


Abstract
Although chromatin folding is known to be of functional importance to control the gene expression program, less is known regarding its interplay with DNA replication. Here, using Circular Chromatin Conformation Capture combined with high-throughput sequencing, we identified megabase-sized self-interacting domains in the nucleus of a human lymphoblastoid cell line, as well as in cycling and resting peripheral blood mononuclear cells (PBMC). Strikingly, the boundaries of those domains coincide with early-initiation zones in every cell types. Preferential interactions have been observed between the consecutive early-initiation zones, but also between those separated by several tens of megabases. Thus, the 3D conformation of chromatin is strongly correlated with the replication timing along the whole chromosome. We furthermore provide direct clues that, in addition to the timing value per se, the shape of the timing profile at a given locus defines its set of genomic contacts. As this timing-related scheme of chromatin organization exists in lymphoblastoid cells, resting and cycling PBMC, this indicates that it is maintained several weeks or months after the previous S-phase. Lastly, our work highlights that the major chromatin changes accompanying PBMC entry into cell cycle occur while keeping largely unchanged the long-range chromatin contacts.
INTRODUCTION
The timely firing of replication origins is essential for most eukaryotic cells to allow the proper duplication of the genetic material during the S-phase. Replication origins are activated according to a specific temporal program that has been studied in various species allowing to distinguish sequences replicated in the early S-phase from sequences replicated later (1–15). The replication timing is mainly cell-type specific, even if some loci exhibit a strong conservation of the replication timing over different cell types (8–14,16). The replication timing is significantly correlated with genomic and epigenetic features (reviewed in (17)). For instance, most of the early-replicated regions are gene-rich, transcriptionally active and localized in open chromatin, while the late-replicated regions are mostly found in silent, gene-poor and closed chromatin (2–8,14,18,19). The replication timing thus reflects several genomic, epigenetic and functional characteristics.
Besides linear features of chromatin, DNA replication has been related to the 3D organization of the nucleus. The replication occurs at discrete foci in the nucleus according to a specific spatiotemporal organization (20,21). Throughout the S-phase, new replication foci de novo assemble immediately next to the previously active ones (22–25), and a substantial segregation between early- and late-replicated loci has been observed (26–29). In addition, from the observation that thousands of replication forks are only found in hundreds of discrete BrdU-labeled foci, it has been proposed that DNA sequences replicating at the same time could gather (27,30–33), possibly through the anchoring of giant chromatin loops on the nuclear skeleton (reviewed in (34–36)). Courbet et al. (37) carried out elegant experiments showing that the size of the nuclear skeleton-anchored loops clearly depends on the velocity of the fork and on the density of initiations. They observed that experimental slowing down of replication forks is compensated for by the mobilization of latent replication origins in the subsequent S-phase, and by the formation of smaller chromatin loops. This argues in favor of a replication-related high-order folding of chromatin. The recent availability of replication timing data in several cell lines now offers the opportunity to test this idea more systematically and to address to which extent a structural counterpart exists to the functional replication domains identified from the replication timing. 3C methodologies (38) have emerged as powerful strategies to analyze in detail the long-range folding of chromatin and by extension, the chromosome organization in the nucleus. Different genome-wide mapping strategies have been developed (39), including circular chromosome conformation capture (4C) for the highly detailed analysis of DNA contacts made by selected loci (40,41), and Hi–C for a more global pairwise comparison of contacts between all genomic loci (42).
Hi–C data have already been compared to the replication timing of related cell lines (13,14). Ryba et al. (14) observed a tight link between the replication timing and the Hi–C correlation matrices (principal eigenvector), showing that sequences localized near each other replicate at similar time. Moreover, Yaffe et al. (13) numbered more interchromosomal interactions than expected between regions having similar replication timing. However, the Hi–C data do not always allow the direct visualization of the chromatin contacts for chosen loci, principally because of insufficient sequencing depth to cover all the combinatorial possibilities. Moreover, the normalization performed in the analysis of Hi–C data (42) gave as much weight to the very-distant interactions (over tens of megabases) as to the more local contacts (below few megabases), despite the fact that the latter are the most frequent ones. Essential points thus remain to be addressed. First, a closer analysis of the local contacts may highlight replication timing-related structural domains in the nucleus. Then, a description of the contacts along the whole chromosome will determine if each locus replicating at the onset of the S-phase can be contacted by a given early-replicated viewpoint. Lastly, the comparison of the contacts in quiescent and cycling cells will address the question of their long-term persistence in resting cells.
Here, we used the 4C methodology (40) in order to identify with high precision the main partners of multiple early- and late-replicated regions and to determine to which extent the 3D organization of chromatin can be linked to the replication timing. For this study, we focused on a large region (20 Mb) of the human genome in which the replication timing presents large fluctuations that are conserved in most examined cell types (Supplementary Figure S1). In particular, these fluctuations define loci replicating earlier than their immediate surroundings (timing peaks), and conversely regions replicating latter than the surroundings (timing valleys). 4C experiments were performed in a lymphoblastoid cell line, in resting and in cycling peripheral blood mononuclear cells (PBMC) using 10 viewpoints in the selected region. We observed, and confirmed by fluorescent in-situ hybridizations (FISH) experiments, that early timing peaks coincide with frontiers limiting the spreading of strong local interactions and delineating megabase-sized 3D-structural domains. In addition, at larger distances, profiles of long-range interactions from timing peaks viewpoints faithfully reproduce the replication timing profile, which sheds a new light on the propensity of early- and late-replicating regions to contact other chromosome regions. These results are robustly observed in cycling and G0-blocked cells, indicating that the observed organization exists in G0 and does not result from the ongoing DNA replication.
MATERIALS AND METHODS
Cell culture
A human lymphoblastoid cell line and PBMC were grown at 37°C with 5% CO2 in RPMI supplemented with 10% fetal bovine serum, glutamax and penicillin/streptomycin. PBMC, which are mostly lymphocytes (43), were isolated from healthy donors using Leucosep® (Greiner bio-one) and Ficoll-Paque PLUS (GE Healthcare) according to manufacturer’s instructions. PBMC stimulation was performed by supplementing the culture medium with 1.5% Phytohemagglutinin (PHA M Form, Life Technology) for 68 h. A million stimulated PBMC were fixed with cold ethanol and stained with propidium iodide (20 µg/ml, 30 min 37°C) for cell-cycle analysis (LSRII, FACSDivaTM, BD Biosciences).
4C protocol and sequencing
Cells were processed for 4C experiment as previously described (40,44,45) with only minor modifications. Cells were dispatched into 7 millions cell aliquots. All aliquots were fixed in phosphate buffered saline 10% SVF and 2% PFA (10 min, RT), lysed in 10 mM Tris, 10 mM NaCl, 0.3% NP-40 and dounced (B pestle). The cross-linked chromatin was digested overnight with 600 U of HindIII (Promega) and ligated with 150 U of T4 DNA ligase (Fermentas). The templates were further cut with 50 U Hin1II (Fermentas) and circularized with 300 U T4 DNA ligase (Fermentas). DNA was purified with phenol extraction followed by an ethanol precipitation, and chromatography (Roche High Pure PCR Product Purification Kit). DNA concentration was measured by fluorimetry (Quant-iT™ dsDNA Assay Kit, Broad Range, Life Technology). For every primer pair used, 10 PCRs were performed on 200 ng input DNA (for primer sequences, see Supplementary Table S1). For primer design, we used a strategy similar to that described in Splinter et al. (44) and Noordermeer et al. (45). PCR products were sequenced using a Solexa Genome Analyser IIx or Solexa Hi-Seq 2000 (76 bp). Reads were first processed to identify the viewpoints and then mapped on the human genome (UCSC hg18 assembly) using Seqmap software (46). Only reads annealing on HindIII restriction sites were kept for subsequent analyses.
4C analyses
The RAW 4C signal consists in the number of reads per restriction site. For local analyses, a running mean was applied on the RAW signal with a window size of 20 restriction sites. To compare local 4C profiles between cell lines (Figure 1C) the running mean signal was divided by the total number of reads corresponding to the chromosome 5. For chromosome-wide and genome-wide analyses, we defined a long-range interaction frequency (LRIF) variable that reinforces the contribution of low-abundance densely packed reads. The LRIF signal is defined as the percentage of restriction sites covered by reads in a 500-kb sliding window (coverage) × the mean number of reads in this sliding window. The sliding step was 100 kb. To compare the LRIF signal between cell lines, its value was divided by the total number of reads on the chromosome 5. Maxima from the LRIF signal were detected using an amplitude threshold-based algorithm (Matlab, The Mathworks). All plots were generated using Matlab. Heat-maps of pairwise distances between 4C peaks were generated by quantifying the percentage of cases where the distances between the 4C peaks are <250 kb. The value of this percentage was encoded using a green–red colormap. Matlab linkage function was used to perform the hierarchical clustering.
Figure 1.

4C interaction profiles associated with replication timing in lymphoblastoid cells (A) MRT of the lymphoblastoid cell line Gm06990, in the portion 150–170 Mb of human chromosome 5 (11). Early (respectively, late) replicating sequences correspond to MRT values ∼0 (respectively, ∼1). The arrowheads indicate the position of the viewpoints used for the 4C analysis. (B) Interaction frequencies profiles of the 10 viewpoints. For each panel, the light gray curve corresponds to the replication timing profile (right y-axis) and the colored filled curve to the 4C interaction frequency profile (left y-axis). The arrowheads indicate the position of the viewpoints; the thin arrows indicate sharp drops in interaction frequency coinciding with early timing peaks; the curly brackets indicate partners of V3 and V4 viewpoints; and the dots indicate preferential partners of P viewpoints coinciding with timing peaks. (C) 4C interaction frequency is highly reproducible from one cell type to another. To normalize for differences in read numbers, the running mean value (B) has been divided by the total number of reads mapped on chromosome 5 (red = lymphoblastoid, green = resting PBMC and blue = PHA-stimulated PBMC). Top, V4 viewpoint; bottom, P1 viewpoint. (D) Timing peaks correspond to zones of sharp changes in 4C data interaction frequency. (Top) The position of timing peaks are marked along the MRT profile in Gm06990 (vertical bars). (Middle) space-scale representation of the 4C interaction frequencies of S- and V-viewpoints. Each horizontal line corresponds to the median value of the interaction frequency computed in sliding windows of size ranging from 200 kb to 1.35 Mb. Due to the very large median-value dynamics, a logarithmic colormap was used (bottom).
FISH experiments
We selected PBMC to perform FISH experiments, thus avoiding ambiguities due to cells in S or G2 phase exhibiting replication doublets. Resting PBMC were swollen in 75 mM KCl (30 min, 37°C), fixed in methanol 3:1 acetic acid, and then spread on microscope glass slides. FISH probes were generated by random priming on BAC clones (Supplementary Table S2) using amino-allyl dUTP. Probes were then coupled with amine-reactive alexa fluorophores (A488, A555 or A647, Life Technology). The specificity of BAC probes was tested by both restriction profile and hybridization on metaphase spreads. Hybridization of denatured probes (80°C, 5 min) on denatured nuclei (73°C, 2 min) were performed overnight at 37°C (in 50% formamide, 2 × SSC, 10% dextran, 10 µg single-stranded DNA, 1–3 µg human CotI). Slides were mounted with fluoromount-G (Southern BioTech) and DAPI was used as DNA counterstain. Images were collected using a wide-field Zeiss Axioimager (63×, Photometrics CoolSNAP HQ camera) and analyzed with Image J. The Rayleigh fits of measured distances 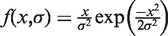 were performed using Matlab (σ corresponds to the shape parameter). The mean square physical distance is defined as
were performed using Matlab (σ corresponds to the shape parameter). The mean square physical distance is defined as 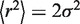 , according to Yokota et al. (47).
, according to Yokota et al. (47).
Mean replication timing profiles and replication U-domains
We used the mean replication timing (MRT) profiles along the complete human genome derived by Baker et al. (48) from Repli-Seq data (11,15). The signal analysis performed in (48) led to the segmentation of about half of the genome into replication timing domains corresponding to U-shaped MRT profiles (late-replicating core surrounded by two early-replicating loci). MRT profiles and U-domains coordinates for Gm06990 and K562 were obtained directly from the authors.
Whole-genome Hi–C chromatin conformation data analysis
We used the spatial proximity maps of the human genome generated using Hi–C method (42). We downloaded 100-kb resolution maps for Gm06990 and K562 cell lines from the GEO web site (GSE18199 binned heatmaps): http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc = GSE18199. Interaction counts between loci on same chromosome were normalized by the mean intrachromosomal interaction count averaged over all pairs of loci separated by the same genomic distance. To properly analyze the behavior of interaction frequencies across timing peaks, we selected well-defined timing peaks that are shared by two consecutive U-shaped replication timing domains (48). We then extracted, from the Hi–C interaction ratio matrices, two 4C-like interaction profiles converging to the shared timing peak: the first one coming from the leftmost U-domain border and the second one coming from the rightmost U-domain border. All 4C-like interaction ratio profiles were finally averaged after rescaling the distances so that the U domain size was 1 (Figure 5, top).
Figure 5.

Hi–C interaction profile across timing peaks. Average Hi–C interaction ratio across timing peaks localized at the border between two consecutive U-domains (top, see ‘Materials and Methods’ section). Bottom, the mean interaction ratio profile as a function of the genomic distance rescaled so that U-domain size be 1. It corresponds to an average over n = 465 (respectively, n = 428) timing peaks in Gm06990 (respectively, K562) cell line.
RESULTS
We used 4C in combination with high-throughput sequencing (4C-seq) (44) to explore the correlation between replication timing and chromosome 3D structure. 4C is used to capture, in the whole genome, the loci interacting with a chosen sequence, namely the viewpoint. We focused on a large (20 Mb) region of human chromosome 5 in which the replication timing measured in Gm06990 lymphoblastoid cell line presents large fluctuations and is highly conserved in different cell types (Supplementary Figure S1). As the replication timing of lymphoblastoid cells appears extremely similar, even when comparing established cell lines from different sources and histories (49), the replication timing of the lymphoblastoid cell line used in this study is very likely close to the one of Gm06990.
The shape of the timing profile in the selected 20 Mb displays timing peaks, i.e. regions replicating earlier than their immediate surroundings, and conversely local timing valleys (replicating later than their surroundings). 4C viewpoints were then unambiguously positioned on some local timing peaks or in timing valleys (Figure 1A). Five viewpoints were selected on early peaks (P1–P5), three in very late valleys (V1, V3 and V4), one in a valley of intermediate timing (mid S-phase, V2) and one in a transition from very early to mid S-phase (S). The local chromatin and genomic features around the viewpoints are shown in Supplementary Figure S2. A mean of 2.6 millions reads per viewpoint was generated in lymphoblastoid cells (Supplementary Table S3). About 76% of the mapped reads are located on chromosome 5 and a much lower read density was observed in other chromosomes (Supplementary Table S4), which is in agreement with the existence of chromosome territories. Similar proportions of intrachromosomal interactions were observed for the other cell types used in the study. Within chromosome 5, most contacts were local and the interaction frequencies rapidly dropped with increasing genomic separation (Supplementary Table S3), as previously reported (39).
Early timing peaks delineate structural self-interacting domains and preferentially contact each other
4C data were locally averaged using a running mean within a window of 20 HindIII sites (∼100 kb). The plots show that interaction frequencies of the valley viewpoints (V1, V2, V3 and V4), as well as of the S viewpoint, quickly decrease to a few percents of the maximum signal over a few megabases distance from the considered viewpoint (Figure 1B). This decrease does not follow a simple pattern; indeed, sharp drops can be seen. Strikingly, most of these drops seem to be localized at, or close to, timing peaks (Figure 1B, arrows). This suggests that contacts between late-replicating regions are somehow limited by intervening timing peaks. To further illustrate the existence of these sharp drops in the 4C profiles, we displayed 4C data using a space-scale representation. Since 4C raw signals present strong deviations from the local mean, we represented the median value in sliding windows of size ranging from 200 kb to 1.35 Mb. The resulting images were obtained by encoding the median values with a logarithmic color map (Figure 1D). It became apparent that 4C signal amplitudes are larger within a domain bordered by two timing peaks, and that clear transitions occur at timing peaks. This analysis confirmed that timing peaks delineate frontiers limiting the DNA contacts of neighboring late-replicating regions, and thus sheds light on the existence of self-interacting chromosomal domains bordered by timing peaks. Interestingly, beyond this local regime, late-replicated loci preferentially interact with other late-replicating regions, as can be seen for the long-range interactions of the V3 and V4 valley viewpoints (Figure 1B, curly brackets).
The interaction frequencies obtained with viewpoints localized on timing peaks (P1–P5) display profiles strongly different from those of the valley viewpoints. P1, P2 and P4 (and to a lesser extent P3) profiles exhibit preferential contacts with nearly every neighboring timing peak, even with those located 10-Mb away (Figure 1B, dots), showing that they loop out the intervening late-replicating domains to contact each other (see also the space-scale representation in Supplementary Figure S3). Although localized on an early peak, the P5 viewpoint does not display specific contacts with other timing peaks, which might be due to specific features of this viewpoint (see ‘Discussion’ section). Interestingly, whereas the contacts between timing valleys are spread over megabases (Figure 1D, curly brackets), the contacts between early timing peaks are restricted to shorter distances (Figure 1D, dots), suggesting the existence of a stronger and more specific anchoring mechanism between early timing peaks than between late regions.
To address whether this replication-related 3D organization of chromatin is also found in non-cycling cells, we performed similar analyses in resting PBMC, and, as a control, in cycling phytohemagglutinin-stimulated PBMC (Supplementary Figure S4). For both cell types, we used the replication timing of Gm06990 cell line as a reference since the timing profile in the region containing the viewpoints is remarkably similar in different cell types (Supplementary Figure S1) and since B- and T-cells have divergent timing on only 4.5% of the genome (49). The local 4C profiles obtained with the three different cell types remarkably superimpose on each other (Figure 1C), arguing for the existence of a robust and common 3D chromatin conformation. In resting and cycling PBMC, we also observed frontiers localized at timing peaks that prevent the contacts of neighboring late-replicated regions (Figure 1C and Supplementary Figures S5 and S6). In addition, using timing peaks as viewpoints, we noticed preferred contacts with every timing peak. This not only confirms the preferential contacts between early-replicated regions as previously observed (13), but more strikingly also shows that the contact frequency profile is strongly correlated with the timing profile in the three related cell types. Moreover, the fact that the local conformation in resting PBMC is similar to the one in cycling cells implies that this correlation exists in G0 and does not result from ongoing DNA replication. It suggests that this organization pattern is conserved all along the cell cycle.
LRIF profiles faithfully reproduce the replication timing profile
In 4C experiments, the measured interaction frequencies have been shown to rapidly drop with increasing genomic distance (50). As a consequence, only few informative ligation events can be captured while analyzing long-range contacts. To collect the long-range contacts, it has been shown that the contribution of isolated reads improperly amplified by PCR should be diminished, and the contribution of low-abundance densely packed reads reinforced (40). Accordingly, we defined a variable called LRIF (see ‘Materials and Methods’ section) that operates such a filtering. While the LRIF signal is synchronous with the coverage and the running mean defined at 500 kb (Supplementary Figure S7), it offers better signal-to-noise ratio, facilitating subsequent analyses.
All along the chromosome, the long-range contacts obtained from V1, V3 and V4 on the one hand, and from P1, P2, P3 and P4 on the other hand, display opposed oscillations in the three cell types tested, i.e. P viewpoints maxima coincided with V viewpoints minima and vice versa (Figure 2A and B and Supplementary Figure S8). To quantify this observation for each viewpoint on the whole chromosome 5, we detected long-range contacts by local maxima in the LRIF signal, and measured the pairwise distances separating each local maximum from the closest local maximum of another viewpoint. As an illustration of the results, the histogram of distances between P4 and P1 peaks is centered at zero, indicating that the long-range contacts of P1 and P4 viewpoints overlap and that P1 and P4 viewpoints indeed interact with the same partners (Figure 2D). On the contrary, the V4–P1 distance distribution is clearly depleted around zero, showing that V4 and P1 have different distant partners (Figure 2D). To depict all the pairwise combinations, we generated a heat-map in which a green–red colormap quantifies overlap of long-range contacts identified from LRIF signals (Figure 2E). The unbiased hierarchical clustering performed revealed the existence of three groups. The first one corresponds to viewpoints on timing peaks P1, P2, P3 and P4 that display LRIF signal in opposition of phase with the second group that includes valley viewpoints V1, V3 and V4. The third group corresponds to V2, S and P5 viewpoints that display intermediate 4C profiles; the hierarchical clustering performed yet tends to position them closer to early viewpoints than to late ones. Similar results were obtained with the three cell lines used in this study, showing the remarkable conservation of the long-range interactions frequencies (Figure 2C and E). Taken together, we conclude that chromosomal domains with opposite replication timing tend to segregate in nuclear space, and they do so even in non-dividing quiescent cells.
Figure 2.

Chromosomal domains with opposite replication timing segregate in the nuclear space. (A) LRIF in lymphoblastoid cells for the 10 viewpoints (see text and Materials and Methods’ section for the definitions). The colored filled curves correspond to the 4C profiles in a region of chromosome 5 centered ∼60 Mb upstream of the viewpoints. The light gray lines correspond to the replication timing in lymphoblastoid cells (11) plotted with the same y-axis as in Figure 1A. (B) Similar analysis performed with resting PBMC. (C) Comparison of LRIF values of the P1 viewpoint in lymphoblastoid cells (red), resting PBMC (green) and PHA-stimulated PBMC (blue). LRIF values have been normalized for differences in read numbers. The upper panel corresponds to the replication timing in lymphoblastoid cells. (D) Distribution of distances between the 4C peaks detected in the LRIF profiles. We detected the 4C peaks and measured the distances between each of them and the closest 4C peak obtained with another viewpoint. All pairwise distances were measured over the whole chromosome 5. Three distance distribution histograms are shown here as examples (V4–P1, V4–V3 and P4–P1). A distribution centered on zero (as P4–P1) indicates that the LRIF maxima of the two viewpoints overlap. (E) To quantify the similarities between LRIF signals from different viewpoints, we calculated the percentage of occurrences where the pairwise distances measured in (D) are <250 kb. This percentage was encoded with a green–red color map. Similar signals are green (V4/V3 and P4/P1), whereas out of phase signals are red (V4/P1). The asterisks correspond to the distribution histograms shown in (D).
We then asked to which extent the long-range partners of every viewpoint coincide with timing peaks. We therefore measured, for each viewpoint and on the whole genome, the distances between every 4C peaks and the closest timing peak. The obtained histograms for lymphoblastoid cells (Figure 3A) clearly show that the long-range contacts of the P4 viewpoint, for example, are close to timing peaks, as the histogram is centered on zero. On the contrary, the histogram for the V3 viewpoint is depleted in values around zero indicating that V3 partners are away from timing peaks. V2 viewpoint exhibits an intermediate situation: the distribution histogram is neither peaked nor depleted around zero. To depict this information for all viewpoints and the three cell lines, a green–red colormap was used to show the coincidence between timing peaks and long-range contacts (Figure 3B). Our results show that the long-range partners of P1, P2, P3 and P4 viewpoints overlap with timing peaks for the whole genome. Conversely, V1, V3 and V4 do not interact with timing peaks.
Figure 3.

Viewpoints localized on local timing peaks mostly interact with other timing-peaked regions. (A) Histograms of the distance between every 4C peak and the closest timing peak on the whole genome. 4C peaks were detected in the whole genome, we then measured the distance between each of them and the closest timing peak. The three histograms represent theses distances measured in lymphoblastoid cells for P4 (left), V3 (middle) and V2 (right) viewpoints. A distribution centered on zero (P4) indicated that the 4C peaks are close to timing peaks. n = number of 4C peaks detected. (B) We computed the percentage of distances <250 kb and represented this value with a green–red color map. 4C signals in phase with the replication timing are green (P4) whereas anti-correlated signals are red (V3).
Overall, we observed that the LRIF profiles correlate genome wide with replication timing. Strikingly, they also reproduce the replication timing signal over very large distances along the chromosome 5, meaning that a given timing peak can interact with any other timing peak of the whole chromosome 5. Interestingly, similar tendency can still be observed on other chromosomes (Supplementary Figure S9), albeit with a much lower interaction strength (about two orders of magnitude). The fact that the interaction frequency profiles tightly follow the replication timing strongly argues in favor of a replication imprint on chromatin organization.
In-situ distance measurements confirm the physical proximity between loci corresponding to early timing peaks
To test whether some of the contacts captured by the 4C-sequencing (4C-Seq), which reflect chromatin folding at the population level, can be observed in single cells, we performed FISH in resting PBMC. We selected BAC probes localized in the 150–170 Mb portion of human chromosome 5. Using three colors FISH, we measured three different sets of physical distances, a first one (EE) corresponding to distances between two local timing peaks, a second set (LL) corresponding to distances between two timing valleys and a third set (LE) corresponding to distances between a peak and a valley (Figure 4A). Results can be depicted by cumulative histograms. For example, as predicted by the 4C results, the d5 (EE) physical distance is smaller than d17 (LE) even if both correspond to a 7.6 Mb genomic length (Figure 4C(3), P < 10−8, Student’s t-test with α-risk of 0.05). Furthermore, the d3 physical distance (EE) is slightly smaller than d1 (LE) and d2 (LE) although d3 corresponds to a much larger DNA fragment than both d1 and d2 (Figure 4C(1)). Likewise, d28, d29 and d30 present similar physical distances although d30 (EE) corresponds to a genomic length twice larger than the LE distances d29 and d30 (Figure 4C(2)). From these three examples, we concluded that d3, d5 and d30, which correspond to physical distances between timing-peaked regions, are smaller than expected if the genomic separation was the only parameter determining the 3D distance in the nucleus.
Figure 4.

FISH distance measurements confirm the physical proximity between early replicated regions. (A) Physical distances measured by three colors FISH in resting PBMC. The arrowheads correspond to the positions of the used probes (Supplementary Table S2), and the line to the measured distances. Three sets of measurements were performed: EE between two timing peaks, LL between two timing valleys and LE between a timing peak and a timing valley. (B) Example of FISH images in resting PBMC nuclei (d1 = GB, d2 = BR, d3 = GR, d28 = BR, d29 = GR, d30 = GB). On both chromosomes, the physical distances separating the gravity centers of the spots were measured in PBMC nuclei. (C) Cumulative distance measurements. (1) d3 (3 Mb) is slightly smaller than d1 (1.9 Mb) and d2 (1.1 Mb, P < 10−3, Student’s t-test). (2) d28, d29, d30 have the same physical distance even if d30 is 5 Mb and d28/d29 are ∼2.5 Mb. (3) d5 is smaller than d17 even if they both cover 7.6 Mb (P < 10−8, Student’s t-test). (D) Relative frequency histograms of d28 distance (2.5 Mb). The green lines correspond to the fit of the frequency histograms by a Rayleigh law (see ‘Materials and Methods’ section). (E) Mean squared physical distance ±95% confidence interval (see ‘Materials and Methods’ section and (47)) as a function of the genomic separation. Red dots, measurement between two timing peaks (EE); blue, between two late regions (LL); green, between a peak and a timing valley (LE). Open dots correspond to distances measured inside a single replication domain; close dots correspond to distances measured between different replication domains. Here, a replication domain is defined by its two timing peak borders surrounding a late-replicated core. P-values shown are evaluated for few EE distances and correspond to pairwise Student’s t-tests with other measured distances with similar (±15%) genomic separation. (F) Physical distances and interaction frequencies between two probes localized within the same replication domain or between two adjacent replication domains. Top: corresponding distances. Middle: as in (E). Bottom: percentage of cases where measured distances are <0.3 µm. P-values correspond to pairwise Student’s t-tests.
It is established that the distance histograms can be easily fitted by a Rayleigh law (Figure 4D) that only depends on a single parameter σ (see ‘Materials and Methods’ section). This Rayleigh parameter perfectly describes the shape of the obtained histograms and can be used to define, for each probe combination, the corresponding mean square physical distance (47). We plotted the mean square physical distance as a function of the genomic separation between the probes (Figure 4E). Only the dots corresponding to EE distances are labeled in Figure 4E, but complete labels and pairwise statistics can be found in Supplementary Figure S10. Among the dots deviating from the mean tendency, most correspond to peak-to-peak distances (d3, d30, d5, d18 and to a lesser extent d6). In particular, d3 is the smallest physical distance, in agreement with 4C results showing that the strongest interaction in PBMC occurs between P3 and P4 peaks (Supplementary Figure S5). This indicates that, for the same genomic separation, the physical distance between two early replicated loci tends to be smaller than the distance between a late and an early replicated locus. Nevertheless, some early-to-early physical distances are not smaller than the mean tendency (d4/d11, d12 and d15). Therefore, if 4C contacts are not necessarily seen by FISH, the FISH results largely agree with those obtained in 4C experiments.
We did not detect spatial proximity between probes in timing valleys (Figure 4E). This may be explained by the fact that contacts between timing valleys are spread over the width of the valleys (Figure 1B, curly brackets), suggesting a more relax interaction pattern between late-replicated regions than between timing peaks. This may also reflect a relative discrepancy between the establishment of contacts (as probed by 4C approach) and the physical proximity seen by 2D-FISH.
The 4C profiles in PBMC indicate that timing peaks are found at frontiers limiting the contacts of neighboring late-replicated regions (Supplementary Figure S5). To test whether this is also observed by FISH, we performed measurements around the P4 timing peak. For comparable genomic separation, physical distances measured across the timing peak tend to be larger than those performed in the valley (Figure 4F, middle). This tendency is supported by the frequency of colocalization between the probes used above in the distance measurements (Figure 4F, bottom). Probes separated by the timing peak less frequently colocalized than probes located in the same timing valley. These observations strengthen the conclusion that a timing peak can limit the interaction capabilities of a late-replicated sequence with other regions which thus contributes to define independent megabase-sized self-interacting structural domains in the nucleus.
DISCUSSION
In the context of DNA replication, the study of structure–function relationships in the nucleus remains challenging. Indeed, some genomic and epigenetic features are of uttermost importance in the definition of replication origin (reviewed in (51)) while replication itself leaves some evolutionary imprints in the genome (16,52,53). Here, we performed a series of 4C-Seq and FISH experiments that reveal to which extent the 3D conformation of chromatin is linked to the replication timing.
At the local scale (1–2 Mb), that is the typical size of a replication timing domain (8,14,48), we found that adjacent timing peaks are prone to physically interact, by looping out the intervening late-replicated sequences (Figure 1B and Supplementary Figures S5 and S6). We recently proposed a segmentation of about half of the genome into U-domains (see ‘Materials and Methods’ section) defined by a late-replicating core surrounded by two timing peaks (48). When averaging the Hi–C data over these U-domains, we recovered the preferential interactions between adjacent timing peaks in Gm06990 cells (Figure 5, left), which corroborates genome wide our conclusions. Moreover, we also noticed similar behavior in the chronic myelogenous leukemia K562 cell line (Figure 5, right), indicating that preferential interactions between adjacent timing peaks are not limited to lymphoid cell types. Nevertheless, our 4C experiments allow to go much further in details since most of the local interactions (Figure 1B, dots) are not detectable in Hi–C matrices (Supplementary Figure S11). In addition to the preferential interaction between adjacent timing peaks, we also observed that frontiers limiting the DNA contacts of late-replicating regions are found at timing peaks. This was observed by 4C (Figure 1 and Supplementary Figures S5 and S6) and by FISH (Figure 4F). Since the timing peaks delineate boundaries of chromosomal domains, we propose to define a structural replication domain by its U-shape consisting in a late-replicated core surrounded by two local early-replicated peaks. Therefore, our definition is complementary to the definition of functional domains based on the replication program (54). As a support for the existence of these domains, we note that the structural frontiers at timing peaks significantly coincide with CCCTC-binding factor or CTCF (Supplementary Figure S11) previously shown to be enriched at the boundaries of chromatin domains (55,56).
At the 10 Mb scale, we also showed preferential interactions between timing peaks (Figures 1B and 4E and Supplementary Figures S5 and S6). Similarly, late-replicated viewpoints (V3, V4) preferentially interact with timing valleys. However, their interactions are spread over almost the entire length of the valley, indicating a relative lack of specific partners (Figure 1B and Supplementary Figures S5 and S6). This might explain why we did not detect any physical proximity by FISH between late-replicated sequences (Figure 4E).
At the scale of entire chromosomes, we demonstrated that chromosomal domains with an opposite replication timing are separated in the nuclear space (Figure 2E) and that an early-replicated (respectively, late-replicated) locus interacts with other timing peaks (respectively, timing valleys; Figures 2A and B and 3). Previous works that compared Hi–C data and replication timing values, already proposed that synchronously replicated sequences interact more frequently than expected (13,14). However, the 4C protocol provides a much higher number of reads for each viewpoint, thereby allowing the direct detection of chromosome-wide interactions. The 4C signals of P1, P2 and P4 viewpoints faithfully reproduce the replication timing along the whole chromosome 5, which demonstrates the astonishing persistence, all over the chromosome, of a timing-linked long-range conformation of chromatin. In addition, our analysis showed that the mid-S-phase replicated P4 peak has a long-range interactions profile similar to those of P1, P2 and P3 (Figure 2A and B and Supplementary Figure S8). This means that it is probably not the precise replication timing value at a given peak that specifies the long-range contacts but rather the timing value relative to its surroundings. The shape of the timing profile, with its local maxima, therefore better predicts the chromatin contacts than the fact of being replicated at the onset of the S-phase. This indicates that, irrespective of its replication timing value, a timing peak may comprise determinants governing the structural conformation of chromatin.
This spatial segregation between early- and late-replicated domains is in agreement with previous results showing the relative absence of interactions between active (early-replicated) and inactive (late-replicated) chromatin, both by 3C-derivated methodologies (40,42,57) and cytological analyses (26–29). The nuclear processes explaining this separation remain to be identified. Insight may come from the P3 viewpoint that is localized in transcriptionally silent chromatin, ∼100-kb away from the nearest expressed gene in PBMC. P3 exhibits in this cell type a positive correlation with the timing, albeit slightly lower than the one measured for P1, P2 and P4 viewpoints that are localized within expressed genes (Figure 3 and Supplementary Figure S2). This might indicate that the influence of transcription and replication probably adds up to enhance the long-range interaction capabilities of selected locus, maybe through the recruitment to spatially coordinated replication and transcription factories (58). This hypothesis is further sustained by the results obtained for P5. This viewpoint lies in a weakly expressed gene (Supplementary Figure S2) and is not located on a sharp timing peak (350-kb shoulder in the replication timing curve; Figure 1). In that context, we assume that the contribution of both transcription and replication are weaker, explaining why P5 interactions with timing peaks cannot be detected beyond the neighboring P4.
Our analysis revealed another striking property of chromatin reorganization while lymphocytes exit quiescence. Quiescent cells and cycling cells are known to have distinct nuclear organization (59–61). For instance, lymphocyte activation is associated with major changes in gene expression (62–66) and major histological modifications of the nucleus with a doubling of nuclear volume and a significant reduction of peripheral heterochromatin (67,68). Despite these important modifications of nuclear architecture, a high similarity in local and long-range chromatin contacts was observed between resting and activated PBMC (Figures 1C and 2C). As PBMC cells were efficiently pushed out of dormancy (Supplementary Figure S4), the absence of differences between resting and activated lymphocytes does not result from an inefficient PBMC activation by PHA. This implies that the histological disappearance of heterochromatin during lymphocyte activation takes place without disruption of the 4C-probed long-range interactions and while most of the neighboring relationships are maintained. The stability of these interactions may rely either on constant structural elements ultimately determined by the genome sequence, either on some nuclear function shared by resting and cycling cells. As a support of the latter, it has been shown that the quiescent C127 cells (mouse mammary epithelial cells), when in vitro replicated, retain a replication timing program similar to the one observed in cycling C127 cells (61).
In summary, using 4C-Seq experiments, we demonstrate that the spatial segregation of early- and late-replicating loci (13,14,26–29) is associated with the establishment of robust chromatin contacts. The 4C interaction profiles, supported by FISH, highlight the existence of megabase-sized self-interacting domains in the nucleus whose boundaries coincide with early-initiation zones. Along the whole chromosome, we also directly observed a considerable correlation between the replication timing profiles and the long-range contact frequencies of every viewpoint, which could not be observed in previous studies (13,14). Moreover, our work shows that it is not the precise replication timing value that specifies the long-range contacts but rather the timing value relative to its surroundings. This chromatin organization scheme is shared by cycling and resting PBMC. This replication-associated structure stably observed in quiescent as well as in activated cells likely reflects some fundamental elements of the chromosome 3D structure that remain to be determined.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Tables 1–4, Supplementary Figures 1–11 and Supplementary References [8,11,14,15,44].
FUNDING
The Agence Nationale de la Recherche (ANR) [REFOPOL, ANR 10 BLANC 1615]; Centre National de la Recherche Scientifique (CNRS) Projects Exploratories Premier Soutien [PEPS 2009]; Fondation pour la Recherche sur le Cancer [ARC ECL 2010R01122]; European Molecular Biology Organization (EMBO) short-term Fellowships [ASTF-381.00-2008]. Funding for open access charge: ANR [REFOPOL, ANR 10 BLANC 1615].
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors are very grateful to D.M. Gilbert, O. Hyrien, H. Julienne and C. Vaillant for stimulating discussions. This work benefited from the expertise of the high throughput sequencing platforms of IMAGIF (Centre de Recherche de Gif—www.imagif.cnrs.fr) and MGX-Montpellier GenomiX (http://www.mgx.cnrs.fr/). The authors also thank the PLATIM (PLAteau Technique d’Imagerie et de Microscopie, IFR128, Lyon FRANCE) and the EFS (Etablissement Français du Sang, Lyon Gerland).
REFERENCES
- 1.Raghuraman MK, Winzeler EA, Collingwood D, Hunt S, Wodicka L, Conway A, Lockhart DJ, Davis RW, Brewer BJ, Fangman WL. Replication dynamics of the yeast genome. Science. 2001;294:115–121. doi: 10.1126/science.294.5540.115. [DOI] [PubMed] [Google Scholar]
- 2.Schübeler D, Scalzo D, Kooperberg C, van Steensel B, Delrow J, Groudine M. Genome-wide DNA replication profile for Drosophila melanogaster: a link between transcription and replication timing. Nat. Genet. 2002;32:438–442. doi: 10.1038/ng1005. [DOI] [PubMed] [Google Scholar]
- 3.White EJ, Emanuelsson O, Scalzo D, Royce T, Kosak S, Oakeley EJ, Weissman S, Gerstein M, Groudine M, Snyder M, et al. DNA replication-timing analysis of human chromosome 22 at high resolution and different developmental states. Proc. Natl Acad. Sci. USA. 2004;101:17771–17776. doi: 10.1073/pnas.0408170101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.MacAlpine DM, Rodríguez HK, Bell SP. Coordination of replication and transcription along a Drosophila chromosome. Genes Dev. 2004;18:3094–3105. doi: 10.1101/gad.1246404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woodfine K, Fiegler H, Beare DM, Collins JE, McCann OT, Young BD, Debernardi S, Mott R, Dunham I, Carter NP. Replication timing of the human genome. Hum. Mol. Genet. 2004;13:191–202. doi: 10.1093/hmg/ddh016. [DOI] [PubMed] [Google Scholar]
- 6.Audit B, Nicolay S, Huvet M, Touchon M, d’Aubenton-Carafa Y, Thermes C, Arneodo A. DNA replication timing data corroborate in silico human replication origin predictions. Phys. Rev. Lett. 2007;99:248102. doi: 10.1103/PhysRevLett.99.248102. [DOI] [PubMed] [Google Scholar]
- 7.Huvet M, Nicolay S, Touchon M, Audit B, d’Aubenton-Carafa Y, Arneodo A, Thermes C. Human gene organization driven by the coordination of replication and transcription. Genome Res. 2007;17:1278–1285. doi: 10.1101/gr.6533407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hiratani I, Ryba T, Itoh M, Yokochi T, Schwaiger M, Chang C-W, Lyou Y, Townes TM, Schübeler D, Gilbert DM. Global reorganization of replication domains during embryonic stem cell differentiation. PLoS Biol. 2008;6:e245. doi: 10.1371/journal.pbio.0060245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Farkash-Amar S, Lipson D, Polten A, Goren A, Helmstetter C, Yakhini Z, Simon I. Global organization of replication time zones of the mouse genome. Genome Res. 2008;18:1562–1570. doi: 10.1101/gr.079566.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Desprat R, Thierry-Mieg D, Lailler N, Lajugie J, Schildkraut C, Thierry-Mieg J, Bouhassira EE. Predictable dynamic program of timing of DNA replication in human cells. Genome Res. 2009;19:2288–2299. doi: 10.1101/gr.094060.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hansen RS, Thomas S, Sandstrom R, Canfield TK, Thurman RE, Weaver M, Dorschner MO, Gartler SM, Stamatoyannopoulos JA. Sequencing newly replicated DNA reveals widespread plasticity in human replication timing. Proc. Natl Acad. Sci. USA. 2010;107:139–144. doi: 10.1073/pnas.0912402107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hiratani I, Ryba T, Itoh M, Rathjen J, Kulik M, Papp B, Fussner E, Bazett-Jones DP, Plath K, Dalton S, et al. Genome-wide dynamics of replication timing revealed by in vitro models of mouse embryogenesis. Genome Res. 2010;20:155–169. doi: 10.1101/gr.099796.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yaffe E, Farkash-Amar S, Polten A, Yakhini Z, Tanay A, Simon I. Comparative analysis of DNA replication timing reveals conserved large-scale chromosomal architecture. PLoS Genet. 2010;6:e1001011. doi: 10.1371/journal.pgen.1001011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ryba T, Hiratani I, Lu J, Itoh M, Kulik M, Zhang J, Schulz TC, Robins AJ, Dalton S, Gilbert DM. Evolutionarily conserved replication timing profiles predict long-range chromatin interactions and distinguish closely related cell types. Genome Res. 2010;20:761–770. doi: 10.1101/gr.099655.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen C-L, Rappailles A, Duquenne L, Huvet M, Guilbaud G, Farinelli L, Audit B, d’Aubenton-Carafa Y, Arneodo A, Hyrien O, et al. Impact of replication timing on non-CpG and CpG substitution rates in mammalian genomes. Genome Res. 2010;20:447–457. doi: 10.1101/gr.098947.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen C-L, Duquenne L, Audit B, Guilbaud G, Rappailles A, Baker A, Huvet M, d’Aubenton-Carafa Y, Hyrien O, Arneodo A, et al. Replication-associated mutational asymmetry in the human genome. Mol. Biol. Evol. 2011;28:2327–2337. doi: 10.1093/molbev/msr056. [DOI] [PubMed] [Google Scholar]
- 17.Farkash-Amar S, Simon I. Genome-wide analysis of the replication program in mammals. Chromosome Res. 2010;18:115–125. doi: 10.1007/s10577-009-9091-5. [DOI] [PubMed] [Google Scholar]
- 18.Thurman RE, Day N, Noble WS, Stamatoyannopoulos JA. Identification of higher-order functional domains in the human ENCODE regions. Genome Res. 2007;17:917–927. doi: 10.1101/gr.6081407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Audit B, Zaghloul L, Vaillant C, Chevereau G, d’Aubenton-Carafa Y, Thermes C, Arneodo A. Open chromatin encoded in DNA sequence is the signature of ‘master’ replication origins in human cells. Nucleic Acids Res. 2009;37:6064–6075. doi: 10.1093/nar/gkp631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakayasu H, Berezney R. Mapping replicational sites in the eucaryotic cell nucleus. J. Cell. Biol. 1989;108:1–11. doi: 10.1083/jcb.108.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Keefe RT, Henderson SC, Spector DL. Dynamic organization of DNA replication in mammalian cell nuclei: spatially and temporally defined replication of chromosome-specific alpha-satellite DNA sequences. J. Cell. Biol. 1992;116:1095–1110. doi: 10.1083/jcb.116.5.1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maya-Mendoza A, Olivares-Chauvet P, Shaw A, Jackson DA. S phase progression in human cells is dictated by the genetic continuity of DNA foci. PLoS Genet. 2010;6:e1000900. doi: 10.1371/journal.pgen.1000900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leonhardt H, Rahn HP, Weinzierl P, Sporbert A, Cremer T, Zink D, Cardoso MC. Dynamics of DNA replication factories in living cells. J. Cell. Biol. 2000;149:271–280. doi: 10.1083/jcb.149.2.271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sporbert A, Gahl A, Ankerhold R, Leonhardt H, Cardoso MC. DNA polymerase clamp shows little turnover at established replication sites but sequential de novo assembly at adjacent origin clusters. Mol. Cell. 2002;10:1355–1365. doi: 10.1016/s1097-2765(02)00729-3. [DOI] [PubMed] [Google Scholar]
- 25.Sadoni N, Cardoso MC, Stelzer EHK, Leonhardt H, Zink D. Stable chromosomal units determine the spatial and temporal organization of DNA replication. J. Cell Sci. 2004;117:5353–5365. doi: 10.1242/jcs.01412. [DOI] [PubMed] [Google Scholar]
- 26.Zink D, Cremer T, Saffrich R, Fischer R, Trendelenburg MF, Ansorge W, Stelzer EH. Structure and dynamics of human interphase chromosome territories in vivo. Hum. Genet. 1998;102:241–251. doi: 10.1007/s004390050686. [DOI] [PubMed] [Google Scholar]
- 27.Ma H, Samarabandu J, Devdhar RS, Acharya R, Cheng PC, Meng C, Berezney R. Spatial and temporal dynamics of DNA replication sites in mammalian cells. J. Cell. Biol. 1998;143:1415–1425. doi: 10.1083/jcb.143.6.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zink D, Bornfleth H, Visser A, Cremer C, Cremer T. Organization of early and late replicating DNA in human chromosome territories. Exp. Cell Res. 1999;247:176–188. doi: 10.1006/excr.1998.4311. [DOI] [PubMed] [Google Scholar]
- 29.Grasser F, Neusser M, Fiegler H, Thormeyer T, Cremer M, Carter NP, Cremer T, Müller S. Replication-timing-correlated spatial chromatin arrangements in cancer and in primate interphase nuclei. J. Cell Sci. 2008;121:1876–1886. doi: 10.1242/jcs.026989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jackson DA, Pombo A. Replicon clusters are stable units of chromosome structure: evidence that nuclear organization contributes to the efficient activation and propagation of S phase in human cells. J. Cell. Biol. 1998;140:1285–1295. doi: 10.1083/jcb.140.6.1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cook PR. The organization of replication and transcription. Science. 1999;284:1790–1795. doi: 10.1126/science.284.5421.1790. [DOI] [PubMed] [Google Scholar]
- 32.Berezney R. Regulating the mammalian genome: the role of nuclear architecture. Adv. Enzyme Regul. 2002;42:39–52. doi: 10.1016/s0065-2571(01)00041-3. [DOI] [PubMed] [Google Scholar]
- 33.Koberna K, Ligasová A, Malínský J, Pliss A, Siegel AJ, Cvacková Z, Fidlerová H, Masata M, Fialová M, Raska I, et al. Electron microscopy of DNA replication in 3-D: evidence for similar-sized replication foci throughout S-phase. J. Cell. Biochem. 2005;94:126–138. doi: 10.1002/jcb.20300. [DOI] [PubMed] [Google Scholar]
- 34.Anachkova B, Djeliova V, Russev G. Nuclear matrix support of DNA replication. J. Cell. Biochem. 2005;96:951–961. doi: 10.1002/jcb.20610. [DOI] [PubMed] [Google Scholar]
- 35.Ottaviani D, Lever E, Takousis P, Sheer D. Anchoring the genome. Genome Biol. 2008;9:201. doi: 10.1186/gb-2008-9-1-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arneodo A, Vaillant C, Audit B, Argoul F, d’Aubenton Carafa Y, Thermes C. Multi-scale coding of genomic information: From DNA sequence to genome structure and function. Phys. Rep. 2011;498:45–188. [Google Scholar]
- 37.Courbet S, Gay S, Arnoult N, Wronka G, Anglana M, Brison O, Debatisse M. Replication fork movement sets chromatin loop size and origin choice in mammalian cells. Nature. 2008;455:557–560. doi: 10.1038/nature07233. [DOI] [PubMed] [Google Scholar]
- 38.Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 39.de Wit E, de Laat W. A decade of 3C technologies: insights into nuclear organization. Genes Dev. 2012;26:11–24. doi: 10.1101/gad.179804.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Simonis M, Klous P, Splinter E, Moshkin Y, Willemsen R, de Wit E, van Steensel B, de Laat W. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C) Nat. Genet. 2006;38:1348–1354. doi: 10.1038/ng1896. [DOI] [PubMed] [Google Scholar]
- 41.Zhao Z, Tavoosidana G, Sjölinder M, Göndör A, Mariano P, Wang S, Kanduri C, Lezcano M, Sandhu KS, Singh U, et al. Circular chromosome conformation capture (4C) uncovers extensive networks of epigenetically regulated intra- and interchromosomal interactions. Nat. Genet. 2006;38:1341–1347. doi: 10.1038/ng1891. [DOI] [PubMed] [Google Scholar]
- 42.Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Delves PJ, Martin SJ, Burton DR, Roitt IM. Roitt’s Essential Immunology. 11th edn. New york: Wiley-Blackwell; 2006. [Google Scholar]
- 44.Splinter E, de Wit E, Nora EP, Klous P, van de Werken HJG, Zhu Y, Kaaij LJT, van Ijcken W, Gribnau J, Heard E, et al. The inactive X chromosome adopts a unique three-dimensional conformation that is dependent on Xist RNA. Genes Dev. 2011;25:1371–1383. doi: 10.1101/gad.633311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Noordermeer D, Leleu M, Splinter E, Rougemont J, Laat WD, Duboule D. The dynamic architecture of Hox gene clusters. Science. 2011;334:222–225. doi: 10.1126/science.1207194. [DOI] [PubMed] [Google Scholar]
- 46.Jiang H, Wong WH. SeqMap: mapping massive amount of oligonucleotides to the genome. Bioinformatics. 2008;24:2395–2396. doi: 10.1093/bioinformatics/btn429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yokota H, van den Engh G, Hearst JE, Sachs RK, Trask BJ. Evidence for the organization of chromatin in megabase pair-sized loops arranged along a random walk path in the human G0/G1 interphase nucleus. J. Cell. Biol. 1995;130:1239–1249. doi: 10.1083/jcb.130.6.1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baker A, Audit B, Chen C-L, Moindrot B, Leleu A, Guilbaud G, Rappailles A, Vaillant C, Goldar A, Mongelard F, et al. Replication fork polarity gradients revealed by megabase-sized U-shaped replication timing domains in human cell lines. PLoS Comput. Biol. 2012;8:e1002443. doi: 10.1371/journal.pcbi.1002443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ryba T, Battaglia D, Chang BH, Shirley JW, Buckley Q, Pope BD, Devidas M, Druker BJ, Gilbert DM. Abnormal developmental control of replication timing domains in pediatric acute lymphoblastic leukemia. Genome Res. 2012;22:1833–1844. doi: 10.1101/gr.138511.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Simonis M, Kooren J, de Laat W. An evaluation of 3C-based methods to capture DNA interactions. Nat. Methods. 2007;4:895–901. doi: 10.1038/nmeth1114. [DOI] [PubMed] [Google Scholar]
- 51.Méchali M. Eukaryotic DNA replication origins: many choices for appropriate answers. Nat. Rev. Mol. Cell. Biol. 2010;11:728–738. doi: 10.1038/nrm2976. [DOI] [PubMed] [Google Scholar]
- 52.Brodie of Brodie EB, Nicolay S, Touchon M, Audit B, d’Aubenton-Carafa Y, Thermes C, Arneodo A. From DNA sequence analysis to modeling replication in the human genome. Phys. Rev. Lett. 2005;94:248103. doi: 10.1103/PhysRevLett.94.248103. [DOI] [PubMed] [Google Scholar]
- 53.Touchon M, Nicolay S, Audit B, Brodie of Brodie E-B, d’Aubenton-Carafa Y, Arneodo A, Thermes C. Replication-associated strand asymmetries in mammalian genomes: toward detection of replication origins. Proc. Natl Acad. Sci. USA. 2005;102:9836–9841. doi: 10.1073/pnas.0500577102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gilbert DM, Takebayashi S-I, Ryba T, Lu J, Pope BD, Wilson KA, Hiratani I. Space and time in the nucleus: developmental control of replication timing and chromosome architecture. Cold Spring Harb. Symp. Quant. Biol. 2010;75:143–153. doi: 10.1101/sqb.2010.75.011. [DOI] [PubMed] [Google Scholar]
- 55.Cuddapah S, Jothi R, Schones DE, Roh T-Y, Cui K, Zhao K. Global analysis of the insulator binding protein CTCF in chromatin barrier regions reveals demarcation of active and repressive domains. Genome Res. 2009;19:24–32. doi: 10.1101/gr.082800.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Handoko L, Xu H, Li G, Ngan CY, Chew E, Schnapp M, Lee CWH, Ye C, Ping JLH, Mulawadi F, et al. CTCF-mediated functional chromatin interactome in pluripotent cells. Nat. Genet. 2011;43:630–638. doi: 10.1038/ng.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sexton T, Yaffe E, Kenigsberg E, Bantignies F, Leblanc B, Hoichman M, Parrinello H, Tanay A, Cavalli G. Three-dimensional folding and functional organization principles of the Drosophila genome. Cell. 2012;148:458–472. doi: 10.1016/j.cell.2012.01.010. [DOI] [PubMed] [Google Scholar]
- 58.Malyavantham KS, Bhattacharya S, Alonso WD, Acharya R, Berezney R. Spatio-temporal dynamics of replication and transcription sites in the mammalian cell nucleus. Chromosoma. 2008;117:553–567. doi: 10.1007/s00412-008-0172-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bridger JM, Boyle S, Kill IR, Bickmore WA. Re-modelling of nuclear architecture in quiescent and senescent human fibroblasts. Curr. Biol. 2000;10:149–152. doi: 10.1016/s0960-9822(00)00312-2. [DOI] [PubMed] [Google Scholar]
- 60.Mehta IS, Amira M, Harvey AJ, Bridger JM. Rapid chromosome territory relocation by nuclear motor activity in response to serum removal in primary human fibroblasts. Genome Biol. 2010;11:R5. doi: 10.1186/gb-2010-11-1-r5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lu J, Li F, Murphy CS, Davidson MW, Gilbert DM. G2 phase chromatin lacks determinants of replication timing. J. Cell. Biol. 2010;189:967–980. doi: 10.1083/jcb.201002002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stentz FB, Kitabchi AE. Transcriptome and proteome expression in activated human CD4 and CD8 T-lymphocytes. Biochem. Biophys. Res. Commun. 2004;324:692–696. doi: 10.1016/j.bbrc.2004.09.113. [DOI] [PubMed] [Google Scholar]
- 63.Argyropoulos C, Nikiforidis GC, Theodoropoulou M, Adamopoulos P, Boubali S, Georgakopoulos TN, Paliogianni F, Papavassiliou AG, Mouzaki A. Mining microarray data to identify transcription factors expressed in naïve resting but not activated T lymphocytes. Genes Immun. 2004;5:16–25. doi: 10.1038/sj.gene.6364034. [DOI] [PubMed] [Google Scholar]
- 64.Hess K, Yang Y, Golech S, Sharov A, Becker KG, Weng N-P. Kinetic assessment of general gene expression changes during human naive CD4 + T cell activation. Int. Immunol. 2004;16:1711–1721. doi: 10.1093/intimm/dxh172. [DOI] [PubMed] [Google Scholar]
- 65.Chtanova T, Newton R, Liu SM, Weininger L, Young TR, Silva DG, Bertoni F, Rinaldi A, Chappaz S, Sallusto F, et al. Identification of T cell-restricted genes, and signatures for different T cell responses, using a comprehensive collection of microarray datasets. J. Immunol. 2005;175:7837–7847. doi: 10.4049/jimmunol.175.12.7837. [DOI] [PubMed] [Google Scholar]
- 66.Grigoryev YA, Kurian SM, Nakorchevskiy AA, Burke JP, Campbell D, Head SR, Deng J, Kantor AB, Yates JR, Salomon DR. Genome-wide analysis of immune activation in human T and B cells reveals distinct classes of alternatively spliced genes. PLoS One. 2009;4:e7906. doi: 10.1371/journal.pone.0007906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Branco MR, Branco T, Ramirez F, Pombo A. Changes in chromosome organization during PHA-activation of resting human lymphocytes measured by cryo-FISH. Chromosome Res. 2008;16:413–426. doi: 10.1007/s10577-008-1230-x. [DOI] [PubMed] [Google Scholar]
- 68.Kyselá K, Philimonenko AA, Philimonenko VV, Janácek J, Kahle M, Hozák P. Nuclear distribution of actin and myosin I depends on transcriptional activity of the cell. Histochem Cell Biol. 2005;124:347–358. doi: 10.1007/s00418-005-0042-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.