-C54) and a long loop-helix-loop (L2-H1-L3) motif (with L2, F25-K32; H1, C33-V40 and L3, R41-S51) inserted between the two anti-parallel β-strands of the central β-sheet (10,11), see Figure 1. Residues that were critical for the specific recognition of invariant bases in major groove DNA were K24 and S52 from the β-sheet, Y50 and S51 from loop L3 and Q3 in the N-terminus (loop L1). In addition, R65 in loop L4 contacted the DNA minor groove (11). In the present work, the DYT6-causing mutations investigated relate to residues located within loops (S6-S21 in loop L1, P26-R29 in loop L2) or located in structured regions (A39 in H1 and F81 in H4), see Figure 1. None of the mutations studied in the present work involve residues that had been shown previously to directly contact DNA bases (11). Instead, they involve residues that are preserved in THAP1 orthologs but are poorly conserved among THAP domain paralogs. All of the DYT6 disease-causing mutant proteins were produced at 37°C as soluble fractions in E. coli and were purified at 4°C using a reproducible protocol (see ‘Materials and Methods’ section). Expression levels of the mutants were compared with that of the wild-type THAP domain (Figure 2A). Most of the proteins were expressed in amounts comparable to that of the wild-type protein with the exception of the S6F protein, which exhibited significantly reduced expression. Furthermore, the P26R protein variant, could not be purified in useable quantity, whatever the temperature used for protein expression (37°C or 16°C), preventing any further biophysical study.
Figure 1.

Sequence and structural mapping of the substituted amino acids in THAP1 (DYT6). (A) The positions of the substituted amino acids are indicated with an arrow along the sequence of the THAP domain. Secondary structure elements are indicated on the top. The four invariant residues and the zinc-coordinating CCCH motifs are shown in red and orange, respectively. Residues marked with an asterisk are those involved in specific DNA recognition (11). (B) The positions of the mutations are mapped onto the molecular surface. (C) The positions of the mutations are mapped onto the 3D structure of the THAP domain interacting with its RRM1 DNA target (11). Orientations are the same than in B. (D) Topology diagram indicating the positions of the mutations.
Figure 2.

Expression and thermostability. (A) The wild-type protein and the THAP domain point mutants (S6F, Y8C, G9C, N12K, D17G, S21T, P26R, R29Q, A39T and F81L) were expressed as described in Materials and Methods section. The arrow indicates their migration band on SDS-PAGE gel; MW, molecular weight markers, NI, before induction with IPTG (B) Plot showing the Tm values of the proteins (ordered by their unfolding temperatures).
Stability and structural integrity of the variant proteins
The thermostability of all mutants was investigated using DSF, which follows temperature-induced changes in the fluorescence of an environmentally sensitive dye (SYPRO® Orange in the present work) that has affinity for hydrophobic regions of a protein. As the protein unfolds upon heating, the dye binds to exposed hydrophobic regions of the protein, leading to a significant increase in fluorescence. The thermal stability of the mutants was compared with that of the wild-type protein, which exhibits a melting Tm value of 46.2 ± 0.6°C at which 50% of the protein is unfolded (Figure 2B). All of the mutant proteins displayed lower thermostability than the wild-type, with unfolding temperatures ranging from 45°C to below 37°C (Table 1 and Figure 2B). In particular, two mutations (N12K and S6F) have a marked effect on protein stability, resulting in a large proportion of unfolded protein (>50%) at physiological temperature.
Table 1.
Unfolding temperatures (Tm) and DNA binding affinities obtained for the wild-type protein and dystonia mutants at 25°C
| DSF | Fluorescence |
ITC |
|||
|---|---|---|---|---|---|
| Tm (°C) | Kd (nM) | Kdmutant/ Kdwt | Kd (nM) | Kdmutant/ Kdwt | |
| WT | 46.2 ± 0.5 | 150 ± 10 | 1 | 540 ± 190 | 1 |
| WT/nsa | nd | 1800 ± 150 | 11.8 | 8928 ± 1280 | 16.5 |
| S21T | 45 ± 0.2 | 420 ± 20 | 2.8 | 3780 ± 900 | 7.0 |
| Y8C | 43.5 ± 0.2 | 230 ± 20 | 1.5 | 510 ± 100 | 0.9 |
| R29Q | 42 ± 0.9 | 680 ± 60 | 4.5 | 1850 ± 500 | 3.4 |
| F81L | 40.4 ± 0.8 | 100 ± 10 | 0.7 | 700 ± 60 | 1.3 |
| A39T | 39.6 ± 0.7 | 400 ± 90 | 2.7 | 4032 ± 1080 | 7.4 |
| G9C | 39.3 ± 0.5 | 390 ± 80 | 2.6 | 920 ± 300 | 1.7 |
| D17G | 38.1 ± 1 | 25 ± 5 | 0.2 | 350 ± 100 | 0.6 |
| S6F | 36.7 ± 0.5 | 660 ± 90 | 4.4 | 3140 ± 1100 | 5.8 |
| N12K | 35.8 ± 0.5 | 80 ± 10 | 0.5 | 600 ± 100 | 1.1 |
| N12K/nsa | nd | 400 ± 65 | 2.7 | 1754 ± 500 | 3.2 |
| P26R | nd | nd | nd | nd | nd |
aWT/ns and N12K/ns: binding to non-specific DNA target. nd: not determined.
NMR spectroscopy was used to investigate the effect of the mutations on the protein structure. In order to dissociate mutation effects from denaturation events, NMR experiments were recorded at 25°C for each of the THAP domain-substituted proteins and 2D 1H-15N HSQC spectra were compared with the spectrum recorded for the wild-type protein (Supplementary Figures S1 and S2). For most of the mutant proteins, the resulting NMR spectra displayed only specific chemical shift changes showing that the overall fold is preserved in any case. The effect of the single-point mutations was probed by NMR CSP to follow the chemical shift changes between the substituted and the wild-type proteins (Figure 3 and Supplementary Figure S3).
Figure 3.

Structural integrity. (Left) Histogram of chemical shift changes (CSPs) measured between the mutant and wild-type proteins as a function of the residue number. Overall CSPs were calculated from the 15N and HN chemical shift changes according to: Δδ = {(ΔδHN)2 + (ΔδN x 0.154)2}1/2 (10). (Right) Histogram of CSPs measured between the substituted and wild-type DNA-bound proteins.
The three mutations displaying Tm values between 42 and 45°C (Y8C, S21T, R29Q) concern residues located in loops of the domain (Figure 1C). Serine 21 is located in loop L1 preceding the first β-strand. It is conserved in THAP1 orthologous proteins but is replaced by threonine in several THAP paralogs. Replacing serine with threonine, a polar and uncharged amino acid, like serine, was therefore expected to be well tolerated. The mutant protein S21T exhibits an HSQC spectrum similar to that recorded for the wild-type protein, showing CSP only for the mutated residue (Figure 3 and Supplementary Figure S1). This result reflects a minor local perturbation induced by the mutation, consistent with the moderate impact on protein stability (Tm value of 45°C). Replacement of Y8 in loop 1, with cysteine gave small CSPs for V40 in the helix and larger changes for some solvent-exposed residues (R42-N44) that occupy positions in the beginning of loop L3, spatially close to Y8 (Figure 3). The substituted residue is not critical for the proper folding and the mutation Y8C resulted in Tm value of 43.5°C (Table 1 and Figure 2B). The R29Q replacement in loop 3 is also relatively well tolerated (Tm value of 42°C). Small changes are observed for few residues in the helix (C33, V40) and for residues E74-V77 in loop L4 (Supplementary Figure S3).
All of the other mutations resulted in Tm differences higher than 5°C compared with the wild-type protein. Among them, the mutations A39T and F81L affect hydrophobic residues that are important for the structure of the THAP domain. In particular, F81 is the last residue of the AVPTIF motif, highly conserved across THAP1 orthologs. It is trapped between residues A39 in the helix and H57 in the CCCH motif (Figure 4A). Its substitution with a hydrophobic and aliphatic amino acid (i.e. leucine) resulted in amide chemical shift changes (>0.5 ppm) for the invariant histidine H57 in the zinc binding site and T79 of the AVPTIF motif (Supplementary Figure S3). Alanine A39 is a short and hydrophobic residue buried in the structure and located in the α-helix. Its replacement by a threonine with a polar side-chain resulted in a large decrease in Tm (Tm shift of 6.6°C). The A39T variant is associated with a good quality HSQC spectrum which displays significant CSPs (between 0.4 and 1.6 ppm) for residues G9 in loop L1, W36 and A38 in the C-terminus of the α-helix and T79 in the AVPTIF motif (Figure 3 and Supplementary Figure S2), reflecting alteration of the hydrophobic packing.
Figure 4.

Close-up views of the THAP domain of THAP1. (A) F81 in the AVPTIF motif sitting between A39 in the helix and H57 in the zinc coordination site. (B) R29 forming a salt-bridge with E35 in the free THAP domain (top) and with E74 in the complex with DNA (bottom), (C) S21 (red) in the THAP–DNA complex providing a polar contact with H23 (pink) and possible water-mediated contacts with the T8 and G9 DNA backbone phosphates (blue).
The two mutations G9C and D17G concern solvent-exposed residues in loop L1 and gave Tm values of 39.3 and 38.1°C, respectively (Table 1 and Figure 2B). Glycine G9 precedes Cys10, which is one of the invariant CCCH zinc coordinating residues. Like it was observed for F81L, G9C substitution resulted in CSPs for the backbone amide proton resonances of two residues, namely H57 in the zinc binding site and T79 in the AVPTIF motif, close to the zinc coordination site (Supplementary Figure S3). Mutation D17G substitutes a solvent-exposed residue located in a region of loop L1 that is turned towards the outside, and that exhibits mobility on a ps-ns time scale (10). Surprisingly, this mutation resulted in chemical shift changes for T79 in the AVPTIF motif and for residues A39-V40 in the α-helix H1 (CSPs < 0.6 ppm), which are not in the vicinity of D17 but rather on the opposite face of the protein (Supplementary Figure S1). One potential explanation could be that the introduction of a glycine residue in place of aspartate D17, resulting in a loss of a negative charge might increase the mobility of loop L1. In summary, the four mutations G9C, D17G, A39T and F81L gave large CSPs for a few residues in the α-helix, for the last histidine of the CCCH motif (H57) and for T79 of the AVPTIF motif. This suggests alteration of the interactions network between the helix, the AVPTIF motif and zinc coordination site, resulting in important fold destabilization (Tm shift above 5°C compared with the wild-type protein).
Finally, the two most unstable mutations, S6F and N12K exhibited Tm values of 36.7°C and 35.8°C, indicating the presence of a large proportion of unfolded protein (>50%) at physiological temperature (Table 1). Insertion of phenylalanine, an aromatic side chain, instead of serine S6 resulted in CSPs (between 0.5 and 1 ppm) for core residues W36-A39 in the helix, a key structural element (Figure 3). Several residues at the end of loop L3, including K46 and T48 and amino acids S51-I53 that are spatially close to S6 in the 3D structure, were also affected. Notably, some of these (T48, S51–S52) participate in DNA recognition (11). The mutation N12K is the most destabilizing one. It maps to an exposed region of loop L1 close to the zinc coordination. Substitution of polar and uncharged asparagine with positively charged lysine gave significant chemical shift changes (∼0.9 ppm) for residues K11-R13 located in loop L1 and surrounding the mutated residue (Supplementary Figure S1). One mutation (P26R) could not be investigated by thermal shift assays and NMR. Proline P26 is one of the four invariant residues in the THAP signature that had been determined to be critical for DNA binding (9). P26 resides in loop L2 and participates with W36 and F81 in maintaining the hydrophobic network (10). Its substitution with a long positively charged amino acid (arginine) is likely to strongly destabilize the hydrophobic core, resulting in fold disruption.
Because the dystonia-causing mutations involve residues located in the DNA-binding domain, we also used NMR spectroscopy to follow the CSPs between the substituted and the wild-type proteins in their DNA-associated state (Figure 3 and Supplementary Figure S3). In many cases, the CSPs profiles exhibited by the DNA-bound substituted proteins were similar to those obtained for the unbound proteins (such as S21T, A39T and F81L). However, several substitutions such as S6F, Y8C and D17G led to larger chemical shift changes for the conserved histidine H57, the last residue of the zinc-coordinating CCCH motif (Figure 3 and Supplementary Figure S3), reflecting structural perturbations at the zinc site in the presence of DNA upon mutations. Notably, no chemical shift change had been observed for H57 in the wild-type domain in complex with DNA (11). In the case of the R29Q mutant, a large chemical shift change (∼0.6 ppm) was observed for asparagine N75 in the presence of DNA. We had previously identified some structural changes caused by the binding of the THAP domain to specific DNA (11). In particular, in the free protein, the arginine side-chain is oriented along the helix, likely forming a salt-bridge with E35 in the helix (Figure 4B). In the complex with DNA, R29 adopts a different orientation that brings it closer to E74 and N75 in loop L4. More precisely, in the bound position, R29 participates in a salt-bridge with the carboxylate side-chain of E74 (Figure 4B), resulting possibly in the stabilization of loop L4. Therefore, the substitution of R29 with a glutamine is likely to prevent the salt-bridge interaction, increasing the mobility of this loop region and destabilizing the beginning of the helix.
DNA binding
In order to address the ability of the proteins to bind DNA, we used ITC and fluorescence quenching, following the intrinsic fluorescence of W36, the single tryptophan, to determine the DNA-binding affinities (Figure 5). Table 1 summarizes the values of the dissociation constants (Kd) for the wild-type protein and the different mutant proteins obtained using these two techniques. The fluorescence assays yielded systematically higher apparent affinities than ITC by several-fold, due to the lower concentration used. However, the relative reductions in DNA-binding compared with the wild-type protein (ratios Kdmutant/Kdwild-type) agreed quite nicely between the two methods (Table 1 and Figure 5C). Five of the mutations (S6F, G9C, S21T, R29Q and A39T) showed a reduction in DNA binding, although the ratios by which affinities are reduced are generally much lower than those previously reported for mutations of residues in the THAP domain of drosophila that are directly involved in DNA recognition (∼15 to 20-fold) (12). Three mutations (Y8C, F81L and N12K) gave binding constants similar to the wild-type (Figure 5C and D). Strikingly, one mutation (D17G) increased the binding affinity, as seen both by ITC (0.6-fold) and fluorescence (0.17-fold in Kd). This variant is clinically distinguishable with an age-of-onset of the symptoms (43 years) that is later than in most of the DYT6-associated cases.
Figure 5.

DNA binding at 25°C. (A) ITC thermodynamic profile obtained for the wild-type protein (15 µM) titrated by DNA (150 µM). The upper isotherm indicates the DNA binding raw data. The lower curve is obtained after integration of individual heat flow signals as function of the DNA/protein molar ratio in the calorimeter cell. (B) Fluorescence titration of W36 (excitation at 295 nm and emission at 324 nm) as a function of increased DNA concentrations, for the wild-type protein (0.5 µM). (C) Histogram of the relative DNA-binding affinities of the mutant proteins determined by ITC. The values reported above the bars indicate the ratios Kdmutant/Kdwild-type. (D) Histogram of the relative DNA-binding affinities of the mutant proteins determined by fluorescence. The proteins are ordered as in Figure 2.
The S6F mutation (c.17C>T nucleotide mutation in exon 1) was discovered in a German family with first symptoms of dystonia in hands, that then developed generalized with cervical dystonia (14). An increase of almost 6-fold in the dissociation constant (4.4-fold by fluorescence) indicates that this mutation reduces substantially DNA binding affinity (Figure 5). As shown by NMR, the substitution S6F gives CSPs for residues T48 and S51 that are involved in DNA recognition (Figure 3), consistent with the observed effect. This defect in DNA binding has not been observed previously with the S6A mutation (10). However, in the present work, the mutation replaces serine S6 with a phenylalanine, much bulkier than an alanine. Mutation S21T (c.61T>A) is a missense mutation, presumably non-recurrent, that was identified in a family with mixed European ancestry. In this family, the arm was the commonly site of onset (13). The S21T mutation decreases binding affinity by 7-fold (ITC) or 3-fold (fluorescence). These results support the previous studies according to which the S21T mutant fails to bind to a THABS-containing TOR1A DNA probe in electrophoretic mobility shift assays and abolishes its interaction with the TOR1A promoter in vivo (23). However, the structure of the DNA–THAP complex did not reveal any direct DNA contact with S21 (11). One potential explanation could be that the mutation destabilizes the polar interaction with His23 located in the centre of the β-sheet, which is crucial for DNA recognition or disrupts possible water-mediated contacts with the phosphate groups of T8 and G9 DNA bases (Figure 4C). Mutation A39T (c.115G>A in exon 2) is a non-recurrent mutation that was identified in a family with European ancestry (13,20). No biochemical data for the A39T variant has been reported in the literature. A more than 7-fold increase in the dissociation constant was found by ITC measurements (∼3-fold increase by fluorescence), reflecting a significant contribution of A39 to the overall DNA-binding affinity (Table 1 and Figure 5). Therefore, together with S6F and S21T, the A39T mutation should be indeed considered pathogenic based on its impact on DNA binding.
Compared to the latter mutations, G9C and R29Q have less consequences on DNA binding (Figure 5C). The G9C variant was identified in a non-Amish family with significant history of dystonia. The mutation is the result of the missense mutation c.25G>T in exon 1, identified in a patient with a multifocal dystonia and who presented an early age of onset in the arm (15). The G9C mutation results in an increase in the dissociation constant by almost 2-fold, in agreement with fluorescence data (2.6-fold, Table 1). These results constitute the first biochemical data reported for this mutant. The R29Q mutation results from the c.86G>A transition and was identified in two sporadic cases suffering from early-onset cervical dystonia and early-onset generalized dystonia (19). Both calorimetric and spectroscopic data show that the R29Q mutation display lower affinity compared with wild-type protein, by 3.4-fold or 4.5-fold, respectively (Figure 5). Consistent with these results, EMSA assays had shown previously that the R29A mutation severely affects DNA binding (10).
Three mutations (Y8C, N12K and F81L) did not change DNA-binding affinity (Table 1 and Figure 5). The Y8C mutation (c.23A>G in exon 1) was identified in a patient with generalized dystonia that started in foot at adolescence onset (14). The dissociation value determined by ITC for the Y8C variant was 510 nM (versus 540 nM obtained for the wild-type protein). These results agree well with fluorescence data (Table 1) and are consistent with previous gel-shift assays (10). Insertion of positively charged lysine instead of asparagine 12 in loop L1 (mutation N12K, c.36C>A) is also not detrimental on DNA binding (Table 1 and Figure 5). Finally, the F81L missense mutation (c.241T>C in exon 2), one of the first reported DYT6 mutations, was initially described as decreasing DNA-binding activity (4). Our calorimetric and fluorescence data indicate that replacing F81 by a leucine does not impair DNA binding in vitro (Figure 5).
Thermodynamic profiles
In addition to the equilibrium binding constant, the ITC method provides a unique means of measuring the heat quantity associated with a binding process required for a complete thermodynamic characterization of the interaction (27,28). The binding constant (Ka) gives the Gibbs free energy change (ΔG) using the relationship, ΔG = –RTln Ka. Figures 5 and 6 show the representative ITC profiles for the wild-type and mutant proteins. In the upper panel, each of the heat bursts corresponds to a single injection of DNA into the protein-containing cell. The isotherm obtained during DNA titration by the wild-type THAP domain of THAP1 shows one binding event with a stochiometry of 1:1 indicating that, in solution one THAP molecule binds to one DNA duplex (Figure 5), in agreement with our previous NMR studies (11). The equilibrium dissociation constant value of 540 ± 190 nM in the Hepes buffer (20 mM Hepes, pH 7.2, 100 mM KCl, 1 mM MgCl2, TCEP 250 µM) agrees reasonably well with the Kd value determined using fluorescence quenching (150 ± 10 nM) and is close to the value of 480 ± 60 nM previously found by fluorescence anisotropy in a buffer consisting of 50 mM Tris, 30 mM NaCl and pH 6.8 (11). The contributions of enthalpy and entropy to the overall free energy change of association are ΔH = –3.7 ± 0.1 kcal/mol and –TΔS ( = ΔG − ΔH) = –4.9 ± 0.3 kcal/mol, respectively, indicating that DNA binding by the wild-type protein is thermodynamically favored and driven both by enthalpy (negative ΔH) and entropy (negative TΔS). As previously described, the wild-type protein forms several specific hydrogen bonds with the invariant bases of the THABS motif (11), and these dominate the negative enthalpy changes. Formation of the complex between the THAP domain and its cognate DNA target is accompanied by the burial of hydrophobic residues and the release of ordered water molecules into the solvent, which result in favourable binding entropy. Overall, the thermodynamic signature obtained for the formation of the specific complex suggests a moderate DNA distorsion and weak DNA bending (29), consistent with the solution structure of the THAP domain interacting with its DNA target (11). The binding of the THAP domain with a DNA fragment that does not contain the THABS motif was investigated using ITC and fluorescence quenching (Table 1). The THAP domain binds its cognate target ≈16-fold more strongly than non-specific DNA (≈12-fold by fluorescence). Moreover, the association of the THAP domain with a non-specific DNA is an entirely entropy driven process at 25°C as the enthalpic component is positive (ΔH = 2.4 ± 0.1 kcal/mol) and is opposed to the binding reaction (Table 2). Specific DNA binding was investigated for the different mutant proteins. In most cases, the introduction of the single-point mutations led to minor differences in ΔG values compared with that of the wild-type protein (Figure 6). This observation is consistent with ratios of relative DNA-binding reductions (Table 1), that do not exceed a factor of 7.4 (ITC) or 4.4 (fluorescence). With the exception of S21T, all of the substituted proteins showed larger releases of heat associated with DNA binding compared with the wild-type protein (Figure 6). Four of them (S6F, Y8C, N12K and A39T) indicate enthalpy changes much larger and more favorable, which drive the binding event (Figure 6). For these mutants, the large enthalpy changes are compensated by weak or unfavorable entropic contributions so that the overall binding free energies do not differ by more than 1.1 kcal/mol from the value obtained for the wild-type protein (Table 2). For the mutant S6F, the large favorable enthalpy change is the exclusive driver for DNA binding, as the mutation results in unfavorable entropy, likely as a result of a loss of conformational freedom associated with the introduction of a bulky amino acid in place of S6. The two mutations Y8C and A39T involve solvation changes at the interface, resulting from the introduction of polar side chain and possible ordering of water molecules in the extra space available following the removal of tyrosine or alanine residues, which contributes unfavorably to ΔS and favorably to ΔH (30). In the case of N12K, introduction of a basic side-chain might favor new electrostatic contacts with the DNA phosphate backbone, resulting in formation of additional enthalpic interactions (van der Waals and hydrogen bonding). However, the enthalpic gain did not improve DNA binding as it is totally compensated by a loss of entropy. The association of the N12K mutant protein with a non-specific sequence was also investigated using ITC and fluorescence at 25°C, revealing a decrease in binding affinity by a factor of 3 (Table 1). In this example, the binding enthalpy is positive and the reaction is entropy driven (Table 2).
Figure 6.

Thermodynamic properties at 25°C obtained from ITC measurements (A) ITC thermodynamic profiles of the THAP domain point mutants (15–20 µM) titrated by DNA (150–200 µM). (B) Plot showing the thermodynamic parameters (ΔG, ΔH and TΔS). The proteins are ordered as in Figure 2.
Table 2.
Thermodynamic parameters of DNA binding for wild-type and DYT6 mutant proteins in Hepes buffer at 25°C
| ΔH (kcal/mol) | −TΔS (kcal/mol) | ΔG (kcal/mol) | |
|---|---|---|---|
| WT | –3.7 ± 0.1 | –4.9 ± 0.3 | –8.5 ± 0.4 |
| WT/nsa | 2.4 ± 0.05 | –9.3 ± 0.01 | –6.9 ± 0.06 |
| S21T | –2.6 ± 0.6 | –4.8 ± 0.7 | –7.4 ± 1.3 |
| Y8C | –7.9 ± 0.2 | –0.6 ± 0.3 | –8.6 ± 0.5 |
| R29Q | –5.3 ± 0.2 | –2.5 ± 0.4 | –7.8 ± 0.6 |
| F81L | –6.3 ± 0.3 | –2.1 ± 0.4 | –8.4 ± 0.7 |
| A39T | –9.2 ± 0.7 | 1.8 ± 0.9 | –7.4 ± 1.6 |
| G9C | –5.6 ± 0.3 | –2.6 ± 0.1 | –8.2 ± 0.4 |
| D17G | –4.7 ± 0.2 | –4.1 ± 0.4 | –8.8 ± 0.6 |
| S6F | –9.7 ± 1.6 | 2.2 ± 1.8 | –7.5 ± 3.4 |
| N12K | –7.5 ± 0.3 | –1.00 ± 0.4 | –8.5 ± 0.7 |
| N12K/nsa | 2.3 ± 0.6 | –10.1 ± 1 | –7.8 ± 1.6 |
| P26R | nd | nd | nd |
aWT/ns and N12K/ns: binding to non-specific DNA target. nd: not determined.
DISCUSSION
The DYT6 dystonia is inherited as an autosomal dominant trait with a reduced penetrance of about 60%. Many of the currently known DYT6-associated mutations fall within the N-terminal THAP DNA-binding domain. However, the residues frequently mutated in DYT6 are not in direct contact with DNA and do not make critical contributions to DNA binding, unlike other transcription factors like the p53 tumor suppressor (11,31). Nevertheless, the mutated residues have been proposed to negatively affect DNA binding ability, establishing transcriptional dysregulation as a cause of primary DYT6 dystonia (2,4). The spectroscopic and calorimetric methods described here demonstrate that many mutations in the THAP domain do not eliminate DNA binding and some even result in stronger DNA binding. The most pronounced effect is the decrease of stability observed for several mutations.
The mutations introduced have a marked effect on protein stability and therefore could decrease the proportion of folded protein
Using NMR, most of the mutated proteins studied were found to retain the global protein folding. DSF was used to examine the thermal stability which has not been reported previously. The wild-type protein exhibits a Tm of 46.2 ± 0.6°C. The rather low intrinsic thermodynamic stability is particularly intriguing, as the importance of maintaining an appropriate range of THAP1 expression for optimal regulation of endothelial cell proliferation has been proposed previously (7). The low stability could help to facilitate rapid degradation for tight regulation of functional THAP1 concentration in the cell, as has been suggested for the p53 tumor suppressor (32). The finding that most of the mutations result in a decrease of the unfolding temperature lowering the Tm to 40°C or less for a few of them, suggests a potential effect in shortening the THAP domain half-time in vivo (33). Two studies have provided some evidence of the negative regulation of TOR1A expression by THAP1 (23,24). As a transcriptional repressor, imbalances in the expression level of folded THAP1 could result in abnormal torsinA protein levels (2,5). However, whether this effect would cause altered cellular integrity and account for DYT6 pathogenesis remains to be determined. Notably, the DNA-binding domain is only one-third of the full-length THAP1 protein. The latter has been proposed to dimerize in vivo, or participate with other partners in protein complexes (11) and it is likely that fine regulation of monomeric folded THAP1 concentration is required for optimal oligomerization of THAP1 on its downstream targets. Therefore, although the stability parameter may only partially account for the apparition of the disease, it should be considered as an additional factor to investigate the pathogenic effects of the mutations.
There is no obvious correlation between protein stability and DNA binding among pathogenic mutations
Of the mutations investigated in the present work, five exhibited a marked reduction in DNA-binding affinity (S6F, G9C, S21T, R29Q and A39T), three had no effect on binding (Y8C, N12K and F81L) and one slightly increased affinity (D17G). Based on our study, there is an apparent lack of correlation between protein stability and DNA binding, as illustrated by the S21T mutant, with reduced DNA-binding affinity and no alteration in protein stability. At the other extreme, the N12K mutant has the most striking effect on protein stability by lowering the Tm by more than 10°C, while it does not modify the overall binding affinity. These two mutations were identified in European family members with the first symptoms of early-onset focal dystonia reported mainly in arm, which developed into generalized or multifocal distributions (13). Only one mutant, Y8C, behaves like the wild-type protein. Though these results would tentatively propose Y8C as a non-pathogenic mutation, it has been reported in a family with generalized dystonia following onset in the foot. Therefore, these three examples show the difficulty of linking the biophysical properties of mutant proteins with associated pathological behaviors.
Furthermore, it has been proposed that, like S21T, some DYT6-associated mutants within the DNA-binding domain, and in particular F81L would result in abrogation of THAP1 binding to TOR1A in vivo (23). In contrast with these studies, we did not observe a reduction of DNA binding by the F81L mutant. However, our studies were performed with a THABS-containing 16-bp DNA sequence much shorter than the DNA fragment used for the electrophoretic mobility shift assays (EMSA) (23). On the other hand, unlike S21T, the F81L mutation lowers protein stability. Therefore, even though these two mutations described in DYT6 patients may lead to similar functional consequences in vivo (i.e. abrogation of THAP1 binding to TOR1A) the molecular mechanisms leading to transcriptional dysregulation of downstream target genes could be different.
Large enthalpy changes are associated with specific DNA binding by the mutated proteins
Our work provides the first thermodynamic data for the association of the THAP domain with its specific DNA target. We found that both favorable enthalpy (ΔH < 0) and entropy (–TΔS < 0) changes drive the binding event, consistent with major-groove recognition (34,35) and formation of hydrogen bonds and van der Waals interactions between the THAP domain and its cognate DNA target. On the other hand, the association of the THAP domain with a non-specific DNA target has a Gibbs energy of binding entirely entropic, as expected when the electrostatic component is the main driving force (35). Surprisingly, with the exception of S21T, reactions of specific DNA recognition by the mutants are more exothermic than observed for the wild-type protein. However, the larger enthalpy changes are accompanied by lower entropic contributions, yielding small changes in the Gibbs binding energies (<1.1 kcal/mol), compared with the wild-type protein. As the binding enthalpy is a measure of binding specificity (van der Waals interactions and hydrogen bonding), the thermodynamic profiles obtained for the variant proteins suggest that differences in specific bonding interactions should be considered. The THAP domain displays a lower affinity for a non-specific target than for its cognate DNA target sequence (≈16-fold). This result is consistent with what has been reported for other sequence-specific DNA-binding proteins (36). Our data showed that the mutation N12K lowers affinity for the non-specific sequence only by a factor of ≈3, compared with the specific target, suggesting that introduction of N12K mutation affects the degree of selectivity. This result illustrates a possible effect of the mutation N12K on DNA-binding specificity. According to these data, in the context of DYT6 dystonia, some of the mutated proteins in the THAP domain, by altering the level of selectivity and therefore binding specificity could fail to target correctly their specific DNA sequence.
In conclusion, THAP1 has recently emerged as the genetic basis for DYT6 dystonia and no clear relationship between genotype and phenotype has emerged so far (20). Since the discovery of the DYT6-THAP1 mutations in 2009, the mutants that fall in the THAP domain were proposed to negatively affect DNA binding. Here, our study shows that most of the mutations identified in the DNA-binding domain retain their ability to bind DNA at permissive temperatures and some of them even bind DNA more strongly. The most striking effect is the decrease of stability observed for many mutations, reducing the already low Tm of the THAP domain to values close to the physiological temperature. Therefore, these data suggest potential effects of the DYT6-associated mutations in shortening the half-time of the THAP domain in vivo, accounting for the disease. However, the lack of any obvious correlation between protein stability, DNA binding and associated phenotype, together with the increasing number of THAP1 mutations detected out of the DNA-binding domain, suggest that additional factors (epigenetic, genetic, stability of mRNA …) might also contribute to the DYT6 disease.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Figures 1–3.
FUNDING
Equipments of microcalorimetry and thermal shift assays have been acquired by the IBiSA Integrated Screening Platform of Toulouse (PICT, IPBS, CNRS - Université de Toulouse). EU structural funds and the Région Midi-Pyrénées are acknowledged for funding the NMR equipment. Funding for open access charge: CNRS and Université de Toulouse.
Conflict of interest statement. None declared.
ACKNOWLEDGMENTS
The authors are grateful to Serges Mazères at IPBS for technical advices with fluorescence measurements and to Deborah Gater at IPBS for critical reading of the manuscript.
REFERENCES
- 1.Schmidt A, Klein C. The role of genes in causing dystonia. Eur. J. Neurol. 2010;17(Suppl. 1):65–70. doi: 10.1111/j.1468-1331.2010.03055.x. [DOI] [PubMed] [Google Scholar]
- 2.Bragg DC, Armata IA, Nery FC, Breakefield XO, Sharma N. Molecular pathways in dystonia. Neurobiol. Dis. 2011;42:136–147. doi: 10.1016/j.nbd.2010.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ozelius LJ, Hewett JW, Page CE, Bressman SB, Kramer PL, Shalish C, de Leon D, Brin MF, Raymond D, Corey DP, et al. The early-onset torsion dystonia gene (DYT1) encodes an ATP-binding protein. Nat. Genet. 1997;17:40–48. doi: 10.1038/ng0997-40. [DOI] [PubMed] [Google Scholar]
- 4.Fuchs T, Gavarini S, Saunders-Pullman R, Raymond D, Ehrlich ME, Bressman SB, Ozelius LJ. Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat. Genet. 2009;41:286–288. doi: 10.1038/ng.304. [DOI] [PubMed] [Google Scholar]
- 5.Ozelius LJ, Bressman SB. Genetic and clinical features of primary torsion dystonia. Neurobiol. Dis. 2011;42:127–135. doi: 10.1016/j.nbd.2010.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roussigne M, Cayrol C, Clouaire T, Amalric F, Girard JP. THAP1 is a nuclear proapoptotic factor that links prostate-apoptosis-response-4 (Par-4) to PML nuclear bodies. Oncogene. 2003;22:2432–2442. doi: 10.1038/sj.onc.1206271. [DOI] [PubMed] [Google Scholar]
- 7.Cayrol C, Lacroix C, Mathe C, Ecochard V, Ceribelli M, Loreau E, Lazar V, Dessen P, Mantovani R, Aguilar L, et al. The THAP-zinc finger protein THAP1 regulates endothelial cell proliferation through modulation of pRB/E2F cell-cycle target genes. Blood. 2007;109:584–594. doi: 10.1182/blood-2006-03-012013. [DOI] [PubMed] [Google Scholar]
- 8.Mazars R, Gonzalez-de-Peredo A, Cayrol C, Lavigne AC, Vogel JL, Ortega N, Lacroix C, Gautier V, Huet G, Ray A, et al. The THAP-zinc finger protein THAP1 associates with coactivator HCF-1 and O-GlcNAc transferase: a link between DYT6 and DYT3 dystonias. J. Biol. Chem. 2010;285:13364–13371. doi: 10.1074/jbc.M109.072579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clouaire T, Roussigne M, Ecochard V, Mathe C, Amalric F, Girard JP. The THAP domain of THAP1 is a large C2CH module with zinc-dependent sequence-specific DNA-binding activity. Proc. Natl Acad. Sci. USA. 2005;102:6907–6912. doi: 10.1073/pnas.0406882102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bessiere D, Lacroix C, Campagne S, Ecochard V, Guillet V, Mourey L, Lopez F, Czaplicki J, Demange P, Milon A, et al. Structure-function analysis of the THAP zinc finger of THAP1, a large C2CH DNA-binding module linked to Rb/E2F pathways. J. Biol. Chem. 2008;283:4352–4363. doi: 10.1074/jbc.M707537200. [DOI] [PubMed] [Google Scholar]
- 11.Campagne S, Saurel O, Gervais V, Milon A. Structural determinants of specific DNA-recognition by the THAP zinc finger. Nucleic Acids Res. 2010;38:3466–3476. doi: 10.1093/nar/gkq053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sabogal A, Lyubimov AY, Corn JE, Berger JM, Rio DC. THAP proteins target specific DNA sites through bipartite recognition of adjacent major and minor grooves. Nat. Struct. Mol. Biol. 2010;17:117–123. doi: 10.1038/nsmb.1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bressman SB, Raymond D, Fuchs T, Heiman GA, Ozelius LJ, Saunders-Pullman R. Mutations in THAP1 (DYT6) in early-onset dystonia: a genetic screening study. Lancet Neurol. 2009;8:441–446. doi: 10.1016/S1474-4422(09)70081-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Houlden H, Schneider SA, Paudel R, Melchers A, Schwingenschuh P, Edwards M, Hardy J, Bhatia KP. THAP1 mutations (DYT6) are an additional cause of early-onset dystonia. Neurology. 2010;74:846–850. doi: 10.1212/WNL.0b013e3181d5276d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao J, Zhao Y, Bastian RW, Perlmutter JS, Racette BA, Tabbal SD, Karimi M, Paniello RC, Wszolek ZK, Uitti RJ, et al. Novel THAP1 sequence variants in primary dystonia. Neurology. 2010;74:229–238. doi: 10.1212/WNL.0b013e3181ca00ca. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Djarmati A, Schneider SA, Lohmann K, Winkler S, Pawlack H, Hagenah J, Bruggemann N, Zittel S, Fuchs T, Rakovic A, et al. Mutations in THAP1 (DYT6) and generalised dystonia with prominent spasmodic dysphonia: a genetic screening study. Lancet Neurol. 2009;8:447–452. doi: 10.1016/S1474-4422(09)70083-3. [DOI] [PubMed] [Google Scholar]
- 17.Bonetti M, Barzaghi C, Brancati F, Ferraris A, Bellacchio E, Giovanetti A, Ialongo T, Zorzi G, Piano C, Petracca M, et al. Mutation screening of the DYT6/THAP1 gene in Italy. Movement disorders. 2009;24:2424–2427. doi: 10.1002/mds.22861. [DOI] [PubMed] [Google Scholar]
- 18.Lohmann K, Uflacker N, Erogullari A, Lohnau T, Winkler S, Dendorfer A, Schneider SA, Osmanovic A, Svetel M, Ferbert A, et al. Identification and functional analysis of novel THAP1 mutations. Eur. J. Hum. Genet. 2012;20:171–175. doi: 10.1038/ejhg.2011.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paisan-Ruiz C, Ruiz-Martinez J, Ruibal M, Mok KY, Indakoetxea B, Gorostidi A, Marti Masso JF. Identification of a novel THAP1 mutation at R29 amino-acid residue in sporadic patients with early-onset dystonia. Movement Disorders. 2009;24:2428–2429. doi: 10.1002/mds.22849. [DOI] [PubMed] [Google Scholar]
- 20.Blanchard A, Ea V, Roubertie A, Martin M, Coquart C, Claustres M, Beroud C, Collod-Beroud G. DYT6 dystonia: review of the literature and creation of the UMD Locus-Specific Database (LSDB) for mutations in the THAP1 gene. Hum. Mutat. 2011;32:1213–1224. doi: 10.1002/humu.21564. [DOI] [PubMed] [Google Scholar]
- 21.Ledoux MS, Xiao J, Rudzinska M, Bastian RW, Wszolek ZK, Van Gerpen JA, Puschmann A, Momcilovic D, Vemula SR, Zhao Y. Genotype-phenotype correlations in THAP1 dystonia: Molecular foundations and description of new cases. Parkinsonism Relat. Disord. 2012;18:414–425. doi: 10.1016/j.parkreldis.2012.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Groen JL, Ritz K, Contarino MF, van de Warrenburg BP, Aramideh M, Foncke EM, van Hilten JJ, Schuurman PR, Speelman JD, Koelman JH, et al. DYT6 dystonia: mutation screening, phenotype, and response to deep brain stimulation. Movement Disorders. 2010;25:2420–2427. doi: 10.1002/mds.23285. [DOI] [PubMed] [Google Scholar]
- 23.Gavarini S, Cayrol C, Fuchs T, Lyons N, Ehrlich ME, Girard JP, Ozelius LJ. Direct interaction between causative genes of DYT1 and DYT6 primary dystonia. Ann. Neurol. 2010;68:549–553. doi: 10.1002/ana.22138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kaiser FJ, Osmanoric A, Rakovic A, Erogullari A, Uflacker N, Braunholz D, Lohnau T, Orolicki S, Albrecht M, Gillessen-Kaesbach G, et al. The dystonia gene DYT1 is repressed by the transcription factor THAP1 (DYT6) Ann. Neurol. 2010;68:554–559. doi: 10.1002/ana.22157. [DOI] [PubMed] [Google Scholar]
- 25.Keller RLJ. 1st edn. Switzerland: Cantina Verlag; 2004. The computer aided resonance assignment tutorial. [Google Scholar]
- 26.Czaplicki J, Cornelissen G, Halberg F. GOSA, a simulated annealing-based program for global optimization of nonlinear problems, also reveals transyears. J. Appl. Biomed. 2006;4:87–94. [PMC free article] [PubMed] [Google Scholar]
- 27.Cliff MJ, Gutierrez A, Ladbury JE. A survey of the year 2003 literature on applications of isothermal titration calorimetry. J. Mol. Recogn. 2004;17:513–523. doi: 10.1002/jmr.714. [DOI] [PubMed] [Google Scholar]
- 28.Chaires JB. Calorimetry and thermodynamics in drug design. Annu. Rev. Biophys. 2008;37:135–151. doi: 10.1146/annurev.biophys.36.040306.132812. [DOI] [PubMed] [Google Scholar]
- 29.Jen-Jacobson L, Engler LE, Jacobson LA. Structural and thermodynamic strategies for site-specific DNA binding proteins. Structure. 2000;8:1015–1023. doi: 10.1016/s0969-2126(00)00501-3. [DOI] [PubMed] [Google Scholar]
- 30.Holdgate GA, Tunnicliffe A, Ward WH, Weston SA, Rosenbrock G, Barth PT, Taylor IW, Pauptit RA, Timms D. The entropic penalty of ordered water accounts for weaker binding of the antibiotic novobiocin to a resistant mutant of DNA gyrase: a thermodynamic and crystallographic study. Biochemistry. 1997;36:9663–9673. doi: 10.1021/bi970294+. [DOI] [PubMed] [Google Scholar]
- 31.Liu Y, Kulesz-Martin M. p53 protein at the hub of cellular DNA damage response pathways through sequence-specific and non-sequence-specific DNA binding. Carcinogenesis. 2001;22:851–860. doi: 10.1093/carcin/22.6.851. [DOI] [PubMed] [Google Scholar]
- 32.Khoo KH, Andreeva A, Fersht AR. Adaptive evolution of p53 thermodynamic stability. J. Mol. Biol. 2009;393:161–175. doi: 10.1016/j.jmb.2009.08.013. [DOI] [PubMed] [Google Scholar]
- 33.Mayer S, Rudiger S, Ang HC, Joerger AC, Fersht AR. Correlation of levels of folded recombinant p53 in Escherichia coli with thermodynamic stability in vitro. J. Mol. Biol. 2007;372:268–276. doi: 10.1016/j.jmb.2007.06.044. [DOI] [PubMed] [Google Scholar]
- 34.Privalov PL, Dragan AI, Crane-Robinson C, Breslauer KJ, Remeta DP, Minetti CA. What drives proteins into the major or minor grooves of DNA? J. Mol. Biol. 2007;365:1–9. doi: 10.1016/j.jmb.2006.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Privalov PL, Dragan AI, Crane-Robinson C. Interpreting protein/DNA interactions: distinguishing specific from non-specific and electrostatic from non-electrostatic components. Nucleic Acids Res. 2011;39:2483–2491. doi: 10.1093/nar/gkq984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nunez N, Clifton MM, Funnell AP, Artuz C, Hallal S, Quinlan KG, Font J, Vandevenne M, Setiyaputra S, Pearson RC, et al. The multi-zinc finger protein ZNF217 contacts DNA through a two-finger domain. J. Biol. Chem. 2011;286:38190–38201. doi: 10.1074/jbc.M111.301234. [DOI] [PMC free article] [PubMed] [Google Scholar]