


Abstract
Bloom (BLM) syndrome is an autosomal recessive disorder characterized by an increased risk for many types of cancers. Previous studies have shown that BLM protein forms a hexameric ring structure, but its oligomeric form in DNA unwinding is still not well clarified. In this work, we have used dynamic light scattering and various stopped-flow assays to study the active form and kinetic mechanism of BLM in DNA unwinding. It was found that BLM multimers were dissociated upon ATP hydrolysis. Steady-state and single-turnover kinetic studies revealed that BLM helicase always unwound duplex DNA in the monomeric form under conditions of varying enzyme and ATP concentrations as well as 3′-ssDNA tail lengths, with no sign of oligomerization being discerned. Measurements of ATPase activity further indicated that BLM helicase might still function as monomers in resolving highly structured DNAs such as Holliday junctions and D-loops. These results shed new light on the underlying mechanism of BLM-mediated DNA unwinding and on the molecular and functional basis for the phenotype of heterozygous carriers of BLM syndrome.
INTRODUCTION
Helicases play essential roles in various aspects of DNA metabolism including DNA replication, repair and recombination (1–5). RecQ family of DNA helicases has been highly conserved during evolution from bacteria to human. Defects in three of the human RecQ members give rise to defined genetic diseases that are characterized with cancer predisposition and/or premature aging. The disorders are Bloom (BLM), Werner (WRN) and Rothmund–Thomson syndromes, caused by loss-of-function mutations in BLM, WRN and RECQ4 helicases, respectively (6,7). The common genetic feature of these diseases is an autosomal recessive trait. The patients carrying two mutated alleles (homozygous carriers) display disease phenotype while the heterozygous carriers clinically appear to be entirely normal (8).
BLM syndrome is a rare human autosomal recessive disorder (9). The patients present severe growth retardation, immunodeficiency, reduced fertility and predisposition to cancer. At the cellular level, the hallmark of BLM syndrome is an elevated rate of sister chromatid exchange (SCE) in homozygous carriers, but this phenomenon is totally absent in heterozygous carriers (10).
The BLM gene product encodes a DNA helicase that functions in homologous recombination repair to prevent genomic instability. The purified BLM protein has been shown to act as a 3′ → 5′ DNA helicase on a variety of different DNA substrates such as forked duplex, Holliday junction, D-loop and G-quadruplex DNA (11–13). BLM is associated with Topoisomerase IIIα, RMI1 and RMI2 to form a complex that is responsible for dissolution of Holliday junction structures to suppress SCE in a reaction that is dependent on BLM helicase activity (14,15).
In a recent survey of patients from the BLM Syndrome Registry, 64 different mutations were identified, among them, 54 cause premature protein-translation termination and 10 missense mutations (16). This study suggests that the majority (84%) of BLM syndrome mutations probably produce truncated proteins that lack nuclear localization signal (NLS) and therefore cannot enter the nuclei to perform their enzymatic functions. The remaining 10 missense mutations (16%) occur at highly conserved amino acid residues within BLM’s DNA helicase domain and its associated C-terminal extended homology region (RecQ-CT), a region that is known to be essential for BLM helicase activity. Two of these missense mutations, Q672R in the helicase domain and C1055S in the RecQ-CT, have been studied in detail and shown to result in impaired ATPase and helicase activities (17,18). The other missense mutations probably have similar effects on BLM activity because they change conserved amino acids. The sequence and structural properties of these mutations explain why the homozygous mutations lead to diseases.
Since BLM syndrome is an autosomal recessive disease, individuals with one normal allele and one mutated allele (i.e. heterozygous carriers) do not display any disease phenotype. Cytological analyses of the cells in BLM syndrome patients did not reveal any apparent anomalies (10). The molecular mechanism by which the theoretical 50% wild-type protein in a cell is enough to maintain the function of BLM is still under investigation. In the case of heterozygous carriers with the truncated mutations, it is possible that only the 50% wild-type protein encoded by the normal allele can enter the nuclei to perform the biological functions, while the 50% NLS-lacking truncated protein has to remain in the cytosol and is finally degraded by proteases. Therefore, the cell functions normally.
The situation, however, is more complicated in the case of heterozygous carriers with missense mutations. Here both wild-type and mutated proteins can be transported into the nuclei. Previous gel filtration and electron microscopy studies indicated that BLM could form hexameric oligomers (19). Thus the inactive mutant may still be capable of interacting with its wild-type version to assemble into a mixed mutant/wild-type complex, whereas the probability to form a wild-type complex will be extremely low. The mixed mutant/wild-type BLM complex may be dysfunctional in activities that require oligomerization. If oligomerization is required for unwinding, then it remains unclear why the disease phenotype fails to manifest in heterozygotes (20,21).
Obviously, the knowledge of quaternary structures of human RecQ helicases in cells and their functional oligomeric states is essential not only for fully understanding the molecular mechanism underlying RecQ helicase-mediated catalytic activities, but also for revealing the pathological basis of human RecQ helicase-deficient disorders. Until now, the oligomeric form of BLM in DNA unwinding has still not been addressed systematically and defined clearly. Note that although previous biochemical characterizations have shown that the helicase core (amino acids 642–1290) of BLM may unwind DNA duplex as monomers (22,23), the results cannot be used to define the oligomeric form of the full-length protein (1417 amino acids) in DNA unwinding because the truncated fragment lacks the N-terminal region (amino acids 1–431) that is essential for BLM oligomerization (24).
In this work, we resorted to dynamic light scattering (DLS) and rapid stopped-flow fluorescence assays to study systematically the oligomeric state of BLM helicase during its catalysis. The results show that BLM helicase underwent dissociation upon ATP hydrolysis and unwound duplex DNA as a monomer under all the different conditions investigated. Furthermore, the helicase might still be in the monomeric form when unwinding complicated DNA substrates such as Holliday junction and G-quadruplex DNA.
MATERIALS AND METHODS
Reagents and buffers
All chemicals were reagent grade and all buffers were prepared in high quality deionized water from a Milli-Q Ultrapure Water Purification Systems (Millipore, France) having resistivity >18.2 MΩ cm. All DNA unwinding and binding assays were performed in reaction buffer A containing 25 mM Tris–HCl (pH 7.5 at 25°C), 50 mM NaCl, 3 mM MgCl2, 1 mM DTT. ATP was from Sigma (USA) and dissolved as a concentrated stock at pH 7.0. The ATP concentration was determined by using an extinction coefficient of 1.54 × 104 cm−1M−1 at 259 nm.
BLM helicase and DNA substrates
The full-length BLM protein was prepared as described (11). The ss/dsDNA substrates used in the unwinding assays had both strands labeled with fluorescein (F) and hexachlorofluorescein (H), respectively. The ssDNA used in the DNA binding assay was labeled with F. The ss/dsDNA, G4 DNA, Holliday junction (HJ) and D-loop used in the ATPase assays had no labeling. The structures and sequences of all substrates are shown in Supplementary Tables S1 and S2. The protein trap used for single-turnover kinetic experiments was 56-nt poly(dT), dT56. All the single-stranded oligonucleotides, with or without fluorescent labels, were from Shanghai Sangon Biological Engineering Technology & Services Co., Ltd (Shanghai, China). The dsDNA was prepared as described (25).
Dynamic light scattering
DLS measurements were performed using a DynaPro NanoStar instrument (Wyatt Technology Europe GmbH, Germany) with a 50-µl cuvette and the scattered light was collected at an angle of 90°. The buffer used for measurements was 50 mM Tris–HCl, pH 8.0, 80 mM NaCl, 1 mM DTT. The protein concentration used was between 0.5 and 0.8 µM for the full-length BLM and 1.5 µM for the helicase core. The concentration of ATP, when it was used, was fixed at 1 mM. The DNA used was a 25-nt ssDNA. The stoichiometry between protein and DNA was kept at 1 in all experiments where DNA was used. A total of 0.1-µm filtered solution was incubated for 5 min at room temperature before collecting data. Auto-correlations were incrementally stored every 10 s at a temperature of 25°C. The data were analyzed with the Dynamics v7.0 software.
DNA unwinding kinetic assays
All the DNA unwinding kinetic assays were carried out using a Bio-Logic SFM-400 mixer with a 1.5 mm × 1.5 mm cell (FC-15, Bio-Logic) and a Bio-Logic MOS450/AF-CD optical system. Fluorescein was excited at 492 nm (2 nm bandwidth) and its emission monitored at 525 nm using a high pass filter with 20 nm bandwidth (D525/20, Chroma Technology Co., USA). All the assays were performed at room temperature.
The kinetic assays were measured in a three-syringe mode. The reacting agents were separately preincubated in reaction buffer A in three syringes (see figure legends for details) and the reaction was initiated by rapid mixing of them. For converting the output data from volts to fraction of DNA unwound, calibration experiments were performed as described (25). All concentrations indicated are after mixing unless noted otherwise.
The kinetic unwinding data curves represented averages of over 10 individual traces. They were analyzed using Bio-Kine (version 4.26, Bio-Logic) with a double-exponential function for those with two unwinding phases (fast and slow) or with the following equation:
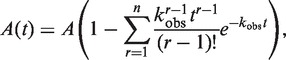 |
(1) |
for those with a negligible slow phase. Equation (1) describes the single-turnover unwinding kinetic process by a helicase in sequential n steps (26). A is the unwinding amplitude, kobs is the observed rate constant for each unwinding step.
ATPase assay
DNA-dependent ATPase activity was measured using a malachite green assay based on a colorimetric estimation of inorganic phosphate produced by ATP hydrolysis (27). Reactions were carried out in reaction buffer A with saturating amounts of structured DNA cofactors, including duplex DNA, Holliday junction, D-loop and G-quadruplex DNA (Supplementary Table S2). Reactions were initiated by adding the enzyme and ATP. Aliquots (5–20 µl) were removed from reactions at 10 min and mixed with 20 µl of 0.1 M EDTA to terminate the reaction. After appropriate dilution, samples (40 µl) were subjected to malachite green assay in a 96-well microplate along with a series of KH2PO4 dilutions ranging from 10 to 140 µM that served for calibration. The ATPase activity (ATPase rate) at each ATP concentration was measured at a protein concentration of 20 nM.
RESULTS
Study of the oligomeric state of BLM by DLS
The previous size-exclusion chromatography and electron microscopy studies have shown that apo BLM may form hexameric ring structures (19). To gain further insight into the oligomeric nature of BLM, we conducted DLS measurements on the full-length BLM as well as, for comparison, on the BLM helicase core (amino acids 642–1290) under various conditions.
The measurements showed that, in the apo state or in the presence of 25-nt ssDNA, the BLM protein was mainly a high-order oligomer, with a hydration radius of ∼10 nm and a calculated mass >700 kDa (Table 1 and Supplementary Figure S1). This oligomer was likely hexameric as observed in previous studies (19). In the presence of ATP, however, BLM existed in two forms, a high-order oligomer and a monomer. More interestingly, in the presence of both ATP and DNA, the high-order BLM oligomer was dissociated and stabilized with a hydration radius of 6.5 nm and a calculated mass of 268 kDa, probably a dimer or monomer.
Table 1.
Parameters for the full-length BLM and the BLM helicase core (amino acids 642–1290) in the absence or presence of ATP and DNA obtained from dynamic light scattering measurements
| Cofactor | Full-length BLM |
BLM helicase core |
||||
|---|---|---|---|---|---|---|
| % Mass | Radius (nm) | Calculated mass (kDa)a | % Mass | Radius (nm) | Calculated mass (kDa)b | |
| apo | 97.2 | 10.1 | 754 | 99.6 | 3.3 | 59.8 |
| DNA | 96.1 | 9.9 | 720 | 99.4 | 3.5 | 60 |
| ATP | 57.8 | 12.7 | 1127 | 99.3 | 2.8 | 34 |
| 42.2 | 4.5 | 112 | ||||
| DNA+ATP | 94.4 | 6.5 | 268 | 99.1 | 3.2 | 48 |
| AMPPNP | 90.5 | 20.3 | 2620 | 98.5 | 3.5 | 60 |
| 9.5 | 4.7 | 108 | ||||
| DNA+AMPPNP | 62.2 | 12.6 | 962 | 98.1 | 3.5 | 60 |
| 37.8 | 8.2 | 380 | ||||
aThe molecular weight of BLM is 159 kDa.
bThe molecular weight of the BLM helicase core is 74.1 kDa.
To further determine whether ATP binding or ATP hydrolysis is responsible for BLM dissociation in the presence of ssDNA, we then performed DLS measurements using the non-hydrolyzable ATP analog AMPPNP. In the presence of AMPPNP alone, BLM mainly existed as high-order oligomers. In the presence of both AMPPNP and ssDNA, about one-third of BLM was dissociated to low-order oligomers (probably trimers) while the rest still existed as high-order oligomers (Table 1 and Supplementary Figure S1). Taken together, the DLS results suggested that it was mainly ATP hydrolysis that triggered the disassembly of high-order BLM oligomers.
In the case of the BLM helicase core, however, the DLS measurements revealed that the protein was always in the monomeric form under all of the different conditions (Table 1). This is consistent with previous biochemical studies showing that the BLM helicase core unwinds duplex DNA as monomers (22,23) and confirms the observation that the N-terminus is essential for BLM oligomerization (24).
To further clarify the nature of the oligomeric state of the full-length BLM in ATP hydrolysis and DNA unwinding, we then studied the DNA unwinding kinetics and ATPase activities of the full-length BLM in the following experiments.
Fork dependence of single-turnover unwinding kinetics
BLM is not able to unwind a blunt-ended duplex DNA but can unwind 3′-ssDNA-tailed and forked duplexes, with the latter being more preferred (12). To determine which substrates to use in our following studies, we first performed a single-turnover unwinding kinetic assay using forked duplexes with a fixed 3′-ssDNA tail and varying 5′-ssDNA tails. The 3′-tail length was chosen as 15-nt as it was previously determined that a BLM monomer has an ssDNA binding-site size of 14 nt (28).
DNA substrate (2 nM) was incubated with an excess of BLM (30 nM) in reaction buffer A, and the unwinding reaction was initiated after rapid mixing with ATP (1 mM) and protein trap (dT56, 5 µM). The BLM concentration was chosen as 30 nM because concentrations >30 nM resulted in a reduction of the unwinding efficiency (data not shown).
It was observed that the unwinding kinetics were clearly characterized by two phases regardless of the 5′-tail length (Figure 1A). The amplitude of the fast phase was independent of the 5′-tail length, whereas that of the slow phase increased linearly with increasing 5′-tail length and thus resulted in an obvious fork-dependent total unwinding efficiency (Figure 1B). The unwinding rates of both phases were insensitive to the tail length (Figure 1C).
Figure 1.

Effect of the 5′-ssDNA tail length on the single-turnover unwinding of forked DNA. The DNA substrates used were forked 16-bp duplexes with a fixed 15-nt 3′-ssDNA tail and varying 5′-ssDNA tails. A total of 2 nM DNA substrate with 30 nM BLM or without BLM (as a control), 1 mM ATP and 5 µM protein trap dT56 were first preincubated, respectively, in three syringes in the reaction buffer A and the unwinding reaction was initiated by rapid mixing of the three solutions. (A) Kinetic time-courses. The solid lines are double-exponential fits of the data. Inset: time courses in the first 1.5 s. (B) Fast-phase, slow-phase and total amplitudes. (C) Rates of the two phases.
For the appearing of two unwinding phases, one possible reason is that, in addition to the BLM helicase pre-bound to the 3′ tail, another BLM helicase might pre-bind to the 5′ tail and somehow unwound the duplex. However, this is ruled out because if one helicase is already bound to the DNA fork through the 3′ tail, it would be difficult for another helicase to bind the 5′ tail due to steric restrictions, especially when the 5′ tail is short or even absent. Another possible reason is that BLM has a higher affinity for a forked duplex DNA and catalyzes DNA unwinding more efficiently when the 5′ tail is long, but this is also ruled out because there should be only one unwinding phase in this case.
Previously, a repetitive unwinding behavior was observed for BLM helicase in single-molecule fluorescence experiments (29). It has been proposed that the BLM helicase may switch strands in the course of DNA unwinding. After strand switching, it translocates 3′ → 5′ along the opposing strand toward the end of the 5′ tail, then releases the 5′ tail and rebinds to the 3′ tail, finally reinitiates a new round unwinding.
We think our results represent the first observation of strand switching and repetitive unwinding of a helicase in ensemble measurements. The fast phase should be contributed by BLM helicase that unwound the duplex completely in a single round, while the slow one might be due to the subsequent repetitive unwinding. As the strand switching of BLM relies on the 5′-ssDNA tail (29), the unwinding amplitude of the slow phase increased with increasing 5′-tail length. The unwinding amplitude of the fast phase, on the other hand, only increased slightly. Thus the reason for forked duplex being unwound more efficiently (12) might be mainly an enhancement of repetitive unwinding by the presence of the 5′ tail.
To further verify the proposed origin for the slow phase, we repeated the single-turnover unwinding assay while adding an 8-nt ssDNA, dA8, upon unwinding initiation. As both the 3′- and 5′-ssDNA tails of the forked substrate were composed entirely of dT, we expected that dA8 would hybridize with the two tails as soon as they were vacated after unwinding initiation and thus the unwinding kinetics would be modified.
Indeed, we observed that the presence of dA8 influenced obviously the unwinding kinetics (Figure 2A). The slow-phase amplitude was reduced in a dA8 concentration-dependent manner while that of the fast-phase was not much affected (Figure 2B). At 2 µM dA8, the former was reduced by ∼50%. The unwinding rates of both phases did not vary significantly (Figure 2C). As a control, we repeated the measurements while using 2 µM dT8. We observed that dT8 caused only slight decreases of both fast- and slow-phase amplitudes (Supplementary Figure S2), probably because the small dT8 molecules disturbed the activity of BLM. The ratio between the two amplitudes was remaining unchanged. This was in contrast with the case of dA8.
Figure 2.

Inhibition effect of dA8 on the single-turnover unwinding of DNA. A forked 16-bp duplex DNA substrate with 15-nt 3′ and 5′ tails was used. The measurements were performed under the same conditions as in Figure 1 except for that dA8 of varying concentrations was added with ATP upon unwinding initiation. (A) Typical kinetic time-courses. The solid lines are double-exponential fits of the data. Inset: time courses in the first 1.5 s. (B) Fast-phase, slow-phase and total amplitudes. (C) Rates of the two phases.
To see if the addition of dA8 destabilized the ss/dsDNA junction and caused the increase of the fast-phase amplitude, we performed the same experiments while replacing BLM with Escherichia coli RecQ (30). As can be seen in Supplementary Figure S3, the unwinding kinetic process of RecQ was characterized only by a single phase (implying no repetitive unwinding occurred) and the amplitude decreased slightly with increasing dA8. This means dA8 did not enhance the DNA unwinding activity of the helicase. We think the slight increase of the fast-phase amplitude in Figure 2B may be due to a dA8-induced reduction of the strand switching probability of BLM.
It should be noted that even though the fork-dependent unwinding behavior of BLM was mainly attributed to the strand-switching and repetitive-unwinding activity of the helicase here, there may exist other possible processes that lead to this behavior (see ‘Discussion’ section). More studies are needed to clarify this issue.
Steady-state unwinding kinetics
To investigate the oligomeric state of BLM in DNA unwinding, we first performed a steady-state unwinding kinetic assay. In such an assay, the helicase concentration should, in principle, be much lower than that of duplex DNA substrate so that the latter is depleted slowly. After initiation by mixing the helicase, ATP and DNA, the helicase may bind to a DNA substrate, unwinds it partially or completely, dissociates from it and then rebinds to another substrate to initiate the next unwinding event. The binding–unwinding–dissociation–rebinding event needs to be repeated many times for each helicase before all DNA substrates are unwound completely.
In the steady-state unwinding reaction, the initial rate would be in linear proportion to the concentration of helicase if it is active as a monomer, and would display a non-linear dependence (sigmoidal) if it requires oligomerization for unwinding. Noting that, if a helicase has a low unwinding processivity, then even though the helicase concentration is comparable to that of the substrate, an unwinding reaction could still be regarded as a steady-state process because of a much prolonged unwinding time. As can be seen in the following, this is just the case for BLM.
In our present assay, the DNA substrate was a 16-bp duplex flanked only by a 15-nt 3′-ssDNA tail, which was chosen because the repetitive unwinding of BLM was shown to be insignificant for it (Figure 1B). Note, however, that repetitive unwinding does not present any problem for the present assay as it is only equivalent to increasing the unwinding efficiency in each binding event, though it could cause difficulty in the data analyses of the other experiments. The DNA trap used was a 16-nt ssDNA that has the same sequence as the short strand of the duplex DNA substrate. As BLM has an intrinsic ssDNA annealing activity (31,32), the DNA trap was used to inhibit BLM-catalyzed reannealing of the two nascent ssDNA in the course of unwinding. As shown in Supplementary Figure S4, its presence indeed enhanced the single-turnover unwinding efficiency.
We performed the steady-state measurements with BLM concentrations ranging from 5 to 30 nM. We observed that the unwinding reaction was far from being completed even at the time of 40 s. As the average reaction time for a single helicase-binding event (i.e. single-turnover unwinding) was <1 s for this substrate (Supplementary Figure S4), the present unwinding reactions could indeed be regarded as steady-state ones.
The unwinding data curves between 0 and 20 s were given in Figure 3A. From exponential fittings we obtained the initial unwinding rate at each BLM concentration (Figure 3B). Clearly, it had a good linear dependence on the BLM concentration, indicating that the active state of BLM might be monomeric under the steady-state conditions.
Figure 3.

DNA unwinding kinetics under steady-state conditions. A total of 60 nM DNA substrate (16-bp duplex with a 15-nt 3′ tail), BLM of varying concentrations with 1 mM ATP, and 20 µM DNA trap were first preincubated, respectively, in three syringes and the reaction was initiated by rapid mixing of them. (A) Typical kinetic time-courses. The solid lines are double-exponential fits of the data. (B) Initial unwinding rate obtained from the fittings. Each data point represents the average value of three independent measurements.
In addition to steady-state assays, pre-steady-state kinetic assays have also been frequently used in studying DNA unwinding activities and the kinetic mechanisms of different helicases. In a pre-steady-state assay, the burst amplitude of DNA unwound is proportional to the concentration of initial productive helicase/DNA complex. For BLM, we observed that the burst amplitude was much lower than expected for a monomeric helicase (Supplementary Figure S5). For example, considering the ∼20% unwinding efficiency (Supplementary Figure S4), ∼8 nM DNA was expected to be unwound in the case of 40 nM BLM, but actually only 1.6 nM DNA was unwound. This implied that BLM bound to DNA as oligomers rather than monomers during the pre-incubation in the absence of ATP, which was consistent with the DLS results (Table 1). Upon addition of ATP, the pre-bound BLM oligomer should be dissociated to monomers and the DNA substrate was unwound by one of them in the burst phase.
It should be noted that there would be no such ‘oligomer-binding’ problem in the above steady-state experiments because DNA, helicase and ATP were mixed simultaneously. In the case of single-turnover kinetic assays, the enzyme concentration was over saturating. Thus even though BLM bound to DNA as oligomers during the preincubation, each DNA molecule would still be bound with a BLM monomer after the pre-bound BLM oligomer was dissociated by ATP.
Single-turnover unwinding kinetics at varying ATP concentrations
It should be mentioned that, in the previous steady-state experiment, if the BLM helicase formed a single and stable oligomeric species in the absence of DNA, and bound and unwound DNA in that oligomeric form, then a linear dependence on BLM concentration could also be observed for the initial unwinding rate. To clarify this issue, we next carried out single-turnover kinetic measurements to study the effect of ATP concentration. In principle, if the helicase is functioning as a monomer, the ATP-concentration dependence of the unwinding rate may be described by the Hill equation with a Hill coefficient of 1. If the helicase is functioning as an oligomer, on the other hand, the corresponding Hill coefficient should be distinct from 1.
In this assay, the concentration of dA8 was chosen as 2.5 µM because the contribution of repetitive unwinding to the total unwinding efficiency could be reduced to a minimum amount with it (<15%, Supplementary Figure S6). As can be seen in the following unwinding curves, the remaining slow phase was negligible as long as the first several seconds of unwinding reaction were concerned. We might therefore suppose that each DNA substrate was unwound by a single BLM monomer and in only one round, and consequently, the unwinding reaction could be regarded as a sequential n-step process described theoretically by Equation (1) (26).
The single-turnover unwinding kinetic data were presented in Figure 4A. By fitting the data curves to Equation (1) with n = 6 (see ‘Kinetic Step Size of BLM in DNA Unwinding’ section), we obtained the unwinding rate constants at different ATP concentrations (Figure 4B). The rate constant increased with increasing ATP concentration, until saturation was reached at >1 mM ATP. It was found that the data could be well fitted with the Hill equation, yielding h = 0.92 ± 0.06 and KM = 20.4 ± 1.7 µM. The Hill coefficient was very close to 1, consistent with a monomeric form of BLM.
Figure 4.

Single-turnover unwinding kinetics at different ATP concentrations. A total of 2 nM DNA substrate (16-bp duplex with a 15-nt 3′ tail) with 30 nM BLM, ATP of varying concentrations with 2.5 µM dA8 and 2 µM DNA trap, and 5 µM dT56 were first preincubated, respectively, and the reaction was initiated by rapid mixing of the three solutions. (A) Typical kinetic time-courses. The solid lines are best fits of the data to Equation (1) with n = 6. (B) Observed unwinding rate constant. The line corresponds to a best fit of the data to the Hill equation, 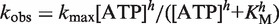 , where h is the Hill coefficient. The fitted values are h = 0.92 ± 0.06 and KM = 20.4 ± 1.7 µM.
, where h is the Hill coefficient. The fitted values are h = 0.92 ± 0.06 and KM = 20.4 ± 1.7 µM.
It should be mentioned that the unwinding efficiency (20%) at 1 mM ATP observed here was somewhat higher than that observed before (Afast = ∼15%, Figure 1B) for the same substrate (16-bp duplex flanked only by a 15-nt 3′ tail). The reason is that an DNA trap was used here to diminish BLM-catalyzed reannealing of the two nascent ssDNA during DNA unwinding.
Inhibition of BLM helicase activity by AMPPNP
As a further evidence for the monomeric form of BLM, we next carried out an AMPPNP inhibition experiment. The measurement was performed under the same conditions as in the previous experiments (Figure 4A). The only difference was that at present, ATP was replaced by a mixture of ATP and AMPPNP, with a varying ratio but a fixed total concentration. We have reasoned that, if BLM unwinds DNA as an oligomer such as a hexamer, the binding of AMPPNP to even one subunit would block the ATPase activities of all the subunits until AMPPNP dissociates from the helicase, because ATP hydrolysis is coordinated between adjacent subunits (33–35). In another word, the helicase activity, especially the unwinding rate, should be very sensitive to AMPPNP at high concentrations.
As shown in Figure 5A, the helicase activity of BLM was indeed inhibited by the presence of AMPPNP. However, the unwinding rate constant was only reduced moderately and monotonously with increasing AMPPNP content in the range of 0–95% (Figure 5B). At 95% AMPPNP, where the ratio between AMPPNP and ATP concentrations was 19, the rate constant only decreased to about half of the value when AMPPNP was absent. The data can be well fitted to Supplementary Equation (5), which is equivalent to the Hill equation with h = 1, with kb = kcat/KM and KM constrained to 20.4 µM as obtained above in Figure 4B, yielding kcat = 19.2 ± 0.5 s−1 and kr = 407 ± 291 s−1. On the contrary, the data do not follow the trend defined by the Hill equation with h = 6 (Figure 5B). This experiment has further shown that BLM functions as a monomer.
Figure 5.

Inhibition of DNA unwinding by AMPPNP. The measurements were performed under single-turnover conditions the same as that in Figure 4 except for that ATP was replaced by a mixture of ATP and AMPPNP with a varying concentration ratio (the total concentration was fixed at 1 mM). (A) Kinetic time-courses. The solid lines are best fits of the data to Equation (1) with n = 6. (B) Unwinding amplitude and observed rate constant. The red and black solid lines correspond to fits of the data to Supplementary Equation (5) and (6), respectively (see the text). The red dashed line represents schematically a variation trend of kcat defined by the Hill equation with h = 6.
As the unwinding rate constant, the unwinding efficiency was also reduced with increasing AMPPNP content, though less significantly (Figure 5B). The reason for the decrease should be that, in the presence of AMPPNP, the probability of the helicase to detach from DNA became higher due to an extended ATP binding time (or reduced effective ATP concentration). By constraining KM, kcat and kr to values obtained above, the data for unwinding efficiency were fitted to Supplementary Equation (6), yielding 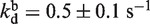 and
and 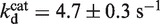 (Figure 5B). These values indicated that BLM bound more tightly to the DNA substrate during the period for ATP and AMPPNP binding than during the period of ATP hydrolysis. This is consistent with the previous dissociation measurements for the BLM helicase core, showing that the helicase has low rates of dissociation from ss/dsDNA in apo and ATP states and higher ones in ADP·Pi and ADP states (23).
(Figure 5B). These values indicated that BLM bound more tightly to the DNA substrate during the period for ATP and AMPPNP binding than during the period of ATP hydrolysis. This is consistent with the previous dissociation measurements for the BLM helicase core, showing that the helicase has low rates of dissociation from ss/dsDNA in apo and ATP states and higher ones in ADP·Pi and ADP states (23).
Single-turnover unwinding kinetics with substrates of different 3′-tail lengths
The above steady-state and single-turnover measurements have established that the BLM helicase unwound DNA as monomers, but the experimental conditions used above might be biased to favor binding of a single BLM monomer to the DNA substrate because it had a 15-nt 3′ tail which was only long enough to accommodate one monomer. To further study the possible oligomeric form of BLM in DNA unwinding, a series of experiments was then performed with 16-bp DNA duplexes flanked by varying 3′-tail lengths. We have expected that, if BLM always functions as a monomer, it would be capable of unwinding all of the different substrates with similar unwinding rates, regardless of the tail length. Otherwise, the unwinding rate would exhibit significant dependence on the tail length.
The experiments were performed under conditions similar to the previous ones (Figure 4A). The only difference was that the 3′-tail length was variable, the ATP concentration was fixed and dA8 was not used because its presence might disturb the ssDNA translocation of trailing helicase molecules at positions away from the ss/dsDNA junction.
Unwinding was observed for all of the DNA substrates (Figure 6A). The efficiency increased obviously with increasing 3′-tail length. As before, the time course for each tail length was biphasic and could be fitted well to a double-exponential function. The amplitude of the fast phase was essentially constant (Figure 6B) and the corresponding rates were similar at all tail lengths (Figure 6C). As only one monomer might be bound to the 10- and 15-nt-tailed substrates, the above phenomenon indicated that the fast phase for each tail length was only contributed by the BLM monomer bound to the ss/dsDNA junction, independent of the trailing monomers.
Figure 6.

Effect of 3′-ssDNA tail on the single-turnover unwinding of DNA. A total of 2 nM DNA substrate (16-bp duplex of varying 3′ tails) with 30 nM BLM, 1 mM ATP with 2 µM DNA trap and 5 µM dT56 were first preincubated, respectively, and the reaction was initiated by rapid mixing of the three solutions. (A) Kinetic time-courses. The solid lines are double-exponential fits of the data. (B) Fast-phase, slow-phase and total amplitudes. (C) Rates of the two phases.
Since the slow unwinding phase was negligible for short-tailed substrates and became more and more significant for longer-tailed substrates (Figure 6B), it should be contributed by the trailing monomers. If dissociation from the substrate occurred for the leading monomer during unwinding, the trailing monomers would continue the unwinding.
Note that, though only one BLM monomer was expected to be responsible for unwinding the 10- and 15-nt-tailed substrates, a slow phase was still observed with these substrates. We think it should be caused by the repetitive unwinding activity, as dA8 was not used here.
Taken together, the above experimental results clearly indicated that BLM unwound DNA as monomers even though the enzyme concentration was saturating and the substrate tail length was long enough to accommodate several monomers. No oligomerization of BLM in DNA unwinding was detected from the observed kinetics.
ATPase activities with different DNA structures
The above results have clearly shown that BLM helicase unwinds duplex DNA as a monomer. However, BLM also resolves different structured DNAs that are formed as transit intermediates during DNA recombination and DNA repair, such as the Holliday junction, D-loop and G-quadruplex DNA (12,13). It is possible that BLM functions as monomers in unwinding duplex DNA, but an oligomer is necessary for resolving the highly structured DNAs.
To clarify this issue, we measured and compared the ATPase activities of BLM in the absence or presence of different DNA structures. Interestingly, the ATPase activity of BLM without DNA substrate was negligibly low, while it was significantly stimulated, to similar extents, by the different substrates ranging from the simple 3′-tailed duplex DNA to the complicated D-loop (Figure 7A and B). Moreover, the dependence of ATPase activity on ATP concentration could be well described by the Hill equation with a Hill coefficient close to 1 in all the cases (Figure 7C and Table 2). These results suggested that BLM might still use the monomeric form in resolving the more complicated DNA structures. Even in the absence of DNA, BLM still hydrolyzed ATP as monomers, though with a much lower rate. It should be noted that at present we cannot exclude the possibility that BLM resolved the complicated DNA structures as non-cooperative multimers. This issue should be addressed in further studies
Figure 7.

ATPase activity in the absence or presence of 3′-tailed ss/dsDNA, G4 DNA, Holliday junction or D-loop. Reactions were carried out in buffer A and quantified as described in ‘Experimental Procedures’ section. (A) Dependence of the ATPase activity (ATPase rate) on ATP concentration in the absence of DNA substrate. The solid line is a best fit of the data to the Hill equation. The obtained parameters of the ATPase reaction, kcat, KM and Hill coefficient, are given below and/or in Table 2. (B) Dependence of the ATPase activity on ATP concentration in the presence of different DNA substrates. The solid lines are best fits of the data to the Hill equation. (C) Hill coefficients obtained from the above fittings.
Table 2.
Steady-state kinetic parameters of the ATPase activity of BLM in the absence or presence of different DNA substrates obtained from fittings of the data in Figure 7A and B
| DNA substrates | kcat (s−1) | KM (µM) | Hill coefficient |
|---|---|---|---|
| No DNA | 0.17 ± 0.03 | 404 ± 56 | 0.98 ± 0.25 |
| ss/dsDNA | 24.0 ± 0.8 | 20.8 ± 2.2 | 0.92 ± 0.20 |
| Holliday junction | 25.6 ± 1.2 | 23.9 ± 1.4 | 1.11 ± 0.13 |
| D-loop | 26.6 ± 1.9 | 22.5 ± 2.4 | 1.09 ± 0.21 |
| G4 | 25.0 ± 1.0 | 22.2 ± 1.7 | 0.93 ± 0.16 |
Kinetic step size of BLM in DNA unwinding
Helicases couple NTP hydrolysis to mechanical movements and unwind duplex DNA or RNA through multiple steps. The step size is an important parameter for characterizing the unwinding behavior of a helicase and is essential for understanding the underlying mechanism. Up to now, the step sizes of many helicases have been determined, ranging from 1 bp (30) to 16 bp (36).
To determine the kinetic step size of BLM, we performed single-turnover unwinding experiments using 15-nt-tailed DNA substrates of different duplex lengths (12–20 bp). The experimental conditions were the same as that in Figure 4A except for that the ATP concentration was fixed at 1 mM and the length of DNA trap (ssDNA) was varying from 12 to 20 nt in accordance with the different substrates.
We observed that the unwinding efficiencies for the 18- and 20-bp substrates were much lower than that for the other shorter substrates (Figure 8A). In the case of the BLM helicase core, the unwinding efficiencies were not so different for different duplexes (23). The reason may be that the reannealing of nascent ssDNA was still significant in the case of BLM even though DNA trap was used. These are consistent with the fact that the full-length BLM has an intrinsic ssDNA annealing activity mediated by the C-terminal residues 1290–1350 (31), which are missing in the BLM helicase core.
Figure 8.

Unwinding of a 15-nt 3′-tailed DNA with varying duplex lengths. The measurements were performed under the same conditions as in Figure 4 except for that the ATP concentration was fixed at 1 mM. (A) Kinetic time courses. The solid lines are best fits of the data to Equation (1) with n = 4, 5, 6, 7 and 8 for the five data curves, respectively, with the corresponding values of kobs as 11.7 ± 0.1, 12.5 ± 0.1, 15.5 ± 0.1, 12.0 ± 0.2 and 13.6 ± 0.2 s−1. (B) Number of steps for unwinding the different duplexes. The straight line was a straight line with a slope of 0.5 bp−1, corresponding to a step size of 2 bp.
By fitting the data curves individually to Equation (1), we obtained the number of steps, n, for each duplex length L (Figure 8A). n increased linearly with the duplex length by following n = (L − 4)/2 (Figure 8B). Thus the kinetic step size of BLM is 2 bp and the number of base pairs at the end of the duplex that melt spontaneously when the helicase approaches is 4 bp. It should be noted that the data curves cannot be fitted well if a kinetic step size of 1 bp is used (Supplementary Figure S7).
The determined step size of BLM is similar to that of E. coli RecQ (1 bp) and the BLM helicase core (1.3 bp) determined before (23,30). From the average value of the observed unwinding rate constant, 13.0 ± 1.5 s−1 and the 2-bp step size, we know that the macroscopic unwinding rate of BLM is 26.0 ± 3.0 bp s−1, which is almost the same as that of the BLM helicase core (∼27 bp s−1) (23) and lower than that of E. coli RecQ (∼50 bp s−1) (30).
DISCUSSION
BLM syndrome, like most autosomal recessive disorders, requires two mutant copies of BLM gene to express the phenotype. The carriers with only one mutant allele appear entirely normal both in clinical observations and at cytogenetic analysis levels (10). Obviously, determination of the oligomeric form of the BLM helicase in DNA unwinding is important both for offering explanations to this kind of phenomena and for understanding the mechanisms and functions of the enzyme at the molecular levels.
In the present work, we provide evidences for the monomeric functional form of the full-length BLM protein by using DLS and fluorometric stopped-flow techniques. Essentially, three new interesting findings are made as follows:
DLS measurements revealed that BLM multimers underwent dissociation upon ATP hydrolysis. Previously, a similar phenomenon was also observed for another RecQ family helicase, human RECQ1, in size exclusion chromatography experiments (37). Interestingly, for RECQ1, it was ATP binding rather than ATP hydrolysis that induced the oligomer disassembly. This difference between the two helicases may be due to the fact that the 1417-amino acid BLM and 649-amino acid RECQ1 have apparently distinct quaternary structures (19,37) and thus the effects of nucleotide-dependent conformational changes of the subunits (i.e. monomers) on their interactions inside an oligomer may be different for the two helicases. This should be addressed in future studies.
BLM was observed to display strand switching and repetitive unwinding behaviors in our kinetic experiments, which confirmed previous single-molecule observations (29). This is a first report that such a phenomenon was observed by ensemble measurements, opening a new avenue to study these behaviors of other helicases with stopped-flow method. Interestingly, these behaviors of BLM do not have no biological significance. As discussed before (29), these activities may be relevant to the processing and recovery of stalled replication forks and to the homologous recombination processes. They may also be used in the displacement of proteins such as hRPA and RAD51. As mentioned before, the strand switching phenomenon was also observed for UvrD in single-molecule measurements using magnetic tweezers (38,39). As both UvrD and BLM have two RecA-like domains typical of superfamilies 1 and 2 helicases (18,40), the underlying mechanisms for strand switching may be similar or the same for the two helicases.
It should be noted that, although the appearance of the second unwinding phase in Figure 1 can be explained by the strand switching and repetitive unwinding model, it can also be explained by the following model. Under the experimental conditions in Figure 1, the enzyme is in excess of DNA. The oligomeric BLM will be bound to the DNA prior to initiation of the reaction. Upon addition of ATP the oligomer may be disrupted but the six or more BLM molecules may be all arranged in the immediate vicinity of the DNA. The addition of a fork may simply allow any of the BLM molecules not ‘chosen’ to make the first attempt at unwinding to remain localized to that particular oligo. Then the second (or third or fourth) BLM molecule can attempt to separate the dsDNA that the first molecule failed to unwind, somewhat similar to the cases in Figure 6 where one DNA substrate could be unwound sequentially by multiple monomers as long as the 3′ tail was long enough to accommodate them. According to this effective localization model, addition of dA8 would also affect the slow phase by sequestering the second strand from the disrupted BLM subunits.
At present we are still incapable of discriminating between the above two models. The different processes described by them may even occur simultaneously. More studies are needed to clarify the mechanism for the fork-dependent unwinding activity of BLM.
The stopped-flow studies further provided several lines of evidence that the functional form of the full-length BLM protein is monomeric: (a) The dependence of steady-state unwinding rate on enzyme concentration is sensitive to the functional species of the helicase. The linear dependence observed (Figure 3) indicated clearly that BLM unwound DNA as a monomer or as a well-defined oligomer that was stable both before and after binding to DNA. However, the latter possibility was excluded by the next single-turnover unwinding measurements at varying ATP concentrations, showing that the ATP-concentration dependence of the unwinding rate constant was well describable by the Hill equation with a Hill coefficient of ∼1 (Figure 4). (b) Study of the inhibition of BLM helicase activity by AMPPNP further supported the monomeric form of the protein by revealing a moderate effect of the ATP analog on DNA unwinding (Figure 5). (c) Note, however, that an oligomerization behavior of BLM might be prohibited in the above experiments as we had used a DNA substrate with a 15-nt 3′ tail that was only long enough to accommodate one single monomer. Thus, we investigated the unwinding kinetics of duplex DNA further with varying 3′-tail ssDNA lengths. Still, no sign of oligomerization in DNA unwinding was found even under the favoring conditions (Figure 6). (d) It is easy to think that BLM might require oligomerization to resolve different structured DNAs such as the Holliday junction, D-loop and G-quadruplex DNA. However, the measurements of the ATPase activities of BLM in the absence or presence of different DNA structures demonstrated that BLM hydrolysed ATP with similar rates in the unwinding of them and the Hill coefficients were close to 1 in all the different cases (Figure 7 and Table 2). Thus BLM might still be in a monomeric form when resolving the structured DNAs.
Taken together, these kinetic studies suggested that BLM functioned as monomers in DNA unwinding under the different conditions investigated. This provides, at the molecular level, a comprehensive explanation to the pathogenesis of both homozygous BLM patients and heterozygous BLM mutation carriers. According to our present results that the hexameric BLM helicase dissociates into monomers during catalysis, it becomes comprehensible that the carrier’s half dose of BLM gene product is sufficient for full BLM function in the maintenance of genomic integrity. Although this is consistent with the clinical observations (20,21), more studies at different levels will be needed to make further confirmations.
What is the structural basis for an apparent hexameric helicase functioning as a monomer during the catalysis? Although the precise 3D structure of BLM needs be determined, the comprehensive molecular modeling based on E. coli RecQ helicase crystal structure (41) and biochemical studies have shown that the arginine finger of BLM for ATP hydrolysis and energy coupling is located at the interface between its two RecA-like domains in a single polypeptide chain (42) and thus its ATPase and helicase activities may not need its oligomerization. However, in the cases of ‘true’ hexameric helicases such as bacteriophage T7 gene 4 (33,43), bovine papillomavirus E1 (44) and the E. coli Rho transcription termination factor (45), the arginine fingers are located at the interfaces between adjacent subunits and thus an oligomerization is absolutely required for their ATPase and helicase activities.
It should be noted that, although all our experiments have revealed a monomeric form of BLM in DNA unwinding, it does not mean that BLM must be monomeric in solution. Indeed, as indicated by the DLS measurements, BLM was oligomeric both in solution and when binding to DNA in the absence of ATP. To further verify this, we studied the equilibrium DNA binding properties of BLM by using a fluorescence anisotropy titration assay (23). We observed the binding was not saturated even at an enzyme concentration of 400 nM (Supplementary Figure S8). We found the data could not be fitted well by a non-cooperative binding model [Equation (1) in Ref. (23)], implying some cooperative binding (or oligomerization) have indeed occurred. This is in contrast with the case of the BLM helicase core, where the ssDNA binding reached saturation at enzyme concentrations as low as ∼25 nM and the whole data could be well fitted by the non-cooperative binding model (23).
Interestingly, like BLM, WRN helicase forms oligomers in solution or when binding to DNA (46), but it also unwinds duplex DNA as monomers (47). The feature that an oligomeric protein dissociates into monomers during catalysis may be common for human RecQ family helicases, at least for BLM and WRN proteins.
So what are the possible functions of BLM oligomerization? Previous studies for different RecQ family helicases have suggested that a helicase oligomerization may be required for its ssDNA annealing activity (31,37,48). For RECQ1, ATP binding favors a smaller oligomeric form and inhibits its annealing activity (37). For BLM, our DLS study revealed that high-order BLM oligomers were dissociated upon ATP hydrolysis while it was previously shown that the ssDNA annealing activity of BLM was inhibited by the presence ATP (31). For WRN, it was shown that the region between the RQC and HRDC domains (amino acids 1072–1150) was involved in higher-order oligomerization of the protein and the deletion of this region strongly inhibited its strand annealing activity (48). In addition to DNA annealing, it was identified for RECQ1 that the N-terminal domain or the higher order oligomer formation promoted by the N terminus was essential for the ability of RECQ1 to disrupt Holliday junctions (49). For BLM, our ATPase measurements suggests that it may use the monomeric form in resolving Holliday junctions, but the possibility that it uses non-cooperative multimers cannot be excluded by the ATPase assay at present. Finally, as the N-terminal domain mediates the interactions of BLM with other proteins such as Rad51 (50) and Topoisomerase III (51,52), another possible function of BLM oligomerization is a self-regulation of these interactions. Further studies are required for giving definite answers.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1 and 2, Supplementary Figures 1–8 and Supplementary Equations 5 and 6.
FUNDING
National Natural Science Foundation of China [10834014]; National Basic Research Program of China [973 Program, 2009CB930704]; Centre National de la Recherche Scientifique (CNRS) (to X.G.X.). Funding for open access charge: CNRS (to X.G.X.).
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We are very grateful to the insightful suggestions of the referees. This work was partly done in the Institut Curie.
REFERENCES
- 1.Matson SW, Bean DW, George JW. DNA helicases - Enzymes with essential roles in all aspects of DNA metabolism. Bioessays. 1994;16:13–22. doi: 10.1002/bies.950160103. [DOI] [PubMed] [Google Scholar]
- 2.Singleton MR, Wigley DB. Modularity and specialization in superfamily 1 and 2 helicases. J. Bacteriol. 2002;184:1819–1826. doi: 10.1128/JB.184.7.1819-1826.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.von Hippel PH. Helicases become mechanistically simpler and functionally more complex. Nat. Struct. Mol. Biol. 2004;11:494–496. doi: 10.1038/nsmb0604-494. [DOI] [PubMed] [Google Scholar]
- 4.Lohman TM, Bjornson KP. Mechanisms of helicase-catalyzed DNA unwinding. Annu. Rev. Biochem. 1996;65:169–214. doi: 10.1146/annurev.bi.65.070196.001125. [DOI] [PubMed] [Google Scholar]
- 5.Patel SS, Picha KM. Structure and function of hexameric helicases. Annu. Rev. Biochem. 2000;69:651–697. doi: 10.1146/annurev.biochem.69.1.651. [DOI] [PubMed] [Google Scholar]
- 6.Bachrati CZ, Hickson ID. RecQ helicases: guardian angels of the DNA replication fork. Chromosoma. 2008;117:219–233. doi: 10.1007/s00412-007-0142-4. [DOI] [PubMed] [Google Scholar]
- 7.Vindigni A, Hickson ID. RecQ helicases: multiple structures for multiple functions? HFSP J. 2009;3:153–164. doi: 10.2976/1.3079540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ellis NA, Groden J, Ye TZ, Straughen J, Lennon DJ, Ciocci S, Proytcheva M, German J. The Blooms-syndrome gene-product is homologous to RecQ helicases. Cell. 1995;83:655–666. doi: 10.1016/0092-8674(95)90105-1. [DOI] [PubMed] [Google Scholar]
- 9.Hickson ID. RecQ helicases: caretakers of the genome. Nat. Rev. Cancer. 2003;3:169–178. doi: 10.1038/nrc1012. [DOI] [PubMed] [Google Scholar]
- 10.German J. Bloom-syndrome - a Mendelian prototype of somatic mutational disease. Medicine. 1993;72:393–406. [PubMed] [Google Scholar]
- 11.Karow JK, Chakraverty RK, Hickson ID. The Bloom’s syndrome gene product is a 3′-5′ DNA helicase. J. Biol. Chem. 1997;272:30611–30614. doi: 10.1074/jbc.272.49.30611. [DOI] [PubMed] [Google Scholar]
- 12.Mohaghegh P, Karow JK, Brosh RM, Bohr VA, Hickson ID. The Bloom’s and Werner’s syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 2001;29:2843–2849. doi: 10.1093/nar/29.13.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bachrati CZ, Borts RH, Hickson ID. Mobile D-loops are a preferred substrate for the Bloom’s syndrome helicase. Nucleic Acids Res. 2006;34:2269–2279. doi: 10.1093/nar/gkl258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mankouri HW, Hickson ID. The RecQ helicase-topoisomerase III-Rmi1 complex: a DNA structure-specific ‘dissolvasome’? Trends Biochem. Sci. 2007;32:538–546. doi: 10.1016/j.tibs.2007.09.009. [DOI] [PubMed] [Google Scholar]
- 15.Singh TR, Ali AM, Busygina V, Raynard S, Fan Q, Du CH, Andreassen PR, Sung P, Meetei AR. BLAP18/RMI2, a novel OB-fold-containing protein, is an essential component of the Bloom helicase-double Holliday junction dissolvasome. Genes Dev. 2008;22:2856–2868. doi: 10.1101/gad.1725108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.German J, Sanz MA, Ciocci S, Ye TZ, Ellis NA. Syndrome-causling mutations of the BLM gene in persons in the Bloom’s syndrome registry. Hum. Mutat. 2007;28:743–753. doi: 10.1002/humu.20501. [DOI] [PubMed] [Google Scholar]
- 17.Neff NF, Ellis NA, Ye TZ, Noonan J, Huang K, Sanz M, Proytcheva M. The DNA helicase activity of BLM is necessary for the correction of the genomic instability of Bloom syndrome cells. Mol. Biol. Cell. 1999;10:665–676. doi: 10.1091/mbc.10.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo RB, Rigolet P, Ren H, Zhang B, Zhang XD, Dou SX, Wang PY, Amor-Gueret M, Xi XG. Structural and functional analyses of disease-causing missense mutations in Bloom syndrome protein. Nucleic Acids Res. 2007;35:6297–6310. doi: 10.1093/nar/gkm536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karow JK, Newman RH, Freemont PS, Hickson ID. Oligomeric ring structure of the Bloom’s syndrome helicase. Curr. Biol. 1999;9:597–600. doi: 10.1016/s0960-9822(99)80264-4. [DOI] [PubMed] [Google Scholar]
- 20.Cleary SP, Zhang W, Di Nicola N, Aronson M, Aube J, Steinman A, Haddad R, Redston M, Gallinger S, Narod SA, et al. Heterozygosity for the BLM(Ash) mutation and cancer risk. Cancer Res. 2003;63:1769–1771. [PubMed] [Google Scholar]
- 21.Baris HN, Kedar I, Halpern GJ, Shohat T, Magal N, Ludman MD, Shohat M. Prevalence of breast and colorectal cancer in Ashkenazi Jewish carriers of Fanconi anemia and Bloom syndrome. Isr. Med. Assoc. J. 2007;9:847–850. [PubMed] [Google Scholar]
- 22.Janscak P, Garcia PL, Hamburger F, Makuta Y, Shiraishi K, Imai Y, Ikeda H, Bickle TA. Characterization and mutational analysis of the RecQ core of the Bloom syndrome protein. J. Mol. Biol. 2003;330:29–42. doi: 10.1016/s0022-2836(03)00534-5. [DOI] [PubMed] [Google Scholar]
- 23.Yang Y, Dou SX, Xu YN, Bazeille N, Wang PY, Rigolet P, Xu HQ, Xi XG. Kinetic mechanism of DNA unwinding by the BLM helicase core and molecular basis for its low processivity. Biochemistry. 2010;49:656–668. doi: 10.1021/bi901459c. [DOI] [PubMed] [Google Scholar]
- 24.Beresten SF, Stan R, van Brabant AJ, Ye T, Naureckiene S, Ellis NA. Purification of overexpressed hexahistidine-tagged BLM N431 as oligomeric complexes. Protein Expr. Purif. 1999;17:239–248. doi: 10.1006/prep.1999.1135. [DOI] [PubMed] [Google Scholar]
- 25.Yang Y, Dou SX, Ren H, Wang PY, Zhang XD, Qian M, Pan BY, Xi XG. Evidence for a functional dimeric form of the PcrA helicase in DNA unwinding. Nucleic Acids Res. 2008;36:1976–1989. doi: 10.1093/nar/gkm1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ali JA, Lohman TM. Kinetic measurement of the step size of DNA unwinding by Escherichia coli UvrD helicase. Science. 1997;275:377–380. doi: 10.1126/science.275.5298.377. [DOI] [PubMed] [Google Scholar]
- 27.Chan KM, Delfert D, Junger KD. A direct colorimetric assay for Ca-2+-stimulated ATPase activity. Anal. Biochem. 1986;157:375–380. doi: 10.1016/0003-2697(86)90640-8. [DOI] [PubMed] [Google Scholar]
- 28.Gyimesi M, Sarlos K, Kovacs M. Processive translocation mechanism of the human Bloom’s syndrome helicase along single-stranded DNA. Nucleic Acids Res. 2010;38:4404–4414. doi: 10.1093/nar/gkq145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yodh JG, Stevens BC, Kanagaraj R, Janscak P, Ha T. BLM helicase measures DNA unwound before switching strands and hRPA promotes unwinding reinitiation. EMBO J. 2009;28:405–416. doi: 10.1038/emboj.2008.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang XD, Dou SX, Xie P, Hu JS, Wang PY, Xi XG. Escherichia coli RecQ is a rapid, efficient, and monomeric helicase. J. Biol. Chem. 2006;281:12655–12663. doi: 10.1074/jbc.M513089200. [DOI] [PubMed] [Google Scholar]
- 31.Cheok CF, Wu L, Garcia PL, Janscak P, Hickson ID. The Bloom’s syndrome helicase promotes the annealing of complementary single-stranded DNA. Nucleic Acids Res. 2005;33:3932–3941. doi: 10.1093/nar/gki712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Machwe A, Xiao LR, Groden J, Matson SW, Orren DK. RecQ family members combine strand pairing and unwinding activities to catalyze strand exchange. J. Biol. Chem. 2005;280:23397–23407. doi: 10.1074/jbc.M414130200. [DOI] [PubMed] [Google Scholar]
- 33.Singleton MR, Sawaya MR, Ellenberger T, Wigley DB. Crystal structure of T7 gene 4 ring helicase indicates a mechanism for sequential hydrolysis of nucleotides. Cell. 2000;101:589–600. doi: 10.1016/s0092-8674(00)80871-5. [DOI] [PubMed] [Google Scholar]
- 34.Crampton DJ, Mukherjee S, Richardson CC. DNA-induced switch from independent to sequential dTTP hydrolysis in the bacteriophage T7 DNA helicase. Mol. Cell. 2006;21:165–174. doi: 10.1016/j.molcel.2005.11.027. [DOI] [PubMed] [Google Scholar]
- 35.Liao JC, Jeong YJ, Kim DE, Patel SS, Oster G. Mechanochemistry of T7 DNA helicase. J. Mol. Biol. 2005;350:452–475. doi: 10.1016/j.jmb.2005.04.051. [DOI] [PubMed] [Google Scholar]
- 36.Serebrov V, Beran RKF, Pyle AM. Establishing a mechanistic basis for the large kinetic steps of the NS3 helicase. J. Biol. Chem. 2009;284:2512–2521. doi: 10.1074/jbc.M805460200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muzzolini L, Beuron F, Patwardhan A, Popuri V, Cui S, Niccolini B, Rappas M, Freemont PS, Vindigni A. Different quaternary structures of human RECQ1 are associated with its dual enzymatic activity. PLoS Biol. 2007;5:157–168. doi: 10.1371/journal.pbio.0050020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dessinges MN, Lionnet T, Xi XG, Bensimon D, Croquette V. Single-molecule assay reveals strand switching and enhanced processivity of UvrD. Proc. Natl Acad. Sci. USA. 2004;101:6439–6444. doi: 10.1073/pnas.0306713101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sun B, Wei KJ, Zhang B, Zhang XH, Dou SX, Li M, Xi XG. Impediment of E. coli UvrD by DNA-destabilizing force reveals a strained-inchworm mechanism of DNA unwinding. EMBO J. 2008;27:3279–3287. doi: 10.1038/emboj.2008.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee JY, Yang W. UvrD helicase unwinds DNA one base pair at a time by a two-part power stroke. Cell. 2006;127:1349–1360. doi: 10.1016/j.cell.2006.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bernstein DA, Zittel MC, Keck JL. High-resolution structure of the E. coli RecQ helicase catalytic core. EMBO J. 2003;22:4910–4921. doi: 10.1093/emboj/cdg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ren H, Dou SX, Rigolet P, Yang Y, Wang PY, Amor-Gueret M, Xi XG. The arginine finger of the Bloom syndrome protein: its structural organization and its role in energy coupling. Nucleic Acids Res. 2007;35:6029–6041. doi: 10.1093/nar/gkm544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crampton DJ, Guo SY, Johnson DE, Richardson CC. The arginine finger of bacteriophage T7 gene 4 helicase: role in energy coupling. Proc. Natl Acad. Sci. USA. 2004;101:4373–4378. doi: 10.1073/pnas.0400968101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Enemark EJ, Joshua-Tor L. Mechanism of DNA translocation in a replicative hexameric helicase. Nature. 2006;442:270–275. doi: 10.1038/nature04943. [DOI] [PubMed] [Google Scholar]
- 45.Thomsen ND, Berger JM. Running in reverse: the structural basis for translocation polarity in hexameric helicases. Cell. 2009;139:523–534. doi: 10.1016/j.cell.2009.08.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Compton SA, Tolun G, Kamath-Loeb AS, Loeb LA, Griffith JD. The Werner syndrome protein binds replication fork and Holliday junction DNAs as an oligomer. J. Biol. Chem. 2008;283:24478–24483. doi: 10.1074/jbc.M803370200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Choudhary S, Sommers JA, Brosh RM. Biochemical and kinetic characterization of the DNA helicase and exonuclease activities of Werner syndrome protein. J. Biol. Chem. 2004;279:34603–34613. doi: 10.1074/jbc.M401901200. [DOI] [PubMed] [Google Scholar]
- 48.Muftuoglu M, Kulikowicz T, Beck G, Lee JW, Piotrowski J, Bohr VA. Intrinsic ssDNA annealing activity in the C-terminal region of WRN. Biochemistry. 2008;47:10247–10254. doi: 10.1021/bi800807n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Popuri V, Bachrati CZ, Muzzolini L, Mosedale G, Costantini S, Giacomini E, Hickson ID, Vindigni A. The human RecQ helicases, BLM and RECQ1, display distinct DNA substrate specificities. J. Biol. Chem. 2008;283:17766–17776. doi: 10.1074/jbc.M709749200. [DOI] [PubMed] [Google Scholar]
- 50.Wu L, Davies SL, Levitt NC, Hickson ID. Potential role for the BLM helicase in recombinational repair via a conserved interaction with RAD51. J. Biol. Chem. 2001;276:19375–19381. doi: 10.1074/jbc.M009471200. [DOI] [PubMed] [Google Scholar]
- 51.Wu L, Davies SL, North PS, Goulaouic H, Riou JF, Turley H, Gatter KC, Hickson ID. The Bloom’s syndrome gene product interacts with topoisomerase III. J. Biol. Chem. 2000;275:9636–9644. doi: 10.1074/jbc.275.13.9636. [DOI] [PubMed] [Google Scholar]
- 52.Hu P, Beresten SF, van Brabant AJ, Ye TZ, Pandolfi PP, Johnson FB, Guarente L, Ellis NA. Evidence for BLM and Topoisomerase III alpha interaction in genomic stability. Hum. Mol. Genet. 2001;10:1287–1298. doi: 10.1093/hmg/10.12.1287. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.