

Abstract
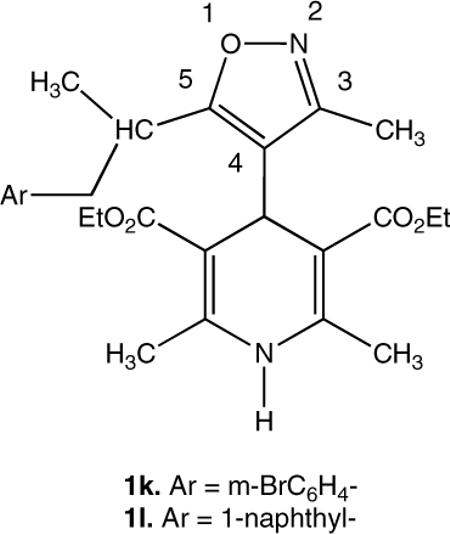
The 4-Isoxazolyl-dihydropyridines (IDHPs) exhibit inhibition of the multidrug-resistance transporter (MDR-1), and exhibit an SAR distinct from their activity at voltage gated calcium channels (VGCC). Among the four most active IDHPs, three were branched at C-5 of the isoxazole, including the most active analog, 1k.
The multidrug-resistance transporter (MDR-1, also known as P-glycoprotein, P-gp) is a member of the ATP-binding cassette (ABC) superfamily of efflux pumps.1,2 Molecules which inhibit this efflux pump are of interest in overcoming multidrug-resistance, and intense efforts are in progress towards developing effective and selective theranostic agents.3,4 Termed multidrug-resistance reversers (MDRRs), several agents have advanced to clinical trials, as an example Tariquidar (XR9576) is presently in several advanced clinical trials in combination with chemotherapeutic agents as an adjuvant against multidrug resistance in cancer.5
4-Aryl-1,4-dihydropyridines (DHPs) that bind the L-type voltage gated calcium channel (VGCC) have been in general medical practice for over three decades,6 The DHP framework serves as a useful scaffold, and more recently DHPs have been recognized as useful ligands of MDR-1.7–10 The information regarding the corresponding structure activity relationship (SAR) at VGCC verses MDR-1 is emerging, and several structural distinctions may eventually lead to practical selectivity. An early observation was that heterocyclic rings in the 4-aryl position of DHPs enhanced the reversal of multidrug resistance and inhibited photoaffinity labeling of P-gp.7 Another interesting observation is that while the (+)-enantiomers of DHPs usually possess superior activity at the VGCC, this enantioselectivity of action is not observed at the MDR-1, with the VGCC distomer being equally effective at reversing drug resistance.11
Replacement of the 4-aryl group of the DHPs with the bioisoteric isoxazole results in robust calcium antagonists,12–19 and we have studied the resulting SAR, conformational dynamics and structural properties of the 4-isoxazolyl-DHPs (IDHPs, Chart 1). Given the bioactivity and unique SAR observed for VGCC activity of IDHPs, we reasoned that they should interact with the MDR-1, based on the common pharmacophore model illustrated in Figure 1 of known MDR-1 ligands nicardipine3 and dexniguldpine10 with an IDHP 1 analog. Furthermore, it is plausible that chemistry developed in our laboratories for the functionalization of isoxazoles could be applied to developing IDHPs with selectivity for MDR-1 over the VGCC.
Chart 1.

Nicardipine, Dexniguldipine and Isoxazolyl-dihydropyridines (IDHP) 1, with numbering.
Figure 1.

SYBYL common pharmacophore model of nicardipine (cyan), dexniguldipine (pink), and an analog of 4-Isoxazolyl-DHP 1k (green).
Chemistry
Known IDHPs were prepared as previously described.12–19 The new IDHPs 1k and 1l were prepared from isoxazolyl-oxazoline 2 using our lateral metalation and electrophilic quenching methodology,20,21 as outlined the Scheme 1 below. The isoxazoyl-oxazoline 2 was deprotonated using n-butyl lithium at low temperature, and electrophilic quenching provided 5-ethyl analog 3, which was used to make both branched aryl analogs 1k and 1l. After the subsequent metalation and electrophilic quenching step, the oxazoline group was deprotected by methylation to the quaternary salt, reduction with sodium borohydride followed by mild aqueous hydrolysis to produce the corresponding aldehydes 5. The Hantzsch procedure for branched aryl examples 1k and 1l required that the reaction be conducted at moderate pressure in an aerosol dispersion tube.
Scheme 1.

Synthesis of branched IDHPs 1k and 1l.
MDR-1 Assay
Screening of IDHPs was performed by the Psychoactive Drug Screening Program (PDSP) of NIMH. The PDSP protocol utilizes live Caco-2 cells, which are derived from human colonic epithelium cells which express MDR-1.22 The assay is based on the passive diffusion of calcein acetoxymethyl ester (Calcein-AM), which is hydrolyzed inside the cell to calcein, which is both fluorescent and negatively charged, and therefore trapped inside the cell. MDR-1 can transport non-fluorescent Calcein-AM from cells, but not hydrolzyed calcein. The assay measures the increase in calcein fluorescence as a function of time using a FlexStation II fluorimeter (Molecular Devices) in 96 well plates in which cells were preincubated with IDHPs (50 M) for 30 minutes, upon which time calcein-AM was added to a final concentration of 150 nM. Fluorescence is monitored over 4 minutes, and each assay was performed in quadruplicate, with a 25 M cyclosporin control. The value from untreated cells is 0% and the slope of the fluorescene is normalized taking the value for cyclosporin as 100%.23
In the event IDHPs exhibit robust MDR-1 activity in the PDSP protocol. An unsubstituted phenyl group at C-3 of the isoxazole provides MDR-1 inhibition (Table 1, Entry 1a), however, both electron donating and withdrawing substituents appeared to lower biological activity (Entries 1b through 1f). The C-5 aryl ethyl series proved to be more promising (Entries 1h to 1l). The most potent VGCC IDHP in this series exhibited only slight MDR-1 activity, however branching at the C-5 position doubled its inhibition activity (Entries 1j verses 1l). The branched bromophenyl analog 1k provided the best activity in this series.
Table 1.
IDHP activity at VGCC and MDR-1.
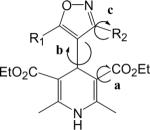
| IDHP | R1 | R2 | VGCC Ki (nM) | VGCC Patch Clamp (M) | Ref. | MDR-1 (% inh.) |
|---|---|---|---|---|---|---|
| 1a | CH3 | C6H5 | 13.9 | 12,13,15 | 48.9 | |
| 1b | CH3 | o-MeO-C6H4 | 17.1 | 19 | 32.8 | |
| 1c | CH3 | 2-MeO-5-Cl-C6H3 | 6.86 | 19 | 15.0 | |
| 1d | CH3 | o-Cl-C6H4 | 9.68 | 19 | 10.9 | |
| 1e | CH3 | m-Cl-C6H4 | 1.98 | 19 | 26.8 | |
| 1f | CH3 | p-Cl-C6H4 | 1.31 | 19 | 11.7 | |
| 1g | i-Pr | C6H5 | a | 14 | 38.4 | |
| 1h | C6H5CH2CH2 | C6H5 | 55.3 | 18 | 27.6 | |
| 1i | p-Biphenyl-CH2CH2 | CH3 | 8.69 | 18 | 18.6 | |
| 1j | 1-naphthyl CH2CH2 | CH3 | 4.1 | 0.47 | 18,19 | 19.0 |
| 1k | m-Br-C6H5CH2(CH3)CH | CH3 | this work | 61.2 | ||
| 1l | 1-naphthyl CH2(CH3)CH | CH3 | this work | 38.3 |
a weak vasodilator in the Langendorff perfused heart assay.
Conformational Dynamics
One unique aspect of the DHPs is their conformational dynamics. Rotation around the isoxazole to DHP bond can give rise to either O-exo or O-endo conformers (Figure 2) which - when the C-3 and/or C-5 substituent is large - gives rise to significantly diverse topologies. The barrier to this process is relatively low, on the order of 30–40 kJ mol−1.17
Figure 2.

Rotation about the single bond between the heterocyclic rings gives rise to significant conformational differences.
Divergent conformations about the isoxazole to DHP bond have also been observed in crystallography, where, for example isoxazole C-5 methyl 1a crystallized O-endo,13 while its C-5 isopropyl analog 1g was observed O-exo.14 Both the C-5 naphthylethyl 1j18 and the branched 2-propylphenyl16 analogs crystallized with the C-5 arylethyl group over the DHP ring (O-endo). In our homology model for VGCC activity of the IDHPs we found that the opposite conformer of C-3 aryl isoxazoles than that found in the solid state (O-exo for 1b-d), with the bulky group pointing away from the DHP ring (O-endo), gave the best explanation for VGCC activity of 1d-f in our SAR.19 Therefore, we usually find it necessary to consider conformational dynamics between the heterocyclic rings of IDHP in interpreting bioactivity.
Molecular Modeling
The molecular models were constructed and visualized with INSIGHT 2000 on a Silicon Graphics Indigo II workstation. The rotational barrier calculations were performed using the torsion force constraint in the Discover module. The torsion was rotated through 360 intervals (one degree increments) using a force constant of 10. After each rotation, the structure was subjected to 1000 steps of minimization, or until a rms value of 0.01 was reached, using the VA09A algorithm. The results of the conformational searches were examined with the Analysis module by constructing a graph of total energy vs. dihedral angle.
The trend observed for the rotational barrier about the heterocyclic rings for the three molecules is 1l < 1k < 1d (Table 2). The studies on 1d have been published, along with VT-NMR studies which indicated the solution barrier was at or below the value calculated for rotation b.19
Table 2.
Calculated Free Energy Barriers for IDHPs (kcal mol−1).
| Rotation | 1l | 1k | 1d | |
|---|---|---|---|---|
|
a | 3.8 | 3.4 | 6.0 |
| b | 29.1 | 16.6 | 15.0 | |
| c | - | - | 25.1 |
Solution Spectra and Nuclear Overhauser
Important structural features of the 4-aryl-DHPs are also maintained among IDHPs. Similar to 4-aryl rings of the most active 4-aryl-DHPs at the VGCC, the isoxazole ring is electron deficient. Comparable degrees of DHP ring pucker exist for both classes. The isoxazole ring similarly bisects that of the DHP ring, as shown in solid-state crystal structures, adopting either the O-endo or O-exo conformation in previous studies with the solid state preference for conformation at the heterocyclic ring juncture dictated by crystal packing forces rather than intrinsic energy differences. Gas phase calculations and solution studies both estimate a barrier to rotation below the energy available at physiological temperature. Thus, the conformation of the small molecule adapts to its macromolecular host.
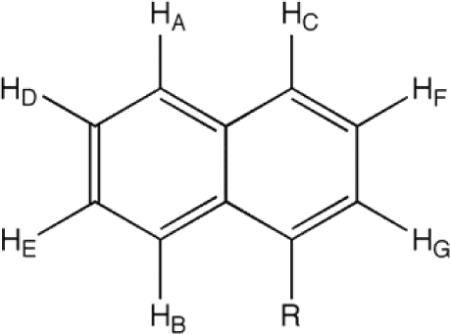
This trend is confirmed in the room temperature 1H NMR spectra for the new compound 1k and 1l at room temperature at 400 or 500 MHz. Two separate signals arise for the C-2 and C-6 methyl groups of the DHP for the naphthyl derivative 1l, with a separation of 0.6 ppm (signals O and P, see Figure 3). It is not possible to distinguish between the effects of diastereotopicity verses conformational constriction, although it seems likely that both exert an effect.
Figure 3.

Nuclear Overhauser spectra of 1l. The O-endo conformation is supported by the nOe signal between the DHP methyls at C-2 and C-6 (O,P) with the naphthyl aryl protons (D) and the DHP C-4 methina (I) and isoxzole C-3 methyl (N) (red circles). The DHP ring C-4 methine (I) and isoxazole C-5' methine (M) correlation is evidence for the O-exo conformer (blue circle).
The calculations indicate lower energy for the O-endo conformation by about 17 kcal for 1l. The 2D NOE experiment, on the other hand, shows the presence of both the O-exo and O-endo conformations, which indicates the actual barrier is at or below the calculated value.
NOE is observed for the DHP C-4 methine (I) and C-3' isoxazole methyl protons (N), which reflects the O-endo conformation (Figure 3). NOE between the same DHP C-4 methine (I) and the C-5' α methine proton (M), in turn, evidences the O-exo conformation. This reveals that rotation about the heterocyclic ring junction, while not completely hindered, is restricted to the extent of causing a non-averaging of the two DHP methyl group protons.
Likewise, a similar, but lesser, effect is observed for the chiral 5-mBr-IDHP derivative 1k. The room temperature 1H NMR shows, again, a non-averaging of the signals for the C-2 and C-6 DHP methyl groups, with a separation this time of only 0.1 ppm (signals L and M, see Figure 4). A similar pattern of nuclear Overhauser correlations again indicate the presence of both conformations at room temperature.
Figure 4.

Nuclear Overhauser spectra of 1k. The O-endo conformation is evidenced by the correlation between the aryl protons (A) and the 2,6-DHP methyls, (L,M) as well as by the C-3 isoxazole methyl group (K) with the DHP C-4 methine proton (F) (red circles). The DHP C-4 methine (F) correlation with the C-5 isoxazole methine (J) can only arise from the O-exo conformer (blue circle).
The common pharmacophore modelling studies of Ecker suggested a branched biaryl motif common to MDR-1 inhibitors.26 A homology model based on the nucleotide binding site of MDR-1 by Reddy suggested a preferred inhibitory conformation wherein both 4-heteroaryl and C3 and C-5 aryl amides adopted a roughly endo relationship,27 which is analogous to the pharmacophore model suggested by Eckert. Based on consideration of the conformational dynamics of IDHPs, and available pharmacophore and homology modelling studies to date, we conclude that for optimal MDR-1 binding IDHPs likely adopt a conformation in which the branched aryl group is preferentially over the DHP moiety, or O-endo for 1k, as illustrated in Figure 1. This leads us to conclude as a working hypothesis that a conformational distinction may exist between VGCC and MDR-1 SAR for the activity of IDHPs.
In summary, the IDHPs exhibit MDR-1 activity with an SAR that is distinct from VGCC activity, and provide an opportunity for further lead optimization. Our working hypothesis is that activity is optimal in the IDHP series for structures which have a branched aryl-alkyl-heterocycle motif. Further analog synthesis and homology modeling studies are underway to test our working hypothesis, and will be reported in due course.
Experimental Section
All chemicals were purchased from Aldrich Chemical Company and used without purification unless otherwise indicated. Solvents were reagent grade and dried just prior to use by standard methods. Melting points were determined in open capillary tubes on a Melt-Temp apparatus and are uncorrected. Radial chromatography was performed on a Harrison Associates chromatotron. Flash chromatography was performed using Baker Flash gel and in house compressed air. NMR spectra were recorded on a Bruker AC200 (200 MHz 1H, 50 MHz 13C), IBM Bruker AC300 (300 MHz 1H, 75 MHz 13C), or Avance Bruker DRX 500 (500 MHz 1H, 125 MHz 13C) spectrometer, at ambient temperature in CDCl3 unless otherwise specified. Chemical shifts are reported downfield from an internal tetramethylsilane (TMS) standard. Mass spectra were obtained on a JEOL AX 505-HA mass spectrometer using EI. Combustion analyses were performed by Desert Analytics, Tucson, AZ.
4-(4', 5'-Dihydro-4',4'-dimethyl-Δ2-oxazolin-2-yl)-3,5-dimethylisoxazole, (2), was prepared as previously described.20
Preparation of 5-ethyl, 3-methyl isoxazole oxazoline (3)
A dry 250-mL round-bottom flask was charged with isoxazolyloxazoline (2) (1.92 g, 9.88 mmol) and THF (100 mL) and cooled to −78°C under N2. A solution of n-BuLi in hexane (2.15 M, 5.1 mL, 10.9 mmol) was added via syringe dropwise. The yellow solution was stirred at −78°C for 2 hours, after which iodomethane (0.7 mL, 11.66 mmol) was added. The reaction was allowed to warm to room temperature overnight, and the solvent removed under vacuum to give a brown oil. The crude product was chromatographed on silica gel (CHCl3) and Kugelrohr distilled (0.6 mm Hg, 67–73°C) to give 1.95 g of 3 as a yellow oil (95%).
Diethyl 2,6-dimethyl-4-[5-(RS-1'-1-naphthyl-prop-2'-yl)-3-methylisoxazol-4-yl]-1,4-dihydropyridine-3,5-dicarboxylate (1l)
To a stirred solution of ethyl isoxazolyl oxazoline (3) from above (1.94 g, 9.30 mmol) in 100 mL dry THF cooled to −78°C under N2 was added 1.05 equiv of 2.15 M n-BuLi (4.5 mL) dropwise. The yellow solution was stirred at −78°C for 2 hours. Then 1-(chloromethyl)naphthalene, that had been just purified on a column of basic aluminum oxide, (1.5 mL, 10.0 mmol) was added dropwise and the reaction allowed to come to room temperature overnight. The mixture was concentrated under vacuum to give a brown oil which was purified by flash chromatography (silica gel, 95% hexane, 5% ETOAc) to give branched isoxazolyl-oxazoline 4l as an 2.72 g amber oil (85%): 1H NMR (500 MHz, CDCl3) δ 1.22 (d, 6H, oxazoline Me's) 1.36 (d, 3H, lateral isoxazole C-5' Me) 2.41 (s, 3H, isoxazole C-3' Me) 3.25–3.30 (q, 1H, diastereotopic CH) and 3.52–3.56 (q, 1H, diastereoptopic CH) 3.58 (d, 1H, oxazoline CH) and 3.81 (d, 1H, other oxazoline CH) 4.15–4.22 (m, 1H, lateral isoxazole C-5' CH) 7.21 (d, 1H, Ar-HG) 7.35–7.38 (t, 1H, Ar-HF) 7.48–7.56 (m, 2H, Ar-HE,HD) 7.73 (d, 1H, Ar-HC) 7.86 (d?, 1H, Ar-HB) 8.24 (d, 1H, Ar-HA); Correlation between carbons and protons in CHn fragments were determined by 1H-13C HMQC and HMBC. Correlations between J coupled protons were determined by 1H-1H COSY.
To a stirred solution of the product 4l (3.60 g, 10.0 mmol) in 90 mL of dry CH2Cl2 was added 1.7 mL (2.45 g, 15.0 mmol) of distilled CF3SO3CH3, and the mixture stirred under N2 until TLC (silica, 80% hexane, 20% EtOAc) showed only baseline material. The mixture was cooled to 0 °C, and a solution of 0.69 g (18.2 mmol) of NaBH4 in 25 mL of 4:1 THF:MeOH was added in one portion. This mixture was stirred at 0 °C for 30 min. Then 5 mL of saturated NH4Cl was added and the mixture allowed to warm to room temperature. Ether, 50 mL, was added, and the layers were separated. The ether layer was washed with saturated NaCl (1 × 25 mL). The combined aqueous layers were extracted with CH2Cl2 (1 × 25 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated to give an orange oil. The crude product was hydrolyzed with 15 mL of 1 M aqueous HCl in 20 mL of 4:1 THF:H2O. The reaction mixture was poured into 50 mL of ether, and the layers were separated. The ether layer was washed with saturated NaHCO3 (3 × 25 mL). Combined aqueous layers were extracted with CH2Cl2 (1 × 25 mL) and the combined organic layers dried over anhydrous Na2SO4, filtered, and concentrated to give the crude aldehyde as a yellow oil. The aldehyde was purified by flash chromatography (silica gel, 80% hexane, 15% EtOAc, 5% CH2Cl2) to give aldehyde 5l as a pale-yellow oil which was used immediately in the next step: 1H NMR (300 MHz, CDCl3) δ 1.47 (d, 3H, lateral isoxazole C-5' Me) 2.29 (s, 3H, isoxazole C-3' Me) 3.39–3.42 (m, 2H, CH2) 3.76–3.82 (m, 1H, CH) 7.01 (d, 1H, Ar-H) 7.20–7.29 (t, 1H, Ar-H) 7.42–7.47 (m, 2H, Ar-H) 7.67 (d, 1H, Ar-H) 7.78 (d, 1H, Ar-H) 7.86 (d, 1H, Ar-H) 9.17 (s, 1H, aldehyde H).
The aldehyde 5l 0.37 g (1.33 mmol) was dissolved in ethanol (10 mL) and transferred to an aerosol dispersion tube to which ethyl acetoacetate (357 mg, 2.75 mmol) and aqueous ammonia (2 mL, 29.6%) were added. The mixture was heated to 100–110 °C for 48 hours, the pressure rising to 30–40 psi. After cooling, the solvents were removed under vacuum to give a brown oil. The crude product was chromatographed on silica gel (CHCl3) and purified by radial chromatography (40% hexane, 20% EtOAc, 10% CH2Cl2), giving 100 mg of 1l as white crystals (15%): mp 144–146 °C; 1H NMR (500 MHz, CDCl3) δ 1.11–1.14 (t, 3H, CH3CH2O-) 1.17–1.19 (t, 3H, CH3CH2O-) 1.20 (d, 3H, lateral isoxazole C-5' Me) 1.59 (s, 3H, DHP-Me) 2.18 (s, 3H, DHP-Me) 2.35 (s, 3H, isoxazole C-3' Me) 3.30–3.35 (m, 2H, lateral isoxazole C-5' CH and diastereotopic CH) 3.53–3.56 (m, 1H, other diastereotopic CH) 3.95–4.00 (m, 2H, CH3CH2O-) 4.05–4.13 (m, 2H, CH3CH2O-) 4.76 (s, 1H, DHP-CH) 5.25 (br s, 1H, NH) 7.15 (d, 1H, Ar-HG) 7.27–7.30 (t, 1H, Ar-HF) 7.43–7.52 (m, 2H, Ar-HE, HD) 7.68 (d, 1H, Ar-HC) 7.85 (d, 1H, Ar-HB) 8.03 (d, 1H, Ar-HA); 13C NMR (125 MHz, CDCl3) δ 10.1, 14.32, 14.38, 19.1, 19.5, 19.9, 28.9, 33.0, 37.0, 59.7, 59.8, 102.1, 102.4, 120.7, 123.5, 125.2, 125.4, 125.8, 126.8, 126.9, 128.9, 131.9, 134.0, 136.0, 142.3, 142.8, 159.8, 166.8, 167.3, 172.7; MS (EI) 502. Anal. Calcd for C30H34N2O5: C, 71.69; H, 6.82; N, 5.57. Found: C, 71.54; H, 6.63; N, 5.30.
Diethyl 2,6-dimethyl-4-[5-(RS-1'-m-bromophenyl-prop-2'-yl)-3-methylisoxazol-4-yl]-1,4-dihydropyridine-3,5-dicarboxylate (1k)
To a stirred solution of ethyl isoxazolyloxazoline-(3) from above (2.69 g, 12.9 mmol) in 100 mL dry THF cooled to −78°C under N2 was added 1.1 equiv of 2.45 M n-BuLi (5.8 mL) dropwise. The yellow solution was stirred at −78°C for 2 hours. Then m-Br-benzylbromide (3.41 g, 13.7 mmol) dissolved in 10 mL dry THF was added dropwise and the reaction allowed to come to room temperature overnight. The mixture was concentrated under vacuum to give a brown oil which was purified by flash chromatography (silica gel, 80% hexane, 20% EtOAc) to give 4.30 g of branched isoxazolyl-oxazoline 4k as a yellow oil (88%): 1H NMR (200 MHz, CDCl3) δ 1.27–1.31 (m, 9H, oxazoline Me's, lateral isoxazole C-5' Me); 2.38 (s, 3H, isoxazole C-3' Me) 2.69–2.80 (m, 1H, diastereotopic CH) and 2.96–3.06 (q, 1H, other diastereoptopic CH) 3.84–3.95 (m, 3H, oxazoline CH2 and lateral isoxazole C-5' CH) 6.97–7.11 (m, 2H, Ar-H) 7.26–7.34 (m, 1H, Ar-H); MS (EI) 376, 378.
To a stirred solution of the product 4k (4.52 g, 12.0 mmol) in 100 mL of dry CH2Cl2 was added 2.0 mL (2.90 g, 17.7 mmol) of distilled CF3SO3CH3, and the mixture stirred under N2 until TLC (silica, 80% hexane, 20% EtOAc) showed only baseline material. The mixture was cooled to 0 °C, and a solution of 0.82 g (21.7 mmol) of NaBH4 in 30 mL of 4:1 THF:MeOH was added in one portion. This mixture was stirred at 0 °C for 30 min. Then 7 mL of saturated NH4Cl was added and the mixture allowed to warm to room temperature. Ether, 50 mL, was added, and the layers were separated. The ether layer was washed with saturated NaCl (1 × 25 mL). The combined aqueous layers were extracted with CH2Cl2 (1 × 25 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated to give an orange oil. The crude product was hydrolyzed with 20 mL of 1 M aqueous HCl in 27 mL of 4:1 THF:H2O. The reaction mixture was poured into 50 mL of ether, and the layers were separated. The ether layer was washed with saturated NaHCO3 (3 × 25 mL). Combined aqueous layers were extracted with CH2Cl2 (1 × 25 mL) and the combined organic layers dried over anhydrous Na2SO4, filtered, and concentrated to give the crude aldehyde 5k as a yellow oil. The aldehyde 5k was purified by flash chromatography (silica gel, 80% hexane, 15% ETOAc, 5% CH2Cl2) to give 1.96 g as a pale-yellow oil (58 %): 1H NMR (200 MHz, CDCl3) δ 1.40 (d, 3H, lateral isoxazole C-5' Me) 2.41 (s, 3H, isoxazole C-3' Me) 2.85–3.07 (m, 2H, CH2) 3.61–3.72 (q, 1H, CH) 6.97 (d, 1H, Ar-H) 7.06–7.13 (t, 1H, Ar-H) 7.22–7.33 (m, 1H, Ar-H) 9.66 (s, 1H, aldehyde H).
The aldehyde 5k 1.96 g (6.36 mmol) was dissolved in ethanol (17 mL) and transferred to an aerosol dispersion tube to which ethyl acetoacetate (1.74 g, 13.3 mmol) and aqueous ammonia (1.5 mL, 29.6%) were added. The mixture was heated to 100–110 °C for 48 hours, the pressure rising to 30–40 psi. After cooling, the solvents were removed under vacuum to give a brown oil. The crude product was chromatographed on silica gel (70% hexane, 30% ETOAc) and purified by radial chromatography (40% hexane, 20% EtOAc, 10% CH2Cl2), giving 600 mg of 1k as white crystals (18%): mp 134–136 °C; 1H NMR (500 MHz, CDCl3) δ 1.13–1.21 (m, 9H, 2 CH3CH2O-, lateral isoxazole C-5' Me) 2.19 (s, 3H, DHP-Me) 2.23 (s, 3H, DHP-Me) 2.33 (s, 3H isoxazole C-3' Me) 2.67–2.72 (q, 1H, lateral isoxazole C-5' CH) 3.03–3.16 (m, 2H, CH2) 4.00–4.14 (m, 4H, 2 CH3CH2O-) 4.79 (s, 1H, DHP-CH) 5.48 (br s, 1H, NH) 6.92 (d, 1H, Ar-H) 7.04–7.07 (t, 1H, Ar-H) 7.24 (t, 1H, Ar-H) 7.27–7.30 (m, 1H, Ar-H); 13C NMR (125 MHz, CDCl3) δ 10.12, 14.37, 14.39, 19.0, 19.6, 19.7, 28.9, 33.8, 40.5, 59.8, 59.9, 102.3, 102.6, 120.7, 122.2, 127.5, 129.1, 129.8, 131.6, 142.2, 142.6, 142.7, 159.7, 166.8, 167.2, 172.1; MS (EI) 528, 530. Anal Calcd for C26H31N2O5Br: C, 58.76; H, 5.88; N, 5.27. Found: C, 58.79; H, 5.96; N, 5.31.
Pharmacophore Computational Modeling
Ligand structures were drawn and energy minimized (Powell method, 0.01 kcal/mol*A gradient termination, MMFF94s force field, MMFF94 charges, 1000 maximum iterations) using the Sybyl modeling program (Tripos, St. Louis, MO, USA). Construction of the overlaying pharmacophore was achieved by assembling the energy minimized structures and merging the collection of structures into the same field. This was followed by energy minimization, molecular dynamics, and an energy minimization simulation. Aggregates for molecular dynamics and minimization simulations were defined as Carbons 2,3,4,5 and 6 of the 1,4-dihydropyridine ring. All ligands were then energy minimized to allow for the lowest energy confirmation of each ligand.
Supplementary Material
Graph 1.

Low energy conformation for 1k for rotation about the isoxazole-DHP bond. Marked as a white cross on the conformational analysis coordinates, upper left hand corner. The isoxazole is parallel to the hypothetical line from N-1 to C-4 of the DHP.
Graph 2.

Saddle point maximum energy conformation for 1k for rotation about the isoxazole-DHP bond. Marked as a white cross on the conformational analysis coordinates, upper left hand corner. The isoxazole is orthogonal to the hypothetical line from N-1 to C-4 of the DHP.
Acknowledgment
The authors thank the NIH for grants GM42029, NS038444, and P20RR015583.
MDR1 data was generously provided by the National Institute of Mental Health's Psychoactive Drug Screening Program (NIMH PDSP), Contract # HHSN-271-2008-00025-C (NIMH PDSP). The NIMH PDSP is Directed by Bryan L. Roth MD, PhD at the University of North Carolina at Chapel Hill and Project Officer Jamie Driscoll at NIMH, Bethesda MD, USA.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
‡Dedicated to the memory of Professor Robert O. Hutchins, peerless mentor and good friend.
References
- 1.Linton KJ. Structure and function of ABC transporters. Physiology. 2007;22:122–130. doi: 10.1152/physiol.00046.2006. [DOI] [PubMed] [Google Scholar]
- 2.(a) Aller SG, Yu J, Ward A, Weng Y, Chittaboina S, Zhuo R, Harrell PM, Trinh YT, Zhang Q, Urbatsch IL, Chang G. Structure of P-glycoprotein Reveals a Molecular Basis for Poly-Specific Drug Binding. Science. 2009;323:1718–22. doi: 10.1126/science.1168750. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Globisch C, Pajeva IK, Wiese M. Identification of Putative Binding Sites of P-glycoprotein Based on its Homology Model. ChemMedChem. 2008;3:280–295. doi: 10.1002/cmdc.200700249. [DOI] [PubMed] [Google Scholar]
- 3.Nobili S, Landini I, Giglioni B, Mini E. Pharmacological Strategies for Overcoming Multidrug Resistance. Current Drug Targets. 2006;7:861–879. doi: 10.2174/138945006777709593. [DOI] [PubMed] [Google Scholar]
- 4.For elegant studies with exciting potential as theranostic agents see: Namanja HA, Emmert D, Davis DA, Campos C, Miller DA, Hrycyna CA, Chmielewski J. Toward Eradicating HIV Reservoirs in the Brain: Inhibiting P-Glycoprotein at the Blood-Brain Barrier with Prodrug Abacavir Dimers. J. Am. Chem. Soc. 2011 doi: 10.1021/ja206867t. ASAP. dx.doi.org/10.1021/ja206867t.. Pires MM, Emmert D, Hrycyna CA, Chmielewski J. Inhibition of P-Glycoprotein-Mediated Paclitaxel Resistance by Reversibly Linked Quinine Homodimers. Mol Pharmacol. 2009;75:92–100. doi: 10.1124/mol.108.050492.. Pires MM, Hrycyna CA, Chmielewski J. Bivalent Probes of the Human Multidrug Transporter P-Glycoprotein. Biochemistry. 2006;45:11695–11702. doi: 10.1021/bi0608109..
- 5.(a) Robey RW, Shukla S, Finley EM, Oldham RK,, Barnett D, Ambudkar SV, Fojo T, Bates SE. Inhibition of P-glycoprotein (ABCB1)- and multidrug resistance-associated protein 1 (ABCC1)-mediated transport by the orally administered inhibitor, CBT-1((R)) Biochem Pharmacol. 2008;3:1302–12. doi: 10.1016/j.bcp.2007.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) [accessed January 20, 2012]; Clinicaltrials.gov, search P-gp,
- 6.Zamponi G. Voltage-Gated Calcium Channels. Landes Bioscience; New York: 2005. [Google Scholar]
- 7.Nogae I, Kohno K, Kikuchi J, Kuwano M, Akiyama S-I, Kiue A, Suzuki K-I, Yoshida Y, Cornwell M, Pastan I, Gottesman M. Analysis of structural features of dihydropyridine analogs needed to reverse multidrug resistance and to inhibit photoaffinity labeling of P-glycoprotein. Biochem. Pharmacol. 1989;38:519–527. doi: 10.1016/0006-2952(89)90393-6. [DOI] [PubMed] [Google Scholar]
- 8.(a) Mehdipour AR, Javindnia K, Hemmateenejad B, Amirghorfran Z, Miri R. Dihydropyridine Derivatives to Overcome Atypical Multidrug Resistance: Design Synthesis, QSAR Studies, and Evaluation of the cytotoxic and Pharmacological Activities. Chem Biol. Drug. Des. 2007;70:337–346. doi: 10.1111/j.1747-0285.2007.00569.x. [DOI] [PubMed] [Google Scholar]; (b) Miri R, Mehdipour A. Dihydropyridines and atypical MDR: A novel perspective of designing general reversal agents for both typical and atypical MDR. Bioorg. Med. Chem. 2008;16:8329–8334. doi: 10.1016/j.bmc.2008.07.025. [DOI] [PubMed] [Google Scholar]
- 9.Shah A, Bariwal J, Molnár J, Kawase M, Motohashi N. Advanced Dihydropyridines as Novel Multidrug Resistance Modifiers and Reversing Agents. Top. Heterocycl. Chem. 2008;15:201–252. [Google Scholar]
- 10.(a) Zhou XF, Shao O, Coburn RA, Morris ME. Quantitative structure-activity and quantitative structure-pharmacokinetics relationship of 1,4-dihydropyridines and pyridines as multidrug resistance modulators. Pharm. Res. 2005;22:1989–1996. doi: 10.1007/s11095-005-8112-0. [DOI] [PubMed] [Google Scholar]; (b) Zhou XF, Zhang L, Tseng E, Scott-Ramsay E, Schentag JJ, Coburn RA, Morris ME. New Aryl 1,4-dihydropyridines and 4-arylpyridines as P-glycoprotein inhibitors. Drug Metab. Dispos. 2005;33:321–328. doi: 10.1124/dmd.104.002089. [DOI] [PubMed] [Google Scholar]
- 11.Hoellt V, Kouba M, Dietel M, Vogt G. Stereoisomers of Calcium Antagonists which Differ Markedly in their Potencies as Calcium Blockers are Equally Effective in Modulating Drug Transport by P-Glycoprotein. Biochem. Pharm. 1992;43:2601–2608. doi: 10.1016/0006-2952(92)90149-d. [DOI] [PubMed] [Google Scholar]
- 12.Natale NR, Quincy DA. Neutral Dichromate Oxidations. Preparation and Utility of Isoxazole Aldehydes. Synth. Commun. 1983;13:817. [Google Scholar]
- 13.Schauer CK, Anderson OP, Natale NR, Quincy DA. Structure of 3,5-Dicarboethoxy-2,6-dimethyl-4-(3'-Phenyl-5'methyl-isoxazol-4'-yl)-1,4-dihydropyridine, A Calcium Antagonist. Acta Crystallogr. 1986;C42:884. [Google Scholar]
- 14.McKenna JI, Schlicksupp L, Natale NR, Willett RD, Maryanoff BE, Flaim SF. Cardioactivity and solid-state structure of two 4-isoxazolyldihydropyridines related to the 4-aryldihydropyridine calcium-channel blockers. J. Med. Chem. 1988;31:473–476. doi: 10.1021/jm00397a035. [DOI] [PubMed] [Google Scholar]
- 15.Natale NR, Triggle DJ, Palmer RB, Lefler BJ, Edwards WD. 4-Isoxazolyl-1,4-dihydropyridines: Biological, Theoretical, and Structural Studies. J. Med. Chem. 1990;33:2255–2259. doi: 10.1021/jm00170a032. [DOI] [PubMed] [Google Scholar]
- 16.Mirzaei YR, Simpson BM, Triggle DJ, Natale NR. Diastereoselectivity in the Lateral Metalation and Electrophilic Quenching of Isoxazolyl- Oxazolines. J. Org. Chem. 1992;57:6271–6279. [Google Scholar]
- 17.Palmer RB, Andro TM, Natale NR, Anderson NH. Conformational Preferences and Dynamics of 4-Isoxazolyl-1,4-dihydropyridine Calcium Channel Antagonists As Determined by Variable Temperature NMR and NOE Experiments. Magn. Reson. Chem. 1996;34:495–504. [Google Scholar]
- 18.Natale NR, Rogers ME, Staples RJ, Triggle DJ, Rutledge A. Lipophilic 4-Isoxazolyl-1,4-dihydropyridines: Synthesis and Structure-Activity Relationships. J. Med. Chem. 1999;42:3087–3093. doi: 10.1021/jm980439q. [DOI] [PubMed] [Google Scholar]
- 19.Zamponi G, Stotz SC, Staples RJ, Rogers TA, Nelson JK, Hulubei V, Blumenfeld A, Natale NR. Unique Structure Activity Relationship of 4-Isoxazolyl-1,4-dihydropyridines. J. Med. Chem. 2003;46:87–96. doi: 10.1021/jm020354w. [DOI] [PubMed] [Google Scholar]
- 20.(a) Natale NR, Niou C-S. A Facile Synthesis of Functionally Complex Isoxazole Derivatives. Tetrahedron Lett. 1984:3943–6. [Google Scholar]; (b) Natale NR, McKenna JI, Niou C-S, Borth M, Hope H. Metalation of Isoxazolyloxazolines, A Facile Route to Functionally Complex Isoxazoles: Utility, Scope and Comparison with Dianion Methodology. J. Org. Chem. 1985;50:5660–6. [Google Scholar]; (c) Niou C-S, Natale NR. Synthesis, Metalation and Electrophilic Quenching of Alkyl-Isoxazole-4-tertiary carboxamides. A Critical Comparison of Three Isoxazole Lateral Metalation Methods. Heterocycles. 1986;24:401–12. [Google Scholar]
- 21.Natale NR, Mirzaei YR. The Lateral Metalation of Isoxazoles. A Review. Org. Prep. Proced. Intl. 1993;25:515–556. [Google Scholar]
- 22.Tiberghien F, Loor F. Ranking of P-glycoprotein substrates and inhibitors by a calcein-AM fluorometry screening assay. Anticancer Drugs. 1996;7:568–578. doi: 10.1097/00001813-199607000-00012. [DOI] [PubMed] [Google Scholar]
- 23.Hamilton G, Cosentini EP, Teleky B, Koperna T, Zacheri J, Riegler M, Feil W, Schiessel R, Wenzi E. The multidrug-resistance modifiers verapamil, cyclosporine A and tamoxifen induce an intracellular acidification in colon carcinoma cell lines in vitro. Anticancer Res. 1993;13:2059–2063. [PubMed] [Google Scholar]
- 24.Crivori P, Reinach B, Pezzetta D, Poggesi I. Computational Models for Identifying Potential P-glycoprotein Substrates and inhibitors. Mol. Pharm. 2006;3:33–44. doi: 10.1021/mp050071a. [DOI] [PubMed] [Google Scholar]
- 25.Dawson RJP, Locher KP. Structure of a bacterial multidrug ABC transporter. Nature. 2006;443:180–185. doi: 10.1038/nature05155. [DOI] [PubMed] [Google Scholar]
- 26.(a) Ecker G, Huber M, Schmid D, Chiba P. The Importance of a Nitrogen Atom in Modulators of Multidrug Resistance. Mol. Pharmacol. 1999;56:791–796. [PubMed] [Google Scholar]; (b) Ecker GF, Schwaha R, Demel M, Chiba P. Pharmacoinformatic approaches to target P-glycoprotein: from inhibitor design to substrate prediction. ACS 235th National Meeting; New Orleans, LA. April 6–10, 2008; Abstract MEDI 161. [Google Scholar]
- 27.Sirisha K, Shekhar MC, Umasankar K, Mahendar P, Sadanandam A, Achaiah G, Reddy VM. Molecular docking studies and in vitro screening of new dihydropyridine derivatives as human MRP1 inhibitors. Bioorg. Med. Chem. 2011;19:3249–3254. doi: 10.1016/j.bmc.2011.03.051. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.