


Abstract
Adenosine deaminases acting on RNA (ADAR1 and ADAR2) are human RNA-editing adenosine deaminases responsible for the conversion of adenosine to inosine at specific locations in cellular RNAs. Since inosine is recognized during translation as guanosine, this often results in the expression of protein sequences different from those encoded in the genome. While our knowledge of the ADAR2 structure and catalytic mechanism has grown over the years, our knowledge of ADAR1 has lagged. This is due, at least in part, to the lack of well defined, small RNA substrates useful for mechanistic studies of ADAR1. Here, we describe an ADAR1 substrate RNA that can be prepared by a combination of chemical synthesis and enzymatic ligation. Incorporation of adenosine analogs into this RNA and analysis of the rate of ADAR1 catalyzed deamination revealed similarities and differences in the way the ADARs recognize the edited nucleotide. Importantly, ADAR1 is more dependent than ADAR2 on the presence of N7 in the edited base. This difference between ADAR1 and ADAR2 appears to be dependent on the identity of a single amino acid residue near the active site. Thus, this work provides an important starting point in defining mechanistic differences between two functionally distinct human RNA editing ADARs.
INTRODUCTION
In recent years, RNA modification processes have become recognized as key to proper cellular function, and dysregulated RNA modification has been shown to lead to human disease. For instance, alternative splicing has been implicated in various diseases such as myotonic dystrophy (1), aberrant transfer RNA modification is associated with two major classes of mitochondrial disease (2), and a lack of certain types of ribosomal RNA modification results in dyskeratosis congenita (3). Another type of post-transcriptional modification is adenosine deamination catalyzed by the ADAR family of enzymes (adenosine deaminases acting on RNA). The ADAR family consists of three enzymes, two with known activity (ADAR1 and ADAR2). These enzymes deaminate adenosine to form inosine, a type of RNA editing. Inosine base pairs with cytosine and is recognized during translation as guanosine, often resulting in codon changes. Aberrant editing has also been correlated with a number of human diseases [e.g. amyotrophic lateral sclerosis, depression, bipolar disorder, dyschromatosis symmetric hereditaria (DSH), Prader-Willi syndrome, cancer, etc. (4–21)].
ADAR1 and ADAR2 have many similarities in terms of their domain structures, catalytic activities and substrate requirements (22–24). However, these two RNA editing adenosine deaminases have distinct biological properties as indicated by their different cellular localization (25–30), the different ways they are regulated (25,31–35) and the different phenotypes displayed by the corresponding knockout mice (36–40). For instance, ADAR1−/− embryos do not survive beyond 12 days post coitus and display a severe defect in hematopoiesis (36–38). On the other hand, ADAR2−/− mice live for 3 weeks after birth and mainly show defects in nervous system function arising from the lack of editing of the glutamate receptor B subunit Q/R site (40). While the RNA-editing substrate(s) responsible for the ADAR1 knockout embryonic lethality is/are unknown at this time, other studies have established the essential function ADAR1 plays in the survival of certain cell types, a function that is not shared with ADAR2 (36–39). Furthermore, the human skin pigmentation disorder DSH is caused by mutations in the adar1 gene and does not appear to involve ADAR2 [see Li et al. (41)]. Finally, ADAR1 has been linked in several different studies to the innate immune response, with both antiviral and proviral roles [see review by Samuel (42)].
It is clear that a full understanding of RNA editing by adenosine deamination requires detailed study of both ADAR1 and ADAR2. Unfortunately, while our knowledge of the ADAR2 structure and catalytic mechanism has grown over the years, our knowledge of ADAR1 has lagged. For many years, both proteins defied attempts at crystallization. Then, in 2005, Macbeth et al. (43) crystallized the catalytic domain of human ADAR2, revealing the placement of various residues within the deaminase active site. No crystal structures have yet been reported for ADAR–RNA complexes. However, prior to the report of the ADAR2 deaminase domain structure, and indeed afterward to complement it, our lab used RNA substrates bearing nucleoside analogs to define structure/activity relationships for the ADAR2 reaction (44–52). These experiments identified key differences between ADAR2 and the nucleoside deaminase ADA (adenosine deaminase) (45), led to a method for mechanism-based trapping of ADAR2 bound to a substrate RNA (49) and allowed us to validate a model for adenosine recognition in the ADAR2 active site (52). In the latter example, docking adenosine monophosphate (AMP) into the ADAR2 active site as determined by X-ray crystallography led to a model for recognition of the edited nucleotide wherein R455 and T375 were in proximity to the adenosine’s N7 and 2′-positions, respectively (Figure 1). Kinetic data generated using RNA substrates bearing adenosine modifications at these positions, along with mutants of ADAR2 at R455 and T375, supported the proposed model (51,52).
Figure 1.

Models of edited nucleotide binding by ADARs. (A) Sequence alignment of ADAR1 and ADAR2 near T375 and R455 of ADAR2. Conserved residues are shown in red, T375 in ADAR2 and N891 in ADAR1 are shown in blue, R455 in ADAR2 and A970 in ADAR1 are shown in purple. (B) Positions of T375 and R455 in the crystal structure of the ADAR2 deaminase domain. AMP has been docked into the structure (43). (C) ADAR1 homology model generated by Phyre2 (protein homology/analogy recognition engine) (56). AMP was docked based upon positioning in the ADAR2 model.
Interestingly, ADAR1 differs from ADAR2 in the identity of these active site residues, suggesting possible differences in the mode of interaction with the edited nucleotide. Based on the sequence alignment, ADAR1 is predicted to have N891 in the position of ADAR2’s T375, and A970 in place of R455 (53) (Figure 1). These differences may indicate a distinct method of recognition of either the sugar (in the case of the Thr/Asn difference) or the base (in the case of the Arg/Ala difference). Testing this idea would require adaptation of our ADAR2 assay to allow site-specific incorporation of nucleoside analogs at a known ADAR1 site such that reaction rates could be determined for adenosine and variants bearing structural changes at the 7- and 2′-positions.
Until now, this type of analysis has not been possible with ADAR1. To allow for efficient site-specific incorporation of nucleoside analogs, the RNA substrate must be amenable to synthesis in two pieces, preferably with the editing site at the 5′-end of one strand short enough to be accessible by chemical synthesis (approximately <50 nt). All previously known ADAR1 sites were too long to make this possible. However, we recently characterized an ADAR1 catalyzed editing event in the pre-mRNA of the DNA repair enzyme NEIL1 (54). The size and stability of the hairpin structure found in the NEIL1 pre-mRNA that supported the ADAR1 reaction suggested it might be a good candidate for incorporation of nucleoside analogs. Here, we describe an 84 nt ADAR1 substrate RNA derived from the NEIL1 message that can be prepared by a combination of chemical synthesis and enzymatic ligation. Incorporation of adenosine analogs into this RNA and subsequent analysis of the rate of ADAR1 catalyzed deamination revealed both similarities and differences in the way the two ADARs recognize the edited nucleotide. Importantly, ADAR1 is more dependent than ADAR2 on the presence of N7 in the edited base such that no product is observed in ADAR1 reactions with 7-deazaadenosine at editing sites. This difference between ADAR1 and ADAR2 appears to be dependent on the identity of a single amino acid residue near the deaminase active site. Thus, this work has provided an important starting point in the process of defining mechanistic differences between two functionally distinct human RNA editing ADARs.
MATERIALS AND METHODS
General biochemical procedures
Unless otherwise stated, all reagents were purchased from commercial sources (Sigma Aldrich or Fisher Scientific) and were used without purification. Reagents for in vitro transcription, in vitro editing, PCR amplification, radiolabeling and preparation of RNAs for splinted ligation were purchased from: Perkin-Elmer Life Sciences: γ-[32P]ATP (6000 Ci/mmol); GE Healthcare: MicroSpin G-25 columns; Promega: RQ1 RNase free DNase, yeast tRNAPhe, RNasin, Access RT-PCR kit, ribonucleotides; New England Biolabs: T4 polynucleotide kinase, T4 DNA ligase, BamHI, acyclonucleotides, deoxynucleotides, VentR® exo(−) DNA Polymerase, RNase Inhibitor, SacI, XbaI, Quick Ligase Kit, DNase I; Sigma Aldrich: Nuclease P1, glycerol and phenol:chloroform; Life Technologies: DNA oligonucleotides; University of Utah DNA/Core Peptide Facility: RNA oligonucleotides; Axxora: 7-deazaadenosine 5′-O-triphosphate; Glen Research: 2′-O-methyladenosine, 8-aza-7-deazaadenosine and 7-deazaadenosine ribonucleoside phosphoramidites; Qiagen: Gel Extraction Kit, PCR Purification Kit. Radioactive gels and TLC plates were imaged using storage phosphor imaging plates from Molecular Dynamics on a Molecular Dynamics 9400 Typhoon phosphorimager. Gels from fluorescent poisoned primer extension were imaged on the same instrument. Data were analyzed using Molecular Dynamics ImageQuant 5.2 software. 4Peaks (Aalsmeer, The Netherlands) and ImageJ software (NIH, Bethesda, MD, USA) (55) were used for quantification of editing using DNA sequencing.
Generation of an ADAR1 homology model
The ADAR1 homology model was generated using Protein Homology/analogy Recognition Engine v2.0 (Phyre2) (56). AMP was added based upon the ADAR2 structural modeling. The model produced by Phyre2 was used as generated with minor adjustments to the N891 side chain conformation to avoid steric clash with the ribose of AMP.
Protein overexpression and purification
For studies on RNA length, human ADAR1 in yeast expression plasmid (YEpTOP2PGAL1) was overexpressed in Saccharomyces cerevisiae and purified as previously described with one modification (57,58). Cells were lysed using a mini bead beater (Biospec Products).
For characterization of the 84 nt RNA made by in vitro transcription, human ADAR1 in yeast expression plasmid (YEpTOP2PGAL1) was overexpressed in S. cerevisiae and purified as previously described (57,58).
For nucleoside analog experiments (both deamination assays and gel shifts), human ADAR1 in yeast expression plasmid (YEpTOP2PGAL1) was overexpressed in S. cerevisiae and purified as previously described (59) with some modifications. The treatment with TEV and the second Ni-NTA column were eliminated, so that the fractions from the first Ni-NTA column were concentrated and dialyzed. For this set of experiments, yeast cells were lysed using a mini bead beater (Biospec Products).
Human ADAR2 and the R455A mutant in yeast expression plasmid (YEpTOP2PGAL1) were overexpressed in S. cerevisiae and purified as previously described (49,57,58).
Synthesis, purification and mass spectrometric analysis of RNA
RNA oligonucleotides were synthesized as described previously, although on a 200 nmol scale (52). The analog-containing RNAs were purified for mass spectrometric analysis by 15% denaturing polyacrylamide gel electrophoresis, visualized by UV shadowing (254 nm light, F254 TLC plate as backing) and extracted from the gel via the crush and soak method at 4°C overnight into 0.5 M NH4OAc containing 0.1 mM EDTA. Polyacrylamide particles were removed using a Centrex filter (0.2 mm) and the solution was desalted using C18 Sep-Pak cartridges. RNA was eluted in 1:1 CH3CN/H2O. The RNA was lyophilized to dryness, resuspended in H2O and quantified by measuring the absorbance at 260 nm. The identities of the analog-containing RNAs were checked by ESI mass spectrometry. Mass spectra were acquired on an LTQ Orbitrap XL mass spectrometer equipped with an electrospray ionization source (ThermoFisher, San Jose, CA, USA), operating in the negative ion mode. Samples were introduced into the source via loop injection at a flow rate of 40 µl/min, in a solvent system of 1:1 methanol: (50 mM hexafluroisopropanol in water, pH adjusted to 7.9 with triethylamine). Mass spectra were acquired using Xcalibur, version 2.0.7 SP1 (ThermoFinnigan), in the range of 600–1800 m/z at a resolution of 30 000 and are an average of 25 scans. The spectra were externally calibrated using the standard calibration mixture. MassLynx software, version 4.1 (Waters, Milford, MA, USA) was used for spectrum processing. ESI mass spectrometry analysis of the RNA containing 2′-O-methyladenosine: calcd: 11 183.6; obsd: 11 183.7. ESI mass spectrometry analysis of RNA containing 2′-deoxyadenosine (dA): calcd: 11 153.6; obsd: 11 153.6. ESI mass spectrometry analysis of RNA containing 8-aza-7-deazaadenosine: calcd: 11 169.6; obsd: 11 169.6. ESI mass spectrometry analysis of the NEIL1 RNA containing 7-deazaadenosine: calcd: 11 168.6; obsd: 11 168.7. ESI mass spectrometry analysis of the GluRB R/G site RNA containing 7-deazaadenosine: calcd: 8451.2; obsd: 8451.2.
The 366, 201, 161 and 84 nt substrates used in the kinetics and truncation studies were prepared by in vitro transcription. The actual length of the 201 nt RNA was 239 nt [201 nt of NEIL1 sequence, plus 38 nt from the plasmid used for in vitro transcription (54)], the 366 nt was 367 nt [366 nt of NEIL1 sequence (chromosome 15: 75 645 905–75 646 270, hg19), plus 5′-G from the T7 promoter], the 161 nt was 162 nt [161 nt of NEIL1 sequence (chromosome 15: 75 645 995–75 646 155, hg19)], plus 5′-G from the T7 promoter] and the 84 nt was 85 nt [84 nt of NEIL1 sequence (chromosome 15: 75 646 036–75 646 119, hg19), plus 5′-G from the T7 promoter]. The 201 nt RNA was prepared as described previously except the product was purified on an 8% denaturing polyacrylamide gel (54). A plasmid was prepared for in vitro transcription of the 84 nt RNA by amplifying a fragment with the primers 84F (GTGTGTGAGCTCTAATACGACTCACTATAGGGCCTGAGCCTGCCCTCT) and 84R (ACACACTCTAGAAGTCCTCCTCCCCGC), digesting both PCR product and pUC19 vector with SacI and XbaI and ligating using the Quick Ligase kit. The plasmid was linearized with XbaI for in vitro transcription. The 366 and 161 nt RNAs were made by in vitro transcription using a PCR product as the template, under conditions described previously (60). The primers were: 366F (TAATACGACTCACTATAGGGCATGGCCGAGTGGGAAGA) and 366R (GACCCCACGTTTCCACCC); and 161F (TAATACGACTCACTATAGGGAGGTCTGGGCCAGGTCTAAC) and 161R (ATGCCATAGCAGCGCA). PCR products were purified by agarose gel electrophoresis. After in vitro transcription from a PCR product, the solution was phenol–chloroform extracted, ethanol precipitated and run through a G-25 column. The sample was DNase digested twice using DNase1. The products were purified by denaturing polyacrylamide gel electrophoresis. Bands were visualized by UV shadowing (254 nm light, F254 TLC plate as backing), and extracted using the crush and soak method overnight at 4°C in 0.5 M NH4OAc, 0.1% SDS and 0.1 mM EDTA. Polyacrylamide particles were removed using a Centrex filter (0.2 mm) and the sample was phenol–chloroform extracted and ethanol precipitated. The sample was lyophilized to dryness and resuspended in 1× TE and 50 mM NaCl. Concentration of RNA was determined by absorbance measurement at 260 nm and then diluted to 180 nM in the same buffer. RNA was refolded by heating at 95°C for 5 min and then slowly cooling to room temperature.
The 45 nt RNA (chromosome 15: 75 646 060–75 646 104, hg19) was chemically synthesized. The RNA was purified, visualized, extracted and prepared for assay as described for the in vitro transcribed RNAs.
A template for in vitro transcription of the 5HT2CR RNA was obtained by BamHI digestion of plasmid SerLIVT as described previously (60). For in vitro transcription of the 329 nt 5HT2CR (no loop) RNA, conditions used were similar to those previously described (60), except that 7-deazaadenosine 5′-O-triphosphate was substituted for adenosine 5′-O-triphosphate in one of the transcription reactions. 7-deazaadenosine 5′-O-triphosphate was concentrated by lyophilization from 10 to 100 mM upon receipt for in vitro transcription.
Preparation of NEIL1 RNA by splinted ligation
The sequences of the RNA oligonucleotides used for splinted ligation were: 5′-AAGGCUACGGGUCAGAGAGCGGGGAGGAGGACUU (called NL34) and 5′-CCUGAGCCUGCCCUCUGAUCUCUGCCUGUUCCUCUGUCCCACAGGGGGCA (called NL845). The analogs were incorporated in place of the adenosine at the 5′-end of NL34. The sequence of the DNA splint was: 5′-GACCCGTAGCCTTTGCCCCCTGTGG (called NL84SP25). For use in the deamination assay, RNA was purified, visualized, extracted and prepared for assay as described for the in vitro transcribed RNAs. RNA concentration was determined by measuring absorbance at 260 nm and calculated using extinction coefficients generated by the Ambion extinction coefficient calculator. The same extinction coefficient was used for A- and analog-containing RNAs. For the preparation of 5′-[32P] end labeled RNA, purified RNA (60 pmol) was treated with γ-[32P]ATP (3 mCi/ml) and T4 polynucleotide kinase (2 U) and incubated at 37°C for 1 h. Unreacted γ-[32P]ATP was removed with a size exclusion G-25 column. Labeled RNA was PhOH:CHCl3 extracted, ethanol precipitated and pelleted via centrifugation.
Splinted ligation was performed by hybridizing labeled NL34, 1 equivalent cold NL845 and 1 equivalent cold NL84SP25 in 1.3× T4 DNA Ligase buffer with 0.04 U/ml RNasin. Sample was heated at 95°C for 5 min and then allowed to slowly cool to room temperature. After cooling, RNasin, rATP and T4 DNA Ligase were added to the mixture to a final concentration of 0.05 U/ml RNasin, 60 mM rATP, 85 U/µl T4 DNA Ligase and 1× T4 DNA Ligase buffer. This was incubated at 30°C for 6 h. After ligation the RNA was again PhOH:CHCl3 extracted, ethanol precipitated, pelleted via centrifugation and then washed with 70% ethanol and pelleted again. The pelleted RNA was dried by lyophilization and re-suspended in water. A DNase digest was performed at 37°C for 1 h and the ligated RNA was purified by running on a 12% polyacrylamide gel. RNA was cut from the gel and isolated using the crush and soak method described above. The isolated oligonucleotides were PhOH:CHCl3 extracted, ethanol precipitated, pelleted via centrifugation and then washed with 70% ethanol and pelleted again. The pelleted RNA was dried by lyophilization and re-suspended in water to a concentration ≤300 nM. The RNA was refolded by heating at 95°C for 5 min and allowing the RNA to slowly cool to room temperature.
Deamination assay
Editing for the NEIL1 deletion study was evaluated as previously with some modifications (54). ADAR1 (50 nM) was mixed with 10 nM RNA in assay buffer containing 15 mM Tris–HCl, pH 7.5, 1.5 mM EDTA, 40 mM KCl, 26 mM NaCl, 5% glycerol, 0.003% Nonidet P-40, 0.5 mM DTT, 160 U/ml RNasin, 0.3 mM BME and 1.0 µg/ml yeast tRNAPhe. Reactions were carried out for 2 h. RT–PCR was carried out using the following primers: for the 366 nt: 366F and 366R; for the 201 nt: RFSL (TGGGTACGAATTCCCCGTACAAGCTT) and 201R (GCGCGCGGATCCATGCCGGTCCTGCAGG); for the 161 nt: 161F and 161R, for the 84 nt: 84FRT (TAATACGACTCACTATAGGGAACTGACTAAACTAGGTGCCACGTCGTGAAAGTCTGACAACCTGAGCCTGCCCTCTGA) and 84R; for the 45 nt: 45F (TAATACGACTCACTATAGGGCCTGTTCCTCTGTCCCA) and 45R (CTCTCTGACCCGTAGCCT) and the Access RT–PCR System (Promega, Madison, WI, USA) according to the manufacturer’s protocol. The extent of editing was determined by DNA sequencing using either a T7 promoter primer (366, 161 and 84 nt) or RFSL (201 nt). Editing was quantified by DNA sequencing for all the RNAs except the 45 nt RNA, for which editing was quantified using the fluorescent poisoned primer extension assay (described below).
Kinetic analysis of the in vitro transcribed 84 nt substrate was evaluated as described for the truncations above. ADAR1 (13, 25, 50, 100 and 200 nM final concentrations) was mixed with 10 nM RNA in assay buffer (see above). After the RT–PCR (described above, except that the forward primer used for the 84 nt RNA was 84F instead of 84FRT), the poisoned primer extension was carried out based on previous work (61) using acycloguanosine triphosphate. The PCR products were phenol–chloroform extracted, ethanol precipitated and pelleted via centrifugation. Samples were resuspended into loading buffer and run on a 15% denaturing polyacrylamide gel. Data were fitted to the equation: [P]t= α[1 − exp(−kobs•t)], where [P]t is the percent edited at time t, α is the fitted reaction endpoint and kobs is the fitted rate constant using KaleidaGraph. Each experiment was carried out in triplicate. For enzyme saturation, kobs was measured as a function of [ADAR1]. The values of apparent Kd and kmax were obtained by fitting the data to the equation: kobs= kmax[ADAR1]/(Kd + [ADAR1]).
Editing of the ligated RNAs was evaluated as previously with some modifications (62). ADAR1 (130 nM final concentration) was mixed with ≤18 nM RNA in assay buffer containing 15 mM Tris–HCl, pH 7.0, 1.5 mM EDTA, 60 mM KCl, 3% glycerol, 0.003% Nonidet P-40, 0.5 mM DTT, 160 U/ml RNasin and 1.0 µg/ml yeast tRNAPhe. Due to the slow rates of deamination of some of the analog-containing RNAs, long reaction times made enzyme denaturation a concern and raised the possibility of artificially low endpoints. Therefore, we chose to analyze the initial, linear portion of the reaction. Reaction rates were obtained by taking the slope of the plot of ln(fraction of substrate) versus time (63). Representative TLC images are shown in the supplementary information (Supplementary Figures S2–S6).
GluRB R/G site RNAs (top strand: 5′–7-deazaAGGUGGGUGGAAUAGUAUAACAAUGU and complementary strand: 5′-AUGUUGUUAUAGUAUCCCACCUACCCU) were purified as described above for mass spectrometric analysis. Editing of the GluRB R/G site RNA was evaluated as previously described (52). A representative TLC image is shown in the Supplementary Data (Supplementary Figure S7). The rate constant for the reaction of ADAR2 R455A with the GluR B R/G site RNA containing 7-deazaadenosine (Table 2) was determined as previously described fitting the rate data using a reaction end point of 80% (as observed with the A-containing substrate) (52).
Table 2.
Single-turnover kinetic parameters for the deamination of adenosine and 7-deazaadenosine by ADAR2 and ADAR2 R455Aa
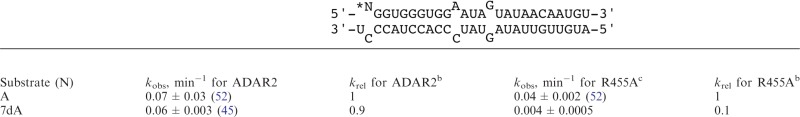 |
N = A or 7-deazaA.
aADAR2 reactions were carried out as described previously (52).
bkrel = kobs for analog/kobs for adenosine.
cData for 7-deazaA were fitted to the equation: [P]t = 0.8[1 − exp(−kobs·t)].
Editing of the transcribed 5HT2CR RNA was evaluated as previously with some modifications (60). ADAR1, ADAR2 or ADAR2 R455A (130 nM final concentration) was mixed with 5 nM RNA in assay buffer containing 15 mM Tris–HCl (pH 7.8 for ADAR1 and pH 7.4 for ADAR2 and ADAR2 R455A), 26 mM KCl, 40 mM potassium glutamate, 1.5 mM EDTA, 0.003% Nonidet P-40, 4% glycerol, 0.6 mM reducing agent (combination of DTT and β-mercaptoethanol), 160 U/ml RNasin and 1.0 µg/ml yeast tRNAPhe. For the reactions with ADAR2 and ADAR2 R455A, the reducing agent was a combination of 0.1 mM β-mercaptoethanol and 0.5 mM DTT. For the reactions with ADAR1, the reducing agent was 0.6 mM DTT. The editing reaction proceeded at 30°C for 2 h. Representative sequencing traces may be found in the Supplementary Data (Supplementary Figure S8).
RESULTS
Generation and characterization of a NEIL1 minimal substrate
In our previous work, we chose to study a section of the NEIL1 pre-mRNA consisting of 100 nt to either side of the recoding site (54). This choice was guided by the predicted secondary structure surrounding the editing site, with additional sequence included to account for other possible structures. To explore further the effect of changing the length of this substrate, we assessed editing of four new RNAs, in addition to the 201 nt substrate analyzed previously: 366, 161, 84 and 45 nt.
Each of the RNAs beyond 84 nt in length supported the ADAR1 reaction with yields of 72–98% under these conditions (Figure 2). A slight decrease in yield was observed for the 45 nt substrate (64 ± 6%) and the best substrate was the 161 nt RNA, which was nearly quantitatively edited by ADAR1 (98 ± 1%). We did not observe an increase in editing at any other adenosines for any of the RNAs. The 84 nt RNA supported more efficient editing than the 45 nt substrate (79% versus 64%) and had the editing site 34 nt from the 3′-end such that it could be prepared by ligation with at least one of the two strands chemically synthesized. Therefore, we chose this substrate for our subsequent studies.
Figure 2.

RNAs of several different lengths were tested in a deamination assay with ADAR1. (A) Relative lengths of RNAs and location within the full NEIL1 pre-mRNA. (B) Editing of the recoding site in RNAs from (A) after 2 h deamination reaction.
We developed a poisoned primer extension assay to monitor ADAR1 editing of the NEIL1 84 nt RNA based upon previous work with seagrass and squid editing sites (61). We used this assay to measure rate constants (kobs) for editing reactions carried out under single-turnover conditions at various concentrations of ADAR1. This allowed us to estimate a maximum rate constant (kmax), apparent affinity constant (Kd) and saturating concentration of ADAR1. We calculated a kmax = 0.03/min and an apparent Kd = 23 nM for this RNA under these conditions and using this method (Figure 3). This analysis also indicated that the RNA was fully saturated with ADAR1 and reached the kmax at concentrations ≥100 nM ADAR1.
Figure 3.

(A) Plot of kobs as a function of ADAR1 concentration for the 84 nt RNA. Editing reaction carried out in 15 mM Tris–HCl, pH 7.5, 1.5 mM EDTA, 40 mM KCl, 26 mM NaCl, 5% glycerol, 0.003% Nonidet P-40, 0.5 mM DTT, 160 U/ml RNasin, 0.3 mM BME and 1.0 µg/ml yeast tRNAPhe. All data points reported are the average ± standard deviation for three experiments. (B) Duplex RNA substrates of ADAR1 used in this study. In the assays with analogs, N represents the site specifically labeled nucleotide, either A or one of the analogs listed in the table (C). The asterisks indicate the 32P-containing phosphodiester. In the assays with in vitro transcribed RNA, N = A and is not radiolabeled.
Comparison of ADAR1 substrates bearing adenosine analogs
Our lab has published a series of studies on the editing of nucleoside analogs by ADAR2 (44–52). Since the sequence analysis suggested differences between ADAR1 and ADAR2 in the active site residues proximal to the adenosine N7 and 2′-positions (Figure 1), we chose to evaluate a subset of analogs in the ADAR1 reaction that varied the functional groups present at these positions [7-deazaadenosine (7dA), 8-aza-7-deazaadenosine (8a7dA), 2′-deoxyadenosine (dA) and 2′-O-methyladenosine (OMe)] (Figure 3). For this purpose, we prepared RNA hairpins based upon the hNEIL1 pre-mRNA derived 84 nt RNA described above using a splinted ligation strategy (64). The structure of the hairpin is shown in Figure 3 with the analogs incorporated at the recoding site. The effect of each modification was evaluated under single-turnover conditions at a saturating concentration of the enzyme. We had previously shown that minor structural changes at the edited adenosine have little effect on the binding affinity of ADAR2 for its RNA substrates (46,48,52). This is due to the fact that the majority of the binding affinity comes from the ADAR2 RNA-binding domain and not from the catalytic domain where the edited nucleotide binds. Using a gel mobility shift assay, we confirmed that each modified RNA is fully bound to ADAR1 under the reaction conditions used here (Supplementary Figure S1 and Supplementary methods).
Similar to the effect observed with ADAR2, the 2′-deoxyadenosine showed a modest decrease in rate, approximately 3-fold slower than adenosine (44) (Table 1). On the other hand, when the hydroxyl group was replaced with a methoxy group, no product was observed in the ADAR1 reaction, indicating a rate at least 50 times slower than adenosine, again similar to that observed with ADAR2 (44) (Table 1).
Table 1.
Single-turnover kinetic parameters for the deamination of adenosine and analogs by ADAR1a
| Substrate (N)b | kobs, min−1c | kreld | krel for ADAR2 | |
|---|---|---|---|---|
| A | 1.0 ± 0.1 × 10−2 | 1 | 1 | |
| dA | 3.0 ± 0.1 × 10−3 | 0.3 | 0.3 (44) | |
| 8a7dA | 2.5 ± 0.2 × 10−3 | 0.25 | 7.6 (52) | |
| 7dA | No reaction | <0.02 | 0.91 (45) | |
| OMe | No reaction | <0.02 | 0.004 (44) |
aADAR1 reactions were carried out with 130 nM enzyme, ≤18 nM RNA substrate in 15 mM Tris–HCl, pH 7.0, 1.5 mM EDTA, 60 mM KCl, 3% glycerol, 0.003% Nonidet P-40, 0.5 mM DTT, 160 U/ml RNasin and 1.0 µg/ml yeast tRNAPhe.
bSubstrate RNA as seen in Figure 3.
ckobs was calculated by taking the slope of the plot of ln(fraction substrate) versus time.
dkrel = kobs for analog/kobs for adenosine.
An RNA substrate containing 8-aza-7-deazaadenosine was deaminated approximately eight times more rapidly than adenosine by ADAR2 (52). This is consistent with previous observations that the 8-aza substitution facilitates covalent hydration of the purine ring, a key step in the deamination mechanism (65,66). However, for ADAR1, the results were significantly different with this analog. The ADAR1 substrate containing 8-aza-7-deazaadenosine was deaminated at a rate 4-fold slower than adenosine (Table 1). In an attempt to determine the source of this difference, we incorporated 7-deazaadenosine at the recoding site. Importantly, this substrate showed no visible deamination product with ADAR1, even when incubated for an extended period (Table 1). This result is in stark contrast to that observed in the ADAR2 reaction, where 7-deazaadenosine is deaminated at a rate nearly equal to that of adenosine (45) (Table 1).
ADAR reactions with RNA transcribed using 7-deazaadenosine triphosphate
The studies described above used an RNA substrate for ADAR1 with different nucleoside analogs incorporated specifically at the editing site. The RNA with 7-deazaadenosine at the editing site showed an unexpectedly low reactivity. To see if this is a general feature of the ADAR1 reaction or specific to this substrate, we wished to analyze a different ADAR1 substrate bearing 7-deazaadenosine. While site-selective modification at other known ADAR1 sites has not been achievable, it is possible to generate ADAR1 substrates via in vitro transcription using analogs of ATP with the caveat that all the adenosines in the RNA, not just the editing site, will be modified with the analog. Given the subtle change in structure between adenosine and 7-deazaadenosine, we believed an ADAR substrate with all adenosine sites replaced with 7-deazaadenosine should maintain enough of the key folded structure to support enzyme binding (67). In addition, 7-deazaadenosine triphosphate is known to efficiently replace ATP in reactions of T7 RNA polymerase and be faithfully incorporated into RNA at A sites (67). Therefore, we transcribed an RNA that is a known substrate for both ADAR1 and ADAR2 using either ATP or 7-deazaadenosine triphosphate. We chose an RNA that is a derivative of the 5HT2CR pre-mRNA we described in an earlier study that has good editing sites for both ADAR1 and ADAR2, albeit at different locations (Figure 4) (60). We observed that ADAR1 editing at the B site (the best editing site for ADAR1 in this RNA) was completely abolished when the adenosines were replaced with 7-deazaadenosine (Figure 4). However, there was a much smaller effect on ADAR2 editing. For instance, ADAR2 editing at the E site, which is deaminated to a similar extent (71 ± 11%) as is the B site by ADAR1 (66 ± 11%), was reduced by less than 2-fold in the RNA containing 7-deazaadenosine (41 ± 3%) (Figure 4). Therefore, the inhibitory effect on ADAR1 observed when the NEIL1 substrate RNA was site specifically modified with 7-deazaadenosine is also seen when the 5HT2CR substrate RNA is modified with 7-deazaadenosine. The small decrease in ADAR2 reactivity observed is most likely a result of effects on stability and/or structure arising from 7-deazaadenosine substitution throughout the RNA.
Figure 4.

Editing of an RNA transcribed in vitro with either ATP or 7-deazaATP. (A) Location of the ‘B’ and ‘E’ editing sites in the context of the 5HT2CR pre-mRNA-derived substrate (60). (B) Extent of editing of this RNA containing A or 7-deazaA with ADAR1, ADAR2 and the ADAR2 R455A mutant. Asterisk indicates no detectable editing.
The R455A mutation of ADAR2 renders it sensitive to the 7-deazaadenosine substitution
There is a high degree of sequence similarity between ADAR1 and ADAR2 in the region surrounding the deaminase active site (Figure 1). However, a key difference is the presence of A970 at a location corresponding to R455 in ADAR2. ADAR2 R455 is on an α-helix near the zinc-containing active site with its side chain directed toward the putative edited nucleotide-binding site (Figure 1). We have shown that the R455A mutation changes ADAR2’s reactivity to adenosine analogs with modifications at the 7-position (52). This observation is consistent with a model of adenosine recognition in the ADAR2 active site where the R455 side chain is near the N7 of the edited nucleotide (Figure 1). Given our observation that ADAR1 is more sensitive to 7-deazaadenosine substitution than is wild-type ADAR2, we wished to determine if this sensitivity is shared by the ADAR2 R455A mutant since its active site may be more ADAR1-like. Since the NEIL1 pre-mRNA is a poor substrate for ADAR2, we used the 5HT2CR pre-mRNA for this analysis. Indeed, when comparing the ADAR2 R455A reactivity on the 5HT2CR pre-mRNA-derived substrate, we find the E site editing to be fully inhibited by 7-deazaadenosine substitution (Figure 4). This stands in contrast to wild-type ADAR2 that edits this site when it is either adenosine (71%) or 7-deazaadenosine (44%) (Figure 4). Furthermore, when we incorporated 7-deazaadenosine site specifically into an ADAR2 substrate RNA, the GluRB R/G site, we found that the rate of deamination by ADAR2 R455A was reduced 10-fold relative to adenosine (Table 2). Wild-type ADAR2 reacts with 7-deazaadenosine in this substrate at a rate nearly identical to its rate with adenosine (Table 2). Unfortunately, study of the ADAR1 A970R mutant has been complicated by poor expression and low activity (data not shown).
DISCUSSION
In this study, comparisons of ADAR1 deamination reactions have been made for a series of substrate analogs obtained by chemo-enzymatic synthesis of the RNA. This approach has been used successfully in the past to investigate structure/activity relationships in the ADAR2 reaction (44–52). This is the first example of using this technique with ADAR1. In the past, we did not have an RNA substrate accessible by chemical synthesis that would allow us to incorporate substrate analogs specifically at an ADAR1 editing site. However, with the discovery of the NEIL1 editing site and deletion studies showing the reaction supported by a relatively short RNA, we were able to design such a substrate.
Upon incorporation of nucleoside analogs at the recoding site of the NEIL1 RNA, we discovered both similarities and differences between ADAR1 and ADAR2. Like ADAR2, ADAR1 cannot tolerate 2′-O-methylation of the edited adenosine, but can tolerate 2′-deoxyadenosine, albeit with a rate reduced by approximately 3-fold (44). In fact, there is a remarkable similarity in relative rates of deamination of adenosines substituted at the 2′-position. Based upon a model of nucleotide recognition in the ADAR2 active site (Figure 1) (43), as well as on data using nucleoside analogs (44, 51), we believe that threonine 375 in ADAR2 hydrogen bonds to the 2′-OH (Figure 4). Homology modeling of ADAR1 positions N891 in the corresponding location (Figure 1) (56). Threonine and asparagine have similar hydrogen bonding properties and polarity, so it is plausible that these residues may make similar contacts with the ribose. In addition, it has been suggested in the literature that 2′-O-methylation, directed by a snoRNA, may function as a mechanism to regulate editing on the 5HT2CR pre-mRNA, highlighting the importance of defining the effect of 2′-O-methylation on editing by ADAR1 and ADAR2 (11,68).
Interestingly, there is a striking difference in how the enzymes accept modifications of the base. ADAR2 tolerates carbon substitution for nitrogen at the 7-position of the ring with virtually no difference in reaction rate (45), and exhibits a 7.6-fold increase in rate with the 8-aza-7-deazaadenosine (52). However, 8-aza-7-deazaadenosine is edited 4-fold slower than adenosine by ADAR1. The lack of detectable deamination product with 7-deazaadenosine suggests that ADAR1, unlike ADAR2, but like ADA, makes an important interaction with N7 of the purine ring (69). While 7-deazaadenosine is not a substrate for ADA (70,71), 8-aza-7-deazaadenosine is a substrate, with a rate 15-fold slower than adenosine (72). For both ADAR1 and ADA, the ability to deaminate 8-aza-7-deazaadenosine could be a result of the increased reactivity of the base containing nitrogen at the 8-position, or it could be a result of the loss of one interaction at the 7-position and a concomitant gain of interaction at the 8-position.
The R455A mutation in ADAR2 was shown in a previous study to result in only a 2-fold decrease in rate of deamination of adenosine (52). The relatively small effect this mutation has on editing adenosine-containing substrates is also shown here in its ability to edit the 5HT2CR RNA E site (Figure 4). This indicates that any beneficial effect R455 may have in catalysis is largely compensated for in the alanine mutant. Yet, this mutant is much more sensitive to the 7-deaza modification at the editing site than is wild-type ADAR2 (Figure 4 and Table 2). While the reason for this dependence requires additional structural and mechanistic studies, it is tempting to speculate that ADAR active sites that bear alanine at this location (wild-type ADAR1, ADAR2 R455A) use a specific contact to N7 during adenosine deamination, perhaps via an ordered water molecule like that observed in the tRNA-modifying adenosine deaminase TadA–RNA complex (73) (Figure 5A). An RNA containing 7-deazaadenosine has not yet been tested with TadA, but it would be intriguing to determine whether this enzyme also requires N7, as suggested by the interactions in the crystal structure. Interestingly, in a functional screen for active mutants of ADAR2 where all possible mutants at position 455 were screened, only small residues (glycine, alanine, serine and threonine) were identified in the active group besides arginine (52). Perhaps these smaller residues allow for the binding of water to substrate N7 in the active site. Hydrogen bonding to N7 can be important in catalysis of adenosine deamination as shown in studies of mouse ADA. In this enzyme, D296 H-bonds to N7 of the adenosine substrate (Figure 5B). Mutation of D296 to alanine substantially reduces substrate binding but also reduces kcat to 0.07% that of wild-type ADA (74). In addition, adenosine monophosphate deaminase (AMPDA) also requires the nitrogen at position 7, as 7-deazaadenosine monophosphate is not a substrate for the form of this enzyme found in rabbit muscle (75). In the crystal structure of the Arabidopsis thaliana AMPDA, D736, a conserved residue that ligates the zinc, also appears well placed to form a hydrogen bonding interaction with N7 (Figure 5C) (76).
Figure 5.

Known interactions with adenine N7 in different enzymes that catalyze adenosine deamination. (A) tRNA adenosine deaminase (TadA), (B) ADA and (C) AMPDA make hydrogen bonding interactions with adenosine. (A) Staphylococcus aureus TadA makes a hydrogen bond to N7 of nebularine through a network of ordered water molecules (73). (B) D296 of murine ADA makes a hydrogen bond to N7 of 1-deazaadenosine (77). (C) D736 of Arabidopsis AMPDA ligates the catalytic zinc, but is also within hydrogen bonding distance of N7 of coformycin 5′-phosphate (76).
For wild-type ADAR2, 7-deazaadenosine remains a good substrate, making it an exception in this group of enzymes. It is possible in this case that the R455 side chain serves to stabilize an SNAr-like reaction transition state not by direct H-bonding to the base, but through a charge–charge interaction (52,78). Such an interaction would not require the presence of N7 in the substrate. A similar such stabilization of a negatively charged transition state by a nearby arginine residue has been observed in several other enzymes, including one stabilizing an SNAr reaction transition state (79–81).
No potent small molecule inhibitors of ADARs are currently known. Such inhibitors would be useful for the study of ADAR function, particularly given the fact that genetic approaches are complicated by the developmental defects of the ADAR knockouts, particularly for ADAR1. The ability to inhibit editing in a time- and dose-dependent manner would be a particularly helpful tool in answering questions regarding the relationship between aberrant editing and disease. Inhibitors that are editing site- or ADAR selective would be useful in this regard. Studies such as described here, where differences in structure/activity relationships are identified, will be useful for the design of inhibitors that can differentiate between ADAR1 and ADAR2. In addition, monitoring editing of RNA containing 7-deazaadenosine may prove to be useful to differentiate reactions of ADAR1 and ADAR2. 7-deazaadenosine (tubercidin) added to culture media is known to lead to efficient incorporation of this analog into cellular RNA (82). Detection of 7-deazainosine at known editing sites after such treatment would imply ADAR2 is the enzyme responsible, since 7-deazaadenosine at these sites would be refractory to deamination by ADAR1. Experiments designed to test this idea are currently underway.
In summary, we have developed an assay to investigate structure/activity relationships for the ADAR1 catalyzed reaction. Using this assay, we have found both similarities and differences between ADAR1 and ADAR2 substrate recognition. The difference in interaction with N7 of the purine ring suggests a difference in the way that these two enzymes catalyze the deamination reaction. Looking forward, the development of this assay will allow us to elucidate additional mechanistic details regarding ADAR1, providing us with a new tool to learn more about this important family of enzymes.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Methods and Supplementary Figures 1–8.
FUNDING
National Institutes of Health (NIH) [GM061115 to P.A.B.]; National Science Foundation in the form of a Graduate Research Fellowship [1148897 to R.A.M.]. Funding for open access charge: NIH [GM061115 to P.A.B.]
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We would like to acknowledge Nicole Schirle for helpful discussions and Dr Cynthia Holsclaw and Dr William Jewell of the UC Davis Mass Spectrometry Facilities for assistance with mass spectrometry analysis.
REFERENCES
- 1.Wang J, Zhang J, Li K, Zhao W, Cui Q. SpliceDisease database: linking RNA splicing and disease. Nucleic Acids Res. 2012;40:D1055–D1059. doi: 10.1093/nar/gkr1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suzuki T, Nagao A, Suzuki T. Human mitochondrial diseases caused by lack of taurine modification in mitochondrial tRNAs. Wiley Interdiscip. Rev. RNA. 2011;2:376–386. doi: 10.1002/wrna.65. [DOI] [PubMed] [Google Scholar]
- 3.Ruggero D, Grisendi S, Piazza F, Rego E, Mari F, Rao PH, Cordon-Cardo C, Pandolfi PP. Dyskeratosis congenita and cancer in mice deficient in ribosomal RNA modification. Science. 2003;299:259–262. doi: 10.1126/science.1079447. [DOI] [PubMed] [Google Scholar]
- 4.Iwamoto K, Kato T. RNA editing of serotonin 2C receptor in human postmortem brains of major mental disorders. Neurosci. Lett. 2003;346:169–172. doi: 10.1016/s0304-3940(03)00608-6. [DOI] [PubMed] [Google Scholar]
- 5.Kawahara Y, Ito K, Sun H, Aizawa H, Kanazawa I, Kwak S. Glutamate receptors: RNA editing and death of motor neurons. Nature. 2004;427:801. doi: 10.1038/427801a. [DOI] [PubMed] [Google Scholar]
- 6.Kwak S, Kawahara Y. Deficient RNA editing of GluR2 and neuronal death in amyotropic lateral sclerosis. J. Mol. Med. 2005;83:110–120. doi: 10.1007/s00109-004-0599-z. [DOI] [PubMed] [Google Scholar]
- 7.Niswender CM, Herrick-Davis K, Dilley GE, Meltzer HY, Overholser JC, Stockmeier CA, Emeson RB, Sanders-Bush E. RNA editing of the human serotonin 5-HT2C receptor: Alterations in suicide and implications for serotonergic pharmacotherapy. Neuropsychopharm. 2001;24:478–491. doi: 10.1016/S0893-133X(00)00223-2. [DOI] [PubMed] [Google Scholar]
- 8.Sodhi MS, Burnet PWJ, Makoff AJ, Kerwin RW, Harrison PJ. RNA editing of the 5-HT(2C) receptor is reduced in schizophrenia. Mol. Psychiatry. 2001;6:373–379. doi: 10.1038/sj.mp.4000920. [DOI] [PubMed] [Google Scholar]
- 9.Vissel B, Royle GA, Christie BR, Schiffer HH, Ghetti A, Tritto T, Perez-Otano I, Radcliffe RA, Seamans J, Sejnowski T, et al. The role of RNA editing of kainate receptors in synaptic plasticity and seizures. Neuron. 2001;29:217–227. doi: 10.1016/s0896-6273(01)00192-1. [DOI] [PubMed] [Google Scholar]
- 10.Kishore S, Stamm S. The snoRNA HBII-52 regulates alternative splicing of the serotonin receptor 2C. Science. 2006;311:230–232. doi: 10.1126/science.1118265. [DOI] [PubMed] [Google Scholar]
- 11.Vitali P, Basyuk E, Le Meur E, Bertrand E, Muscatelli F, Cavaillé J, Huttenhofer A. ADAR2-mediated editing of RNA substrates in the nucleolus is inhibited by C/D small nucleolar RNAs. J. Cell Biol. 2005;169:745–753. doi: 10.1083/jcb.200411129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Morabito MV, Abbas AI, Hood JL, Kesterson RA, Jacobs MM, Kump DS, Hachey DL, Roth BL, Emeson RB. Mice with altered serotonin 2C receptor RNA editing display characteristics of Prader-Willi syndrome. Neurobiol. Dis. 2010;39:169–180. doi: 10.1016/j.nbd.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Doe CM, Relkovic D, Garfield AS, Dalley JW, Theobald DEH, Humby T, Wilkinson LS, Isles AR. Loss of the imprinted snoRNA mbii-52 leads to increased 5htr2c pre-RNA editing and altered 5HT2CR-mediated behaviour. Hum. Mol. Genet. 2009;18:2140–2148. doi: 10.1093/hmg/ddp137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Silberberg G, Lundin D, Navon R, Ohman M. Deregulation of the A-to-I RNA editing mechanism in psychiatric disorders. Hum. Mol. Genet. 2012;21:311–321. doi: 10.1093/hmg/ddr461. [DOI] [PubMed] [Google Scholar]
- 15.Miyamura Y, Suzuki T, Kono M, Inagaki K, Ito S, Suzuki N, Tomita Y. Mutations of the RNA-specific adenosine deaminase gene (DSRAD) are involved in dyschromatosis symmetrica hereditaria. Am. J. Hum. Genet. 2003;73:693–699. doi: 10.1086/378209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cenci C, Barzotti R, Galeano F, Corbelli S, Rota R, Massimi L, Di Rocco C, O'Connell MA, Gallo A. Down-regulation of RNA editing in pediatric astrocytomas: ADAR2 editing activity inhibits cell migration and proliferation. J. Biol. Chem. 2008;283:7251–7260. doi: 10.1074/jbc.M708316200. [DOI] [PubMed] [Google Scholar]
- 17.Galeano F, Leroy A, Rossetti C, Gromova I, Gautier P, Keegan LP, Massimi L, Di Rocco C, O'Connell MA, Gallo A. Human BLCAP transcript: new editing events in normal and cancerous tissues. Int. J. Cancer. 2010;127:127–137. doi: 10.1002/ijc.25022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ma CH, Chong JH, Guo Y, Zeng HM, Liu SY, Xu LL, Wei J, Lin YM, Zhu XF, Zheng GG. Abnormal expression of ADAR1 isoforms in Chinese pediatric acute leukemias. Biochem. Biophys. Res. Commun. 2011;406:245–251. doi: 10.1016/j.bbrc.2011.02.025. [DOI] [PubMed] [Google Scholar]
- 19.Paz N, Levanon EY, Amariglio N, Heimberger AB, Ram Z, Constantini S, Barbash ZS, Adamsky K, Safran M, Hirschberg A, et al. Altered adenosine-to-inosine RNA editing in human cancer. Genome Res. 2007;17:1586–1595. doi: 10.1101/gr.6493107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dominissini D, Moshitch-Moshkovitz S, Amariglio N, Rechavi G. Adenosine-to-inosine RNA editing meets cancer. Carcinogenesis. 2011;32:1569–1577. doi: 10.1093/carcin/bgr124. [DOI] [PubMed] [Google Scholar]
- 21.Gurevich I, Tamir H, Arango V, Dwork AJ, Mann JJ, Schmauss C. Altered editing of serotonin 2C receptor pre-mRNA in the prefrontal cortex of depressed suicide victims. Neuron. 2002;34:349–356. doi: 10.1016/s0896-6273(02)00660-8. [DOI] [PubMed] [Google Scholar]
- 22.Melcher T, Maas S, Herb A, Sprengel R, Seeburg PH, Higuchi M. A mammalian RNA editing enzyme. Nature. 1996;379:460–464. doi: 10.1038/379460a0. [DOI] [PubMed] [Google Scholar]
- 23.Doyle M, Jantsch MF. New and old roles of the double-stranded RNA-binding domain. J. Struct. Biol. 2003;140:147–153. doi: 10.1016/s1047-8477(02)00544-0. [DOI] [PubMed] [Google Scholar]
- 24.Lehmann KA, Bass BL. Double-stranded RNA adenosine deaminases ADAR1 and ADAR2 have overlapping specificities. Biochemistry. 2000;39:12875–12884. doi: 10.1021/bi001383g. [DOI] [PubMed] [Google Scholar]
- 25.Patterson JB, Samuel CE. Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase. Mol. Cell. Biol. 1995;15:5376–5388. doi: 10.1128/mcb.15.10.5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Poulsen H, Nilsson J, Damgaard CK, Egebjerg J, Kjems J. CRM1 mediates the export of ADAR1 through a nuclear export signal within the Z-DNA binding domain. Mol. Cell. Biol. 2001;21:7862–7871. doi: 10.1128/MCB.21.22.7862-7871.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Desterro JMP, Keegan LP, Lafarga M, Berciano MT, O'Connell M, Carmo-Fonseca M. Dynamic association of RNA-editing enzymes with the nucleolus. J. Cell Sci. 2003;116:1805–1818. doi: 10.1242/jcs.00371. [DOI] [PubMed] [Google Scholar]
- 28.Sansam CL, Wells KS, Emeson RB. Modulation of RNA editing by functional nucleolar sequestration of ADAR2. Proc. Natl Acad. Sci. USA. 2003;100:14018–14023. doi: 10.1073/pnas.2336131100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eckmann CR, Neunteufl A, Pfaffstetter L, Jantsch MF. The human but not the Xenopus RNA-editing enzyme ADAR1 has an atypical nuclear localization signal and displays the characteristics of a shuttling protein. Mol. Biol. Cell. 2001;12:1911–1924. doi: 10.1091/mbc.12.7.1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Strehblow A, Hallegger M, Jantsch MF. Nucleocytoplasmic distribution of human RNA-editing enzyme ADAR1 is modulated by double-stranded RNA-binding domains, a leucine-rich export signal, and a putative dimerization domain. Mol. Biol. Cell. 2002;13:3822–3835. doi: 10.1091/mbc.E02-03-0161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.George CX, Samuel CE. Characterization of the 5'-flanking region of the human RNA-specific adenosine deaminase ADAR1 gene and identification of an interferon-inducible ADAR1 promoter. Gene. 1999;229:203–213. doi: 10.1016/s0378-1119(99)00017-7. [DOI] [PubMed] [Google Scholar]
- 32.George CX, Samuel CE. Human RNA-specific adenosine deaminase ADAR1 transcripts possess alternative exon 1 structures that initiate from different promoters, one constitutively active and the other interferon inducible. Proc. Natl Acad. Sci. USA. 1999;96:4621–4626. doi: 10.1073/pnas.96.8.4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawakubo K, Samuel CE. Human RNA-specific adenosine deaminase (ADAR1) gene specifies transcripts that initiate from a constitutively active alternative promoter. Gene. 2000;258:165–172. doi: 10.1016/s0378-1119(00)00368-1. [DOI] [PubMed] [Google Scholar]
- 34.Marcucci R, Brindle J, Paro S, Casadio A, Hempel S, Morrice N, Bisso A, Keegan LP, Del Sal G, O'Connell MA. Pin1 and WWP2 regulate GluR2 Q/R site RNA editing by ADAR2 with opposing effects. EMBO J. 2011;30:4211–4222. doi: 10.1038/emboj.2011.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Desterro JMP, Keegan LP, Jaffray E, Hay RT, O'Connell MA, Carmo-Fonseca M. SUMO-1 modification alters ADAR1 editing activity. Mol. Biol. Cell. 2005;16:5115–5126. doi: 10.1091/mbc.E05-06-0536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hartner JC, Schmittwolf C, Kispert A, Müller AM, Higuchi M, Seeburg PH. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J. Biol. Chem. 2004;279:4894–4902. doi: 10.1074/jbc.M311347200. [DOI] [PubMed] [Google Scholar]
- 37.Hartner JC, Walkley CR, Lu J, Orkin SH. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat. Immunol. 2009;10:109–115. doi: 10.1038/ni.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang Q, Miyakoda M, Yang W, Khillan J, Stachura DL, Weiss MJ, Nishikura K. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J. Biol. Chem. 2004;279:4952–4961. doi: 10.1074/jbc.M310162200. [DOI] [PubMed] [Google Scholar]
- 39.XuFeng R, Boyer MJ, Shen H, Li Y, Yu H, Gao Y, Yang Q, Wang Q, Cheng T. ADAR1 is required for hematopoietic progenitor cell survival via RNA editing. Proc. Natl Acad. Sci. USA. 2009;106:17763–17768. doi: 10.1073/pnas.0903324106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000;406:78–81. doi: 10.1038/35017558. [DOI] [PubMed] [Google Scholar]
- 41.Li M, Yang L, Li C, Jin C, Lai M, Zhang G, Hu Y, Ji J, Yao Z. Mutational spectrum of the ADAR1 gene in dyschromatosis symmetrica hereditaria. Arch. Dermatol. Res. 2010;302:469–476. doi: 10.1007/s00403-010-1039-2. [DOI] [PubMed] [Google Scholar]
- 42.Samuel CE. ADARs: viruses and innate immunity. Curr. Top. Microbiol. Immunol. 2012;353:163–195. doi: 10.1007/82_2011_148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Macbeth MR, Schubert HL, Vandemark AP, Lingam AT, Hill CP, Bass BL. Inositol hexakisphosphate is bound in the ADAR2 core and required for RNA editing. Science. 2005;309:1534–1539. doi: 10.1126/science.1113150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yi-Brunozzi HY, Easterwood LM, Kamilar GM, Beal PA. Synthetic substrate analogs for the RNA-editing adenosine deaminase ADAR-2. Nucleic Acids Res. 1999;27:2912–2917. doi: 10.1093/nar/27.14.2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Easterwood LM, Véliz EA, Beal PA. Demethylation of 6-O-methylinosine by an RNA-editing adenosine deaminase. J. Am. Chem. Soc. 2000;122:11537–11538. [Google Scholar]
- 46.Stephens OM, Yi-Brunozzi HY, Beal PA. Analysis of the RNA-editing reaction of ADAR2 with structural and fluorescent analogues of the GluR-B R/G editing site. Biochemistry. 2000;39:12243–12251. doi: 10.1021/bi0011577. [DOI] [PubMed] [Google Scholar]
- 47.Yi-Brunozzi HY, Stephens OM, Beal PA. Conformational changes that occur during an RNA-editing adenosine deamination reaction. J. Biol. Chem. 2001;276:37827–37833. doi: 10.1074/jbc.M106299200. [DOI] [PubMed] [Google Scholar]
- 48.Véliz EA, Easterwood LM, Beal PA. Substrate analogues for an RNA-editing adenosine deaminase: mechanistic investigation and inhibitor design. J. Am. Chem. Soc. 2003;125:10867–10876. doi: 10.1021/ja029742d. [DOI] [PubMed] [Google Scholar]
- 49.Haudenschild BL, Maydanovych O, Véliz EA, Macbeth MR, Bass BL, Beal PA. A transition state analogue for an RNA-editing reaction. J. Am. Chem. Soc. 2004;126:11 213–11219. doi: 10.1021/ja0472073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Maydanovych O, Beal PA. C6-substituted analogues of 8-azanebularine: probes of an RNA-editing enzyme active site. Org. Lett. 2006;8:3753–3756. doi: 10.1021/ol061354j. [DOI] [PubMed] [Google Scholar]
- 51.Jayalath P, Pokharel S, Véliz E, Beal PA. Synthesis and evaluation of an RNA editing substrate bearing 2'-deoxy-2'-mercaptoadenosine. Nucleos. Nucleot. Nucl. 2009;28:78–88. doi: 10.1080/15257770902736459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pokharel S, Jayalath P, Maydanovych O, Goodman RA, Wang SC, Tantillo DJ, Beal PA. Matching active site and substrate structures for an RNA editing reaction. J. Am. Chem. Soc. 2009;131:11882–11891. doi: 10.1021/ja9034076. [DOI] [PubMed] [Google Scholar]
- 53.Kim U, Wang Y, Sanford T, Zeng Y, Nishikura K. Molecular cloning of cDNA for double-stranded RNA adenosine deaminase, a candidate enzyme for nuclear RNA editing. Proc. Natl Acad. Sci. USA. 1994;91:11457–11461. doi: 10.1073/pnas.91.24.11457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yeo J, Goodman RA, Schirle NT, David SS, Beal PA. RNA editing changes the lesion specificity for the DNA repair enzyme NEIL1. Proc. Natl Acad. Sci. USA. 2010;107:20715–20719. doi: 10.1073/pnas.1009231107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Abramoff MD, Magalhães PJ, Ram SJ. Image processing with ImageJ. Biophot. Int. 2004;11:36–42. [Google Scholar]
- 56.Kelley LA, Sternberg MJE. Protein structure prediction on the Web: a case study using the Phyre server. Nat. Protocols. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- 57.Ley HL., III . Editing of Hepatitis Delta Virus Antigenomic RNA by Recombinant Human Adenosine Deaminases That Act on RNA. 2001. PhD Dissertation, University of Utah, Salt Lake City, UT. [Google Scholar]
- 58.Macbeth MR, Lingam AT, Bass BL. Evidence for auto-inhibition by the N terminus of hADAR2 and activation by dsRNA binding. RNA. 2004;10:1563–1571. doi: 10.1261/rna.7920904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eggington JM, Greene T, Bass BL. Predicting sites of ADAR editing in double-stranded RNA. Nat. Commun. 2011;2:319. doi: 10.1038/ncomms1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schirle NT, Goodman RA, Krishnamurthy M, Beal PA. Selective inhibition of ADAR2-catalyzed editing of the serotonin 2c receptor pre-mRNA by a helix-threading peptide. Org. Biomol. Chem. 2010;8:4898–4904. doi: 10.1039/c0ob00309c. [DOI] [PubMed] [Google Scholar]
- 61.Roberson LM, Rosenthal JJC. An accurate fluorescent assay for quantifying the extent of RNA editing. RNA. 2006;12:1907–1912. doi: 10.1261/rna.166906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maydanovych O, Easterwood LM, Cui T, Véliz EA, Pokharel S, Beal PA. Probing adenosine-to-inosine editing reactions using RNA-containing nucleoside analogs. Methods Enzymol. 2007;424:369–386. doi: 10.1016/S0076-6879(07)24017-0. [DOI] [PubMed] [Google Scholar]
- 63.Fedor MJ, Uhlenbeck OC. Kinetics of intermolecular cleavage by hammerhead ribozymes. Biochemistry. 1992;31:12042–12054. doi: 10.1021/bi00163a012. [DOI] [PubMed] [Google Scholar]
- 64.Moore MJ, Sharp PA. Site-specific modification of pre-mRNA: the 2'-hydroxyl groups at the splice sites. Science. 1992;256:992–997. doi: 10.1126/science.1589782. [DOI] [PubMed] [Google Scholar]
- 65.Erion MD, Reddy MR. Calculation of relative hydration free energy differences for heteroaromatic compounds: use in the design of adenosine deaminase and cytidine deaminase inhibitors. J. Am. Chem. Soc. 1998;120:3295–3304. [Google Scholar]
- 66.Albert A. Covalent Hydration in Nitrogen Heterocycles. Adv. Heterocycl. Chem. 1976;20:117–143. [Google Scholar]
- 67.Wieczorek A, Dinter-Gottlieb G, Gottlieb PA. Evidence that total substitution of adenine with 7-deazaadenine in the HDV antigenomic ribozyme changes the kinetics of RNA folding. Bioorg. Med. Chem. Lett. 1994;4:987–994. [Google Scholar]
- 68.Cavaillé J, Buiting K, Kiefmann M, Lalande M, Brannan CI, Horsthemke B, Bachellerie JP, Brosius J, Hüttenhofer A. Identification of brain-specific and imprinted small nucleolar RNA genes exhibiting an unusual genomic organization. Proc. Natl Acad. Sci. USA. 2000;97:14 311–14316. doi: 10.1073/pnas.250426397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wilson DK, Rudolph FB, Quiocho FA. Atomic structure of adenosine deaminase complexed with a transition-state analog: understanding catalysis and immunodeficiency mutations. Science. 1991;252:1278–1284. doi: 10.1126/science.1925539. [DOI] [PubMed] [Google Scholar]
- 70.Frederiksen S. Specificity of adenosine deaminase toward adenosine and 2'-deoxyadenosine analogues. Arch. Biochem. Biophys. 1966;113:383–388. doi: 10.1016/0003-9861(66)90202-5. [DOI] [PubMed] [Google Scholar]
- 71.Seela F, Xu K. Pyrazolo[3,4-d]pyrimidine ribonucleosides related to 2-aminoadenosine and isoguanosine: synthesis, deamination and tautomerism. Org. Biomol. Chem. 2007;5:3034–3045. doi: 10.1039/b708736e. [DOI] [PubMed] [Google Scholar]
- 72.Bennett LL, Allan PW, Smithers D, Vail MH. Resistance to 4-aminopyrazolo (3,4-d) pyrimidine. Biochem. Pharmacol. 1969;18:725–740. doi: 10.1016/0006-2952(69)90043-4. [DOI] [PubMed] [Google Scholar]
- 73.Losey HC, Ruthenburg AJ, Verdine GL. Crystal structure of Staphylococcus aureus tRNA adenosine deaminase TadA in complex with RNA. Nat. Struct. Mol. Biol. 2006;13:153–159. doi: 10.1038/nsmb1047. [DOI] [PubMed] [Google Scholar]
- 74.Sideraki V, Mohamedali KA, Wilson DK, Chang Z, Kellems RE, Quiocho FA, Rudolph FB. Probing the functional role of two conserved active site aspartates in mouse adenosine deaminase. Biochemistry. 1996;35:7862–7872. doi: 10.1021/bi952920d. [DOI] [PubMed] [Google Scholar]
- 75.Zielke CL, Suelter CH. Substrate specificity and aspects of deamination catalyzed by rabbit muscle 5′-adenylic acid aminohydrolase. J. Biol. Chem. 1971;246:1313–1317. [PubMed] [Google Scholar]
- 76.Han BW, Bingman CA, Mahnke DK, Bannen RM, Bednarek SY, Sabina RL, Phillips GN. Membrane association, mechanism of action, and structure of Arabidopsis embryonic factor 1 (FAC1) J. Biol. Chem. 2006;281:14939–14947. doi: 10.1074/jbc.M513009200. [DOI] [PubMed] [Google Scholar]
- 77.Wilson DK, Quiocho FA. A pre-transition-state mimic of an enzyme: X-ray structure of adenosine deaminase with bound 1-deazaadenosine and zinc-activated water. Biochemistry. 1993;32:1689–1694. doi: 10.1021/bi00058a001. [DOI] [PubMed] [Google Scholar]
- 78.Luo M, Schramm VL. Transition state structure of E. coli tRNA-specific adenosine deaminase. J. Am. Chem. Soc. 2008;130:2649–2655. doi: 10.1021/ja078008x. [DOI] [PubMed] [Google Scholar]
- 79.Phillips MA, Fletterick R, Rutter WJ. Arginine 127 stabilizes the transition state in carboxypeptidase. J. Biol. Chem. 1990;265:20692–20698. [PubMed] [Google Scholar]
- 80.Phillips MA, Hedstrom L, Rutter WJ. Guanidine derivatives restore activity to carboxypeptidase lacking arginine-127. Protein Sci. 1992;1:517–521. doi: 10.1002/pro.5560010406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gildenhuys S, Dobreva M, Kinsley N, Sayed Y, Burke J, Pelly S, Gordon GP, Sayed M, Sewell T, Dirr HW. Arginine 15 stabilizes an S(N)Ar reaction transition state and the binding of anionic ligands at the active site of human glutathione transferase A1-1. Biophys. Chem. 2010;146:118–125. doi: 10.1016/j.bpc.2009.11.003. [DOI] [PubMed] [Google Scholar]
- 82.Acs G, Reich E, Mori M. Biological and Biochemical Properties of the Analogue Antibiotic Tubercidin. Proc. Natl Acad. Sci. USA. 1964;52:493–501. doi: 10.1073/pnas.52.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.