


Abstract
p32 is an evolutionarily conserved and ubiquitously expressed multifunctional protein. Although p32 exists at diverse intra and extracellular sites, it is predominantly localized to the mitochondrial matrix near the nucleoid associated with mitochondrial transcription factor A. Nonetheless, its function in the matrix is poorly understood. Here, we determined p32 function via generation of p32-knockout mice. p32-deficient mice exhibited mid-gestation lethality associated with a severe developmental defect of the embryo. Primary embryonic fibroblasts isolated from p32-knockout embryos showed severe dysfunction of the mitochondrial respiratory chain, because of severely impaired mitochondrial protein synthesis. Recombinant p32 binds RNA, not DNA, and endogenous p32 interacts with all mitochondrial messenger RNA species in vivo. The RNA-binding ability of p32 is well correlated with the mitochondrial translation. Co-immunoprecipitation revealed the close association of p32 with the mitoribosome. We propose that p32 is required for functional mitoribosome formation to synthesize proteins within mitochondria.
INTRODUCTION
Mitochondria are essential organelles that are present in virtually all eukaryotic cells. The primary function of mitochondria is ATP production via the oxidative phosphorylation (OXPHOS) pathway. Additionally, mitochondria perform crucial roles in many other metabolic, regulatory and developmental processes. The circular 16.5-kb human mitochondrial DNA (mtDNA) molecule encodes 2 rRNAs, 22 tRNAs and 13 proteins that are members of the respiratory chain (1–3). All other proteins, including the mitochondrial translation apparatus, are derived from nuclear genes and are imported from the cytoplasm. In addition to core components of the small and large subunits of mitochondrial ribosomes (mitoribosomes), many other factors are required for translation initiation, elongation and termination in mitochondria (4).
Mammalian cells contain up to thousands of copies of mtDNA, which are organized in nucleoids (5,6). Mitochondrial transcription factor A (TFAM) is a key activator of mitochondrial transcription, and is a participant in mitochondrial genome replication to maintain mtDNA (7). It may also be a primary factor for packaging mtDNA in the nucleoid (8). Nucleoids may dynamically change their structure and distribution within mitochondria that are undergoing fission and fusion and are involved in various dynamic processes including mitochondrial replication, transcription and translation (9). Previously, we isolated p32 as a protein associated with TFAM (10).
Transcription from both the heavy-strand promoter (HSP) and light-strand promoter (LSP) generates polycistronic molecules that almost cover the entire H- and L-strand, respectively. It is generally accepted that tRNAs are excised from long polycistronic primary transcripts to generate mature messenger RNAs (mRNAs) and ribosomal RNAs (rRNAs); this is called the tRNA punctuation model (11–13). During or immediately after cleavage of tRNAs, mRNAs are polyadenylated by a mitochondrial poly(A) polymerase. Indeed, in the case of some mRNAs, poly(A) tail addition is necessary to form a stop codon at the end of the open reading frame and may also be necessary for stabilization of some RNAs (14–16). Thus, nine monocistronic and two dicistronic mRNA transcripts are formed. Mitochondrial mRNAs do not contain 5′-modifications (13), and to date there has been no report of a canonical mitochondrial poly(A)-binding protein. Bioinformatic analyses revealed no obvious candidate, although several metabolic mitochondrial enzymes have been shown to be capable of binding RNA and poly(A) sequences (17,18). Another important post-transcriptional process is mRNA transfer to the mitoribosome, which is required to control RNA levels and translational activity. However, the proteins involved and the exact mechanism are yet to be revealed.
Protein–RNA interactions play essential roles in post-transcriptional control of gene expression including splicing, nuclear-cytoplasmic transport, localization, quality control, mRNA degradation and translational regulation (19). Numerous mitochondrial RNA-binding proteins without known RNA-binding domains have also been identified (18). AUH (AU RNA-binding protein /enoyl-CoA hydratase), which forms a doughnut-like structure composed of two trimers, is highly positively charged because of lysine residues in its α-helix H1 that is located on the edge of the cleft between the trimers. A mutational analysis showed that the lysine residues in the α-helix H1 are essential for the RNA-binding activity of AUH (20,21).
p32 [complement component 1, q subcomponent-binding protein (C1qBP); also called gC1qR or HABP1] was first isolated from a membrane preparation of Raji cells and originally copurified with the pre-mRNA splicing factor SRSF1 (also called SF2 and ASF) in human HeLa cells (22). The p32 protein is a doughnut-shaped trimer, which primarily localizes to the mitochondrial matrix, but has also been reported to be present at the cell surface, in the nucleus and cytosol, as well as within secretory granules (23–26). p32 is synthesized as a pre protein and is processed by proteolytic cleavage of the N-terminal amino acids containing the mitochondrial signal sequence (27). The pre-protein form of p32 is only expressed in germ cells (28), which clearly justifies the predominant localization of p32 in the mitochondrial matrix.
Human p32 interacts with various molecules including hyaluronic acids, human immunodeficiency virus Tat protein, complement 1q, proapoptotic factor HRK and tumor suppressor ARF (29–31). These observations suggest that p32 may be a multifunctional chaperone protein (32). HABP1/p32/C1qBP represented as, synonym in Human Genome, exhibits structural plasticity influenced by ionic environment due to asymmetric charge distribution. The presence of salt stabilizes the compact trimeric conformation and can interact with hyaluronan only (33). Thus, one of the ligand of this protein is hyaluronan, a complex mucopolysaccharide which is well known for its regulatory role during embryonic development (34). p32/HABP1 expression in Schizosaccharomyces pombe induces growth inhibition and morphological abnormalities such as elongation, multinucleation and aberrant cell septum formation in several strains, implying a role for this protein in cell-cycle progression and cytokinesis (35). However, the primary physiological role of p32 in mammalian cells is unclear, particularly in the mitochondrial matrix, despite its predominant localization there.
Previously, we identified a Saccharomyces cerevisiae homolog of human p32, mam33, which localized to the mitochondrial matrix. Mam33-deficient yeast cells are significantly defective in maintenance of the mitochondrial genome and show impairment of mitochondrial ATP synthesis. The growth impairment is restored by the introduction of human p32 cDNA, which demonstrates the evolutionarily conserved function of p32 homologs among eukaryotes. Taken together, we propose that both human p32 and yeast mam33 reside in the mitochondrial matrix and play an important role in maintaining mitochondrial OXPHOS (27). Very recently, p32-knockdown cells exhibited reduced synthesis of mtDNA-encoded OXPHOS polypeptides and were less tumorigenic in vivo (36).
To explore the role of the p32 protein in vivo, particularly in the mitochondrial matrix, we generated mice with a homozygous disruption of the p32 gene. We show that p32 inactivation causes mid-gestation lethality of knockout embryos and defects in OXPHOS, because of severe impaired protein synthesis of mtDNA-encoded protein. Here, we propose that the mitochondrial matrix protein p32 functions as an essential RNA-binding factor in mitochondrial translation, and is indispensable for embryonic development.
MATERIALS AND METHODS
Animals
Animals were mated overnight, and females were examined for a vaginal plug the following morning. At noon of that day, vaginal plug detection was recorded as embryonic day (E) 0.5. Mouse experiments were performed in accordance with the guidelines of the animal ethics committee of Kyushu University Graduate School of Medicine, Japan.
Immunoblotting
Briefly, cells were lysed with lysis buffer (50 mM Tris–HCl, pH 7.5, 1 mM EDTA, 150 mM NaCl and 0.5% NP-40) and then subjected to immunoblotting as described elsewhere (37). Signals were visualized with horseradish peroxidase (HRP)-conjugated anti-rabbit IgG and an enhanced chemiluminescence reagent (GE Healthcare, Piscataway, NJ). Chemiluminescence was recorded and quantified with a chilled charge-coupled device camera (LAS1000plus).
Immunofluorescence imaging of mouse embryonic fibroblasts
Immunofluorescence was carried out according to established techniques. Briefly, mouse embryonic fibroblasts (MEFs) were incubated in the presence of 500 nM MitoTracker Red (Invitrogen) for 20 min. Cells were fixed and permeabilized, then incubated with a 1:200 dilution of anti-p32 serum in PBS/1% bovine serum albumin (BSA) for 1 h. Glass slides were mounted using Superfrost (Matsunami). Fluorescence images were obtained using a confocal laser microscope (Nikon).
Antibodies
Polyclonal antibodies against mouse p32, HA, TFAM, LRPPRC and VDAC were raised in our laboratory. Antibodies against COXI, COXIII, NDUFA9, SDHA, UQCRFS1, ATP synthase and COXVa were purchased from Invitrogen. Antibodies against β-actin, MRPS22, MRPS29 and MRPL3 were purchased from Sigma, Proteintech Group Inc, BD Biosciences and Abcam, respectively. Alexa 488-conjugated anti-rabbit and anti-mouse IgG for fluorescence microscopy, Alexa 568-conjugated anti-mouse IgG for fluorescence microscopy of paraffin-embedded tissue sections, HRP-conjugated anti-mouse IgG and diaminobenzidine (DAB) for BrdU staining were all purchased from Nichirei.
MEF culture and cell proliferation assay
SV40 large T antigen-immortalized MEFs were generated from E14 p32flox/flox C57BL/6 embryos by standard methods. MEFs and HeLa cell were cultured in Dulbecco’s modified Eagle’s medium (DMEM) (1000 mg/l glucose) supplemented with 10% FBS at 37°C in a humidified atmosphere with 5% CO2. For cell proliferation assay, MEF cells (1 × 104) were seeded in triplicate in 35 mm dishes and cultured in DMEM (1000 mg/l glucose) plus dialyzed 10% fetal bovine serum (FBS) without pyruvate. Cells were trypsinized and counted daily for up to 96 h using a Coulter Counter (Beckman Coulter). Pyruvate (1 mM), uridine (0.2 mM) and glucose (3500 mg/l) were added on Days 0 and 4. We used dialyzed FBS to remove small molecules such as uridine and pyruvate.
RNA band-shift assays
RNA electrophoretic mobility shift assays (REMSAs) were carried out according to established techniques. Briefly, a synthesized oligonucleotide probe (DNA or RNA) was end-labeling in the presence of [γ-32P]ATP by a T4 polynucleotide kinase. To form RNA–protein complexes, the indicated amount of purified His-p32 was incubated with the 32P-labeled oligonucleotide probe at 25°C for 30 min in binding buffer [10 mM HEPES, pH 7.6, 3 mM MgCl2, 20 mM KCl, 1 mM dithiothreitol (DTT), 50 U RNase inhibitor (Toyobo) and 5% glycerol]. Heparin (5 μg) was added for a further 10 min incubation to prevent non-specific binding. Samples were electrophoresed in a 6% non-denaturing polyacrylamide gel in Tris–borate buffer. Gels were dried and visualized using a BAS2500 (Fuji).
Sucrose gradient analysis of mitochondrial ribosomes
MEFs were solubilized in a non-ionic detergent (1% lauryl maltoside). Total cell lysates (2 mg) were loaded onto a 15–30% sucrose density gradient in 20 mM Tris–HCl, pH 7.5, 150 mM NaCl and 1 mM CaCl2, and then centrifuged at 100 000g for 3 h at 4°C in a swinging bucket rotor (SW 40.1; Beckman Coulter). After separation, 17 fractions were precipitated by 10% trichloroacetic acid and washed in acetone, and then the entire fraction was resolved by SDS–polyacrylamide gel electrophoresis (PAGE).
ATP quantification
Cellular ATP was quantified using an ATP-determination kit according to the manufacturer’s instructions (Promega). Briefly, four MEFs, plated at equal densities, were lysed in passive lysis buffer (Promega). Equal volumes of cell lysate were added to the standard reaction solution, and luminescence was measured and normalized to the protein amount in each lysate. The values used were in the linear range of the assay as determined by a standard curve.
Oxygen consumption assay
Oxygen consumption was measured as described elsewhere (38). MEFs were trypsinized, PBS washed twice and then permeabilized with 0.1 μg/ml digitonin. The optimum incubation time for permeabilization (50 s for MEFs) was determined for each cell line as the shortest time after which 99% of cells were trypan blue-positive. Cells were placed into the reaction chamber of a Clark-type electrode (Hansatech), and oxygen concentrations were measured in 1 ml volumes at 37°C with substrates (glutamate + malate, succinate + glycerol-3-phosphate; Sigma) and inhibitors according to standard protocols (39). Oxygen consumption was represented as the mean ± SD, nanomoles O2 consumed per minute per cell.
Expression constructs
The expression construct containing mouse p32 cDNA was generated by standard methods. Site-directed mutagenesis to generate p32 mutants K89A and K93A was performed using methods described elsewhere (37). cDNAs of wild-type and mutant K89A/K93A p32 were cloned into the BamHI/XhoI sites of the expression vector pcDNA3 (Invitrogen).
Immunoprecipitation using anti-HA antibodies
Immunoprecipitation (IP) was carried out according to established techniques (37). Doxycycline-induced and non-induced HeLa (p32-HA) cells (1 × 108 cells) and MEF cells were homogenized and centrifuged at 900 g for 10 min. The supernatant (adjusted to 10% Percoll) was overlaid on a discontinuous Percoll density gradient (4 ml of 40% and 4 ml of 20% Percoll buffer; GE Healthcare) in a 12 ml centrifugation tube. After centrifugation at 60 000g for 1 h using a SW41-Ti rotor (Beckman Coulter), the mitochondrial phase located in the middle of the tube was collected. Two to three milligrams of mitochondrial protein were solubilized in 1 ml IP buffer (10 mM Tris–HCl, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1% NP-40 and 0.1% SDS) containing 40 µl of beads coated with anti-HA or anti-p32 antibody. After 12 h of rotation, the beads were washed four times with IP buffer and eluted with 0.1 M glycine (pH 2.5).
Pulse-labeling of mitochondrial translation products
Mitochondrial translation products were pulse-labeled in vitro with [35S]-(methionine and cysteine) (GE Healthcare). In experiments where the label was chased, cells were incubated for 6 min in 100 µg/ml emetine or 250 µg/ml chloramphenicol prior to labeling for 60 min. Labeled cells were then rinsed with an Hypotonic buffer (10 mM Tris–HCl, pH 7.4, 150 mM MgCl2, 10 mM KCl). After centrifugation at 1150g for 5 min, cell pellets were resuspended in loading buffer consisting of 93 mM Tris–HCl, pH 6.7, 7.5% glycerol, 1% SDS, 0.25 mg/ml bromophenol blue and 3% mercaptoethanol. The total lysate was then subjected to 15% SDS-PAGE for 3 h at 180 V. Gels were measured using a BAS2500 (Fuji).
RNA co-IP with p32-HA protein
Briefly, HeLa whole cell lysates induced by doxycycline were first precleared with 50 µg rabbit IgG (Bio-Rad) for 15 min at 4°C, followed by binding to anti-HA-coated agarose beads or anti-LRPPRC antibody with protein G agarose for 12 h at 4°C. Then, p32-HA was eluted using glycine (pH 2.5), and RNA was extracted with an RNAeasy kit (Qiagen). The eluted RNA was treated with 100 U DNase I at 37°C for 20 min, and then the RNA was purified again with an RNAeasy kit.
Activity of respiratory mitochondrial complex
MEFs were lysed in a hypotonic buffer (2.5 mM Tris–HCl, pH 7.5 and 2.5 mM MgCl2) on ice for 15 min, and then sonicated for 15 s (25% output, duty cycle; TAITEC) to measure respiratory complex activity.
To detect complex I activity, spectrophotometric assays were performed to evaluate NADH:Q oxidoreductase activity at 30°C by monitoring the decrease in the absorbance of nicotinamide adenine dinucleotide reduced form (NADH) at 340 nm, as described elsewhere (40). Briefly, 10 μM decylubiquinone (artificial electron acceptor), 2 μg antimycin A (complex III inhibitor), 5 mM sodium azide (NaN3) (complex IV inhibitor) and 0.5 mg whole-cell lysate were mixed in 1 ml standard reaction medium (2.5 mM MgCl2 and 50 mM inorganic phosphate, pH 7.3). The reaction was initiated with 100 μM NADH. The activity of the enzyme was determined by the difference of absorbance with and without 1.25 μg rotenone (complex I inhibitor). Data were expressed as nmol NADH oxidized/min/μg protein.
Succinate dehydrogenase (complex II) activity was determined spectrophotometrically at 30°C (41). Briefly, the reaction was initiated with 50 μM dichlorophenolindophenol (DCPIP; used as an artificial electron acceptor) in 1 ml standard reaction medium supplemented with 20 mM succinate, 2 μg rotenone, 2 μg antimycin A, 5 mM NaN3 and 0.1 mg whole-cell lysate.
Cytochrome c reductase (complex III) activity was evaluated spectrophotometrically at 30°C by monitoring the increase in absorbance at 550 nm of cytochrome c. Briefly, the reaction was initiated by the addition of 5 mM reduced decylubiquinone to 1 ml standard reaction medium supplemented with 2 μg rotenone, 5 mM NaN, 60 μM cytochrome c and cell lysate. The reaction was stopped by addition of 2 μg Antimycin A.
Cytochrome c oxidase (complex IV) activity was determined spectrophotometrically at 550 nm at 30°C (42). Briefly, the reaction was initiated by the addition of 50 μg whole-cell lysate to 1 ml standard reaction medium supplemented with 20 μM ferrocytochrome c.
mRNA quantification
RT of 1 µg total RNA was performed with random hexamer primers using SuperScript II RT (Invitrogen) according to the manufacturer’s instructions. The expression of mitochondrial genes was detected by qPCR with a thermal cycler (StepOne plus; Applied Biosystems). PCR primers are listed in Supplementary Table S1.
Measurement of mitochondrial membrane potential
Mitochondrial membrane potential (Δψm) was estimated using a JC-1 probe (Invitrogen). This reagent is a highly reliable, cationic and mitochondria-specific fluorescent dye, which becomes concentrated in mitochondria in proportion to the membrane potential, because it is highly lipophilic. Increasing amounts of dye accumulate in mitochondria with increasing Δψm and ATP-generating capacity. The dye is present as monomers at lower concentrations (green fluorescence), but at higher concentrations forms J-aggregates (red fluorescence). The ratio of the fluorescence at 590 nm to that at 527 nm represents the relative Δψm value. Fluorescence was measured at the two wavelengths by a FACS Caliber (Becton Dickinson).
RESULTS
Disruption of p32 causes embryonic lethality
To experimentally address the function of p32 in a physiological in vivo context, we used gene targeting to generate p32-deficient mice. We constructed a gene replacement vector that introduced a gene cassette, consisting of the neomycin resistance gene (Neo) and flanking loxP sites upstream and downstream of exon 3, into the endogenous p32 gene (Supplementary Figure S1A). After injection of embryonic stem cells harboring the targeted allele, germline transmission was confirmed by Southern blotting and PCR analysis (Supplementary Figure S1B and S1C). Heterozygous p32+/− mice were generated by breeding p32fl/+ mice with a Cre transgenic mouse strain (EII-Cre) allowing universal expression of Cre-recombinase in all tissues. Intercrosses of p32+/− mice revealed no viable p32−/− offspring (Supplementary Figure S1D and Table 1). To determine the time at which the p32 mutant became lethal, we examined embryos from p32+/− intercrosses at various developmental stages. In contrast to wild-type embryos, the growth of p32−/− embryos appeared retarded as early as E10.5. Most p32−/− embryos had been resorbed by E11.5, and therefore p32−/− embryos died between E10.5 and E11.5.
The phenotype of p32−/− embryos included markedly shrunken, poorly developed, pale and anemic organs (Figure 1A). p32 protein was absent from p32−/− embryos. In E10.5 p32−/− embryos, BrdU incorporation revealed that cells did not proliferate in various tissues including the brain, heart and liver. These data suggest that growth retardation, smaller size of organ and severe anemia account for the embryonic lethality of the p32 mutation.
Table 1.
p32 deficiency causes embryonic lethality
| Genotype |
Disintegrated or resorbed | Total | |||
|---|---|---|---|---|---|
| +/+ | +/− | −/− | |||
| E8.5 | 8 | 19 | 5 | 5 | 32 |
| E9.5 | 17 | 35 | 15 | 17 | 67 |
| E10.5 | 5 | 16 | 7 | 10 | 28 |
| E11.5 | 4 | 15 | 6 | 4 | 25 |
| E12.5 | 1 | 9 | 2 | 3 | 12 |
| E13.5 | 3 | 21 | 1 | 2 | 25 |
| Newborn | 21 | 41 | 0 | 62 | |
Embryos at the indicated stages were isolated from intercrosses of heterozygous mice, and the total numbers (n) of intact and disintegrated or resorbed embryos were counted.
Figure 1.
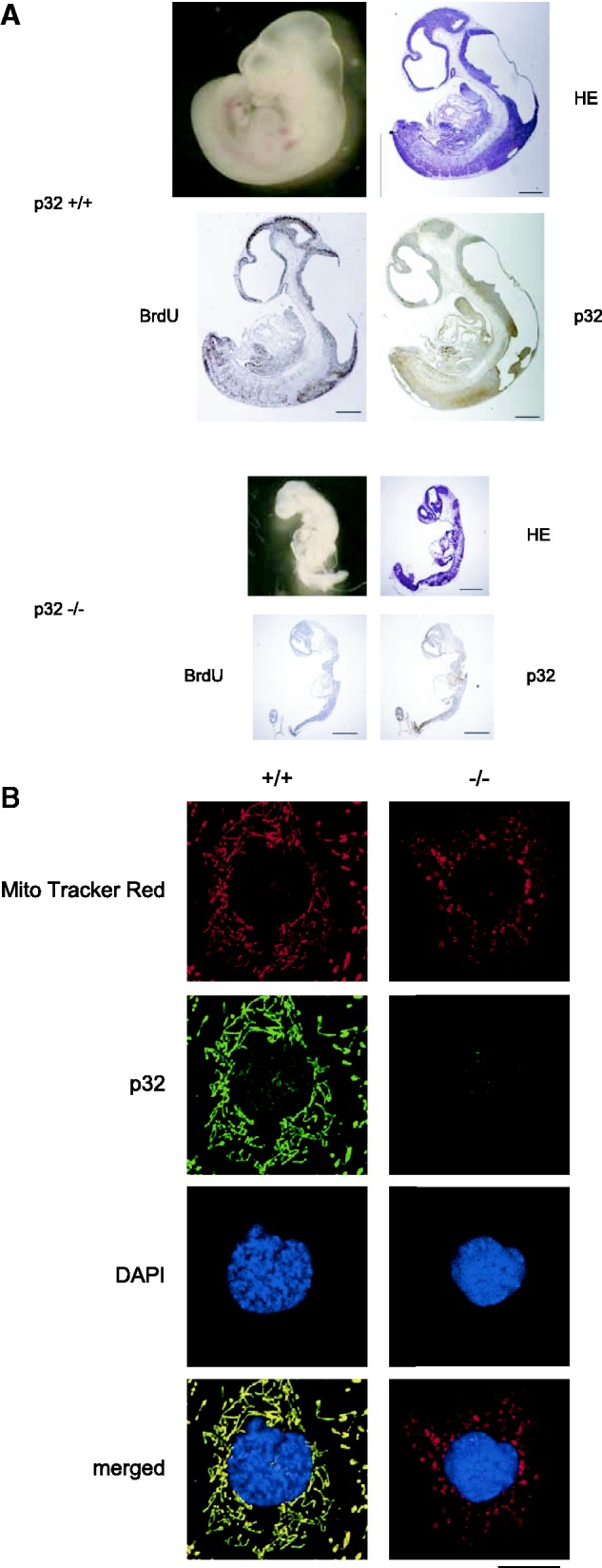
Disruption of p32 causes embryonic lethality. (A) Whole mounts of E10.5 p32−/− embryos and histological analysis of p32−/− embryos compared with those of wild-type p32+/+ littermates. Photomicrographs (upper left) and hematoxylin and eosin (HE) staining (upper right) are shown. Pregnant mice were injected with BrdU at 2 h prior to dissection. Sections of p32+/+ and p32−/− embryos at E10.5 were stained with BrdU (lower left) or an anti-p32 antibody (lower right). In vivo labeling with BrdU revealed arrest of proliferation in E10.5 embryos. Scale bars: 500 µm. (B) Immunofluorescence staining of p32 and MitoTracker Red in wild-type and p32-knockout cells. Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). MEFs were treated with MitoTracker Red (500 nM) for 20 min before p32 staining. Cells were fixed, fluorescently stained by anti-p32 antibodies, and then analyzed by confocal microscopy. The lower panel shows merged images. Scale bar: 20 μm.
Establishment of p32-knockout MEFs
To define the functional consequences of p32 deletion, MEFs were isolated from homozygous p32flox/flox embryos and infected with a adenovirus encoding Cre-recombinase, which led to efficient deletion of p32 in vitro. We also immortalized p32−/− cells by transfection with a plasmid vector encoding the SV40 large T antigen. Western blot analysis confirmed the absence of p32 in p32−/− cells and half wild-type p32 levels in p32+/− cells (Supplementary Figure S2). p32−/− MEFs exhibited an enlarged and flattened cell morphology compared with that of p32+/+ MEFs, suggesting that loss of p32-induced cell morphological change.
Abnormal mitochondrial morphology in p32−/− MEFs
Maintenance of proper mitochondrial morphology is critical for the function of this organelle. The basic morphology of mitochondria in cells is a dynamic tubulovesicular reticulum. Mitochondrial morphology changes dynamically as a result of a balance in the fusion and fission occurring in response to cellular energy demands, differentiation and pathological conditions (43,44). Therefore, we determined the effect of p32 depletion on the morphology of mitochondria using Mitotracker Red in p32+/+ and p32−/− MEF cell lines. In wild-type cells, p32 colocalized to the tubulovesicular mitochondria. In contrast, upon loss of p32, the tubular mitochondrial organization shifted to a punctate, granular shape (Figure 1B). This result suggests that the loss of p32 exhibited tubulovesicular morphological change.
Establishment of mitochondrial re-expression of p32
To directly confirm that p32 regulated mitochondrial function, we expressed mitochondria-targeted and cytoplasmic forms of p32 in p32−/− MEFs. We obtained stable p32−/− MEFs that expressed a low level of mitochondrially targeted p32 (immature) (Supplementary Figure S3A). The recombinant p32 was the same size as endogenous p32, suggesting that we successfully restored p32 expression in knockout cells, albeit at low levels. The cDNA construct encoded a p32 protein lacking the first 71 amino acids, which produced a cytoplasm-only p32. The expression level of this cytoplasm-only p32 was significantly lower than that of the mitochondria-targeted p32 (Supplementary Figure S3B). We were able to observe the cytoplasmic p32 only after inhibition of proteasomal degradation with MG-132 (Supplementary Figure S3C). These data suggest that cytoplasmic p32, which is unable to localize to the mitochondria, is extremely unstable in MEFs.
Loss of p32 reduced complex activity
We first sought to investigate a possible role for p32 in OXPHOS. The electron transport chain consists of five multi-subunit enzymatic complexes formed from the products of both nuclear and mitochondrial genes. We measured the activities of the electron transport chain complexes by spectroscopic assays that revealed the activities of complexes I, III and IV were strongly reduced in p32−/− cells, while the activity of complex II was unchanged (Figure 2A). Interestingly, complex II is the only OXPHOS complex encoded exclusively by nuclear DNA. Re-expression of p32 in p32−/− cells partially restored complex I, III and IV activities. These finding suggested that p32 might be involved in mtDNA-related protein function because complexes I, III and IV include proteins encoded by mtDNA.
Figure 2.


Reduced mitochondrial respiratory activities in p32−/− MEFs. (A) The activity of each complex was measured using cell lysates as described in ‘Materials and Methods’ section. Reduced enzymatic activities of mitochondrial complexes I, III and IV in p32-knockout cells were observed. In contrast, complex II activity was not decreased. Re-expression of p32 in p32−/− cells restored the enzymatic activities of complexes I, III and IV. The results represent the mean ± SD of three independent experiments. *P < 0.05; **P < 0.01 versus controls (p32+/+ versus p32−/− and p32−/− versus p32−/−::p32). (B) Oxygen consumption by digitonin-permeabilized wild-type (p32+/+) and knockout (p32−/−) MEFs. Glutamate and malate (glu+mal) respiration depends on the activities of complexes I, III and IV; glycerol-3-phosphate and succinate (G3P+suc) respiration depends on the activities of complexes III and IV. Rotenone (a complex I inhibitor) and Antimycin A (a complex III inhibitor) completely inhibit O2 consumption at each step. Data show the mean ± SD of triplicate experiments and **P < 0.01 versus controls (p32+/+ versus p32−/−). (C) Decreased mitochondrial membrane potential in p32-knockout MEFs. MEFs were stained with the fluorescent dye JC-1 and then analyzed by flow cytometry at 527 and 590 nm. The fluorescence ratio of JC1 dimer (Red)/JC-1 monomer (Green) is shown. Dissipation of the membrane potential with CCCP was used as a control. (D) Effects of 2-deoxy-d-glucose (2DG) (20 mM) and oligomycin (10 μM) on intracellular ATP content in MEFs. The ATP content in untreated MEFs is presented in lane 1. The ATP concentration of untreated MEFs (lane 1) was subtracted from that of 2DG-treated MEFs (lane 2) for assessment of glycolytic ATP production (lanes 1 and 2). The ATP concentration in 2DG-treated cells (lane 2) was subtracted from that in cells treated with 2DG + oligomycin (lane 3) for mitochondrial ATP production (lanes 2–3). Wild-type (p32+/+), p32-knockout (p32−/−), re-expressed p32 (p32−/−::p32) and vector only-transfected (p32−/−::vector) MEFs were used. The ATP response was measured using a Luminescence ATP assay kit in a 96-well plate. Data show the mean ± SD of triplicate experiments. (E) Cell proliferation monitored in MEFs. Upper panel: the diamonds, squares, triangles and circles represent wild-type MEFs (p32+/+), p32-knockout MEFs (p32−/−), knockout MEFs with reintroduced p32 cDNA (p32−/−::p32) and knockout MEFs with introduced vector-only cDNA (p32−/−::vector), respectively. Lower panel: pyruvate (1 mM) was added to wild-type and p32−/− MEFs. The cell number was counted at 24, 48, 72 and 96 h after seeding. Here, we used DMEM (1000 mg/l glucose) supplemented with 10% dialyzed FBS without pyruvate.
Mammalian p32 is required for oxidative respiration
To further test mitochondrial OXPHOS function, we measured cellular respiration by polarographic assays of digitonin-permeabilized cells. Oxygen electrode studies showed a 3-fold decrease in oxygen consumption dependent on complexes I + III + IV (glutamate + malate) and complexes III + IV (succinate + G3P) in p32-knockout MEFs compared with those of wild-type MEFs (Figure 2B), indicating that p32 is required for mitochondrial respiration. Decreased respiration in p32-knockout MEFs suggests a reduction in functional respiratory complexes.
p32-knockout MEFs show reduced mitochondrial membrane potential and ATP production
We suspected that p32-mediated inhibition of the mitochondrial respiratory chain may affect the mitochondrial membrane potential (Δψm). The Δψm value was measured using JC-1, a fluorescent dye sensitive to mitochondrial membrane potential. The Δψm values of p32-knockout MEFs were lower than those of wild-type MEFs (Figure 2C). Carbonyl cyanide 3-chlorophenylhydrazone (CCCP)-treated cells served as a positive control for depolarization of the mitochondrial membrane.
Because mitochondria are the cellular powerhouses for energy production, we measured ATP levels in wild-type and p32−/− MEFs. In wild-type cells, the inhibition of glycolysis by 2-deoxy-glucose (2DG) only slightly decreased the ATP level (Figure 2D, upper set, p32+/+, lane 2) probably because pyruvate in DMEM supported mitochondrial OXPHOS. After oligomycin, an inhibitor of complex V, was added together with 2DG, the cellular ATP level was strongly decreased (Figure 2D, upper set, p32+/+, lane 3), indicating that wild-type MEFs were largely dependent on mitochondrial ATP production (see lower set and legends for calculation). The total ATP level was rather higher in p32-knockout MEFs than in wild-type MEFs (Figure 2D, upper set, p32+/+, lane 1 and p32−/−, lane 1). A marked decrease in mitochondrial ATP production by 2DG was observed in the knockout MEFs (upper set, p32−/−, lane 2) and further addition of oligomycin marginally decreased the ATP production (upper set, p32−/−, lane 3), indicating that p32-knockout MEFs mostly depends on glycolytic ATP production. The decrease in mitochondrial ATP production may be over-compensated by glycolytic ATP production that was estimated by 2DG-sensitive cellular ATP (Figure 2D, lower set table). Re-expression of p32 in p32−/− cells restored mitochondrial ATP production, but not the introduction of an empty vector, indicating that p32 is required for efficient ATP production via OXPHOS.
Retarded proliferation of p32−/− MEFs
To assess potential defects in proliferation caused by mitochondrial OXPHOS dysfunction, we cultured cells at a standard concentration of glucose (1000 mg/l) without pyruvate, which allowed easier observation of the effects of glucose on OXPHOS. We dialyzed FBS to remove small molecules such as uridine and pyruvate. As shown in Figure 2E, proliferation of p32-knockout MEFs was strongly retarded compared with that of wild-type cells. Re-expression of p32 in p32−/− MEFs rescued the growth retardation, indicating that p32 is important for normal cell proliferation.
Cells depleted of mtDNA would require high glucose, pyruvate and uridine for proliferation. To determine whether the addition of high glucose, pyruvate or uridine could rescue the proliferation of p32−/− MEFs, we added each of these supplements separately. Only pyruvate (1 mM) was able to rescue the proliferation of p32−/− MEFs (Figure 2E, lower panel); addition of high glucose (3500 mg/l) and uridine (0.2 mM) did not rescue proliferation (data not shown). Taken together with no decrease of cellular ATP in p32−/− MEFs (Figure 2D), these results suggest that impaired regeneration of NAD+ (probably by complex I) is particularly critical for the retardation of p32−/− MEF proliferation.
No decreased mtDNA copy number and RNA expression
We measured the mtDNA copy number because the observed defects were likely related to mtDNA. The normal copy number of mtDNA was confirmed by qPCR, which excluded defects of mtDNA replication and/or repair as a cause of compromised protein synthesis (Figure 3A). To determine whether p32 affected the transcription of mtDNA, total RNA was isolated from cells and was assessed by qRT–PCR (Figure 3B). The steady-state level of 12S and 16S rRNA did not show a significant change between wild-type and p32−/− MEFs. No mtDNA-encoded mRNAs showed a decrease in p32-knockout MEFs compared with those in wild-type cells, although the levels of ND1 and ND6 transcripts were increased two-fold in knockout cells. The RNA expression levels in p32-re-expressed cells were also not significantly changed compared with those in wild-type cells (Figure 3B). Thus, p32 knockout did not cause a decrease in mtDNA-encoded transcripts. These results suggest that p32 deficiency does not affect the amounts of mtDNA or mRNAs.
Figure 3.

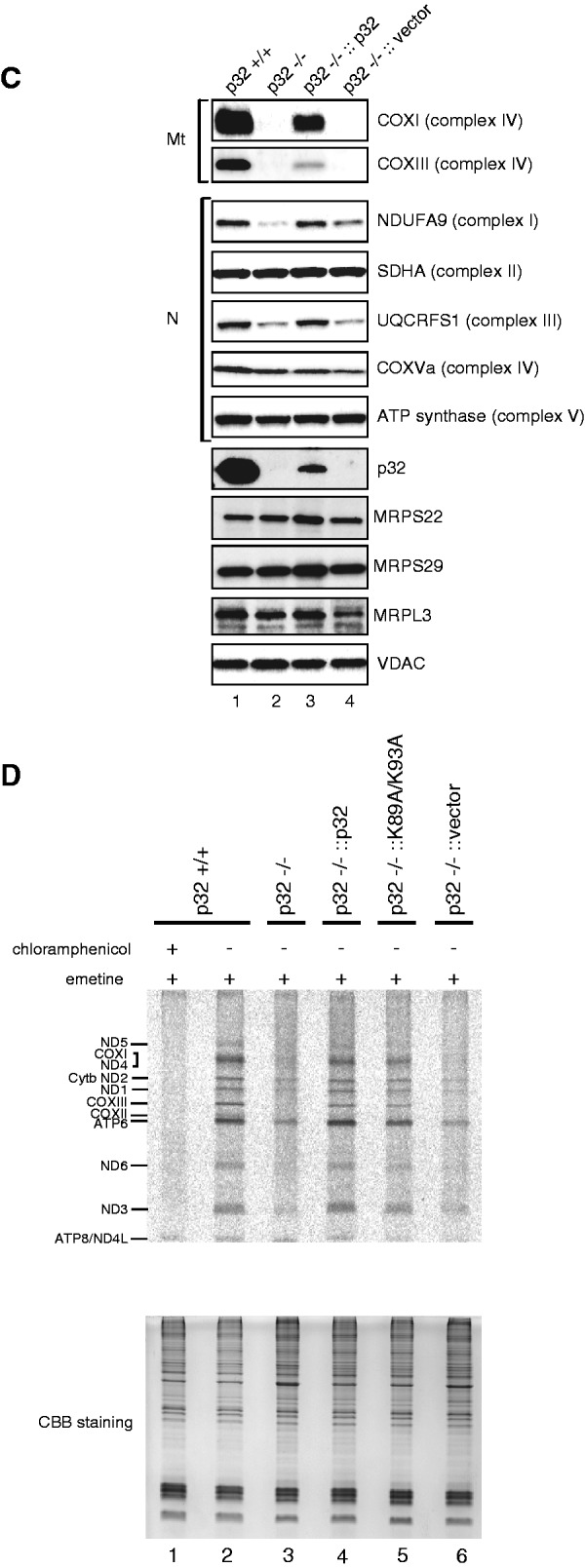
MtDNA copy number, RNA and protein expression in p32−/− MEFs. (A) The amount of mtDNA per cell was estimated based on the ratio of mtDNA/nuclear (n)DNA (ND2/AT-III). The relative mtDNA amount between p32−/− and p32+/+ cells is shown. (B) Real-time PCR quantification of mitochondrial gene transcript levels isolated from wild-type (p32+/+) MEFs, p32-knockout (p32−/−) MEFs, and knockout MEFs with reintroduced p32 cDNA (p32−/−::p32). Two nuclear-encoded RNA species (β-actin and 18S rRNA) were also quantified. Data were normalized to the expression level in wild-type p32+/+ MEFs for each RNA species. Data show the mean ± SD of triplicate experiments and *P < 0.05; versus controls (p32+/+ versus p32−/−). (C) Expression of mitochondrial proteins. Crude mitochondria were prepared from equal amounts of MEFs. Ten micrograms of protein for each sample was loaded. Blots were incubated with the indicated antibodies. Lane 1, wild-type MEFs; lane 2, p32-knockout MEFs; lane 3, knockout MEFs with reintroduced p32 cDNA; lane 4, knockout MEFs with introduced vector-only cDNA. Mt: OXPHOS protein encoded by mitochondria DNA; N: OXPHOS protein encoded by the nucleus. (D) In vivo mitochondrial translation was performed for 60 min using the cell lysates. The products were labeled during the reaction with a mixture of [35S]-methionine and [35S]-cysteine in the presence of emetine and/or chloramphenicol, and then detected by autoradiography after SDS–PAGE (upper panel). Deficient translation was observed in p32-knockout cells (lane 3). Equal loading was confirmed by CBB staining following exposure (lower panel). Lanes 1 and 2, wild-type MEFs; lane 3, p32-knockout MEFs; lane 4, knockout MEFs with reintroduced p32 cDNA; lane5, knockout MEFs with introduced p32::K89A/K93A mutant; lane 6, knockout MEFs with vector-only cDNA. The protein indicated are representative proteins based on their molecular weight.
Knockout of p32 decreases mitochondria- and nuclear-encoded proteins in mitochondria
mtDNA encodes 13 polypeptides/subunits of the mitochondrial electron respiratory chain. To determine why depletion of p32 caused a respiratory defect, we measured the expression of several members of the respiratory chain by western blotting. Knockout of p32 resulted in a decrease in COXI and COXIII, subunit I and III of complex IV, which is encoded by mtDNA (Figure 3C). The loss of nuclear-encoded complex III subunit (UQCRFS1) may reflect the instability of the complex, which arises from general loss of mtDNA-encoded components, as shown later (Figure 3D). NDUFA9 (complex I) was slightly decreased in knockout cells. ATP synthase (complex V) barely changed. Re-expression of p32 in p32-knockout MEFs (p32−/−::p32) restored mtDNA-encoded COXI, COXIII and nuclear-encoded NDUFA9, and UQCRFS1, but introduction of the vector only (p32−/−::vector) did not rescue the levels of these proteins. There were no differences in the expression level of the complex II 70-kDa subunit (SDHA) or in the levels of mitochondrial proteins VDAC, mitochondrial ribosomal protein (MRP) S29 and S22, again indicating that p32 knockout specifically affects the levels of complexes I, III and IV.
Reduced mitochondrial translation rate
Next, to investigate the defect in mitochondrial protein synthesis, wild-type and knockout MEFs were pulse-labeled with a mixture of [35S]-methionine and [35S]-cysteine. To distinguish between proteins synthesized in the cytoplasm and mitochondria, cells were pre-treated with emetine and/or chloramphenicol, which are specific inhibitors of cytoplasmic and mitochondrial protein synthesis, respectively. Electrophoresis of whole-protein extracts from emetine-treated cells showed radioactive bands in the control (Figure 3D, lane 2). This profile clearly showed the products of mitochondrial protein synthesis. The synthesis of these proteins was completely inhibited by chloramphenicol (lane 1), confirming mitochondrial protein synthesis. This mitochondrial protein synthesis was strikingly reduced in p32-deficient cells (lane 3). This reduced protein synthesis was not caused by a reduction of mitochondrial mass, because there were no differences in the levels of mitochondrial proteins VDAC, MRPS29 and MRPL3, as shown in Figure 3C, which indicated that the mitochondrial mass in p32-deficient cells was normal. Re-expression of p32 in p32-deficient MEFs partially recovered the translation (lane 4). These results suggest that p32 deficiency results in a defect of mitochondrial protein synthesis.
Depletion of p32 affects mitoribosome distribution
Protein synthesis within mitochondria is performed by 55S mitoribosomes, which consist of a small 28S subunit and a large 39S subunit. There were no differences in the levels of mitoribosomal proteins such as MRPS29, MRPS22 and MRPL3, indicating that the total quantity of mitoribosomal subunits in p32-deficient cells was not largely affected (Figure 3C). To clarify the role of p32 in mitochondrial translation, we analyzed sedimentation profiles in a sucrose density gradient (Figure 4A and Supplementary Figure S4). The small and large subunits were traced with antibodies against MRPS29 and MRPL3, respectively, and were compared between wild-type, p32-knockout and p32-re-expressed MEFs. Similar levels of the 28S small subunit (fractions 5–6) and 39S large subunit (fractions 7–8) were found in the MEFs of these three cell lines. The 55S mitoribosome was mainly distributed in fractions 9 and 10 observed as the second peaks in MRPS29 and MRPL3 plots (Figure 4B). However, in p32−/− cells, 55S formation were decreased. In p32-re-expressed cells, 55S ribosome formation was slightly recovered (fractions 9–10). The mitoribosomes broadly spanning the heavier fractions (fractions 11–16) may have represented translating multi-mitoribosomes or aggregated form. After treatment with EDTA in wild-type p32 MEF cells, 55S mitoribosome and heavier ribosome (fractions 11–16) were decreased and resemble as the p32−/− cells pattern (Supplementary Figure S4). These results suggested that p32 deficiency barely affected mitoribosome biogenesis (monosome formation), but did affect the formation of functioning 55S mitoribosomes. Though p32 mainly existed in the lighter fractions 1–4, it was also found in fractions 5–16. Considering that p32 is an abundant and multi-associated protein, p32 may functionally interact with the mitoribosome, although it is not integrated into it. Taken together, we theorize that p32 depletion somehow affects the formation of a functioning mitoribosome.
Figure 4.


Reduction of p32 results in decreased mitoribosomes. (A) Upper panels: Sedimentation analysis of mitoribosomal particles by centrifugation via a linear 15–30% sucrose density gradient. Fraction numbers are indicated. The migration of mitoribosomal particles in wild-type (+/+), p32-knockout (−/−) MEFs and p32 re-expressed (−/−::p32) MEFs was determined by immunoblotting with antibodies against MRPL3 (39S large subunit), MRPS29 (28S small subunit) and p32. Arrows indicate peaks of optical density for the S value markers. Asterisk (*) shows non-specific band of MRPL3 and p32 at top fraction (1–2). (B) Representative blots were analyzed densitometrically. The signal intensity of the protein in each fraction was plotted. The maximum value was 100% for each protein level.
p32 binds to mitochondrial mRNA
Protein–RNA interactions play essential roles in post-transcriptional control of gene expression, including in mRNA degradation and translational regulation. p32 was originally copurified with the pre-mRNA splicing factor SRSF1 from human HeLa cells (22). In addition, p32 was designated a mitochondrial RNA-binding protein by proteomic studies (18). These studies suggest an RNA-binding ability of p32. To test whether p32 was able to bind mitochondrial mRNA, hemagglutinin-tagged p32 (p32-HA) protein was expressed in HeLa cells and immunoprecipitated from total cell lysates. RNA was extracted from the immunoprecipitate (pellet). The p32-bound mRNAs were directly quantified by qRT–PCR and northern blotting (Figure 5 and Supplementary Figure S5). After induction of p32-HA, a substantial enrichment of the immunoprecipitate was observed for all 11 mitochondrial transcripts analyzed, but cytoplasmic mRNAs (MAPK6 and β-actin) were not enriched (Figure 5A, left panel), confirming that p32 was able to interact with mitochondrial mRNA in vivo. After IP using the anti-p32 antibody and p32+/+ MEFs, we also observed that mitochondrial mRNAs, including ND1 and ND3 mRNAs, were enriched (Figure 5A and B), suggesting that p32 was also bound to mitochondrial mRNA in mouse cells. Northern blotting showed that ND1 and COX1 mRNAs were detected at the same size after IP of p32, but cytoplasmic β-actin mRNA were not detected, which suggests that p32 was bound to these intact mitochondrial mRNAs (Figure 5C). We observed that the LRPPRC which RNA binding to mtRNA were reported (45), was not bound to mitochondrial mRNA in this experimental condition (Figure 5A, right panel) or TFAM which bind to mtDNA was also not bound (data not shown). Enrichment of 12S and 16S rRNAs was also observed (Figure 5A). There are no mtDNA in IP sample, because of no detection of PCR product without reverse transcriptase treatment (data not shown). 12S and 16S rRNAs were likely to be enriched via ribosome binding because western blotting of the eluates from the IPs showed that large and small mitoribosomal subunit proteins, MRPS22 and MRPL3, were co-immunoprecipitated with p32-HA (Figure 7). Taken together, these data are consistent with p32-association with all mitochondrial mRNAs in the mitochondrial matrix.
Figure 5.

p32 interaction with RNA and the mitoribosome. (A) p32 is associated with mitochondrial RNA in HeLa cells. p32-HA expression in HeLa cells was induced with doxycycline (DOX+) or not (DOX–). HA-tagged p32 was immunoprecipitated (left panel). LRPPRC was also immunoprecipitated using the LRPPRC antibody and confirmed the IP by western blotting (right panel). The each RNA levels were measure by qPCR and the ratio of each RNA level (LRPPRC/IgG) is shown for mitochondria- and nuclear-encoded genes. (B) In MEF cells, p32 were immunoprecipitated using the p32 antibody. Then RNA was extracted from the immunoprecipitated samples and each RNA species was measured by qPCR as described in the materials and methods. The ratio of each RNA level (p32/ IgG) is shown for mitochondria- and nuclear-encoded genes. (C) Co-IP of RNA visualized on northern blot analysis in HeLa cells. RNA from whole-cell lysates (lanes 1 and 2) and immunoprecipitants with anti-HA (lanes 3 and 4) were loaded on gels. Northern blot analysis was performed using ND1, COX1 and β-actin probes.
Figure 7.

p32 interact with several ribosomal protein. p32-HA expression in HeLa cells was induced with doxycycline (DOX+) or not (DOX–). Lysed mitochondria were immunoprecipitated with anti-HA agarose (left panel), and a lysate of p32+/+ MEFs was immunoprecipitated with an anti-p32 antibody (right panel) followed by western blotting with antibodies for the indicated proteins.
p32 binds to RNA oligonucleotides in vitro
Because p32 deficiency did not reduce mtDNA transcripts or affect biogenesis of mitoribosomes (Figures 3C and 4A), and p32 interacted with all mitochondrial mRNAs (Figure 5A and Figure 5B), we considered that p32 may bind to mRNAs via their poly(A) tail or in a sequence-independent manner. To test this hypothesis, we investigated the interaction of recombinant p32 protein with several RNA oligonucleotides in vitro. A REMSA showed that recombinant p32 protein clearly bound to a 22-mer poly(A) RNA in a dose-dependent manner (Figure 6A, lanes 1–7), while control glutathione-S-transferase (GST) protein and recombinant histidine (His)-tagged TFAM protein did not, indicating that this interaction was not mediated by the GST- or His-tag (Figure 6A, lanes 14-19). An interaction was not observed with a single-stranded, 22-mer poly(A) DNA oligonucleotide (Figure 6B, lanes 13–15), indicating that the p32 interaction was RNA-specific. The interaction with the 22-mer poly(U) RNA oligonucleotide was very weak (lanes 7–9). We also observed that recombinant p32 protein clearly bound to random 14- and 40-mer RNAs in a dose-dependent manner (Figure 6B, lanes 19–21 and 25–27), indicating that the p32 interaction was not sequence-specific. Consistently, a competition assay showed that the retarded band was more strongly competed with by unlabeled 40-mer RNA than by 22-mer poly(A) RNA and 14-mer RNA (Figure 6C), indicating that p32 bound to RNA with a length dependency. The retarded band was not competed with by unlabeled poly(A) DNA (Figure 6C). These results suggest that p32 binds to mitochondrial mRNAs in a manner dependent on length, but not sequence.
Figure 6.



p32 binds to RNA oligonucleotides in vitro. (A) Upper: diagram of alignment in the α helix I of Homo sapiens, Mus musculus, Pan troglodytes, Canis lupus, Bos taurus and Rattus norvegicus p32. K89 and K93 in mouse p32 are shown as bold. Lower: purified wild-type and mutant p32, but not recombinant TFAM-His and GST proteins, bind to poly(A) oligonucleoside. Protein–poly(A) RNA complexes were separated by a REMSA using a 6% native polyacrylamide gel. An arrow indicates free probe. Recombinant wild-type and mutant proteins (2–20 μM) were incubated with 22-mer poly(A) RNA and then separated by a REMSA. Purified p32 bind to poly(A) oligonucleotide prefer to mutant p32. (B) The indicated amount of wild-type His-p32 and mutant His-p32 (K89A/K93A) fusion protein was incubated with 32P-labeled random 14-mer RNA, 40-mer RNA, 22-mer poly(A) RNA, 22-mer poly(U) RNA or 22-mer poly(A) DNA oligonucleotides at 25°C for 30 min. p32–oligonucleotide complexes were separated by a electrophoretic REMSA using a 6% native polyacrylamide gel. The arrow indicates free probe. (C) Competition assays were performed by adding unlabeled RNA or DNA. The indicated fold amount of unlabeled RNA or DNA (0.2×, 1.0× or 5.0× relative to the 32P-labeled probe) was incubated with recombinant p32 (4 μM) and 32P-labeled poly(A) RNA; the complexes were separated by native polyacrylamide gel electrophoresis. The arrow indicates free probe. The lower panel indicates the intensity of the complex band. The 100% value represents the intensity of the complex of recombinant p32 and 32P-labeled poly(A) RNA. (D) Expression of mitochondrial proteins. Cell lysates were prepared from equal amounts of wild-type MEFs, p32-knockout MEFs and MEFs with reintroduced wild-type p32 cDNA, vector-only cDNA and mutant K89A/K93A p32 cDNA. Blots were incubated with antibodies for the indicated proteins.
Lysine acetylation is a reversible post-translational protein modification. In many proteins, the acetylation plays a key role in regulating gene expression and protein function. A global analysis of lysine acetylation revealed that lysines 91 and 95 of human p32 were acetylated (46). These basic amino acid residues in the α helix H1, which is located on the edge of p32, may be able to provide electrostatic interactions with the RNA phosphate groups, raising a possibility of the involvement of the two lysines in the RNA binding. Therefore, we focused on the corresponding lysines in mouse p32, i.e. helix 1 K89 and K93 (Figure 6A, lanes 8–13). We constructed a double mutant, K89A/K93A, in which the two lysine residues were replaced by alanine. The RNA-binding activities of His-fusion wild-type and mutant p32 proteins to 22-mer poly(A) RNA, random 14-mer and random 40-mer RNAs were analyzed by a REMSA (Figure 6B, lanes 4–6, 10–12, 22–24 and 28–30). Mutant p32 showed a very weak RNA–protein complex band. The dissociation constant (Kd) of the complex for wild-type p32 was estimated to be 6 μM by a mobility-shift assay. However, the p32 lysine mutant diminished poly(A) affinity up to Kd 20 μM (Figure 6A). This mutant p32 retained the same intact trimer structure as wild-type p32 as revealed by size exclusion chromatography (Supplementary Figure S6). These results suggest that the lysine residues in α helix H1 are involved in this RNA binding.
To examine the importance of the RNA-binding activity of p32 in mitochondrial translation, we re-expressed wild-type and K89A/K93A mutant p32 in p32-knockout MEFs. The two p32 proteins were expressed to a similar degree (Figure 6D, lanes 3 and 5 in the uppermost panel). p32 deficiency caused defects of mitochondrial COXI and COXIII (lanes 1 and 2). Re-expression of wild-type p32 restored COXI and COXIII (lane 3). However, re-expression of the lysine mutant p32 restored COXI and COXIII much more weakly than did the wild-type p32 (lane 5). There were no differences in the expression levels of SDHA and β-actin, indicating that the lysine residues of p32 partially affected the COXI and COXIII protein level. We also performed in vivo labeling using the K89A/K93A mutant and observed that translation synthesis of the p32 mutant was less than the re-expressed wild-type p32 (Figure 3D, lanes 4 and 5). The COXI restoration ability of the K89A/K93A mutant was well correlated with its RNA-binding ability (Figure 6A and D). These results suggest that the RNA-binding ability of p32 is important for mitochondrial protein synthesis in mammalian cells.
Proteins associated with human p32
To further examine the association of p32 with translating mitoribosomes, we sought proteins that interacted with p32 in mitochondria. We immunoprecipitated p32-HA-associated proteins with anti-HA antibodies after cross-linking reactions in HeLa cells (Supplementary Figure S7). The cross-linked proteins that were identified by LC–MS/MS are listed in Table 2. This analysis revealed many mitoribosomal proteins, supporting the role of p32 in the translation. In addition, many proteins that were related to nucleoids, RNA-binding and translation machinery were identified by LC-MS/MS. To confirm the association of p32 with ribosomal proteins, we carried out IP and western blotting using p32-HA-overexpressing HeLa cells and p32+/+ MEFs. Ribosomal proteins including MRPS22, MRPS29 and MRPL3 were detected in the immunoprecipitates. However, VDAC were not immunoprecipitated with anti-p32 antibodies (Figure 7). These results suggest that p32 is associated with mitoribosomes in conjunction with all mitochondrial mRNAs for mitochondrial protein synthesis.
Table 2.
List of proteins identified in complex with p32
| Function | Gene | ||||
|---|---|---|---|---|---|
| 28S ribosomal | MRPS10 | MRPS2 | MRPS22 | MRPS23 | MRPS25 |
| MRPS26 | MRPS27 | MRPS28 | DAP3 | MRPS30 | |
| MRPS34 | MRPS5 | MRPS9 | MRPS7 | ||
| 39S ribosomal | MRPL12 | MRPL18 | MRPL19 | MRPL21 | MRPL22 |
| MRPL38 | MRPL4 | MRPL44 | MRPL45 | MRPL46 | |
| Chaperone | HSPD1 | HSPA9 | HSPA8 | TRAP1 | |
| Translation | ERAL1 | TSFM | TUFM | ||
| RNA binding | LRPPRC | PTCD3 | |||
| Protease | LONP1 | CLPX | |||
| Transcription | TFB2M | TFAM | |||
| Others | PHB | PHB2 | |||
DISCUSSION
We have shown that p32 is essential for mouse embryonic development, based on the following evidence: (i) p32 is ubiquitously expressed in whole-mouse embryonic tissue; (ii) embryos lacking p32 are shrunken, show significantly arrested development and die at approximately E10.5; (iii) mitochondria in p32-deficient MEFs show morphological aberrations, whereas other organelles appear to be unaffected. These results suggest that mitochondrial p32 is a key molecule in mouse embryonic development.
The function of p32 in mitochondria was investigated in knockout MEFs: p32−/− cells. The knockout cells show severely impaired activities of electron transfer chain complexes I, III and IV but not complex II, leading strong support to requirement of p32 for expression of the mitochondrial genome. We showed that p32 deficiency impairs protein synthesis, but does not cause loss of mtDNA and transcripts. We speculated that p32 can bind to mitochondrial mRNA, but p32 is not involved in stabilizing mitochondrial mRNA. There are many mitochondria RNA-binding protein such as PTCD1∼4 and LRPPRC which might be involved in stabilizing the mt-mRNA, suggesting that loss of p32 protein have no effect on the steady-state levels of mRNAs. Thus, p32 appears to be specifically required for mitochondrial protein synthesis. In sucrose gradient centrifugation and western blot experiments, p32-knockout cells exhibited normal mitoribosomal subunit assembly, but decreased 55S mitoribosomes, suggesting that p32 is not required for the subunit assembly, but is required for correct functioning of mitoribosomes with mRNA. Co-IP assays showed that p32 interacts with all mitochondrially encoded mRNAs, at least in part via poly(A) binding, and localizes close to the mitoribosome.
In this study, we obtained stable p32−/− MEFs that expressed a low level of mitochondrially targeted p32 (Supplementary Figure S3). Even at low-level expression in p32−/− MEFs, re-expression of p32 in p32−/− cells partially restored complex activity, mitochondrial ATP production, cell proliferation and protein translation, suggesting that a small amount of p32 rescues the mitochondrial respiratory function via mitochondrial translational regulation. We successfully expressed p32 protein with the same plasmid up to a similar level of endogenous p32 in a human cancer cell line (data not shown), suggesting that the mechanism of p32 expression in MEFs is different from that in human cancer cells.
The Shine-Dalgarno sequence in the 5′-untranslated region of prokaryotes and the 7-methylguanylate cap structure in eukaryotic cell cytoplasm facilitate ribosome binding and direct the ribosome to the start codon. In contrast, mitochondrial mRNAs do not possess 5′-untranslated nucleotides or a cap structure (13,47). The small subunit of mitoribosomes appears to tightly bind mitochondrial mRNA in a sequence-independent manner without initiation factors or an initiation tRNA(48). However, the exact mechanisms of mRNA binding to mitoribosomes are unclear. Because of the unusual characteristics of mitochondrial mRNAs, it is thought that the mRNA ‘entry gate’ on the small subunit recognizes the unique unstructured 5′-sequence of mitochondrial mRNA (49,50). Here, we observed that p32 is required for the association of mitochondrial mRNAs with mitoribosomes (Figures 5 and 7). One possible mechanism is that p32 mediates the binding of mRNA to the small subunit and consequently enhance 55S formation. In prokaryotes, it is believed that initiation of leaderless mRNA occurs in intact 70S ribosomes. In p32−/− cells, the 55S mitoribosome peak was strongly reduced (Figure 4B, upper panel). Hence, another possible mechanism is that p32 contributes to guiding this unique 5′-leaderless mRNA to mitoribosomes for initiation of translation in part by enhancing the mitoribosome formation or stabilizing the mitoribosome.
We observed that the total ATP level was significantly higher in p32-knockout MEFs than in wild-type MEFs (Figure 2D), and the total ATP level is almost the same as glycolytic ATP production in p32−/− MEFs. The knockdown of RelA, the dominant NF-κB transactivating subunit, markedly enhances glucose consumption and lactate production in MEFs under normal culture conditions. RelA-deficient cells also exhibit decreased oxygen consumption and cell survival, although they show increased ATP levels (51), the same as p32−/− MEFs, suggesting that p32- and RelA-deficient cells show increased ATP levels due to the high activity of aerobic glycolysis.
Using in vivo labeling, we showed that re-expression of p32 in p32-deficient MEFs partially recovers the translation (Figure 3D, lane 4). However, the ratio of each protein is almost the same as the wild-type (lane 2), indicating that mtDNA-encoded protein synthesis is equally and generally suppressed and restored in p32−/− cells and in p32-resotred cells, respectively. Re-introduction of p32 into p32−/− cells well rescues complex I activity, but cannot compensate the reduced activity of complexes III and IV to a similar extent. This discrepancy may be due to the number or function of the assembly factors of each complex.
Mitochondrial RNA-binding proteins identified in previous proteomic studies include mitochondrial ribosomal proteins, tRNA synthetases, AUH, p32, LRPPRC (leucine-rich PPR motif-containing protein) and ribonuclease H1. Ponamarev et al. performed a proteomic analysis of bovine mitochondrial proteins with affinity to polyadenylate or polyuridylate (18). Of the 64 identified proteins, 51 possess a defined mitochondrial function, including 6 known RNA-binding proteins such as AUH. That study also showed by affinity purification that mitochondrial adenylate kinase 3, AUH and carnitine acetyltransferase bound poly(A), but not poly(U), while p32 bound poly(U) but not poly(A). However, our in vitro cell-free experiments demonstrated that purified recombinant p32 preferentially binds poly(A) rather than poly(U) RNA. The reason for this discrepancy is currently unknown. p32 may bind poly(U) via another protein in a crude system.
Disruption of hyaluronan synthetase 2 gene by generation Has−/− transgenic mice also results in abnormalities during midgestation with severe cardiac and vascular deformities, having similar phenotypic changes that takes place with p32/gC1qR/HABP1-deficient mice (52). Apoptosis induction by mitochondrial accumulation of HABP1 in fibroblast cell line with the generation of ROS and inhibition in complex I and mitochondrial dysfunction is supporting the OXPHOS regulation of p32 (53).
Acetylation of lysine is a reversible post-translational modification, which neutralizes the positive charge of this amino acid and modulates protein function in diverse ways. It plays a key role in the regulation of gene expression via the modification of core histone tails by histone acetyltransferases and deacetylases. A global analysis of lysine acetylation showed that p32 is acetylated at lysines 91 and 95 in human cell lines (46), suggesting a possible role of the lysine acetylation in p32 function and mitochondrial mRNA regulation.
Three p32 molecules form a doughnut-shaped quaternary structure with a sizable central channel and an unusual asymmetric charge distribution on the surface. This structure is highly positively charged because of the Lys residues in the α helix H1, which are located at the edge of the cleft of p32. A row of Lys residues in the α helix H1 on the surface of the molecule is conserved between human and mouse p32. The distances between these Lys residues are similar to those between the RNA phosphate groups, raising the possibility that the lysine residues continuously bind to single-stranded RNA. The alanine mutation of the two lysines in fact decreased the RNA-binding activity of p32 (Figure 6). Combined with their structural position, the lysine pair in p32 could cooperatively contribute to binding a region of single-stranded RNA. These lysine residues are conserved only among the mammals (Figure 6A) and not conserved in Caenorhabditis elegans or S. cerevisiae (23). Hence, the two lysine residues K89/K93 of the mouse p32 might play an indirect or additional role in RNA binding and resultantly in mitochondrial translation. At present, it remains yet to be identified which amino acids of p32 directly bind to RNA. However, the present lysine mutation results clearly support the importance of RNA-binding ability of p32 for the mitochondrial translation.
Mammalian mitochondria contain their own genome that is almost fully transcribed from both strands, which generates polycistronic RNA units that are processed and matured. Mitochondrial mRNAs are modified by oligo- or polyadenylation at their 3′-termini, but the exact function of this post-transcriptional addition is unclear. The role of polyadenylation in transcription may involve mRNA stability (54). To analyze the function of polyadenylation in mitochondria, Wydro et al. manipulated the mitochondrial mRNA poly(A) tail by targeting a cytosolic poly(A)-modifying enzyme (PABP1) to mitochondria. The observed decline in mitochondrial translation is likely due to a dominant negative action of mtPABP1 via disruption of essential protein–protein interactions in the poly(A) extension. These results indicate that poly(A) normally interacts with endogenous components that promote translation. However, coating of the poly(A) tail by mtPABP1 did not lead to transcript decay, but caused a marked inhibition of mitochondrial translation. It was also shown that removal of the 3′-adenylyl extensions results in a variable effect on mRNA steady-state levels, increasing ND1, ND2 and ND6 mRNAs or decreasing COX1 and COX2, which suggests that the mitochondrial RNA degradosome is involved in these changes. Those data are consistent with endogenous RNA-binding factor(s) such as p32 interacting with poly(A) to optimize mitochondrial protein synthesis and increase mRNA.
p32 is a very acidic protein with a calculated isoelectric point of 4 (23). In contrast, the one side of the doughnut-shape is much less negatively charged. This polarity in charge distribution clearly suggests asymmetric functional roles for the two sides of the protein. These conserved surface features are very likely to be important for protein–protein interactions and ligand binding. We observed that various proteins, such as mitoribosomal proteins, are associated with p32. In Escherichia coli, the majority of ribosomal proteins in both 30S and 50S subunits are basic. The acidic side of p32 may be involved in protein–protein interactions, such as those with the mitoribosome, while the other side of p32 is involved in binding RNA via its α helix H1.
RNA chaperones are proteins that non-specifically interact with RNA and promote RNA folding by either resolving non-native conformations or impeding their formation (55). Based on our findings that p32 shows significant RNA binding and stimulates translation, we speculate that RNA chaperoning is a major activity of p32. We suggest that the putative RNA chaperone activity of p32 contributes to RNA rearrangement during the early phase of translation initiation. A chaperoning function may be important for p32 to transport RNAs to the mitoribosome.
The RNA-binding ability is well correlated with the mitochondrial protein restoration ability in p32−/− MEFs. Thus, the RNA-binding activity of p32 may be critically important for proper and efficient translation in the mitochondrial matrix. p32 may not only guide mRNA to the mitoribosomes, but also markedly contribute to efficient initiation and/or elongation reactions, because mitoribosomes in the heavier fractions strongly decreased in p32−/− MEFs. Taken together, we provide the first demonstration that p32 plays a vital role in mitochondrial homeostasis and fetal development.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online: Supplementary Table 1, Supplementary Figures 1–7 and Supplementary Methods.
FUNDING
Grants-in-Aid for Scientific Research from the Ministry of Education, Science, Technology, Sports, and Culture of Japan (MEXT) [#19209019 and #21590337]. Funding for open access charge: MEXT.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors would like to acknowledge the technical expertise of the Support Center for Education and Research, Kyushu University, and Masami Takade for electron microscopy.
REFERENCES
- 1.Anderson S, Bankier AT, Barrell BG, de Bruijn MH, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- 2.Kang D, Hamasaki N. Mitochondrial transcription factor A in the maintenance of mitochondrial DNA: overview of its multiple roles. Ann. N Y Acad. Sci. 2005;1042:101–108. doi: 10.1196/annals.1338.010. [DOI] [PubMed] [Google Scholar]
- 3.Asin-Cayuela J, Gustafsson CM. Mitochondrial transcription and its regulation in mammalian cells. Trends Biochem. Sci. 2007;32:111–117. doi: 10.1016/j.tibs.2007.01.003. [DOI] [PubMed] [Google Scholar]
- 4.Rorbach J, Soleimanpour-Lichaei R, Lightowlers RN, Chrzanowska-Lightowlers ZM. How do mammalian mitochondria synthesize proteins? Biochem. Soc. Trans. 2007;35:1290–1291. doi: 10.1042/BST0351290. [DOI] [PubMed] [Google Scholar]
- 5.Chen XJ, Butow RA. The organization and inheritance of the mitochondrial genome. Nat. Rev. Genet. 2005;6:815–825. doi: 10.1038/nrg1708. [DOI] [PubMed] [Google Scholar]
- 6.Kucej M, Butow RA. Evolutionary tinkering with mitochondrial nucleoids. Trends Cell. Biol. 2007;17:586–592. doi: 10.1016/j.tcb.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 7.Parisi MA, Clayton DA. Similarity of human mitochondrial transcription factor 1 to high mobility group proteins. Science. 1991;252:965–969. doi: 10.1126/science.2035027. [DOI] [PubMed] [Google Scholar]
- 8.Ohno T, Umeda S, Hamasaki N, Kang D. Binding of human mitochondrial transcription factor A, an HMG box protein, to a four-way DNA junction. Biochem. Biophys. Res. Commun. 2000;271:492–498. doi: 10.1006/bbrc.2000.2656. [DOI] [PubMed] [Google Scholar]
- 9.Shadel GS. Expression and maintenance of mitochondrial DNA: new insights into human disease pathology. Am. J. Pathol. 2008;172:1445–1456. doi: 10.2353/ajpath.2008.071163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kanki T, Ohgaki K, Gaspari M, Gustafsson CM, Fukuoh A, Sasaki N, Hamasaki N, Kang D. Architectural role of mitochondrial transcription factor A in maintenance of human mitochondrial DNA. Mol. Cell. Biol. 2004;24: 9823–9834. doi: 10.1128/MCB.24.22.9823-9834.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Clayton DA. Replication and transcription of vertebrate mitochondrial DNA. Annu. Rev. Cell. Biol. 1991;7:453–478. doi: 10.1146/annurev.cb.07.110191.002321. [DOI] [PubMed] [Google Scholar]
- 12.Ojala D, Montoya J, Attardi G. tRNA punctuation model of RNA processing in human mitochondria. Nature. 1981;290:470–474. doi: 10.1038/290470a0. [DOI] [PubMed] [Google Scholar]
- 13.Montoya J, Ojala D, Attardi G. Distinctive features of the 5'-terminal sequences of the human mitochondrial mRNAs. Nature. 1981;290:465–470. doi: 10.1038/290465a0. [DOI] [PubMed] [Google Scholar]
- 14.Montoya J, Lopez-Perez MJ, Ruiz-Pesini E. Mitochondrial DNA transcription and diseases: past, present and future. Biochim. Biophys. Acta. 2006;1757:1179–1189. doi: 10.1016/j.bbabio.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 15.Tomecki R, Dmochowska A, Gewartowski K, Dziembowski A, Stepien PP. Identification of a novel human nuclear-encoded mitochondrial poly(A) polymerase. Nucleic Acids Res. 2004;32:6001–6014. doi: 10.1093/nar/gkh923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nagaike T, Suzuki T, Katoh T, Ueda T. Human mitochondrial mRNAs are stabilized with polyadenylation regulated by mitochondria-specific poly(A) polymerase and polynucleotide phosphorylase. J. Biol. Chem. 2005;280:19721–19727. doi: 10.1074/jbc.M500804200. [DOI] [PubMed] [Google Scholar]
- 17.Nakagawa J, Waldner H, Meyer-Monard S, Hofsteenge J, Jeno P, Moroni C. AUH, a gene encoding an AU-specific RNA binding protein with intrinsic enoyl-CoA hydratase activity. Proc. Natl Acad. Sci. USA. 1995;92:20512055. doi: 10.1073/pnas.92.6.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ponamarev MV, She YM, Zhang L, Robinson BH. Proteomics of bovine mitochondrial RNA-binding proteins: HES1/KNP-I is a new mitochondrial resident protein. J. Proteome Res. 2005;4:43–52. doi: 10.1021/pr049872g. [DOI] [PubMed] [Google Scholar]
- 19.Lukong KE, Chang KW, Khandjian EW, Richard S. RNA-binding proteins in human genetic disease. Trends Genet. 2008;24:416–425. doi: 10.1016/j.tig.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 20.Kurimoto K, Fukai S, Nureki O, Muto Y, Yokoyama S. Crystal structure of human AUH protein, a single-stranded RNA binding homolog of enoyl-CoA hydratase. Structure. 2001;9:1253–1263. doi: 10.1016/s0969-2126(01)00686-4. [DOI] [PubMed] [Google Scholar]
- 21.Kurimoto K, Kuwasako K, Sandercock AM, Unzai S, Robinson CV, Muto Y, Yokoyama S. AU-rich RNA-binding induces changes in the quaternary structure of AUH. Proteins. 2009;75:360–372. doi: 10.1002/prot.22246. [DOI] [PubMed] [Google Scholar]
- 22.Petersen-Mahrt SK, Estmer C, Ohrmalm C, Matthews DA, Russell WC, Akusjarvi G. The splicing factor-associated protein, p32, regulates RNA splicing by inhibiting ASF/SF2 RNA binding and phosphorylation. EMBO J. 1999;18:1014–1024. doi: 10.1093/emboj/18.4.1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jiang J, Zhang Y, Krainer AR, Xu RM. Crystal structure of human p32, a doughnut-shaped acidic mitochondrial matrix protein. Proc. Natl Acad. Sci. USA. 1999;96:3572–3577. doi: 10.1073/pnas.96.7.3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matthews DA, Russell WC. Adenovirus core protein V interacts with p32–a protein which is associated with both the mitochondria and the nucleus. J. Gen. Virol. 1998;79(Pt 7):1677–1685. doi: 10.1099/0022-1317-79-7-1677. [DOI] [PubMed] [Google Scholar]
- 25.van Leeuwen HC, O'Hare P. Retargeting of the mitochondrial protein p32/gC1Qr to a cytoplasmic compartment and the cell surface. J. Cell Sci. 2001;114:2115–2123. doi: 10.1242/jcs.114.11.2115. [DOI] [PubMed] [Google Scholar]
- 26.Soltys BJ, Kang D, Gupta RS. Localization of P32 protein (gC1q-R) in mitochondria and at specific extramitochondrial locations in normal tissues. Histochem. Cell. Biol. 2000;114:245–255. doi: 10.1007/s004180000191. [DOI] [PubMed] [Google Scholar]
- 27.Muta T, Kang D, Kitajima S, Fujiwara T, Hamasaki N. p32 protein, a splicing factor 2-associated protein, is localized in mitochondrial matrix and is functionally important in maintaining oxidative phosphorylation. J. Biol. Chem. 1997;272:24363–24370. doi: 10.1074/jbc.272.39.24363. [DOI] [PubMed] [Google Scholar]
- 28.Bharadwaj A, Ghosh I, Sengupta A, Cooper TG, Weinbauer GF, Brinkworth MH, Nieschlag E, Datta K. Stage-specific expression of proprotein form of hyaluronan binding protein 1 (HABP1) during spermatogenesis in rat. Mol. Reprod. Dev. 2002;62:223–232. doi: 10.1002/mrd.10135. [DOI] [PubMed] [Google Scholar]
- 29.Rubinstein DB, Stortchevoi A, Boosalis M, Ashfaq R, Ghebrehiwet B, Peerschke EI, Calvo F, Guillaume T. Receptor for the globular heads of C1q (gC1q-R, p33, hyaluronan-binding protein) is preferentially expressed by adenocarcinoma cells. Int. J. Cancer. 2004;110:741–750. doi: 10.1002/ijc.20105. [DOI] [PubMed] [Google Scholar]
- 30.Sunayama J, Ando Y, Itoh N, Tomiyama A, Sakurada K, Sugiyama A, Kang D, Tashiro F, Gotoh Y, Kuchino Y, et al. Physical and functional interaction between BH3-only protein Hrk and mitochondrial pore-forming protein p32. Cell Death Differ. 2004;11:771–781. doi: 10.1038/sj.cdd.4401418. [DOI] [PubMed] [Google Scholar]
- 31.Itahana K, Zhang Y. Mitochondrial p32 is a critical mediator of ARF-induced apoptosis. Cancer Cell. 2008;13:542–553. doi: 10.1016/j.ccr.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Storz P, Hausser A, Link G, Dedio J, Ghebrehiwet B, Pfizenmaier K, Johannes FJ. Protein kinase C [micro] is regulated by the multifunctional chaperon protein p32. J. Biol. Chem. 2000;275:24601–24607. doi: 10.1074/jbc.M002964200. [DOI] [PubMed] [Google Scholar]
- 33.Jha BK, Salunke DM, Datta K. Structural flexibility of multifunctional HABP1 may be important for regulating its binding to different ligands. J. Biol. Chem. 2003;278:27464–27472. doi: 10.1074/jbc.M206696200. [DOI] [PubMed] [Google Scholar]
- 34.Spicer AP, Tien JY. Hyaluronan and morphogenesis. Birth Defects Res. C Embryo Today. 2004;72:89–108. doi: 10.1002/bdrc.20006. [DOI] [PubMed] [Google Scholar]
- 35.Mallick J, Datta K. HABP1/p32/gC1qR induces aberrant growth and morphology in Schizosaccharomyces pombe through its N-terminal alpha helix. Exp. Cell Res. 2005;309:250–263. doi: 10.1016/j.yexcr.2005.05.025. [DOI] [PubMed] [Google Scholar]
- 36.Fogal V, Richardson AD, Karmali PP, Scheffler IE, Smith JW, Ruoslahti E. Mitochondrial p32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol. Cell. Biol. 2010;30:1303–1318. doi: 10.1128/MCB.01101-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uchiumi T, Ohgaki K, Yagi M, Aoki Y, Sakai A, Matsumoto S, Kang D. ERAL1 is associated with mitochondrial ribosome and elimination of ERAL1 leads to mitochondrial dysfunction and growth retardation. Nucleic Acids Res. 2010;38:5554–5568. doi: 10.1093/nar/gkq305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Claypool SM, Oktay Y, Boontheung P, Loo JA, Koehler CM. Cardiolipin defines the interactome of the major ADP/ATP carrier protein of the mitochondrial inner membrane. J. Cell Biol. 2008;182:937–950. doi: 10.1083/jcb.200801152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hofhaus G, Shakeley RM, Attardi G. Use of polarography to detect respiration defects in cell cultures. Methods Enzymol. 1996;264:476–483. doi: 10.1016/s0076-6879(96)64043-9. [DOI] [PubMed] [Google Scholar]
- 40.Estornell E, Fato R, Pallotti F, Lenaz G. Assay conditions for the mitochondrial NADH:coenzyme Q oxidoreductase. FEBS Lett. 1993;332:127–131. doi: 10.1016/0014-5793(93)80498-j. [DOI] [PubMed] [Google Scholar]
- 41.Trounce IA, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. 1996;264:484–509. doi: 10.1016/s0076-6879(96)64044-0. [DOI] [PubMed] [Google Scholar]
- 42.Brautigan DL, Ferguson-Miller S, Margoliash E. Mitochondrial cytochrome c: preparation and activity of native and chemically modified cytochromes c. Methods Enzymol. 1978;53:128–164. doi: 10.1016/s0076-6879(78)53021-8. [DOI] [PubMed] [Google Scholar]
- 43.Ishihara N, Nomura M, Jofuku A, Kato H, Suzuki SO, Masuda K, Otera H, Nakanishi Y, Nonaka I, Goto Y, et al. Mitochondrial fission factor Drp1 is essential for embryonic development and synapse formation in mice. Nat. Cell Biol. 2009;11:958–966. doi: 10.1038/ncb1907. [DOI] [PubMed] [Google Scholar]
- 44.Alirol E, Martinou JC. Mitochondria and cancer: is there a morphological connection? Oncogene. 2006;25:4706–4716. doi: 10.1038/sj.onc.1209600. [DOI] [PubMed] [Google Scholar]
- 45.Ruzzenente B, Metodiev MD, Wredenberg A, Bratic A, Park CB, Camara Y, Milenkovic D, Zickermann V, Wibom R, Hultenby K, et al. LRPPRC is necessary for polyadenylation and coordination of translation of mitochondrial mRNAs. EMBO J. 2012;31:443–456. doi: 10.1038/emboj.2011.392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- 47.Grohmann K, Amairic F, Crews S, Attardi G. Failure to detect "cap" structures in mitochondrial DNA-coded poly(A)-containing RNA from HeLa cells. Nucleic Acids Res. 1978;5:637–651. doi: 10.1093/nar/5.3.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liao HX, Spremulli LL. Interaction of bovine mitochondrial ribosomes with messenger RNA. J. Biol. Chem. 1989;264:7518–7522. [PubMed] [Google Scholar]
- 49.Sharma MR, Koc EC, Datta PP, Booth TM, Spremulli LL, Agrawal RK. Structure of the mammalian mitochondrial ribosome reveals an expanded functional role for its component proteins. Cell. 2003;115:97–108. doi: 10.1016/s0092-8674(03)00762-1. [DOI] [PubMed] [Google Scholar]
- 50.Jones CN, Wilkinson KA, Hung KT, Weeks KM, Spremulli LL. Lack of secondary structure characterizes the 5' ends of mammalian mitochondrial mRNAs. RNA. 2008;14:862–871. doi: 10.1261/rna.909208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mauro C, Leow SC, Anso E, Rocha S, Thotakura AK, Tornatore L, Moretti M, De Smaele E, Beg AA, Tergaonkar V, et al. NF-kappaB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat. Cell Biol. 2011;13:1272–1279. doi: 10.1038/ncb2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Camenisch TD, Spicer AP, Brehm-Gibson T, Biesterfeldt J, Augustine ML, Calabro A, Jr, Kubalak S, Klewer SE, McDonald JA. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J. Clin. Invest. 2000;106:349–360. doi: 10.1172/JCI10272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chowdhury AR, Ghosh I, Datta K. Excessive reactive oxygen species induces apoptosis in fibroblasts: role of mitochondrially accumulated hyaluronic acid binding protein 1 (HABP1/p32/gC1qR) Exp. Cell Res. 2008;314:651–667. doi: 10.1016/j.yexcr.2007.10.033. [DOI] [PubMed] [Google Scholar]
- 54.Wydro M, Bobrowicz A, Temperley RJ, Lightowlers RN, Chrzanowska-Lightowlers ZM. Targeting of the cytosolic poly(A) binding protein PABPC1 to mitochondria causes mitochondrial translation inhibition. Nucleic Acids Res. 2010;38:3732–3742. doi: 10.1093/nar/gkq068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Herschlag D. RNA chaperones and the RNA folding problem. J. Biol. Chem. 1995;270:20871–20874. doi: 10.1074/jbc.270.36.20871. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.